

ABSTRACT
Soil microbiome modification may alter system function, which may enhance processes like bioremediation. In this study, we filled microcosms with gamma-irradiated soil that was reinoculated with the initial soil or cultivated bacterial subsets obtained on regular media (REG-M) or media containing crude oil (CO-M). We allowed 8 weeks for microbiome stabilization, added crude oil and monoammonium phosphate, incubated the microcosms for another 6 weeks, and then measured the biodegradation of crude oil components, bacterial taxonomy, and functional gene composition. We hypothesized that the biodegradation of targeted crude oil components would be enhanced by limiting the microbial taxa competing for resources and by specifically selecting bacteria involved in crude oil biodegradation (i.e., CO-M). Postincubation, large differences in taxonomy and functional gene composition between the three microbiome types remained, indicating that purposeful soil microbiome structuring is feasible. Although phylum-level bacterial taxonomy was constrained, operational taxonomic unit composition varied between microbiome types. Contrary to our hypothesis, the biodegradation of C10 to C50 hydrocarbons was highest when the original microbiome was reinoculated, despite a higher relative abundance of alkane hydroxylase genes in the CO-M microbiomes and of carbon-processing genes in the REG-M microbiomes. Despite increases in the relative abundances of genes potentially linked to hydrocarbon processing in cultivated subsets of the microbiome, reinoculation of the initial microbiome led to maximum biodegradation.
IMPORTANCE In this study, we show that it is possible to sustainably modify microbial assemblages in soil. This has implications for biotechnology, as modification of gut microbial assemblages has led to improved treatments for diseases like Clostridium difficile infection. Although the soil environment determined which major phylogenetic groups of bacteria would dominate the assemblage, we saw differences at lower levels of taxonomy and in functional gene composition (e.g., genes related to hydrocarbon degradation). Further studies are needed to determine the success of such an approach in nonsterile environments. Although the biodegradation of certain crude oil fractions was still the highest when we inoculated with the diverse initial microbiome, the possibility of discovering and establishing microbiomes that are more efficient in crude oil degradation is not precluded.
INTRODUCTION
Oil production, oil spills, and the storage of oily wastes by the petroleum industry have led to massive releases of petroleum hydrocarbons into the environment, making them some of the most ubiquitous environmental pollutants (1). Conventional decontamination technologies such as excavation (i.e., dig and dump), incineration, and chemical treatment of contaminants are costly and may further disrupt disturbed ecosystems (2, 3). When effective, bioremediation is a cheaper and more sustainable approach to decontamination. Bioaugmentation, the addition of targeted microbial isolates to nonsterile soils, has had various impacts on bioremediation and has generally not increased the abundance of the added organisms over the long term (4–8). In contrast to single isolate additions, high-throughput sequencing now allows us to explore the extent to which we can modify complex soil microbiomes. Studies in medical research have clearly linked changes in microbiome composition with health and disease, and gut microbiome modification is now used to treat certain human health conditions (9, 10). As a result, it is worth exploring whether soil microbiome modification may also be an effective approach for improving plant, animal, and/or soil health (11).
Unlike the biotic gut environment, the soil matrix is unlikely to be substantially altered by microbial activity in the short term, restricting the extent to which introduced microbes can modify their surroundings. For instance, soil organic matter can have molecular turnover times of 4 to ∼300 years (12), whereas large metabolite shifts in the gut (a biotic microbial habitat) were observed just 2 weeks after gut microbiome transplantation in mice (13). Biotic constraints such as plant root exudates (14), plant immune signaling (15), and competition from native microorganisms (4) can play important roles in shaping soil microbiomes, but, in general, soil bacterial composition follows predictable patterns based on abiotic constraints, such as physicochemical soil parameters (16–19). These constraints complicate soil microbiome manipulation and may restrict our ability to alter microbiome trajectories.
Some complex microbial functional traits (e.g., photosynthesis and nitrogen fixation) are strongly linked to specific phylogenetic groups and decreases in microbial diversity have been linked to declines in denitrification, nitrification, and methane oxidation (20, 21). For widespread functions like simple carbon substrate degradation, redundancy within the microbiome may mean similar functioning by very different microbial assemblages, reducing the importance of microbial diversity. Nevertheless, the relationship between microbial diversity and carbon use is unclear. Some studies performed with low numbers of cultured microorganisms (1 to 95 isolates) suggest that productivity increases with higher diversity (22–24), while others show that reductions in the diversity of soil microbial assemblages can lead to increased processing of organic compounds (20, 25–27).
To investigate how soil microbiome modification might affect soil function, we examined how targeted subsets of a soil microbiome would affect the degradation of certain crude oil components. Microcosms containing a gamma-irradiated soil were reinoculated with the initial soil (INIT) microbiome or one of two simplified and specialized subsets of that microbiome: all bacteria cultivated on regular media (REG-M) or all bacteria cultivated on media containing crude oil (CO-M). After allowing the inoculated microbiomes to stabilize, we added crude oil and assessed the biodegradation of total C10 to C50 hydrocarbons and polycyclic aromatic hydrocarbon (PAH) compounds, bacterial taxonomy, and functional gene composition after 6 weeks of incubation. We hypothesized that the biodegradation of the paraffin and aromatic fractions in crude oil would be enhanced by reducing the number of microbial taxa competing for resources and by specifically selecting a diverse array of bacteria cultivated on crude oil-based media. We also examined whether incubation of the microbiomes in soil would lead to convergence in taxonomic and functional profiles, due to the strong controlling influence of abiotic soil parameters.
MATERIALS AND METHODS
Experimental setup.
The experimental design is illustrated graphically in Fig. S1 in the supplemental material. In May 2014, the top 20 cm of soil was collected from a grass-covered site in Varennes, QC, Canada (45°41′56″N, 73°25′43″W). The collected soil, while uncontaminated, was harvested from an area adjacent to a former petroleum refinery. To the best of our knowledge, this area has never been contaminated but has likely received microbial migrants that have been exposed to petroleum contaminants. A portion of the collected soil was dried at 50°C for 5 days, sieved through 2-mm mesh to remove root fragments and stones, and then sterilized with a minimum gamma irradiation exposure of 50 kGy (Nordion Gamma Centre of Excellence, Laval, QC, Canada) as was done by van Elsas et al. (28). Sterile soil was used as the matrix for soil microcosms, which were seeded with three different microbiomes derived from the initial soil: (i) the initial unsterilized soil (INIT) (5% of microcosm soil volume as was done by Degens [26]), (ii) all bacteria isolated on standard media (REG-M), or (iii) all bacteria isolated on media containing crude oil as a potential carbon and energy source (CO-M). Ten replicates per microbiome type were used. We extracted 116.9 ng DNA/g gamma-irradiated soil and assumed that the soil was mostly, or entirely, sterile, since inoculation of a 10−1 dilution of gamma-irradiated soil onto tryptic soy agar (TSA) plates produced no colonies after 2 weeks; inoculation of the initial soil led to growth over the entire surface of the plates (see Fig. S2 in the supplemental material). Soil parameters pre- and poststerilization are provided in Table 1.
TABLE 1.
Soil parameters before and after sieving plus gamma irradiation
| Soil parameter | Presieving/sterilization | Postsieving/sterilization |
|---|---|---|
| Humidity (%) | 4.8 | 2.9 |
| pH | 7.28 | 6.72 |
| Volatile organic matter at 550°C (%) | 6.7 | 7.4 |
| Total Kjeldahl nitrogen (mg/kg) | 2,400 | 2,600 |
| CEC (cmol+/kg)a | 24 | 25 |
CEC, cation-exchange capacity.
Bacterial isolation and microcosm preparation.
Bacteria were isolated on five culture media types, categorized as either regular (standard) media or crude oil media (media compositions are provided in Table 2). The two types of regular media were a complex medium (TSA) rich in carbon and a limited medium containing M9 salts (M9) and a small amount of a single carbon source (M9 plus glucose). The three types of crude oil media were prepared by mixing crude oil (Gulf of Mexico, Montreal pipeline) to a final volume of 5% with cooling culture media containing either TSA, M9 plus glucose, or M9 alone. Crude oil was mixed 2:1 with acetone before addition to culture media in order to create a more homogeneous mixture, and acetone was allowed to volatilize under sterile conditions as was done by Zafra et al. (29). The addition of crude oil to warm media may have sped up the volatilization of certain compounds (and may not perfectly reflect the crude oil added to soil microcosms), but likely most of those that are of interest in bioremediation (i.e., persistent compounds) were retained. Bacteria were grown on each medium at three dilutions and were incubated in the dark at 25°C for 2, 7, 14, and 28 days in order to maximize the diversity of the isolated bacteria. Inoculations were staggered so that the final day of each incubation period was the same for all petri dishes. Seven replicates were used per type of culture medium, per dilution, and per incubation period, for a total of 420 petri dishes.
TABLE 2.
Composition of the regular and crude oil media (based on 1 liter of medium) used to isolate bacterial assemblages from the initial soila
| Soil component and dilution | Regular medium |
Crude oil medium |
|||
|---|---|---|---|---|---|
| M9-glucose | TSA | M9-crude oil | M9-glucose-crude oil | TSA-crude oil | |
| Agar (g) | 15 | 15 | 15 | 15 | 15 |
| TSBb (g) | 30 | 30 | |||
| M9 salts (ml)c | 200 | 200 | 200 | ||
| MgSO4, 1 M (ml) | 2 | 2 | 2 | ||
| CaCl2, 1 M (μl) | 100 | 100 | 100 | ||
| Micronutrients (ml)d | 1 | 1 | 1 | ||
| Glucose, 10% (ml) | 10 | 10 | |||
| Acetone-crude oil mix (10:20) (ml) | 30 | 30 | 30 | ||
| Soil dilution (100 μl) | 10−4 to 10−6 | 10−5 to 10−7 | 10−1 to 10−3 | 10−4 to 10−6 | 10−5 to 10−7 |
Each medium contained 100 mg/liter of cycloheximide. Cycloheximide, glucose, and acetone-crude oil mix were added after autoclaving. Soil dilutions for each medium were selected based on pretrial observations of which dilutions yielded between 10 and 300 colonies per petri dish. Each petri dish was inoculated with 100 μl of a specific soil dilution.
TSB, tryptic soy broth.
Na2HPO4-7H2O (64 g/liter), KH2PO4 (15 g/liter), NaCl (2.5 g/liter), NH4Cl (5 g/liter).
MnSO4-4H2O (6 g/liter), ZnSO4-7H2O (2.65 g/liter), KCl (1.5 g/liter), CuSO4-5H2O (130 mg/liter), Na2MoO4-2H2O (2 mg/liter).
Colonies were scraped off 40 petri dishes for each of the five types of culture media using plastic spreaders (minimum of three petri dishes per incubation time) and suspended in saline solution (0.85% NaCl). A total of 30 ml of each suspension containing bacterial colonies was added directly to 150 g of sterile soil, resulting in 20% soil moisture. We measured the DNA concentration of the initial unsterilized soil and that of each of the five media types immediately after inoculation into sterilized soil in order to inoculate each treatment with the same final amount of DNA and equal proportions of the appropriate media types (e.g., 1:1 of M9 plus glucose and TSA in REG-M). Under sterile conditions, 10 microcosms (250-ml autoclaved flasks stoppered with foam plugs) per treatment were filled with 70 g of soil preinoculated with one of the three microbiome types. Microcosms were incubated at 25°C for 8 weeks to allow microbiomes to stabilize as done by Degens (26), and microbiome growth (estimated by DNA quantification) had mostly leveled off at the end of the stabilization period. Then microcosms were spiked with crude oil to a concentration of 6,000 mg/kg soil. Monoammonium phosphate (MAP) was added as a nutrient supplement to a concentration of 50 mg/kg soil, and water saturation was adjusted to 20%. Microcosms were incubated for another 6 weeks at 25°C in the dark before measurement of degradation of C10 to C50 hydrocarbons and PAHs and analysis of the bacterial taxonomic and functional profiles in each microbiome type. A set of three sterile microcosms was also incubated for 6 weeks at 25°C to account for abiotic loss of crude oil components.
Analysis of crude oil degradation.
For all microcosms, C10 to C50 hydrocarbons (sum of all hydrocarbon compounds with chain lengths from C10 to C50) were quantified at Maxxam Analytics (Montreal, QC, Canada) with the MA. 416 − C10-C50 1.0 method used by the Centre d'Expertise en Analyze Environnementale du Québec (30). Briefly, hydrocarbons are extracted with hexane, and samples are read using gas chromatography with a flame ionization detector (GC-FID). PAH analysis of a subset of the samples (5 from each microbiome type) was performed at AGAT Laboratories (Saint-Laurent, QC, Canada) with the MA. 400 − HAP 1.1 method used by the Centre d'Expertise en Analyze Environnementale du Québec (31). In this case, a variety of PAH compounds are quantified from extracts using gas chromatography-mass spectrometry (GC-MS) in selected ion monitoring (SIM) mode. Note that these two analyses only target a portion of the compounds present in crude oil.
We also spiked three microcosms directly with crude oil without incubation in order to provide reference values for overall degradation. These baseline samples were also analyzed for C10 to C50 hydrocarbons and PAHs. Sterile noninoculated controls (n = 3) were rerun after the initial experiment to estimate abiotic loss of hydrocarbons, following a concern with contamination in our initial sterile microcosms. These samples were run under the same conditions, and degradation was compared with separate baseline samples that were frozen at the outset of the incubation (the same starting material and equivalent addition of crude oil). We only performed C10 to C50 analyses on the sterile controls, since the baseline PAH content was extremely low at 9.2 mg/kg dry soil. As a result, PAH analyses are reported as supplemental information only. Detailed raw data for all PAH compounds detected are provided in Table S1 in the supplemental material.
DNA isolation, amplification, and Illumina MiSeq sequencing.
Soil samples were collected following homogenization with a sterile spatula. Total soil DNA was isolated from 250-mg soil subsamples using the Mo Bio PowerSoil DNA isolation kit and quantified using a Qubit dsDNA HS assay kit and Qubit fluorometer (Life Technologies, Burlington, ON, Canada). Initial PCR amplifications were performed on an Eppendorf Mastercycler ProS (Eppendorf, Mississauga, ON, Canada) using 1:10 diluted DNA extracts, and subsequent processing for sequencing was performed following mainly the Illumina 16S Metagenomic Sequencing Library Preparation guide (part no. 15044223 rev. B). The 16S V3 and V4 regions were amplified with the universal bacterial primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) (32), containing the required Illumina adaptors at the 5′ end of the primer sequences (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′ for the forward primer and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′ for the reverse primer). Initial reactions were performed in 25-μl volumes using 200 μM deoxynucleoside triphosphates (dNTPs), 400 nM each primer, 2.5 μl of 10× Taq buffer, and 1 U of HotStar Taq and the following cycling conditions: 15 min at 95°C, 25 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, and a final elongation step of 5 min at 72°C. PCR products were cleaned using NucleoMag NGS clean-up and size select beads (Macherey-Nagel, Bethlehem, PA). Unique codes were added to each sample by amplifying 2.5 μl of the purified PCR product with 2.5 μl of each Nextera XT Index primer (Illumina Inc., San Diego, CA, USA), 12.5 μl of 2× Kapa HiFi HotStart ReadyMix, and 5 μl of water (total volume of 25 μl per sample) using the following conditions: 3 min at 98°C, 8 cycles of 30 s at 98°C, 30 s at 55°C, and 30 s at 72°C, with a final elongation step of 5 min at 72°C. PCR products were cleaned a second time with NucleoMag beads, quantified using a Qubit fluorometer (Life Technologies, Burlington, ON, Canada), and combined in an equimolar ratio. This final product was run out on a 1.2% agarose gel to isolate the DNA band at the expected size and purified using the PureLink quick gel extraction kit (Life Technologies). The eluted pool was sequenced on an Illumina MiSeq system using the 600-cycle MiSeq reagent kit v.3, following the manufacturer's recommendations. A total of 10,223,352 reads across 114 libraries were obtained following paired-end merging.
Illumina HiSeq for shotgun metagenomics.
DNA extracts from the end of the incubation were selected from five preassigned microcosms per microbiome type and from three extracts from the initial unmodified soil. Extracted DNA was sheared to a mean size of 200 bp in 130-μl AFA Fiber Snap-Cap microTUBEs (Covaris, Woburn, MA, USA) using the M220 focused ultrasonicator (Covaris) with a default program. Sample preparation of sheared DNA for Illumina HiSeq 2500 2 × 150-bp sequencing was performed using the Ovation Ultralow system V2 1-96 (NuGen, San Carlos, CA, USA), following the manufacturer's instructions. Eighteen samples were multiplexed across 2 HiSeq lanes at the McGill University and Genome Québec Innovation Centre (Montreal, QC, Canada). A total of 164,216,968 paired-end reads were recovered. A summary of the HiSeq sequence characteristics is provided in Table S2 in the supplemental material.
Bioinformatic analyses.
In mothur v.1.32.1 (33), paired-end reads were merged using the command make.contigs. Primers were removed with trim.seqs (pdiffs, 2; maxambig, 0), and the group sequences matching the trimmed fasta file were obtained with list.seqs followed by get.seqs. To reduce the file size for compatibility with the 32-bit version of USEARCH (34), we performed singleton removal in mothur instead of USEARCH. The fasta file was reduced to only unique sequences (unique.seqs), singletons were removed (split.abund, cutoff, 1), and the fasta file was then repopulated with all of the original sequences, minus those identified as singletons. The single fasta file was then split into separate files for each sample to facilitate naming in QIIME (MacQIIME v.1.8.0) (35), and the command add_qiime_labels.py was used to name and merge individual files. Next, we followed the steps described in the Brazilian Microbiome Project (BMP) pipeline (36) to produce operational taxonomic units (OTUs) at 97% similarity (starting at step 4; http://www.brmicrobiome.org/#!16S-profiling-pipeline-new-illumina/czxl). Taxonomic classifications were made using the QIIME-formatted Greengenes (gg_13_8) 16S rRNA gene database (37). The resulting OTU table was uploaded to R v.3.0.2 (38) for further analyses. We also projected functional profiles from our 16S rRNA gene data using PICRUSt (39). These were compared with our metagenomic data sets and allowed us to predict changes in major functional gene groups over time, as well as in the bacteria cultured on each of the five media types.
Paired-end reads from HiSeq libraries were imported to MG-RAST v.3.5 (40), merged, and uploaded with default parameters (except that dereplication was deselected) for functional and taxonomic annotation. Tables corresponding to the hierarchical levels of interest from the SEED subsystem annotations (SEED level 1 and level 3) were downloaded from the Metagenome Analysis subpage and uploaded to R for further analyses. To examine the relative abundance of alkane hydroxylases in our samples, we produced an All Annotations table using GenBank annotations in the MG-RAST analysis platform, searched “alkane” under “function,” and grouped all alkane hydroxylase annotations.
Statistical analyses.
We focused our analyses on week 0 (pre-crude oil spike) and week 6 (end of incubation), omitting samples collected at week 3, since the OTU-level compositions of the microbiomes were very similar at week 3 and week 6 (see Fig. S3 in the supplemental material). All principal-coordinate analyses (PCoA), Bray-Curtis distances, and diversity indices were calculated in the R package vegan (41). Proportional Venn diagrams were created using EulerApe (42) to visualize the OTUs shared between the three microbiome types. Taxonomic bubble plots were created using the R package PBSmodelling (43).
To examine whether there was persistence in the taxonomic structure despite the observed convergence in high-level taxonomy, we reduced our set of OTUs to the 137 that were shared between all microbiome types at all time points (i.e., initial soil/culture media, poststabilization, and post-crude oil incubation). At each time point, we normalized the abundance of each OTU across all treatment means to 1 (i.e., mean abundance in REG-M plus mean abundance in CO-M plus mean abundance in INIT = 1) and then projected these ratios onto ternary plots, created with the R package vcd (44), coloring each OTU by the microbiome type in which it was most abundant in the initial inoculum. Our aim was to determine whether, independent of changes in the overall abundance of an OTU, an OTU would remain more or less abundant as a result of its initial abundance.
One-way analysis of variance (ANOVA) tests (for crude oil degradation and targeted functional gene categories related to carbon processing) were performed with the lm function followed by the Tukey honestly significant difference (HSD) test in the R package stats. STAMP v.2.0.9 (45) was used to perform multiple comparisons of the relative abundance of functional gene categories and OTU abundance across microbiome types using the Benjamini-Hochberg false discovery rate (FDR) correction (46) and to produce principal-component analysis (PCA) plots, scatterplots, and extended error bar plots. A coinertia analysis (R package ade4) was performed to analyze the relationships between the 16S rRNA gene data set and the functional metagenomic data set (SEED subsystems annotations level 3).
Accession number(s).
MiSeq and HiSeq data have been deposited in the NCBI Sequence Read Archive and are available under the project number SRP073489. HiSeq data are also available through MG-RAST (4625855.3 to 4625856.3, 4626132.3 to 4626140.3, 4626321.3 to 4626325.3, and 4626327.3 to 4626328.3).
RESULTS AND DISCUSSION
In this study, we reintroduced the original microbiome of a soil, as well as two cultivated microbiome subsets, to a sieved, dried, and gamma-irradiated version of the initial soil. Details on the structures of the inocula for the three microbiome types and remnant DNA in the gamma-irradiated soil are provided in the supporting information and Fig. S4 and S5 in the supplemental material. Our design allowed us to avoid the confounding effects of microbiome origin, differences in soil parameters, and competition with established microbes, while still performing targeted microbiome manipulation within a soil matrix. After 14 weeks in soil (8 weeks of stabilization plus 6 weeks of metabolic interaction with crude oil and MAP), we compared taxonomy and functional gene composition for each microbiome type and the capacity of each to degrade the components of spiked crude oil.
We expected that a simplified microbiome, obtained exclusively on bacterial growth media containing crude oil, would enhance crude oil biodegradation in soil by selecting mainly bacteria involved in this process. No significant difference in C10 to C50 degradation was observed between the cultured microbiomes, while C10 to C50 degradation was significantly higher in microcosms inoculated with the source soil, even though the relative abundances of genes related to alkane degradation and carbon processing were higher in the CO-M and REG-M microbiomes, respectively. This appears to be true for the absolute abundances of these genes as well, since the amount of DNA extracted from the INIT microbiomes at week 6 was similar to that extracted from the CO-M microbiomes and less than that extracted from the REG-M microbiomes. There is increasing recognition that the relative abundances of functional genes for a biogeochemical process and the rate of the actual process can be weakly linked (47). It is likely to be even more difficult to identify strong links when a process is widespread throughout the microbiome, since organisms possessing the genes may be inactive or contributing little to the overall process rates. Here, only a few OTUs increased significantly in relative abundance after addition of crude oil and MAP, supporting this possibility.
Microbiome trajectories following addition to soil and incubation with crude oil.
Shannon diversity changed significantly between week 0 and week 6 for each microbiome type (paired t tests, P < 0.05), while evenness was only significantly changed in the INIT microbiomes (paired t test, P < 0.05). As was observed across a range of soil types treated with diesel (18), Shannon diversity of 16S rRNA genes declined slightly in REG-M and CO-M microbiomes following incubation with crude oil and MAP (Fig. 1a). Diversity (and evenness) actually increased in the INIT microbiomes, while the Chao1 richness estimate declined in all microbiome types (Fig. 1a). The Bray-Curtis distance between microbiome types at the phylum level shows microbiome convergence after 8 weeks of stabilization in the gamma-irradiated soils and further convergence after 6 weeks of incubation with crude oil, despite large phylum-level differences in the microbiome inocula (Fig. 1b and d). In contrast, OTU composition remained distinct across microbiome types, with only slight decreases in the Bray-Curtis distance relative to the starting microbiome composition (Fig. 1c and e). The CO-M microbiome remained the most stable over time (Fig. 1c), likely indicating that the CO-M media screened for bacteria that were able to use crude oil as a substrate.
FIG 1.

Shifts in microbiome structure through time by microbiome type. (a) Metrics for operational taxonomic unit (OTU) richness, diversity, and evenness in the three microbiome types, both before and after incubation with crude oil. Bars represent standard errors or estimated standard errors in the case of the Chao richness estimator. Principal-coordinate analysis (PCoA) ordinations of Bray-Curtis dissimilarity at the phylum (b) and OTU (c) levels. Shapes colored in white represent the means of replicate DNA extractions from the initial inocula. For the REG-M and CO-M inocula, they represent the means of the two and three media types used to produce them, respectively, as bacteria from each media type were added in equal amounts. Bar charts showing mean Bray-Curtis distances between microbiome types and to the median of all samples at each time point are shown at both the phylum (d) and OTU (e) levels. INIT, sterile soil reinoculated with the initial soil; REG-M, sterile soil reinoculated with bacteria cultured on regular media; CO-M, sterile soil reinoculated with bacteria cultured on media containing crude oil; week 0, after 8 weeks stabilization in soil; week 6, after 6 weeks incubation with crude oil.
Phylum-level convergence was mostly linked to Actinobacteria dominance of the microbiome (Fig. 2a). Despite large initial differences in the phylum-level composition of the microbiome types, each was dominated by Actinobacteria following incubation in the sterile soil. At week 0 (pre-crude oil spike), Actinobacteria represented 79.4% of sequences on average across microbiome types and 86.6% on average at week 6. This highlights the seemingly powerful constraints of abiotic soil parameters on bacterial taxonomy, supported by many environmental surveys of soil bacterial composition (e.g., 16–19). Although we use a single soil substrate in this study, the abiotic environment appeared to lead to taxonomic convergence, and soil type has been shown to influence the trajectories of bacterial assemblages across a range of soil types following diesel contamination (18). The nature of the soil environment appears to determine whether petroleum contamination will lead to dominance by Actinobacteria or Proteobacteria (18).
FIG 2.

Shifts in taxonomy through time by microbiome type. (a) Bubble plots showing the mean relative abundances of the most abundant phyla and the 20 most abundant Actinobacteria OTUs (across week 0 and week 6 combined) by microbiome type. Bubbles are shown for the initial inocula, week 0 (pre-crude oil), and week 6 (post-crude oil). Actinobacteria are highlighted to indicate that this phylum converged strongly by week 6 across microbiome types and was by far the dominant bacterial group. The abundance of Actinobacteria at week 6 did not differ significantly across microbiome types, while those of all other phyla and genera shown did (P < 0.05; multiple ANOVA with P value correction using the Benjamini-Hochberg false discovery rate). (b) Dot plot with 95% confidence intervals showing changes in OTU abundance from week 0 to week 6 for each microbiome type. The OTUs displayed are those with a relative abundance that increased significantly between time points by at least 1% in at least one microbiome type. The abundance of each OTU for each microbiome type at week 0 is indicated in a bar plot at the bottom of the figure. (c) Ternary plots of OTUs shared between all three microbiome types and across all time points to demonstrate treatment fidelity over time. The relative abundance of each OTU across microbiome types was normalized to 1 at each time point, so that the comparison represents the abundance of an OTU in relation to those of the other microbiome types, as opposed to other OTUs within a microbiome type. Colors represent the inoculum in which the OTU had the highest relative abundance (INIT, blue; REG-M, gold; CO-M, red). OTUs that are closer to the points of the triangle have higher relative abundances in the microbiome type represented by that point than the other microbiome types, whereas points close to the center have similar relative abundances in all microbiome types. (d) Proportion of total sequences for each microbiome type (INIT, blue; REG-M, gold; CO-M, red) at each time point that are classified as one of the 137 OTUs that were shared across time points and across microbiome types. INIT, sterile soil reinoculated with the initial soil; REG-M, sterile soil reinoculated with bacteria cultured on regular media; CO-M, sterile soil reinoculated with bacteria cultured on media containing crude oil.
However, the relative abundances of the 20 most abundant genera within the Actinobacteria (between week 0 and week 6) varied across microbiome types, despite being present in all three (Fig. 2a). A very small number of OTUs were disproportionately favored by crude oil and MAP addition, with different taxa responding in each microbiome type. In fact, only 8 OTUs across all phyla significantly increased in relative abundance by ≥1% from week 0 to week 6 (Benjamini-Hochberg corrected P < 0.05) (Fig. 2b). In the CO-M microcosms, two OTUs identified as Arthrobacter and Rhodococcus were the largest responders, increasing by ∼23% and 8%, respectively. In the INIT and REG-M microcosms, a different Rhodococcus OTU increased by averages of >10 and >30%, respectively, while an OTU classified as Mycobacterium responded positively in both (∼5 and 11%, respectively). Of the genera shown in Fig. 2, Arthrobacter, Rhodococcus, Mycobacterium, and Nocardioides are all identified as degraders of hydrocarbon compounds in the Minnesota Biocatalysis/Biodegradation Database, while both Williamsia and members of the Alcaligenaceae have been isolated using hydrocarbons as a sole carbon and energy source (48, 49). All were 98 to 100% similar to sequences in the NCBI nucleotide database that were derived from taxa living in organic-contaminated environments. Since different OTUs responded to crude oil and MAP in each microbiome type, despite the presence of all OTUs in each, biotic interactions may play an important role in controlling which taxa thrive, rather than additives leading to microbiome convergence. Interactions may also simply be influenced by the initial relative abundance of the responding OTUs.
Targeted microbiome selection with addition to soil has potential in biotechnology, but to become a useful tool, the microbiomes that persist over time should roughly resemble those added to the soil. When we analyzed the normalized relative abundance of OTUs shared between the three microbiome types (i.e., relative abundance per OTU across treatments), we found that OTUs tended to remain most abundant in the treatment in which they were initially present at a higher relative abundance (Fig. 2c), even if the relative abundance of that OTU declined across all treatments on average. In fact, after 8 weeks of stabilization in soil, 85.4% of shared OTUs were the most abundant in the microbiome type in which they were most abundant in the source material (initial soil or combined media), and this was true of 81.8% of OTUs after another 6 weeks of incubation with crude oil and MAP. At the end of the incubation with crude oil, 89.1% of shared OTUs remained most abundant in the microbiome type in which they were most abundant at the end of the initial stabilization period. This is important for the future of microbiome manipulation in soils, because, despite the impact of soil on taxonomy, the relative abundance of OTUs can be purposefully modified, at least in the absence of an indigenous microbiome.
Although many OTUs were not considered in this analysis, the shared OTUs represented the majority of sequences for all microbiome types at week 0 and week 6 (Fig. 2d). Well-characterized hydrocarbon-degrading genera represented a lower proportion of sequences in the INIT than in REG-M or CO-M microbiomes (see Fig. S6 in the supplemental material; see also the genera listed in Table S3 in the supplemental material), although these genera represented far more of the sequences in each microbiome type than in the source soil, and were more abundant at week 6 than week 0 (see Fig. S6 in the supplemental material).
Divergence in functional gene profiles.
Despite taxonomic convergence at the phylum level, the functional gene content of the microbiome types differed with respect to gene categories related to carbon processing. This shows that, despite the constraints of the soil matrix on taxonomy, we can create microbiomes in a single environment that vary with respect to functional potential. Although it is well known that the link between taxonomy and function is not as strong in microorganisms as in macroorganisms, due to processes such as lateral gene transfer (50), it was interesting to observe that key functional differences in microbiomes were maintained over 14 weeks in a common environment. We also show that a large difference in taxonomic diversity did not lead to substantial differences in the overall functional profiles of the INIT and CO-M microbiomes, highlighting the high degree of functional redundancy that exists within complex soil microbial assemblages.
The functional profiles of all microbiome types differed substantially from those of the source soil (see Fig. S7 to S9 in the supplemental material). PCA revealed redundancy in functional potential between the INIT and CO-M microbiomes, which clustered closely and were distinct from the REG-M and source soil profiles (Fig. 3a). This is in contrast to the 16S rRNA gene data when analyzed at the OTU level, in which the INIT and REG-M microbiomes at week 6 clustered most closely together (Fig. 1c and the 16S rRNA gene data from shotgun sequencing in MG-RAST, which are not shown here). A coinertia analysis showed that the OTU tables from our 16S rRNA data and the functional gene abundance tables (SEED subsystem annotations level 3) were more closely related than would be expected by chance (see Fig. S10 in the supplemental material) but also emphasized the differential clustering of microbiome types in the taxonomic and functional data. There was no significant difference in functional categories identified per sequence in the INIT and CO-M microbiomes or in the source soil (see Table S2 in the supplemental material), despite an estimate of roughly twice as many 16S rRNA gene OTUs in the INIT microbiome as in the CO-M microbiome at week 6 (Fig. 1a).
FIG 3.

Functional profiles of the three microbiome types at week 6 and from the initial source soil, based on shotgun metagenomic sequencing. (a) Principal-component analysis (PCA) ordination on SEED subsystem functional profiles produced by MG-RAST (level 3). (b) Regression of Shannon diversity of level 3 SEED functions against Shannon diversity of bacterial OTUs. Colors are as in panel a. (c, d) Scatterplots comparing the relative abundances of functional categories between INIT and CO-M and INIT and REG-M microbiomes. A comparison between REG-M and CO-M is not shown, due to the high degree of similarity between the CO-M and INIT profiles. Lines extending from points in the scatterplots indicate standard deviations. (e) Histogram and confidence intervals of functions found to be significantly different between INIT and REG-M, with an effect size (difference in relative abundance) of at least 0.25. These functions are found within the orange-colored area of panel d. There were no significant differences of this size between INIT and CO-M. (f) Relative abundances of functional categories related to carbon processing across microbiome types and within the initial source soil. The top three histograms are level 1 categories from SEED subsystem annotations, whereas the bottom histogram (alkane hydroxylases) was created by combining all GenBank alkane hydroxylase annotations using the All Annotations search function in MG-RAST. INIT, sterile soil reinoculated with the initial soil; REG-M, sterile soil reinoculated with bacteria cultured on regular media; CO-M, sterile soil reinoculated with bacteria cultured on media containing crude oil.
The Shannon diversity of bacterial OTUs was linearly correlated (R2 = 0.295, P = 0.018) with the Shannon diversity of SEED level 3 functional categories, although the curve was much steeper for the REG-M microcosms, which had lower functional diversity than the other microbiome types (Fig. 3b). The relative abundance of SEED level 3 categories was very similar between the CO-M and INIT microbiomes (Fig. 3c), but not between REG-M and INIT microbiomes, in which a number of categories, mostly related to the metabolism of different carbon compounds, were notably higher in the REG-M microbiomes (Fig. 3d and e). We compared certain SEED level 1 categories related to carbon processing, which we expected were important in crude oil biodegradation. The relative abundances of “carbohydrates,” “fatty acids, lipids, and isoprenoids,” and “metabolism of aromatic compounds” were significantly higher in the REG-M microbiomes than in all others and significantly lower in the initial soil microbiome (Fig. 3f). We grouped all functional annotations of alkane hydroxylases (involved in the biodegradation of straight-chain petroleum hydrocarbons), and these were significantly higher in the CO-M microbiomes than in all others (Fig. 3f). The addition of MAP is also likely to have influenced the microbiomes, and we observed small but significant increases in the relative abundance of N metabolism genes in all microbiomes compared to those in the source soil and small but significant decreases in genes related to P metabolism (see Fig. S7 to S9 in the supplemental material). However, we should also note that these findings are based on the abundance of known and classifiable genes. We showed that the cultivated microbiome subsets contained a higher proportion of known and cultivated hydrocarbon-degrading genera than did the initial microbiome (see Fig. S6 in the supplemental material; see also the genera listed in Table S3 in the supplemental material). As a result, we may be underestimating the proportion of important functional genes in the initial microbiome, since more unclassified and uncultivated microorganisms are present.
We also used PICRUSt metagenome projections (39) to predict metagenome functional content from 16S rRNA gene data, in order to see how the proportion of biodegradation genes may have varied over time and across media types. We looked at shifts in the KEGG orthology functional category Xenobiotics Biodegradation and Metabolism and found that at week 6 after spiking with crude oil and MAP and in the initial soil samples, the PICRUSt projections inflated the counts relative to what we observed in the sequenced metagenomes (data not shown) but still presented the same general pattern. The relative abundance of genes in this category was projected to be 4 to 4.5% higher in the bacteria sequenced from the crude oil plus M9 media than in any of the other media types and was projected to be higher on average than that in any of the microbiome types at week 0 or week 6, with the exception of REG-M at week 6 (see Fig. S11 in the supplemental material). A PCA also showed media type separation based on projected functional profiles, and the functional profiles of all microbiome types shifted from those of the initial soil and the four media types containing either TSA or glucose at week 0 toward the functional profile of the crude oil plus M9 medium type by week 6 (Fig. 4). Despite this, the taxonomic composition of the most abundant OTUs in each microbiome type at week 6 did not resemble that of the crude oil plus M9 medium (see Fig. S12 in the supplemental material).
FIG 4.

PCA plot produced using STAMP analysis of PICRUSt-projected functional profiles (level 3, KEGG orthology). This plot shows that the week 6 profiles group together with the profiles from the crude oil plus M9 media, whereas the pre-crude oil profiles group together with the profiles from all other media types (TSA and glucose, with or without crude oil added).
Functional redundancy in microbiome subsets.
Defining functional redundancy in the soil microbiome is challenging. Although taxonomy has sometimes been correlated with function (18, 51, 52), these correlations tend to be confounded by differences in soil conditions. When starting with a single microbial strain, increasing microbial diversity is likely to increase carbon processing (as observed by Bell et al. [22]) since there is a high chance of complementarity. In hyperdiverse soil microbiomes, however, most forms of metabolism will likely be present, and the pool of genes for potential horizontal gene transfer is much larger. We observed nearly identical functional gene profiles in the INIT and CO-M microbiomes (when classified to level 3 of the SEED subsystems), even though the CO-M microbiome consisted only of cultivable taxa and was estimated to contain around half as many OTUs. It needs to be emphasized, however, that these similarities are driven primarily by basic metabolic functions, which are overrepresented in functional databases; many specialized functions within the INIT microbiomes are likely not present in the CO-M microbiomes.
In terms of actual degradation, we observed significantly more degradation of C10 to C50 hydrocarbons in the INIT microbiomes (PANOVA = 0.0117, η2 = 0.281) (Fig. 5). Although the total concentration of PAH compounds in the crude oil was low (9.2 mg/kg on average), there was significantly more PAH degradation (PANOVA = 0.0338, η2 = 0.431) in the INIT microbiomes than in the REG-M microbiomes (see Fig. S13 in the supplemental material). These differences may be related to the presence of uncultivated microbes, including eukaryotes, which would not have been captured using bacterial culture media. A previous study demonstrated that bacteria and fungi were responsible for 82% and 13% of hexadecane degradation, respectively (53), and the high efficiency of degradation in the cultured microbiome subsets reinforces the importance of bacteria in hydrocarbon degradation, especially when PAH compounds are not abundant. A diverse microbiome, while functionally similar to a less diverse microbiome, might also adapt better to multiple environments and better tolerate changing environmental conditions (e.g., increasing salt concentrations and warming [54]). However, we must also consider the possibility that the lab-cultivated submicrobiomes are less adapted to the soil environment than the initial microbiome. This might be due to declines in basic ecological traits not related to crude oil degradation or a reduced ability to interact efficiently with coexisting microbes.
FIG 5.
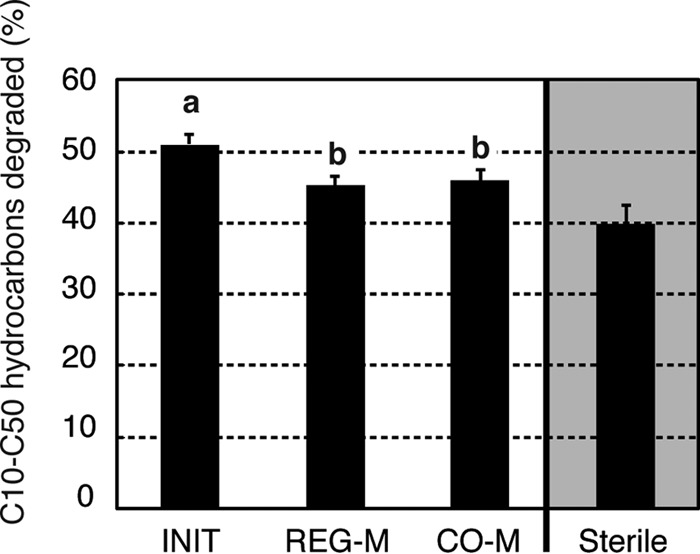
Percent degradation of C10 to C50 hydrocarbons after 6 weeks of microcosm incubation following the addition of crude oil and monoammonium phosphate. Bars represent standard errors. Different letters over columns indicate significant differences between treatments based on a Tukey HSD post hoc test following one-way ANOVA. The sterile (noninoculated) controls were rerun after the initial experiment, as the original controls experienced contamination. The incubation conditions were the same, and the C10 to C50 loss was compared to that of paired baseline samples that were frozen at the outset of this incubation. For treatments, n = 10, and for sterile controls, n = 3. INIT, sterile soil reinoculated with the initial soil; REG-M, sterile soil reinoculated with bacteria cultured on regular media; CO-M, sterile soil reinoculated with bacteria cultured on media containing crude oil.
Culturing without isolation can produce diverse and functionally distinct microbiomes.
New methods allow increasingly high-throughput culturing of individual microbial strains and can even select variant individuals from a single species with particular functional abilities (55). Hand-selected isolates might potentially be combined into designer soil inocula, but the interactions between these isolates in situ will be difficult to predict, as will the consistency of their functions, phenotypes, and mobile genomes. In addition, microbial strains that grow and evolve together can be more productive as a group, in part because some adapt to using the by-products of coexisting species (56).
In this study, we show that diverse soil microbiome subsets with distinct taxonomic and functional profiles can be quickly obtained through bulk culturing on standard media. Isolation followed by scraping and sequencing appears to capture a high percentage of OTUs compared to that for traditional colony picking, although we cannot determine how many were derived from remnant DNA in the sterile soil, as the same OTUs may have been present in both the culture media and the sterile soil. This was also observed by Shade et al. (57), who captured ∼21% of all soil bacterial OTUs using only one type of cultivation medium, compared to the five types used in this study (we observed 22.4% and 33.8% of all observed OTUs in our REG-M and CO-M inocula, respectively, as shown in Fig. S4 in the supplemental material). Although less sophisticated, this approach is of interest for a few reasons: (i) microbial inocula adapted to local conditions and to each other can be developed for any target soil; (ii) government regulations often complicate the introduction of nonnative species to soils; and (iii) small changes in media composition might capture microbiome subsets with different functional phenotypes (e.g., r- and K-strategists, salt-tolerant microbes, and phenanthrene degraders). This method should prove complementary to other complex microbiome selection approaches and will help provide a range of inocula for testing the persistence of microbiomes introduced to soil.
In conclusion, as has been shown through environmental surveys, this study demonstrates the strong controlling influence of soil abiotic conditions on bacterial phylum-level taxonomy but less so at the OTU level. Microbiome composition at the OTU level was roughly maintained throughout a 14-week incubation, demonstrating that it is possible to purposefully structure the soil microbiome to some extent. Interestingly, OTUs that responded positively to crude oil and MAP spiking varied by microbiome type, even though the responding OTUs were present in all microbiome types. These microbiomes were maintained under sterile conditions, so further studies are needed to determine whether differentially seeded microbiomes can resist invasion by the initial soil community or whether convergence in structure and function is inevitable without modification of the physical soil environment. It will also be necessary to track microbiome structure beyond the 14-week interval used in this study, to determine whether convergence increases with time. Finally, this study shows that the initial soil microbiome more efficiently degraded added crude oil than did a presumably specialized bacterial assemblage selected on crude oil media, although this might vary by soil type and soil history.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the GenoRem Project, which is funded by Génome Québec and Genome Canada. T.H.B. was also supported by a grant from the Fonds de Recherche du Québec-Nature et Technologies.
We thank Stéphane Daigle for his assistance with the experimental design and Fahad Al-Otaibi for his assistance with soil analyses.
The GenoRem project contains several industrial partners, but these partners have in no way influenced or modified the contents of the manuscript or the analysis of the results presented.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01327-16.
REFERENCES
- 1.Samanta SK, Singh OV, Jain RK. 2002. Polycyclic aromatic hydrocarbons: environmental pollution and bioremediation. Trends Biotechnol 20:243–248. doi: 10.1016/S0167-7799(02)01943-1. [DOI] [PubMed] [Google Scholar]
- 2.Glass DJ. 1999. Current market trends in phytoremediation. Int J Phytoremediation 1:1–8. doi: 10.1080/15226519908500001. [DOI] [Google Scholar]
- 3.Hu GJ, Li JB, Zeng GM. 2013. Recent development in the treatment of oily sludge from petroleum industry: a review. J Hazard Mater 261:470–490. doi: 10.1016/j.jhazmat.2013.07.069. [DOI] [PubMed] [Google Scholar]
- 4.Thompson IP, van der Gast CJ, Ciric L, Singer AC. 2005. Bioaugmentation for bioremediation: the challenge of strain selection. Environ Microbiol 7:909–915. doi: 10.1111/j.1462-2920.2005.00804.x. [DOI] [PubMed] [Google Scholar]
- 5.Afzal M, Yousaf S, Reichenauer TG, Kuffner M, Sessitsch A. 2011. Soil type affects plant colonization, activity and catabolic gene expression of inoculated bacterial strains during phytoremediation of diesel. J Hazard Mater 186:1568–1575. doi: 10.1016/j.jhazmat.2010.12.040. [DOI] [PubMed] [Google Scholar]
- 6.Tyagi M, da Fonseca MMR, de Carvalho CCCR. 2011. Bioaugmentation and biostimulation strategies to improve the effectiveness of bioremediation processes. Biodegradation 22:231–241. doi: 10.1007/s10532-010-9394-4. [DOI] [PubMed] [Google Scholar]
- 7.van Veen JA, van Overbeek LS, van Elsas JD. 1997. Fate and activity of microorganisms introduced into soil. Microbiol Mol Biol Rev 61:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomassin-Lacroix EJM, Eriksson M, Reimer KJ, Mohn WW. 2002. Biostimulation and bioaugmentation for on-site treatment of weathered diesel fuel in Arctic soil. Appl Microbiol Biotechnol 59:551–556. doi: 10.1007/s00253-002-1038-0. [DOI] [PubMed] [Google Scholar]
- 9.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. 2013. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 4:125–135. doi: 10.4161/gmic.23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuentes S, van Nood E, Tims S, Heikamp-de Jong I, ter Braak CJF, Keller JJ, Zoetendal EG, de Vos WM. 2014. Reset of a critically disturbed microbial ecosystem: faecal transplant in recurrent Clostridium difficile infection. ISME J 8:1621–1633. doi: 10.1038/ismej.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller UG, Sachs JL. 2015. Engineering microbiomes to improve plant and animal health. Trends Microbiol 23:606–617. doi: 10.1016/j.tim.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 12.Bol R, Poirier N, Balesdent J, Gleixner G. 2009. Molecular turnover time of soil organic matter in particle-size fractions of an arable soil. Rapid Commun Mass Spectrom 23:2551–2558. doi: 10.1002/rcm.4124. [DOI] [PubMed] [Google Scholar]
- 13.Martin FPJ, Wang Y, Yap IKS, Sprenger N, Lindon JC, Rezzi S, Kochhar S, Holmes E, Nicholson JK. 2009. Topographical variation in murine intestinal metabolic profiles in relation to microbiome speciation and functional ecological activity. J Proteome Res 8:3464–3474. doi: 10.1021/pr900099x. [DOI] [PubMed] [Google Scholar]
- 14.Chaparro JM, Sheflin AM, Manter DK, Vivanco JM. 2012. Manipulating the soil microbiome to increase soil health and plant fertility. Biol Fertil Soils 48:489–499. doi: 10.1007/s00374-012-0691-4. [DOI] [Google Scholar]
- 15.Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, Malfatti S, del Rio TG, Jones CD, Tringe SG, Dangl JL. 2015. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349:860–864. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- 16.Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc Natl Acad Sci U S A 104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell TH, Yergeau E, Maynard C, Juck D, Whyte LG, Greer CW. 2013. Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. ISME J 7:1200–1210. doi: 10.1038/ismej.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranjard L, Dequiedt S, Prévost-Bouré NC, Thioulouse J, Saby NPA, Lelievre M, Maron PA, Morin FER, Bispo A, Jolivet C, Arrouays D, Lemanceau P. 2013. Turnover of soil bacterial diversity driven by wide-scale environmental heterogeneity. Nat Commun 4:1434. doi: 10.1038/ncomms2431. [DOI] [PubMed] [Google Scholar]
- 20.Griffiths BS, Ritz K, Bardgett RD, Cook R, Christensen S, Ekelund F, Sørensen SJ, Bååth E, Bloem J, de Ruiter PC, Dolfing J, Nicolardot B. 2000. Ecosystem response of pasture soil communities to fumigation-induced microbial diversity reductions: an examination of the biodiversity-ecosystem function relationship. Oikos 90:279–294. doi: 10.1034/j.1600-0706.2000.900208.x. [DOI] [Google Scholar]
- 21.Philippot L, Spor A, Hénault C, Bru D, Bizouard F, Jones CM, Sarr A, Maron PA. 2013. Loss in microbial diversity affects nitrogen cycling in soil. ISME J 7:1609–1619. doi: 10.1038/ismej.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK. 2005. The contribution of species richness and composition to bacterial services. Nature 436:1157–1160. doi: 10.1038/nature03891. [DOI] [PubMed] [Google Scholar]
- 23.Gravel D, Bell T, Barbera C, Bouvier T, Pommier T, Venail P, Mouquet N. 2011. Experimental niche evolution alters the strength of the diversity-productivity relationship. Nature 469:89–92. doi: 10.1038/nature09592. [DOI] [PubMed] [Google Scholar]
- 24.Ho A, de Roy K, Thas O, De Neve J, Hoefman S, Vandamme P, Heylen K, Boon N. 2014. The more, the merrier: heterotroph richness stimulates methanotrophic activity. ISME J 8:1945–1948. doi: 10.1038/ismej.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fournier G, Fournier JC. 1993. Effect of microbial competition on the survival and activity of 2,4-D-degrading Alcaligenes xylosoxidans subsp. denitrificans added to soil. Lett Appl Microbiol 16:178–181. doi: 10.1111/j.1472-765X.1993.tb01389.x. [DOI] [Google Scholar]
- 26.Degens BP. 1998. Decreases in microbial functional diversity do not result in corresponding changes in decomposition under different moisture conditions. Soil Biol Biochem 30:1989–2000. doi: 10.1016/S0038-0717(98)00071-6. [DOI] [Google Scholar]
- 27.Bell TH, Yergeau E, Juck D, Whyte LG, Greer CW. 2013. Alteration of microbial community structure affects diesel degradation in an Arctic soil. FEMS Microbiol Ecol 85:51–61. doi: 10.1111/1574-6941.12102. [DOI] [PubMed] [Google Scholar]
- 28.van Elsas JD, Chiurazzi M, Mallon CA, Elhottova D, Kristufek V, Salles JF. 2012. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc Natl Acad Sci U S A 109:1159–1164. doi: 10.1073/pnas.1109326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zafra G, Absalon AE, Cuevas MD, Cortes-Espinosa DV. 2014. Isolation and selection of a highly tolerant microbial consortium with potential for PAH biodegradation from heavy crude oil-contaminated soils. Water Air Soil Pollut 225:1826. doi: 10.1007/s11270-013-1826-4. [DOI] [Google Scholar]
- 30.Centre d'Expertise en Analyse Environnementale du Québec. 2002. Dosage des hydrocarbures pétroliers C10 à C50 dans les sols et les sédiments. MA. 416 – C10-C50 1.0. Ministère de l'Environnement du Québec, Québec, QC, Canada. [Google Scholar]
- 31.Centre d'Expertise en Analyse Environnementale du Québec. 2016. Détermination des hydrocarbures aromatiques polycycliques: dosage par chromatographie en phase gazeuse couplée à un spectromètre de masse. MA. 400 – HAP 1.1, Rév. 4. Ministère du Développement durable, de l'Environnement et de la Lutte Contre Les Changements Climatiques, Québec, QC, Canada. [Google Scholar]
- 32.Herlemann DPR, Labrenz M, Jurgens K, Bertilsson S, Waniek JJ, Andersson AF. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 35.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pylro VS, Roesch LFW, Morais DK, Clark IM, Hirsch PR, Tótola MR. 2014. Data analysis for 16S microbial profiling from different benchtop sequencing platforms. J Microbiol Methods 107:30–37. doi: 10.1016/j.mimet.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 37.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 39.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R, Beiko RG, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oksanen J, Blanchet GB, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2015. vegan: community ecology package. R package version 2.3-0. https://cran.r-project.org/web/packages/vegan/vegan.pdf.
- 42.Micallef L, Rodgers P. 2014. eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS One 9:e101717. doi: 10.1371/journal.pone.0101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schnute JT, Couture-Beil A, Haigh R, Kronlund AR, Boers N. 2015. PBSmodelling: GUI tools made easy: interact with models and explore data. R package version 2.67.266. https://cran.r-project.org/web/packages/PBSmodelling/index.html.
- 44.Meyer D, Zeileis A, Hornik K. 2015. vcd: visualizing categorical data. R package version 1.4-1. https://cran.r-project.org/web/packages/vcd/index.html.
- 45.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. 2014. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 57:289–300. [Google Scholar]
- 47.Rocca JD, Hall EK, Lennon JT, Evans SE, Waldrop MP, Cotner JB, Nemergut DR, Graham EB, Wallenstein MD. 2015. Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J 9:1693–1699. doi: 10.1038/ismej.2014.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obuekwe CO, Al-Jadi ZK, Al-Saleh ES. 2009. Hydrocarbon degradation in relation to cell-surface hydrophobicity among bacterial hydrocarbon degraders from petroleum-contaminated Kuwait desert environment. Int Biodeter Biodegr 63:273–279. doi: 10.1016/j.ibiod.2008.10.004. [DOI] [Google Scholar]
- 49.Kubota K, Koma D, Matsumiya Y, Chung SY, Kubo M. 2008. Phylogenetic analysis of long-chain hydrocarbon-degrading bacteria and evaluation of their hydrocarbon-degradation by the 2,6-DCPIP assay. Biodegradation 19:749–757. doi: 10.1007/s10532-008-9179-1. [DOI] [PubMed] [Google Scholar]
- 50.Boon E, Meehan CJ, Whidden C, Wong DHJ, Langille MGI, Beiko RG. 2014. Interactions in the microbiome: communities of organisms and communities of genes. FEMS Microbiol Rev 38:90–118. doi: 10.1111/1574-6976.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allison SD, Martiny JBH. 2008. Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci U S A 105:11512–11519. doi: 10.1073/pnas.0801925105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R. 2012. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6:1007–1017. doi: 10.1038/ismej.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song HG, Pedersen TA, Bartha R. 1986. Hydrocarbon mineralization in soil: relative bacterial and fungal contribution. Soil Biol Biochem 18:109–111. doi: 10.1016/0038-0717(86)90111-2. [DOI] [Google Scholar]
- 54.Awasthi A, Singh M, Soni SK, Singh R, Kalra A. 2014. Biodiversity acts as insurance of productivity of bacterial communities under abiotic perturbations. ISME J 8:2445–2452. doi: 10.1038/ismej.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang BL, Ghaderi A, Zhou H, Agresti J, Weitz DA, Fink GR, Stephanopoulos G. 2014. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nat Biotechnol 32:473–478. doi: 10.1038/nbt.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawrence D, Fiegna F, Behrends V, Bundy JG, Phillimore AB, Bell T, Barraclough TG. 2012. Species interactions alter evolutionary responses to a novel environment. PLoS Biol 10:e1001330. doi: 10.1371/journal.pbio.1001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shade A, Hogan CS, Klimowicz AK, Linske M, McManus PS, Handelsman J. 2012. Culturing captures members of the soil rare biosphere. Environ Microbiol 14:2247–2252. doi: 10.1111/j.1462-2920.2012.02817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.