

ABSTRACT
β-N-Acetylhexosaminidases have attracted interest particularly for oligosaccharide synthesis, but their use remains limited by the rarity of enzyme sources , low efficiency, and relaxed regioselectivity of transglycosylation. In this work, genes of 13 β-N-acetylhexosaminidases, including 5 from Bacteroides fragilis ATCC 25285, 5 from Clostridium perfringens ATCC 13124, and 3 from Bifidobacterium bifidum JCM 1254, were cloned and heterogeneously expressed in Escherichia coli. The resulting recombinant enzymes were purified and screened for transglycosylation activity. A β-N-acetylhexosaminidase named BbhI, which belongs to glycoside hydrolase family 20 and was obtained from B. bifidum JCM 1254, possesses the bifunctional property of efficiently transferring both GalNAc and GlcNAc residues through β1-3 linkage to the Gal residue of lactose. The effects of initial substrate concentration, pH, temperature, and reaction time on transglycosylation activities of BbhI were studied in detail. With the use of 10 mM pNP-β-GalNAc or 20 mM pNP-β-GlcNAc as the donor and 400 mM lactose as the acceptor in phosphate buffer (pH 5.8), BbhI synthesized GalNAcβ1-3Galβ1-4Glc and GlcNAcβ1-3Galβ1-4Glc at maximal yields of 55.4% at 45°C and 4 h and 44.9% at 55°C and 1.5 h, respectively. The model docking of BbhI with lactose showed the possible molecular basis of strict regioselectivity of β1-3 linkage in β-N-acetylhexosaminyl lactose synthesis.
IMPORTANCE Oligosaccharides play a crucial role in many biological events and therefore are promising potential therapeutic agents. However, their use is limited because large-scale production of oligosaccharides is difficult. The chemical synthesis requires multiple protecting group manipulations to control the regio- and stereoselectivity of glycosidic bonds. In comparison, enzymatic synthesis can produce oligosaccharides in one step by using glycosyltransferases and glycosidases. Given the lower price of their glycosyl donor and their broader acceptor specificity, glycosidases are more advantageous than glycosyltransferases for large-scale synthesis. β-N-Acetylhexosaminidases have attracted interest particularly for β-N-acetylhexosaminyl oligosaccharide synthesis, but their application is affected by having few enzyme sources, low efficiency, and relaxed regioselectivity of transglycosylation. In this work, we describe a microbial β-N-acetylhexosaminidase that exhibited strong transglycosylation activity and strict regioselectivity for β-N-acetylhexosaminyl lactose synthesis and thus provides a powerful synthetic tool to obtain biologically important GalNAcβ1-3Lac and GlcNAcβ1-3Lac.
INTRODUCTION
Oligosaccharides are widely distributed in nature and play a crucial role in many biological events, such as cell structure modulation, cell-cell recognition and communication, and cell-microbe/toxin interaction and adhesion; therefore, they are promising and important potential therapeutic agents (1–3). However, their use is limited because of the difficulty of large-scale production of oligosaccharides. Their chemical synthesis requires multiple protecting group manipulations to control the regio- and stereospecificity of glycosidic bonds, which hinders the efficient production of oligosaccharides. Enzymatic synthesis can produce oligosaccharides in one step by using two major classes of enzyme, namely, glycosyltransferases (EC 2.4) and glycosidases (EC 3.2.1). Glycosyltransferases, the natural enzymes for specific synthesis of oligosaccharides and polysaccharides, require expensive nucleoside sugars as glycosyl donors and generally exhibit strict acceptor specificity. In contrast, glycosidases, which are enzymes that normally hydrolyze carbohydrates, can use low-cost, simple glycosides as glycosyl donors and show a broad acceptor specificity for glycosylation or transglycosylation reactions in vitro; thus, the use of glycosidases is a more effective practical approach for large-scale synthesis of oligosaccharides (4, 5).
β-N-Acetylhexosaminidases (EC 3.2.1.52) are an important class of glycosidases that catalyze hydrolysis of terminal N-acetyl-β-d-hexosamine (HexNAc), N-acetyl-β-d-galactosamine (GalNAc), or N-acetyl-β-d-glucosamine (GlcNAc) from various oligosaccharides and glycoconjugates. These enzymes are widely distributed in microorganisms, plants, and animals, and they play crucial roles in nature (6). For instance, they can cleave a GalNAc residue from GM2 gangliosidose, and their deficiency will cause lysosomal storage diseases (7, 8). They are also involved in bacterial cell wall recycling, fungal cell wall chitin lysis, and the insect life cycle (9–11). Multiple β-N-acetylhexosaminidases can be produced for utilization of chitin, intestinal mucosal glycans, and milk oligosaccharides by some intestinal bacteria, such as Bacteroides fragilis, Clostridium perfringens, and Bifidobacterium bifidum (12–14).
Based on amino acid homology, β-N-acetylhexosaminidases are classified into glycoside hydrolase (GH) families 3, 20, 84, and 123 (15–21). Among them, GH3 enzymes generally employ a double-displacement mechanism, in which a pair of acidic residues serve as catalytic acid/base and nucleophile residues (15, 16). In contrast, GH20, GH84, and GH123 enzymes employ the substrate-assisted mechanism in which the 2-acetamido group of the substrate acts as a nucleophile to form an oxazoline intermediate for catalysis (17–21). A large number of bifunctional β-N-acetylhexosaminidases from the GH20 family show hydrolysis activity toward both GalNAc- and GlcNAc-containing substrates (17, 18), whereas GH3, GH84, and GH123 enzymes specifically hydrolyze GalNAc- or GlcNAc-containing substrates (15, 16, 19–21).
β-N-Acetylhexosaminidases have recently attracted increasing interest owing to their ability to catalyze glycosyl transfer to form β-N-acetyl-d-hexosaminide. β-N-Acetylhexosaminidases in the GH20 family have been successfully used to synthesize β-N-acetylhexosaminyl oligosaccharides, such as GlcNAcβ1-4GlcNAcβ1-4ManNAc, GalNAcβ1-4GlcNAcαUDP, (GlcNAcβ1-4)3GlcNAc, and 6′-sulfodisaccharides (e.g., GlcNAcβ1-4Glcα-OAll, GlcNAcβ1-3/1-6Galα-OAll, and GlcNAcβ1-4GlcNAc) (22–25). However, the available enzyme sources for synthesis remain limited. Most of the reported β-N-acetylhexosaminidases with transglycosylation ability are obtained from fungi, especially those belonging to the genera Aspergillus (22–24, 26–29), Penicillium (28, 30), and Talaromyces (31, 32), and only three are obtained from the bacteria Nocardia orientalis and Serratia marcescens YS-1 (33–35). Moreover, most β-N-acetylhexosaminidases with transglycosylation ability display low glycosyl transfer efficiency at 1% to 10% yield, as well as poor regioselectivity, resulting in isomer production with multiple glycosyl linkages that are difficult to isolate (33, 35, 36). Only two fungal β-N-acetylhexosaminidases can catalyze transfer of both GalNAc and GlcNAc residues for synthesis (24, 25).
GalNAc- and GlcNAc-containing oligosaccharides are widely distributed in microorganisms, plants, and animals, and they play important biological roles. They are present in sugar structures of gangliosides, N-glycan, mucin-type O-glycan, blood group antigens, tumor-associated glycans, milk oligosaccharides, and chitin (12, 37, 38). β-N-Acetylhexosaminyl lactose (HexNAc-Lac) is a class of important oligosaccharides. GalNAcβ1-3Lac shares oligosaccharide constituents with the blood group P-related glycan, which inhibits respiratory pathogens (39, 40). GlcNAcβ1-3Lac is the sugar unit of many cancer-related carbohydrate antigens, such as KH-1 adenocarcinoma antigen, N3 antigens associated with gastrointestinal cancer, and primary acute myeloid leukemia antigen (41, 42), which may serve as a potential biomarker and a target for therapy. In addition, GlcNAcβ1-3Lac is the backbone structure of one group of human milk oligosaccharides (HMOs) and thus can be elongated with sugar modifications to produce HMOs (43) that can be used as prebiotics (12). However, access to these trisaccharides at large scale remains a challenge. Nevertheless, GlcNAcβ1-3Lac has been synthesized by β-N-acetylhexosaminidase at a low yield of less than 10% (33, 35, 36, 44), whereas the synthesis of GalNAcβ1-3Lac by this enzyme has not yet been reported.
In this work, an efficient and regioselective β-N-acetylhexosaminidase, named BbhI, with transglycosylation ability and isolated from B. bifidum JCM 1254 was obtained by screening 13 enzymes. BbhI showed strict regioselectivity of transglycosylation at 3-OH of galactose residues in lactose, resulting in high yields of GalNAcβ1-3Lac and GlcNAcβ1-3Lac. This work is the first to demonstrate the synthesis of GalNAcβ1-3Lac by a glycosidase. The molecular basis of strict regioselectivity of β1-3 linkage in β-N-acetylhexosaminyl lactose synthesis was subsequently studied via sequence analysis and model docking of BbhI with lactose.
MATERIALS AND METHODS
Materials.
p-Nitrophenyl 2-acetamido-2-deoxy-β-d-galactopyranoside (pNP-β-GalNAc), p-nitrophenyl 2-acetamido-2-deoxy-β-d-glucopyranoside (pNP-β-GlcNAc), and the other nitrophenyl glycosides were purchased from Sigma (St. Louis, MO, USA). High-performance liquid chromatography (HPLC)-grade acetonitrile was supplied by Honeywell Burdick & Jackson (Muskegon, MI, USA). Ni2+ Sepharose high performance was obtained from GE Healthcare (Uppsala, Sweden). Bio-Gel P2 was purchased from Bio-Rad Laboratories (Hercules, CA, USA). Other chemicals used were of analytical grade. Restriction endonucleases were purchased from New England BioLabs. T4 DNA ligase was purchased from TaKaRa Corporation (Tokyo, Japan). FastPfu Fly DNA polymerase was obtained from Transgen (China).
Strains and media.
B. bifidum JCM 1254 was grown anaerobically at 37°C in a medium containing 10 g of glucose, 5 g of peptone, 5 g of yeast extract, 4 g of K2HPO4, 5 g of sodium acetate, 0.2 g of MgSO4, 6.8 g of ascorbic acid, and 0.4 g of cysteine hydrochloride in 1,000 ml of water (pH 7.0). B. fragilis ATCC 25285 was grown anaerobically at 37°C in a medium containing 5 g of yeast extract, 20 g of peptone, 5 g of NaCl, 60 g of glucose, 5 mg of hemin, and 0.5 mg of vitamin K1 in 1,000 ml of water (pH 7.0). C. perfringens ATCC 13124 was grown anaerobically at 40°C in medium containing 5 g of yeast extract, 5 g of peptone, 5 g of sodium acetate, 0.2 g of MgSO4, 4 g of K2HPO4, 0.4 g of cysteine hydrochloride, and 6.8 g of ascorbic acid in 1,000 ml of water (pH 7.0). Anaerobic culture was performed in a Forma anaerobic system (Thermo) under a mixture of nitrogen-hydrogen-carbon dioxide at a 84.9:10:5.1 (vol/vol/vol) ratio.
Escherichia coli strains DH5α and BL21(DE3) were grown at 37°C in LB medium containing 5 g of yeast extract, 10 g of peptone, and 7 g of NaCl in 1,000 ml of water (pH 7.0). The medium for the E. coli cells containing pET-21b(+) plasmid was supplemented with ampicillin (50 μg/ml). The pET-21b(+) plasmid (Novagen) was used to construct an expression vector with His tag.
Screening of β-N-acetylhexosaminidases for transglycosylation.
Ten putative and three known β-N-acetylhexosaminidase genes deposited in the Carbohydrate-Active enZYmes database (http://www.cazy.org/) were obtained by PCR. Among them, seven genes encoding the truncated enzymes BF0669, BF1807, BF4033, CPF0184, CPF1487, BbhI, and BbhII without putative signal peptides and/or transmembrane regions (see Table S1 in the supplemental material) were amplified. The designed primers are listed in Table 1. The primers for the genes of BF0669, BF0953, BF1807, BF1811, and BF4033 enzymes were designed based on the genome sequence of B. fragilis ATCC 25285 (GenBank accession no. CR626927.1); the primers for the genes of CPF1103, CPF1238, CPF0184, CPF1487, and CPF1473 enzymes were designed based on the genome sequence of C. perfringens ATCC 13124 (GenBank accession no. CP000246.1); the primers for the genes of BbhI, BbhII, and BbhIII enzymes were designed based on the reported β-N-acetylhexosaminidase gene sequences of B. bifidum JCM 1254; the GenBank accession numbers for these gene sequences are AB504521.1, AB504522.1, and AB542715.1, respectively.
TABLE 1.
Primers used in this study
| Enzyme source | Primer | DNA sequence (5′→3′)a | Restriction enzyme |
|---|---|---|---|
| B. fragilis ATCC 25285 | |||
| BF0669 | BF0669F | CGATCATATGCCCAAACCTTTTGTGATTCC | NdeI |
| BF0669R | CGCGCTCGAGTTTCTTATAAACCTTCAGAT | XhoI | |
| BF0953 | BF0953F | CGCGCATATGAAAAAAGGAATTACTCT | NdeI |
| BF0953R | CGATCTCGAGATATACTTCTATTTCGTCCA | XhoI | |
| BF1807 | BF1807F | ATATCATATGCCGGAACCGCAAAAGTTTACC | NdeI |
| BF1807R | ATATCTCGAGCTTCGTTTTGTCGGGCGAAT | XhoI | |
| BF1811 | BF1811F | CGCGGGATCCGATGAGAAATCTTTTTAAAAT | BamHI |
| BF1811R | CGCGCTCGAGATTCAACGTAATTTCATCTA | XhoI | |
| BF4033 | BF4033F | ATATCATATGCAGCAAGGAGTGACACAATGTG | NdeI |
| BF4033R | ATATCTCGAGCAACGAATTCAGTATTTTGTTCGC | XhoI | |
| C. perfringens ATCC 13124 | |||
| CPF1103 | CPF1103F | CGCCATATGAGAGTTAAATTAGTTGGAT | NdeI |
| CPF1103R | CCCTCGAGCCAACTTAAAATATTTTGTGTT | XhoI | |
| CPF1238 | CPF1238F | CGCGCATATGCATTTAATACCAAGACCAA | NdeI |
| CPF1238R | CCGCTCGAGACCCCTTAAATATGAGCACA | XhoI | |
| CPF0184 | CPF0184F | CGGCTAGCGAAAACTTTTATGAAATATATCCAA | NheI |
| CPF0184R | CGCGCTCGAGATTTAGTATTCTATGATTTAT | XhoI | |
| CPF1487 | CPF1487F | CTAGGATCCGAATGGCAGCAAAGAAACAAAG | BamHI |
| CPF1487R | CCGCTCGAGAGTGTTTGGTAAATTACCCTC | XhoI | |
| CPF1473 | CPF1473F | CGCCATATGAAAAAAGATACTACTC | NdeI |
| CPF1473R | CCGCTCGAGCAAGGTTTCCATAGTTAAT | XhoI | |
| B. bifidum JCM 1254 | |||
| BbhI | BbhI F | AGTCAAGCTTAGCGATGACAATCTTGCACT | HindIII |
| BbhI R | ATATCTCGAGCTTGGCGACCTCGTCAGGCG | XhoI | |
| BbhII | BbhII F | ATATGTCGACGCGGCAGCGGCGGAATCATC | NheI |
| BbhII R | ATATCTCGAGACCGGTCTCGGCGACGACAT | XhoI | |
| BbhIII | BbhIII F | CGCGCATATGGTGCAGTATCGATACTGTTGT | NdeI |
| BbhIII R | ATATCTCGAGTGAACGCAGGGCGTCGGCA | XhoI |
Restriction enzyme sites are underlined.
The genomic DNAs of B. fragilis ATCC 25285, C. perfringens ATCC 13124, and B. bifidum JCM 1254 were extracted and used as templates for PCR. The PCR conditions were the following: a hot start at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 55°C to 65°C for 30 s, 72°C for 1 kb/min, and a final step at 72°C for 10 min. The PCR products were purified, ligated into pET-21b vector, and transformed into E. coli BL21(DE3). The proper transformants were grown at 37°C in LB medium containing ampicillin (50 μg/ml). The recombinant enzymes were induced by addition of isopropyl-1-thio-β-d-galactoside when the cell density reached 0.6 to 0.8 at 600 nm. After continuous cultivation for 3 to 5 h, the cells were harvested and disrupted by ultrasonic treatment. The lysates were centrifuged, and the enzymes were purified from the suspension through Ni2+ chelation chromatography.
Transglycosylation activities of the purified recombinant enzymes were detected by incubating the enzymes with 20 mM pNP-β-GalNAc or pNP-β-GlcNAc as donor and 200 mM lactose as acceptor. The reactions were stopped by heating at 100°C for 10 min and detected by thin-layer chromatography (TLC). The recombinant β-N-acetylhexosaminidase obtained from B. bifidum JCM 1254, named BbhI, is the desired enzyme that displays excellent properties for transglycosylation.
Enzyme and protein assays.
The activity of β-N-acetylhexosaminidase was measured by adding 20 μl of 1.5 and 0.025 mg/ml enzyme solutions to 60 μl of 2 mM pNP-β-GalNAc and pNP-β-GlcNAc, respectively. The reaction was performed at 37°C for 10 min and stopped by adding 120 μl of 1 M Na2CO3 solution. The amount of p-nitrophenol released was measured at 405 nm. One unit of enzyme activity is defined as the amount of enzyme that produced 1 μmol p-nitrophenol per min under the assay conditions. Assays for the other nitrophenyl glycosides were performed under the same conditions. The protein content of the sample was measured by the Bradford assay protocol using bovine serum albumin (BSA) as a standard (45).
Determination of the stereoselectivity of BbhI.
BbhI (0.01 U/ml) was incubated with pNP-β-GlcNAc (1 mM) dissolved in D2O at room temperature, followed by detection of the reaction mixture to record the 1H nuclear magnetic resonance (NMR) spectra by using Agilent DD2-600 spectrometer at 600 MHz at 2, 10, 30, 60, 120, 180, and 240 min. Data were processed using VnmrJ 4.0 and MestReNova software.
Synthesis of HexNAc-Lac by BbhI.
The reactions for the synthesis of HexNAc-Lac were performed by incubating BbhI with pNP-β-HexNAc dissolved in dimethyl sulfoxide (DMSO) as the glycosyl donor and lactose as the acceptor. The enzyme was used at concentrations of 2 mg/ml (0.06 U/ml) for pNP-β-GalNAc and 0.02 mg/ml (0.12 U/ml) for pNP-β-GlcNAc. The effects of lactose concentration (50, 100, 200, 300, 400, and 500 mM) on HexNAc-Lac synthesis were measured using 25 mM glycosyl donor at pH 7.0 and 37°C for 7.5 h (pNP-β-GalNAc) or 2.5 h (pNP-β-GlcNAc). The effects of the concentration of glycosyl donor (5, 10, 15, 20, 25, and 30 mM) were measured using 400 mM lactose at pH 7.0 and 37°C for 7.5 h (pNP-β-GalNAc) or 2.5 h (pNP-β-GlcNAc). The effects of pH were examined at 19 different pH values (acetate buffer at 3.6, 4.0, 4.6, 5.0, 5.4, and 5.8; phosphate buffer at 5.8, 6.0, 6.5, 7.0, 7.5, and 8.0; Tris-HCl buffer at 8.0, 8.5, and 9.0; glycine buffer at 9.0, 9.5, 10.0, and 10.7; Na2HPO4-NaOH buffer at 10.7, 11.0, 11.5, and 12.0) at 37°C by using 400 mM lactose in the presence of 10 mM pNP-β-GalNAc for 7.5 h or 20 mM pNP-β-GlcNAc for 2.5 h. The effects of temperature and reaction time were investigated using 400 mM lactose and 10 mM pNP-β-GalNAc or 20 mM pNP-β-GlcNAc at pH 5.8 at six different temperatures (35°C, 40°C, 45°C, 50°C, 55°C, and 60°C), followed by interval sampling within 14 h. All reactions were stopped by heating at 100°C for 10 min, and the remaining donor and resulting products were detected by HPLC. The yield of the product was defined as the ratio of the concentration of the synthesized glycoside product (millimolar) to the initial concentration of donor (millimolar). The ratio of the transglycosylation and hydrolysis activity (RT/H) was calculated by dividing the concentration (millimolar) of the synthesized product by the concentration (millimolar) of the hydrolyzed donor.
Isolation of HexNAc-Lac synthesized by BbhI.
The reaction mixture was separated by a Bio-Gel P2 (Bio-Rad, CA, USA) column (1.5 by 100 cm) with distilled water as an eluent. The eluted fractions were collected and subjected to sugar determination by TLC. The corresponding eluents were pooled and lyophilized to dry powder.
HPLC and TLC analyses.
HPLC was performed by an Agilent 1200 series equipped with an Acchrom XAmide analysis column (4.6 by 250 mm) at 30°C. Samples were eluted with 72% (vol/vol) acetonitrile at a flow rate of 1.0 ml/min and detected through a UV detector (G1314B) at 210 nm. TLC was performed by loading samples on silica gel 60 F254 plates (Merck, Germany). The developing solvent was a mixture of n-butanol–ethanol–water (5:3:2, vol/vol/vol). Sugars on the TLC plate were detected by spraying with diphenylamine-aniline-phosphoric acid reagent and heating at 86°C for 10 min.
MS and NMR analyses.
Mass spectra were recorded on a Shimadzu liquid chromatography-mass spectrometry ion trap time of flight (LCMS-IT-TOF) instrument (Kyoto, Japan) equipped with an electrospray ionization (ESI) source in positive/negative ion mode at a resolution of 10,000 full width at half-maximum. 1H and 13C NMR spectra were recorded on an Agilent DD2-600 spectrometer at room temperature at 600 MHz for 1H and at 150 MHz for 13C. Chemical shifts were expressed in parts per million (ppm) downfield from the internal tetramethylsilane of D2O. Chemical shifts and coupling constants were calculated from a first-order analysis of the spectra. Assignments were fully supported by homo- and heteronuclear correlation two-dimensional (2D) techniques, including correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), and heteronuclear multiple band correlation (HMBC) experiments, by following standard Agilent pulse programs.
Molecular modeling and docking.
Homology modeling of BbhI was performed using PHYRE2, and the structure of BbLNBase obtained from B. bifidum JCM 1254 (PDB entry 4H04) served as the template. BbhI shares 33% sequence identities with the model BbLNBase. The 3D structures of protein and lactose were first prepared using SYBYL-X 1.1. The lactose was subsequently docked into the active sites of BbhI using Genetic Optimization of Ligand Docking (GOLD) 3.0.1 under standard settings. The top-ranked model from the GOLD analysis was apparently catalytically plausible. The docking results were visualized with PyMol 1.3.
RESULTS
Screening of β-N-acetylhexosaminidases for transglycosylation.
Genes of 13 β-N-acetylhexosaminidases belonging to GH20, GH84, and GH123 were cloned from B. fragilis ATCC 25285, C. perfringens ATCC 13124, and B. bifidum JCM 1254 and then successfully expressed in E. coli. The recombinant enzymes were purified (see Fig. S1 in the supplemental material) and incubated with pNP-β-HexNAc as a glycosyl donor and lactose as an acceptor to detect transglycosylation activity. The results showed that 11 enzymes exhibited transglycosylation activities, but not BF1807, obtained from B. fragilis ATCC 25285, and CPF1103, obtained from C. perfringens ATCC 13124 (Table 2). Five GH20 enzymes, namely, BF0669, BF1811, CPF1238, BbhI, and BbhII, transferred both GalNAc and GlcNAc residues to lactose, whereas two GH20 enzymes (BF0953 and BbhIII), two GH84 enzymes (CPF0184 and CPF1487), and two GH123 enzymes (BF4033 and CPF1473) only transferred one kind of residue from pNP-β-HexNAc (see Fig. S1). Interestingly, BbhI from B. bifidum JCM 1254, an enzyme in the GH20 family, exhibited strict regioselectivity toward lactose without isomer production from both glycosyl donors. BbhI also exhibited the highest transglycosylation activity regardless of the kind of glycosyl donor. Hence, this enzyme was chosen for further studies.
TABLE 2.
Screening of transglycosylation ability of various β-N-acetylhexosaminidases
| Enzyme source | GenBank accession no. | Enzyme | GH family | Molecular mass (kDa) | Transglycosylation activitya |
|
|---|---|---|---|---|---|---|
| pNP-β-GalNAc | pNP-β-GlcNAc | |||||
| B. fragilis NCTC 9343 | CAH06414.1 | BF0669 | 20 | 71 | +b | +b |
| CAH06695.1 | BF0953 | 20 | 91 | − | +c | |
| CAH07506.1 | BF1807 | 20 | 74 | − | − | |
| CAH07510.1 | BF1811 | 20 | 86 | +b | +b | |
| CAH09708.1 | BF4033 | 123 | 66 | +b | − | |
| C. perfringens ATCC 13124 | ABG83419.1 | CPF1103 | 20 | 75 | − | − |
| ABG83624.1 | CPF1238 | 20 | 71 | +c | +b | |
| ABG83307.1 | CPF0184 | 84 | 179 | − | +c | |
| ABG84775.1 | CPF1487 | 84 | 126 | − | +c | |
| ABG82546.1 | CPF1473 | 123 | 68 | +c | − | |
| B. bifidum JCM 1254 | BAI94822.1 | BbhI | 20 | 170 | +c | +c |
| BAI94823.1 | BbhII | 20 | 107 | +b | +b | |
| BAI94829.1 | BbhIII | 20 | 86 | − | +b | |
The transglycosylation reaction using pNP-β-GalNAc or pNP-β-GlcNAc as glycosyl donors and lactose as acceptor. Symbols: +, transglycosylation ability could be detected; −, no transglycosylation ability could be detected.
Multiple products.
Single product.
Figure S1 shows that the recombinant BbhI reached an electrophoresis purity at a subunit molecular mass of ∼170 kDa, consistent with the molecular mass (170.1 kDa) deduced from its nucleotide sequence. The specific activities for pNP-β-GalNAc and pNP-β-GlcNAc were 0.03 and 6 U/mg, respectively. BbhI was highly active at pH 5.0 to 6.0 and stable at pH 4.0 to 11.0. The enzyme showed an optimal temperature at approximately 45°C to 55°C, and it was stable below 55°C (see Fig. S2 in the supplemental material). With respect to substrate specificity for hydrolysis, BbhI was active when pNP-β-GalNAc or pNP-β-GlcNAc was used. The result showed that no activity existed toward the other substrates with α-linkages or without GalNAc or GlcNAc in the glycon moieties, including pNP-α- or β-d-galactopyranoside, pNP-α- or β-d-glucopyranoside, pNP-α-l-fucopyranoside, pNP-β-d-fucopyranoside, pNP-α- or β-d-mannopyranoside, pNP-2-acetylamino-2-deoxy-α-d-galactopyranoside, and pNP-2-acetylamino-2-deoxy-α-d-glucopyranoside.
Determination of the stereoselectivity of BbhI.
The enzyme BbhI was incubated with pNP-β-GlcNAc, and variation in anomeric configurations of the released GlcNAc was detected by real-time recording of 1H NMR spectra of the reaction mixture. As shown in Fig. 1, the signal peaks at 5.15, 5.02, and 4.53 ppm represented the anomeric proton signals of pNP-β-GlcNAc, α-GlcNAc, and β-GlcNAc, respectively. The initial hydrolysis product obtained within 30 min was obviously β-GlcNAc, confirming the β-stereoselective property of the enzyme. When the reaction time was prolonged, β-GlcNAc underwent mutarotation to yield α-GlcNAc, and the conversion finally reached equilibrium after 4 h. These results clearly indicated that BbhI operated via a retaining mechanism commonly employed in classical GH20 enzymes.
FIG 1.
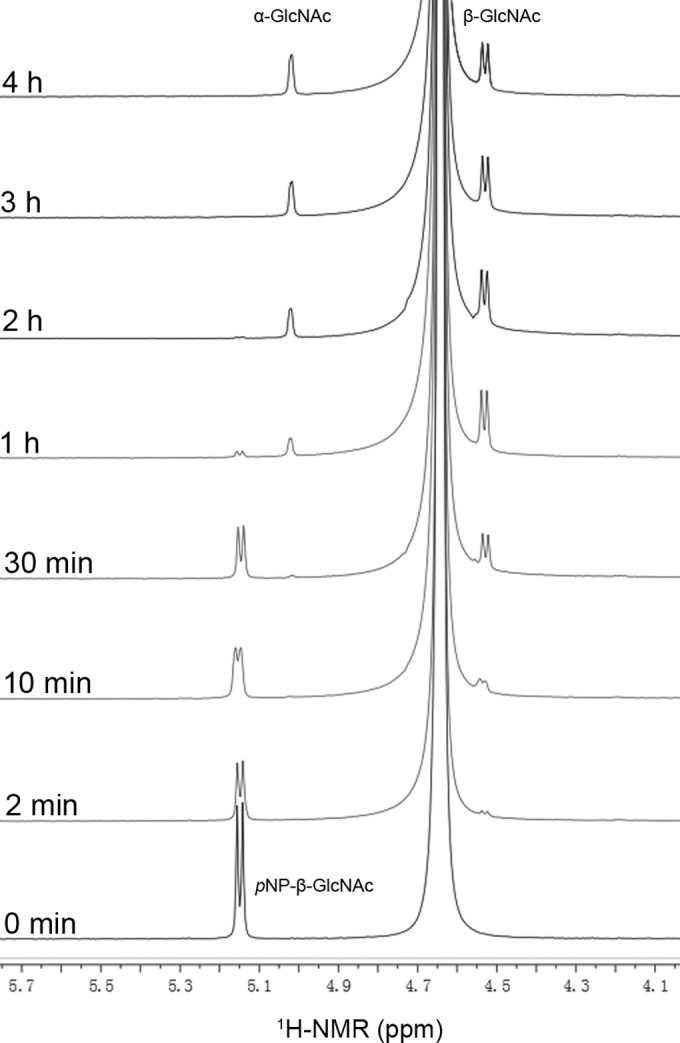
Time course of the enzymatic cleavage of pNP-β-GlcNAc.
HexNAc-Lac synthesis by BbhI.
Transglycosylation reactions were performed by incubating BbhI with pNP-β-HexNAc and lactose under various conditions. Figure 2A and C shows the effects of acceptor concentration on product yields. When either pNP-β-GalNAc or pNP-β-GlcNAc was used as the glycosyl donor and the lactose concentrations were increased from 50 mM to 400 mM, the product yields increased and then tended to stabilize. The maximum product yields at 38.1% and 28.8% for pNP-β-GalNAc and pNP-β-GlcNAc, respectively, were obtained at 400 mM acceptor. Thus, subsequent reactions were performed at 400 mM lactose. Figure 2B and D shows the effects of donor concentration on product yields. When pNP-β-GalNAc was used as the donor, the yield was slightly enhanced when the donor concentration was increased from 5 mM to 10 mM. The product yield was 43.9% at 5 mM donor and reached a maximum yield of 45% at 10 mM donor. Continuous increase in donor concentration from 10 mM to 30 mM reduced product yields. When the concentration of pNP-β-GlcNAc as donor increased from 5 mM to 20 mM, the product yields were remarkably improved. The maximum yield of 37.3% was obtained at 20 mM donor, and then the product yields decreased. Thus, subsequent reactions were performed using 10 mM pNP-β-GalNAc or 20 mM pNP-β-GlcNAc as the glycosyl donor.
FIG 2.

Effects of substrate concentrations on the yields of GalNAc-Lac (A and B) and GlcNAc-Lac (C and D) synthesized by BbhI. The enzyme was used at concentrations of 2 mg/ml (0.06 U/ml) for pNP-β-GalNAc and 0.02 mg/ml (0.12 U/ml) for pNP-β-GlcNAc. The lactose concentrations (50 to 500 mM) were tested at pH 7.0 and 37°C in the presence of 25 mM pNP-β-GalNAc for 7.5 h (A) or 25 mM pNP-β-GlcNAc for 2.5 h (C). The donor concentrations were tested by incubation with 400 mM lactose at pH 7.0 and 37°C, in the presence of pNP-β-GalNAc (5 to 30 mM) for 7.5 h (B) or pNP-β-GlcNAc (5 to 30 mM) for 2.5 h (D). Data points represent the means ± standard deviations (SD) from three replicates.
The pH value also strongly affected product formation (Fig. 3). When pNP-β-GalNAc was used as donor, the product yields were greater than 40% over the pH range of 4.6 to 10.0, and the maximum yield of 51.3% was obtained at pH 5.8. As for reaction mixtures using pNP-β-GlcNAc as the donor, the pattern of pH effects was similar to that in pNP-β-GalNAc, and the product yields also reached a maximum of 38.3% at pH 5.8.
FIG 3.
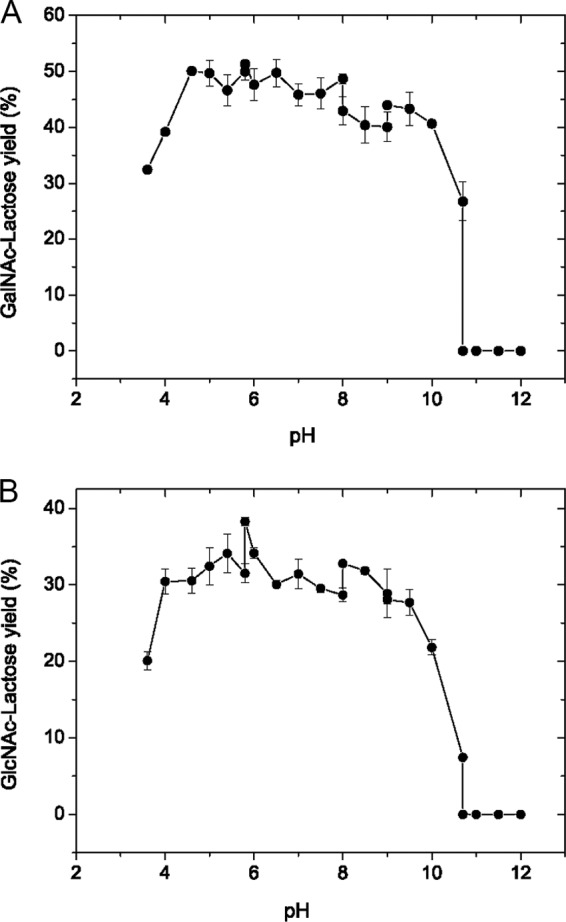
Effects of pH on the yields of GalNAc-Lac (A) and GlcNAc-Lac (B) synthesized by BbhI. The enzyme was used at concentrations of 2 mg/ml (0.06 U/ml) for pNP-β-GalNAc and 0.02 mg/ml (0.12 U/ml) for pNP-β-GlcNAc. Reactions were carried out at 37°C by incubating the enzyme with 400 mM lactose in the presence of 10 mM pNP-β-GalNAc for 7.5 h (A) or 20 mM pNP-β-GlcNAc for 2.5 h (B) in buffers from pH 3.6 to 12.0. Data points represent the means ± SD from three replicates.
The effects of temperature and reaction time were studied by measuring time curves at different temperatures. As shown in Fig. 4A, the reaction temperature markedly affected GalNAc-Lac formation. As the temperature increased from 35°C to 50°C, the time required for product yields to peak decreased. However, when the temperature was increased to 55°C and 60°C, the peak values appeared slowly. The maximal product yield reached 55.4% at 45°C and 4 h. In contrast, GlcNAc-Lac formed considerably faster. Figure 4B shows that the increase in temperature from 35°C to 55°C noticeably accelerated the reaction, making the peak values of the product appear quickly. The maximal product yield reached 44.9% at 55°C and 1.5 h. For all temperatures tested, the products accumulated first and then gradually decreased as a result of hydrolysis.
FIG 4.
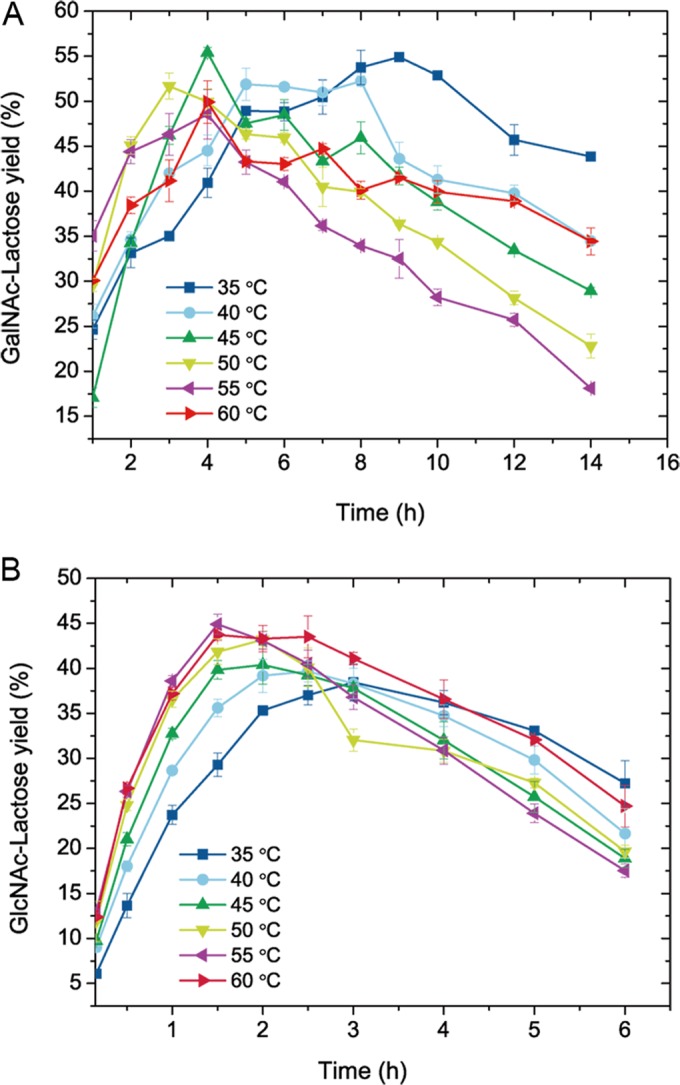
Effects of temperature and reaction time on the yields of GalNAc-Lac (A) and GlcNAc-Lac (B) synthesized by BbhI. The enzyme was used at concentrations of 2 mg/ml (0.06 U/ml) for pNP-β-GalNAc and 0.02 mg/ml (0.12 U/ml) for pNP-β-GlcNAc. Reactions were carried out at pH 5.8 by incubation of the enzyme with 400 mM lactose in the presence of 10 mM pNP-β-GalNAc (A) or 20 mM pNP-β-GlcNAc (B). Data points represent the means ± SD from three replicates.
Based on the comprehensive results of the above-described examination, the optimal conditions for GalNAc-Lac synthesis include an initial concentration of 10 mM pNP-β-GalNAc and 400 mM lactose in phosphate buffer at pH 5.8 and an incubation at 45°C for 4 h. The optimal conditions for GlcNAc-Lac synthesis were the following: an initial concentration of 20 mM pNP-β-GlcNAc and 400 mM lactose in phosphate buffer, pH 5.8, and an incubation at 55°C for 1.5 h. Under optimal conditions, 5.54 mM GalNAc-Lac and 8.98 mM GlcNAc-Lac were synthesized and 3.62 mM pNP-β-GalNAc and 6.14 mM pNP-β-GlcNAc were hydrolyzed, demonstrating that the RT/H were 1.53 for GalNAc-Lac and 1.46 for GlcNAc-Lac.
Isolation and identification of HexNAc-Lac.
The synthesis reactions were performed under optimal conditions. The resulting products were purified using Bio-Gel P2 column chromatography and analyzed by MS and NMR spectroscopy (see Fig. S3 to S14 in the supplemental material). From 10 ml of reaction mixture, 49 mg of GlcNAc-Lac and 30 mg of GalNAc-Lac were obtained.
The positive-ion ESI mass spectrum of GalNAc-Lac showed a peak of [M + H]+ ion at m/z 546.2, consistent with the molecular mass of GalNAc-Lac (545). The complete structural characterization was retrieved from the results of 2D NMR involving COSY, HSQC, and HMBC spectra, all of which were used to assign the chemical shifts and configurations of the sugar residues present in GalNAc-Lac (Table 3). The proton signal of the double peak at δ 4.45 ppm in the 1H NMR spectrum and the carbon signal at δ 103.2 ppm in the 13C spectrum were deduced to be H-1 and C-1 of the GalNAc residue, which was further confirmed by their correlation signals in the HSQC spectrum. The anomeric proton and carbon signals of the Gal residue were detected at δ 4.27 ppm in the 1H NMR spectrum and δ 102.8 ppm in the 13C spectrum. The protons at δ 4.50 and 5.05 ppm, coupled with the carbon at δ 95.6 and 91.7 ppm in the HSQC spectrum, respectively, were eventually identified as H-1 signals from the Glc residue as β and α configurations (α:β = 35:65). Cross peaks were found between C-1 (δ 103.2 ppm) of the GalNAc residue and H-3 (δ 3.55 ppm) of the Gal residue in the HMBC spectrum. This result, together with the coupling constant of H-1 of GalNAc (J = 8.58 Hz), confirmed the β1-3 linkage between GalNAc and Gal residues. Similarly, overlapped signals were found between C-1 (δ 102.8 ppm) of the Gal residue and H-4 (δ 3.45 ppm) of the Glc residue in the HMBC spectrum. This finding, together with the coupling constant of H-1 of Gal (J = 7.98 Hz), revealed the β1-4 linkage between Gal and Glc residues. All of these results demonstrated that the chemical structure of GalNAc-Lac was GalNAcβ1-3Galβ1-4Glc.
TABLE 3.
1H and 13C NMR data assignment of HexNAc-Lac
| Compound and residue | Chemical shifts (δ, ppm) |
|||||
|---|---|---|---|---|---|---|
| H-1, C-1 | H-2, C-2 | H-3, C-3 | H-4, C-4 | H-5, C-5 | H-6, C-6 | |
| GalNAc-Lac | ||||||
| β-GalNAc | 4.45 | 3.77 | 3.57 | 3.78 | 3.42 | 3.60 |
| 103.2 | 52.4 | 74.7 | 67.6 | 70.0 | 60.9 | |
| β-Gal | 4.27 | 3.43 | 3.55 | 3.96 | 3.50 | 3.60 |
| 102.8 | 74.7 | 81.7 | 68.4 | 74.9 | 60.9 | |
| β-Glc | 4.50 | 3.09 | 3.46 | 3.42 | 3.42 | 3.60,a 3.78b |
| 95.6 | 73.6 | 74.2 | 78.1 | 70.0 | 60.0 | |
| β-Glc | 5.05 | 3.39 | 3.64 | 3.45 | 3.76 | 3.68,a 3.79b |
| 91.7 | 71.0 | 71.3 | 78.1 | 70.7 | 60.0 | |
| GlcNAc-Lac | ||||||
| β-GlcNAc | 4.48 | 3.59 | 3.40 | 3.29 | 3.32 | 3.78,a 3.63b |
| 102.8 | 55.5 | 73.5 | 75.5 | 69.6 | 60.0 | |
| β-Gal | 4.24 | 3.44 | 3.56 | 3.95 | 3.40 | 3.69 |
| 102.8 | 74.6 | 81.9 | 68.2 | 73.5 | 60.0 | |
| β-Glc | 4.46 | 3.11 | 3.48 | 3.46 | 3.66 | 3.62,a 3.56b |
| 95.6 | 73.7 | 74.2 | 78.2 | 71.3 | 60.8 | |
| β-Glc | 5.02 | 3.41 | 3.55 | 3.48 | 3.78 | 3.72,a 3.59b |
| 91.7 | 71.0 | 74.7 | 78.2 | 70.0 | 60.3 | |
Chemical shift for H-6a.
Chemical shift for H-6b.
The positive-ion ESI mass spectrum of GlcNAc-Lac showed a peak of [M + H]+ ion at m/z 546.2, consistent with the molecular mass of GlcNAc-Lac (545). Anomeric proton signals of GlcNAc and Gal residues appeared at δ 4.48 and 4.24 ppm in the 1H NMR spectrum. They were coupled with anomeric carbon signals overlapped at δ 102.8 ppm in the HSQC spectrum. The protons at δ 4.46 and 5.02 ppm correlated with the carbon at δ 95.6 and 91.7 ppm, respectively, in the HSQC spectrum and were eventually identified as H-1 signals from the Glc residue as β and α configurations (α:β = 35:65). The β1-3 linkage between GlcNAc and Gal residues was determined by the existence of cross peaks between C-1 (δ 102.8 ppm) of the GlcNAc residue and H-3 (δ 3.56 ppm) of the Gal residue in the HMBC spectrum, together with a large coupling constant of H-1 of GlcNAc (J = 8.87 Hz). The β1-4 linkage of Gal and Glc residues was confirmed based on the overlapped signals between C-1 (δ 102.8 ppm) of the Gal residue and H-4 (δ 3.48 ppm) of the Glc residue in the HMBC spectrum, together with the coupling constant of H-1 of Gal (J = 7.8 Hz). Thus, the GlcNAc-Lac structure was completely characterized as GlcNAcβ1-3Galβ1-4Glc.
Sequence analysis and homology modeling of BbhI.
Glycosidases generally produce multiple isomers during transglycosylation. To probe the mechanism of strict regioselectivity of β1-3 linkage in β-N-acetylhexosaminyl lactose synthesis, we subjected BbhI to sequence alignment, molecular modeling, and docking analysis.
The results of multiple-sequence alignments revealed that the key residues for enzyme activity were highly conserved among various β-N-acetylhexosaminidases (Fig. 5A). GH20 enzymes contain a pair of highly conserved catalytic residues, namely, Asp-Glu; Glu acts as an acid/base residue, whereas Asp serves as a nucleophile assistant residue that assists the substrate 2-acetamido group to act as a nucleophile (15). In the case of BbhI, the residues Asp714 and Glu715 were predicted as nucleophile assistant and catalytic acid/base amino acids, whereas the residues His647, Trp743, Trp769, and Trp850 can be considered essential for enzyme activity based on comparison with the corresponding residues of other reported β-N-acetylhexosaminidases (18, 46). The putative 3D model of BbhI suggests that these predicted residues are responsible for enzyme activity (Fig. 5B to E). Clearly, BbhI shared the common characteristic of all known structures of GH20 enzymes, that is, the (β/α)8-barrel architecture of the catalytic domain (see Fig. S15 in the supplemental material). The predicted catalytic Asp714 and Glu715, located at the catalytic cavity, as well as four aromatic residues (His647, Trp743, Trp769, and Trp850), apparently formed a hydrophobic pocket used to position lactose in the active site (Fig. 5C and E). The docking result (Fig. 5D) further showed that Glu715 may form an H-bond with 3-OH of the Gal residue of lactose. Tyr795 and Asp852 may interact with 6-OHs of Gal and Glc residues, respectively, whereas Asp714 is likely to participate in H-bonding with 4-OH of Gal and 3-OH of Glc residues (Fig. 5D).
FIG 5.

Multiple-sequence alignments and docking models of BbhI with lactose. (A) Partial result of multiple alignments of GH20 β-N-acetylhexosaminidases from B. bifidum JCM 1254 (BbhI; GenBank accession no. BAI94822.1), B. bifidum JCM 1254 (BbLNBase; GenBank accession no. ABZ78855.1), Arthrobacter aurescens TC1 (AAur; GenBank accession no. ABM09106.1), and Paenibacillus sp. strain TS12 (Hex1; GenBank accession no. BAI63641.1) using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/). The possible nucleophile assistant residue (Asp714) and acid/base residue (Glu715) are boxed. The four aromatic residues (H647, W743, W769, and W850) that seemed to form a hydrophobic pocket are indicated by arrows. (B) Putative 3D structure of BbhI (gray) docking with lactose (green). (C) Enlarged image of the dotted-line labeled domain in panel B that contains active cavity. (D) BbhI-lactose interaction. Catalytic residues are shown in pink, while other residues that also form an H-bond with lactose are shown in yellow. The interactions between residues and lactose are indicated by blue dotted lines. (E) BbhI-lactose interaction. Aromatic residues (yellow) formed a hydrophobic pocket around lactose (green).
DISCUSSION
Oligosaccharide synthesis has attracted attention worldwide either for theoretical studies of its functions in vivo or for its practical applications in food and medical industries (47–49). β-N-Acetylhexosaminidases have shown great potential in enzymatic synthesis of functional glycans. In this work, we describe a β-N-acetylhexosaminidase, BbhI, obtained from B. bifidum JCM 1254 that exhibits strong transglycosylation activity and strict regioselectivity for β-N-acetylhexosaminyl lactose synthesis. This enzyme is a powerful synthetic tool that can transfer both GalNAc and GlcNAc residues through β1-3 linkage to the Gal residue of lactose to obtain biologically important GalNAcβ1-3Lac and GlcNAcβ1-3Lac at relatively high yields.
In glycosidase-catalyzed synthesis, transglycosylation and hydrolysis by the enzyme both affect the yield of glycoside product. Excessive acceptor concentration can increase formation of glycoside product. Moreover, addition of organic solvents can increase the solubility of a substrate and reduce water activity, promoting transglycosylation over hydrolysis. Other conditions, such as reaction time, temperature, and pH, can also affect product yield (5). In this work, the final concentration of 20% (vol/vol) DMSO was used for the synthesis reaction by BbhI. The effects of substrate concentration, pH, temperature, and reaction time on the yield of the transglycosylation product were studied, and the optimal conditions for the maximum yields of GalNAc-Lac and GlcNAc-Lac were obtained. The RT/H of BbhI was 1.53 for GalNAc-Lac or 1.46 for GlcNAc-Lac under optimal conditions, consistent with the reported range of glycosidase catalysis at 0.2 to 2.15 (50–52). Site-directed mutation of a thermostable β-glucosidase obtained from Thermotoga neapolitana increased the transferase/hydrolase ratio by 7-fold (50). Thus, the RT/H of BbhI can be improved in the future through molecular evolution.
Synthesis of GlcNAcβ1-3Galβ1-4Glc and its derivatives by glycosidase has been reported, but the reaction yields are rather low (less than 10%). The β-N-acetylhexosaminidase from Aspergillus oryzae formed GlcNAcβ1-3Galβ1-4Glc at 0.36% yield through reverse hydrolysis using GlcNAc and lactose at 45°C for 4 days (44). The β-N-acetylhexosaminidase from N. orientalis transfers GlcNAc from GlcNAc2 to methyl-β-lactoside, pNP-β-Lac, and pNP-β-LacNAc to form GlcNAcβ1-3Galβ1-4Glc-OMe, GlcNAcβ1-3Galβ1-4Glc-β-pNP, and GlcNAcβ1-3Galβ1-4GlcNAc-β-pNP at 3.4%, 1.9%, and 1.5% yields at 40°C for 20, 20, and 12 h, respectively (33, 35). Two β-N-acetylhexosaminidases, namely, HEX1 and HEX2, transfer GlcNAc from GlcNAc2 to lactose to form GlcNAcβ1-3Galβ1-4Glc at 2% and 8% yields, respectively, at 25°C for 2 h (36). In this study, BbhI transferred GlcNAc from pNP-β-GlcNAc to lactose to form GlcNAcβ1-3Galβ1-4Glc at a yield of 44.9% at 55°C for 1.5 h. This enzymatic strategy showed higher product yield and shorter reaction time than those of the enzymatic methods discussed above, providing an efficient approach for enzymatic synthesis of GlcNAcβ1-3Galβ1-4Glc.
To the best of our knowledge, GalNAcβ1-3Galβ1-4Glc synthesis by glycosidase has not yet been reported. Only one β-1-3-N-acetylgalactosaminyltransferase (EC 2.4.1.79) from human plasma is available for the formation of GalNAcβ1-3Galβ1-4Glc at 0.4 μM yield by using 2.5 μM UDP-GalNAc as donor and 125 μM lactose as acceptor at 37°C for 3 days. This reaction required high-cost UDP-GalNAc (100 mg for $2,425; Sigma) as donor (53). BbhI can synthesize GalNAcβ1-3Galβ1-4Glc at a yield of 55.4% at 45°C for 4 h using pNP-β-GalNAc (250 mg for $443; Sigma) as donor, which incurs a lower cost than using UDP-GalNAc. BbhI is the first glycosidase that can transfer GalNAc residue to Gal in lactose through the β1-3 linkage, providing a novel and efficient method to obtain GalNAcβ1-3Galβ1-4Glc.
Two fungal β-N-acetylhexosaminidases can catalyze transfer of both GalNAc and GlcNAc residues for synthesis. With the use of pNP-β-GalNAc and pNP-β-GlcNAc as donors, the β-N-acetylhexosaminidase from Trichoderma harzianum can transfer both GalNAc and GlcNAc residues to UDP-GlcNAc to form GalNAcβ1-4GlcNAcαUDP and GlcNAcGlcNAcαUDP (regioselectivity not identified) at 22% and 1.9% yields at 30°C for 8 and 14 h, respectively (25). In addition, when pNP-β-GalNAc and pNP-β-GlcNAc were used as donors, the β-N-acetylhexosaminidase from A. oryzae can transfer both GalNAc and GlcNAc residues to GlcNAcβ1-4ManNAc to form GalNAcβ1-4GlcNAcβ1-4ManNAc and GlcNAcβ1-4GlcNAcβ1-4ManNAc at 41% and 36% yields at 37°C for 2 h and 50 min, respectively (24). In this work, BbhI transferred both GalNAc and GlcNAc residues from pNP-β-GalNAc and pNP-β-GlcNAc to lactose to form GalNAcβ1-3Galβ1-4Glc and GlcNAcβ1-3Galβ1-4Glc with 55.4% and 44.9% yields at 45°C for 4 h and at 55°C for 1.5 h, respectively. We used 10 ml of reaction mixture and obtained 49 mg of GlcNAc-Lac and 30 mg of GalNAc-Lac. Given that the preparation of the enzyme is easy and simple, this synthetic approach can be used for commercial applications given the availability of large quantities of donor substrates.
The stereoselectivity of glycosidases in transglycosylation are strict for either α or β configurations in the anomeric center, whereas their regioselectivity is relatively flexible, usually leading to regioisomers that are difficult to purify. The regioselectivity of glycosidases mainly depends on two main factors: the sources of the enzymes and the kinds of substrates (5, 54, 55). The β-N-acetylhexosaminidase from A. oryzae synthesizes GlcNAcβ1-3/1-6Galβ1-4Glc via reverse hydrolysis by using GlcNAc and lactose (44). Two β-N-acetylhexosaminidases from the soil-derived metagenomic library form GlcNAcβ1-3Galβ1-4Glc and GlcNAcβ1-4Galβ1-4Glc using GlcNAc2 as donor and lactose as acceptor (36). The β-N-acetylhexosaminidase from N. orientalis transfers GlcNAc from GlcNAc2 to methyl-β-lactoside to form GlcNAcβ1-3/1-6Galβ1-4Glc-OMe and Galβ1-4(GlcNAcβ1-6) Glc-OMe, as well as to pNP-β-LacNAc to form GlcNAcβ1-3/1-6Galβ1-4GlcNAc-β-pNP and Galβ1-4(GlcNAcβ1-6) GlcNAc-β-pNP (33, 35). This work found that BbhI exhibits strict regioselectivity of β1-3 linkage in transglycosylations toward lactose using pNP-β-GalNAc and pNP-β-GlcNAc as donors. The strict regioselectivity of BbhI for transglycosylation was speculated to be related to the characteristic 3D structures of the enzyme.
The strict regioselective synthesis by BbhI can possibly be explained by the results of multiple-sequence alignments and by the docking model. In the catalytic cavity of BbhI, the equatorial 3-OH of Gal in lactose appeared within the H-bonding distance from Glu715, indicating that this 3-OH group can be activated by Glu715 and is positioned appropriately for the subsequent nucleophilic attack on the anomeric carbon of HexNAc to achieve the regioselectivity of β1-3 linkage between HexNAc and Gal in lactose. Additionally, Asp714 is located within H-bond distances of the 4-OH of Gal and 3-OH of Glc, and two H-bonds (Tyr795 to 6-OH of Gal and Asp852 to 6-OH of Glc) seemed to exist, probably causing the 3-OH of Gal residue in lactose rather than other hydroxyls to be oriented toward the anomeric carbon of HexNAc. Moreover, lactose nestled in a hydrophobic pocket was formed by four aromatic residues (His647, Trp743, Trp769, and Trp850) and stabilized by π-π stacking interaction with His647; these phenomena could account for the regioselectivity of β1-3 linkage in β-N-acetylhexosaminyl lactose synthesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Major State Basic Research Development Program of China (973 Program) (no. 2012CB822102), National High Technology Research and Development Program of China (863 Program) (no. 2012AA021504), National Major Scientific and Technological Special Project for Significant New Drugs Development (no. 2012ZX09502001-005), the Key Grant Project of Chinese Ministry of Education (no. 313033), and the Science and Technology Development Project of Shandong Province (no. 2014GSF121006 and 2015GSF121004).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01325-16.
REFERENCES
- 1.Dube DH, Bertozzi CR. 2005. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discov 4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 2.Babu P. 2014. Glycans in regeneration. ACS Chem Biol 9:96–104. doi: 10.1021/cb400784j. [DOI] [PubMed] [Google Scholar]
- 3.Lloyd DH, Viac J, Werling D, Rème CA, Gatto H. 2007. Role of sugars in surface microbe-host interactions and immune reaction modulation. Vet Dermatol 18:197–204. doi: 10.1111/j.1365-3164.2007.00594.x. [DOI] [PubMed] [Google Scholar]
- 4.Trincone A, Giordano A. 2006. Glycosyl hydrolases and glycosyltransferases in the synthesis of oligosaccharides. Curr Org Chem 10:1163–1193. doi: 10.2174/138527206777698075. [DOI] [Google Scholar]
- 5.Zeuner B, Jers C, Mikkelsen JD, Meyer AS. 2014. Methods for improving enzymatic trans-glycosylation for synthesis of human milk oligosaccharide biomimetics. J Agric Food Chem 62:9615–9631. doi: 10.1021/jf502619p. [DOI] [PubMed] [Google Scholar]
- 6.Slámová K, Bojarová P, Petrásková L, Křen V. 2010. β-N-Acetylhexosaminidase: what's in a name…? Biotechnol Adv 28:682–693. doi: 10.1016/j.biotechadv.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Mahuran D. 1999. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta 1455:105–138. doi: 10.1016/S0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 8.Wendeler M, Sandhoff K. 2009. Hexosaminidase assays. Glycoconj J 26:945–952. doi: 10.1007/s10719-008-9137-5. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Q, Li H, Merdek K, Park JT. 2000. Molecular characterization of the β-N-acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J Bacteriol 182:4836–4840. doi: 10.1128/JB.182.17.4836-4840.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rast DM, Baumgartner D, Mayer C, Hollenstein GO. 2003. Cell wall-associated enzymes in fungi. Phytochemistry 64:339–366. doi: 10.1016/S0031-9422(03)00350-9. [DOI] [PubMed] [Google Scholar]
- 11.Hogenkamp DG, Arakane Y, Kramer KJ, Muthukrishnan S, Beeman RW. 2008. Characterization and expression of the β-N-acetylhexosaminidase gene family of Tribolium castaneum. Insect Biochem Mol Biol 38:478–489. doi: 10.1016/j.ibmb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Bode L. 2012. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22:1147–1162. doi: 10.1093/glycob/cws074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen HC, Chang CC, Mau WJ, Yen LS. 2002. Evaluation of N-acetylchitooligosaccharides as the main carbon sources for the growth of intestinal bacteria. FEMS Microbiol Lett 209:53–56. doi: 10.1111/j.1574-6968.2002.tb11108.x. [DOI] [PubMed] [Google Scholar]
- 14.Martens EC, Chiang HC, Gordon JI. 2008. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vocadlo DJ, Withers SG. 2005. Detailed comparative analysis of the catalytic mechanisms of β-N-acetylglucosaminidases from families 3 and 20 of glycoside hydrolases. Biochemistry 44:12809–12818. doi: 10.1021/bi051121k. [DOI] [PubMed] [Google Scholar]
- 16.Litzinger S, Fischer S, Polzer P, Diederichs K, Welte W, Mayer C. 2010. Structural and kinetic analysis of Bacillus subtilis N-acetylglucosaminidase reveals a unique Asp-His dyad mechanism. J Biol Chem 285:35675–35684. doi: 10.1074/jbc.M110.131037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miwa M, Horimoto T, Kiyohara M, Katayama T, Kitaoka M, Ashida H, Yamamoto K. 2010. Cooperation of β-galactosidase and β-N-acetylhexosaminidase from bifidobacteria in assimilation of human milk oligosaccharides with type 2 structure. Glycobiology 20:1402–1409. doi: 10.1093/glycob/cwq101. [DOI] [PubMed] [Google Scholar]
- 18.Ito T, Katayama T, Hattie M, Sakurama H, Wada J, Suzuki R, Ashida H, Wakagi T, Yamamoto K, Stubbs KA, Fushinobu S. 2013. Crystal structures of a glycoside hydrolase family 20 lacto-N-biosidase from Bifidobacterium bifidum. J Biol Chem 288:11795–11806. doi: 10.1074/jbc.M112.420109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. 2005. O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. J Biol Chem 280:25313–25322. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 20.He Y, Macauley MS, Stubbs KA, Vocadlo DJ, Davies GJ. 2010. Visualizing the reaction coordinate of an O-GlcNAc hydrolase. J Am Chem Soc 132:1807–1809. doi: 10.1021/ja9086769. [DOI] [PubMed] [Google Scholar]
- 21.Sumida T, Fujimoto K, Ito M. 2011. Molecular cloning and catalytic mechanism of a novel glycosphingolipid-degrading β-N-acetylgalactosaminidase from Paenibacillus sp. TS12. J Biol Chem 286:14065–14072. doi: 10.1074/jbc.M110.182592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uzawa H, Zeng X, Minoura N. 2003. Synthesis of 6′-sulfodisaccharides by b-N-acetylhexosaminidase-catalyzed transglycosylation. Chem Commun (Cambridge) 1:100–101. [DOI] [PubMed] [Google Scholar]
- 23.Singh S, Packwood J, Samuel CJ, Critchley P, Crout DH. 1995. Glycosidase-catalysed oligosaccharide synthesis: preparation of N-acetylchitooligosaccharides using the β-N-acetylhexosaminidase of Aspergillus oryzae. Carbohydr Res 279:293–305. doi: 10.1016/0008-6215(95)00302-9. [DOI] [PubMed] [Google Scholar]
- 24.Aboitiz N, Cañada FJ, Husakova L, Kuzma M, Kren V, Jiménez-Barbero J. 2004. Enzymatic synthesis of complex glycosaminotrioses and study of their molecular recognition by hevein domains. Org Biomol Chem 2:1987–1994. doi: 10.1039/B401037J. [DOI] [PubMed] [Google Scholar]
- 25.Nieder V, Kutzer M, Kren V, Gallego RG, Kamerling PJ, Elling L. 2004. Screening and characterization of β-N-acetylhexosaminidases for the synthesis of nucleotide-activated disaccharides. Enzyme Microb Technol 34:407–414. doi: 10.1016/j.enzmictec.2003.11.017. [DOI] [Google Scholar]
- 26.Ogata M, Zeng X, Usui T, Uzawa H. 2007. Substrate specificity of N-acetylhexosaminidase from Aspergillus oryzae to artificial glycosyl acceptors having various substituents at the reducing ends. Carbohydr Res 342:23–30. doi: 10.1016/j.carres.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Lakshmanan T, Loganathan D. 2005. Enzymatic synthesis of N-glycoprotein linkage region disaccharide mimetics using β-N-acetylhexosaminidases from Aspergillus oryzae and Vigna radiate. Tetrahedron Asymmetry 16:255–260. doi: 10.1016/j.tetasy.2004.11.016. [DOI] [Google Scholar]
- 28.Weignerová L, Vavrušková P, Pišvejcová A, Thiem J, Křen V. 2003. Fungal β-N-acetylhexosaminidases with high β-N-acetylgalactosaminidase activity and their use for synthesis of β-GalNAc-containing oligosaccharides. Carbohydr Res 338:1003–1008. doi: 10.1016/S0008-6215(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 29.Rauvolfová J, Kuzma M, Weignerová L, Fialová P, Přikrylová V, Pišvejcová A, Macková M, Křen V. 2004. β-N-Acetylhexosaminidase-catalysed synthesis of non-reducing oligosaccharides. J Mol Catal B Enzym 29:233–239. doi: 10.1016/j.molcatb.2003.10.008. [DOI] [Google Scholar]
- 30.Husáková L, Riva S, Casali M, Nicotra S, Kuzma M, Hunková Z, Křen V. 2001. Enzymatic glycosylation using 6-O-acylated sugar donors and acceptors: beta-N-acetylhexosaminidase-catalysed synthesis of 6-O, N, N′-triacetylchitobiose and 6′-O, N, N′-triacetylchitobiose. Carbohydr Res 331:143–148. doi: 10.1016/S0008-6215(01)00027-1. [DOI] [PubMed] [Google Scholar]
- 31.Slámová K, Gažák R, Bojarová P, Kulik N, Ettrich R, Pelantová H, Sedmera P, Křen V. 2010. 4-Deoxy-substrates for β-N-acetylhexosaminidases: how to make use of their loose specificity. Glycobiology 20:1002–1009. doi: 10.1093/glycob/cwq058. [DOI] [PubMed] [Google Scholar]
- 32.Bojarová P, Slámová K, Křenek K, Gažák R, Kulik N, Ettrich R, Pelantová H, Kuzma M, Riva S, Adámek D, Bezouška K, Křen V. 2011. Charged hexosaminides as new substrates for β-N-acetylhexosaminidase-catalyzed synthesis of immunomodulatory disaccharides. Adv Synth Catal 353:2409–2420. doi: 10.1002/adsc.201100371 (Author Correction, 356: 259, doi:.) [DOI] [Google Scholar]
- 33.Murata T, Tashiro A, Itoh T, Usui T. 1997. Enzymatic synthesis of 3′-O- and 6′-O-N-acetylglucosaminyl-N-acetyllactosaminide glycosides catalyzed by β-N-acetyl-D hexosaminidase from Nocardia orientalis. Biochim Biophys Acta 1335:326–334. doi: 10.1016/S0304-4165(96)00152-3. [DOI] [PubMed] [Google Scholar]
- 34.Kurakake M, Goto T, Ashiki K, Suenaga Y, Komaki T. 2003. Synthesis of new glycosides by transglycosylation of N-acetylhexosaminidase from Serratia marcescens YS-1. J Agric Food Chem 51:1701–1705. doi: 10.1021/jf020965x. [DOI] [PubMed] [Google Scholar]
- 35.Matahira Y, Tashiro A, Sato T, Kawagishi H, Usui T. 1995. Enzymic synthesis of lacto-N-triose II and its positional analogues. Glycoconj J 12:664–671. doi: 10.1007/BF00731263. [DOI] [PubMed] [Google Scholar]
- 36.Nyffenegger C, Nordvang RT, Zeuner B, Łężyk M, Difilippo E, Logtenberg MJ, Schols HA, Meyer AS, Mikkelsen JD. 2015. Backbone structures in human milk oligosaccharides: trans-glycosylation by metagenomic β-N-acetylhexosaminidases. Appl Microbiol Biotechnol 99:7997–8009. doi: 10.1007/s00253-015-6550-0. [DOI] [PubMed] [Google Scholar]
- 37.Marionneau S, Cailleau-Thomas A, Rocher J, Le Moullac-Vaidye B, Ruvoën N, Clément M, Le Pendu J. 2001. ABH and Lewis histo-blood group antigens, a model for the meaning of oligosaccharide diversity in the face of a changing world. Biochimie 83:565–573. doi: 10.1016/S0300-9084(01)01321-9. [DOI] [PubMed] [Google Scholar]
- 38.Fuster MM, Esko JD. 2005. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer 5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 39.Thomas R, Brooks T. 2006. Attachment of Yersinia pestis to human respiratory cell lines is inhibited by certain oligosaccharides. J Med Microbiol 55:309–315. doi: 10.1099/jmm.0.46102-0. [DOI] [PubMed] [Google Scholar]
- 40.Thomas R, Brooks T. 2004. Common oligosaccharide moieties inhibit the adherence of typical and atypical respiratory pathogens. J Med Microbiol 53:833–840. doi: 10.1099/jmm.0.45643-0. [DOI] [PubMed] [Google Scholar]
- 41.Danishefsky SJ, Allen JR. 2000. From the laboratory to the clinic: a retrospective on fully synthetic carbohydrate-based anticancer vaccines. Angew Chem Int Ed Engl 39:836–863. doi:. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Wen L, Ma X, Chen Z, Yu Y, Zhu J, Wang Y, Liu Z, Liu H, Wu D, Zhou D, Li Y. 2012. High expression of lactotriaosylceramide, a differentiation-associated glycosphingolipid, in the bone marrow of acute myeloid leukemia patients. Glycobiology 22:930–938. doi: 10.1093/glycob/cws061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeuner B, Nyffenegger C, Mikkelsen JD, Meyer AS. 2016. Thermostable β-galactosidases for the synthesis of human milk oligosaccharides. Nat Biotechnol 33:355–360. [DOI] [PubMed] [Google Scholar]
- 44.Matsuo I, Kim S, Yamamoto Y, Ajisaka K, Maruyama JI, Nakajima H, Kitamoto K. 2003. Cloning and overexpression of β-N-acetylglucosaminidase encoding gene nagA from Aspergillus oryzae and enzyme-catalyzed synthesis of human milk oligosaccharide. Biosci Biotechnol Biochem 67:646–650. doi: 10.1271/bbb.67.646. [DOI] [PubMed] [Google Scholar]
- 45.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 172:248–254. [DOI] [PubMed] [Google Scholar]
- 46.Sumida T, Ishii R, Yanagisawa T, Yokoyama S, Ito M. 2009. Molecular cloning and crystal structural analysis of a novel β-N-acetylhexosaminidase from Paenibacillus sp. TS12 capable of degrading glycosphingolipids. J Mol Biol 392:87–99. [DOI] [PubMed] [Google Scholar]
- 47.Sears P, Wong CH. 2001. Toward automated synthesis of oligosaccharides and glycoproteins. Science 291:2344–2350. doi: 10.1126/science.1058899. [DOI] [PubMed] [Google Scholar]
- 48.Seeberger PH, Werz DB. 2007. Synthesis and medical applications of oligosaccharides. Nature 446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 49.Driguez PA, Potier P, Trouilleux P. 2014. Synthetic oligosaccharides as active pharmaceutical ingredients: lessons learned from the full synthesis of one heparin derivative on a large scale. Nat Prod Rep 8:980–989. [DOI] [PubMed] [Google Scholar]
- 50.Lundemo P, Adlercreutz P, Karlsson EN. 2013. Improved transferase/hydrolase ratio through rational design of a family 1 β-glucosidase from Thermotoga neapolitana. Appl Environ Microbiol 79:3400–3405. doi: 10.1128/AEM.00359-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ha SJ, Seo DH, Jung JH, Cha J, Kim TJ, Kim YW, Park CS. 2009. Molecular cloning and functional expression of a new amylosucrase from Alteromonas macleodii. Biosci Biotechnol Biochem 73:1505–1512. doi: 10.1271/bbb.80891. [DOI] [PubMed] [Google Scholar]
- 52.Abdul Manas NH, Pachelles S, Mahadi NM, Illias RM. 2014. The characterisation of an alkali-stable maltogenic amylase from Bacillus lehensis G1 and improved malto-oligosaccharide production by hydrolysis suppression. PLoS One 9:e106481. doi: 10.1371/journal.pone.0106481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeya A, Hosomi O, Shimoda N, Yazawa S. 1992. Biosynthesis of the blood group P antigen-Like GalNAcβ1-3Galβ1-4GlcNAc/Glc structure: a novel N-acetylgalactosaminyltransferase in human blood plasma. J Biochem 112:389–395. [DOI] [PubMed] [Google Scholar]
- 54.Linde D, Macias I, Fernández-Arrojo L, Plou FJ, Jiménez A, Fernández-Lobato M. 2009. Molecular and biochemical characterization of a beta-fructofuranosidase from Xanthophyllomyces dendrorhous. Appl Environ Microbiol 75:1065–1073. doi: 10.1128/AEM.02061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rauvolfová J, Weignerová L, Kuzma M, Přikrylová V, Macková M, Pišvejcová A, Křen V. 2004. Enzymatic synthesis of N-acetylglucosaminobioses by reverse hydrolysis: characterisation and application of the library of fungal β-N-acetylhexosaminidases. J Mol Catal B Enzym 29:259–264. doi: 10.1016/j.molcatb.2004.02.007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.