

Abstract
Barrett’s esophagus is the major risk factor for esophageal adenocarcinoma. It has a low but non-neglectable risk, high surveillance costs and no reliable risk stratification markers. We sought to identify early biomarkers, predictive of Barrett’s malignant progression, using a meta-analysis approach on gene expression data. This in silico strategy was followed by experimental validation in a cohort of patients with extended follow up from the Instituto Português de Oncologia de Lisboa de Francisco Gentil EPE (Portugal). Bioinformatics and systems biology approaches singled out two candidate predictive markers for Barrett’s progression, CYR61 and TAZ. Although previously implicated in other malignancies and in epithelial-to-mesenchymal transition phenotypes, our experimental validation shows for the first time that CYR61 and TAZ have the potential to be predictive biomarkers for cancer progression. Experimental validation by reverse transcriptase quantitative PCR and immunohistochemistry confirmed the up-regulation of both genes in Barrett’s samples associated with high-grade dysplasia/adenocarcinoma. In our cohort CYR61 and TAZ up-regulation ranged from one to ten years prior to progression to adenocarcinoma in Barrett’s esophagus index samples. Finally, we found that CYR61 and TAZ over-expression is correlated with early focal signs of epithelial to mesenchymal transition. Our results highlight both CYR61 and TAZ genes as potential predictive biomarkers for stratification of the risk for development of adenocarcinoma and suggest a potential mechanistic route for Barrett’s esophagus neoplastic progression.
Introduction
Barrett’s esophagus (BE) is a premalignant metaplastic condition originated by the replacement of the normal squamous epithelium (NE) of the esophagus with a specialized columnar epithelial type that displays mixed gastric and intestinal characteristics [1]. BE is the major risk factor for the development of esophageal adenocarcinoma (EA) [2] and may progress to EA, through a low-grade to high-grade dysplasia (HGD) sequence. EA is the cancer with the fastest rising incidence in high-income countries [3] and has poor prognosis, with a high-related mortality and morbidity. Based on an estimated annual cancer risk of 0.5%, international guidelines universally recommend periodic endoscopic surveillance with a systematized biopsy protocol [4]. However, data reviewed on recent international guidelines estimates that BE risk of progression is very low (0.12%-0.33% patients/year [5, 6]. This fueled a running controversy on the costs/benefits of routine surveillance [7]. Apart from this debate, biopsy-based identification and grading of dysplasia in BE specimens is still the gold standard method to identify BE patients at risk of neoplastic progression [4], despite all the problems associated with such practice: costly, invasive, subjective dysplasia grading, biopsy sampling errors, unnecessary biopsying of low risk BE patients. Thus, a major current need in BE clinical management is of better methods and predictive biomarkers to stratify patients with an increased risk of disease progression [8], ideally early and/or biopsy-independent.
Recent high-throughput molecular studies have been instrumental for the enhanced understanding of many molecular events driving the BE "metaplasia-dysplasia-carcinoma" sequence [9, 10]. Despite all the resulting knowledge prompting the evaluation of >200 novel candidate biomarkers as predictors of progression (reviewed in [11]), none has yet reached routine clinical practice [12]. Molecular biomarkers of BE that predict progression to malignancy are still needed because their usage, alone or in combination with other biomarkers can facilitate more cost-effective surveillance. However due to the low progression rate of non-dysplastic BE few patients are available per discovery study. High-throughput molecular studies using robust sample sizes are scarce and thus published biomarker studies typically include very small patient cohorts. To maximize the discovery of new progression biomarkers, publicly available data needs to be used and combined into larger meta-cohorts. In particular the mining and re-analysis of existent gene expression data can be of great value, even if merging of distinct datasets may produce noisy predictions because such predictions can then be validated in patient cohorts.
In the present study, we set to define early molecular biomarkers predictive of BE progression to malignancy, through the usage of an innovative bioinformatics framework applied to publicly available global transcriptome data associated with BE to EA progression. In-silico generated prediction were validated in a cohort of patients under surveillance for more than ten years. We shown that CYR61 and TAZ are up-regulated in BE index biopsies (negative for dysplasia) from patients that progress to cancer (P-BE), years before the development of EA as compared to index biopsies from BE patients that did not progress (non P-BE). To our knowledge, this is the first study to show that molecular changes associated with features of the epithelial to mesenchymal transition (EMT) invasive phenotype, usually detectable in late EA progression, can occur remarkably early in at risk BE mucosa. These changes are observable also at the protein level and show promise of clinical utility.
Materials and Methods
Public data collection, pre-processing and graphical display
We mined Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo) [13, 14] or directly asked the authors for public microarray datasets on BE transcriptomes according to the criteria: 1) existence of clinical information on EA presence/absence at BE sample collection and 2) microarray experiments performed in the Affymetrix® Human Genome U133A microarray platform (HGU133a). Three datasets on HGU133a were retrieved: Kimchi et al. [15], Stairs et al. [16] and Watts et al. [17]. Kimchi et al. [15] study contained 8 BE samples adjacent to EA and thus these were classified as progressed BE (P-BE) plus 8 paired EA samples. Both Stairs et al. [16] and Watts et al. [17] series contained BE samples (n = 7 and n = 18, respectively) that were negative for dysplasia/EA at the time of collection and thus were classified as non-progressed (nonP-BE). In addition, the Watts et al. [17] dataset also contained EA samples but from distinct individuals of the nonP-BE samples.
Data analysis was performed with R Statistical Computing software [14] complemented with Bioconductor [18] packages. Heatmaps and Venn-diagrams were plotted using gplots (http://CRAN.R-project.org/package=gplots) and VennDiagram (http://CRAN.R-project.org/ package=VennDiagram) packages, respectively. Affy [19] and frma [20] packages were respectively used for raw data uploading and pre-processing and for frozen robust multi-array (fRMA) normalization. The R script used is available upon request.
Differential expression analysis
We have used a Bayesian differential expression analysis (DEA) approach implemented in the R package limma [21] to define differentially expressed genes. Threshold for selection of differentially expressed probe sets was set to a B-statistic parameter Lods (already adjusted for multiple testing) ≥5 and a log2 ratio ≥ +0.58 or ≤- 0.58. The very conservative Lods>5 was based on DEA results between EA samples from Kimchi et al. [15] and Watts et al. [17] datasets, where no significant DE probe sets are expected, to control for inter-dataset variability noise.
Barcode analysis
Probe set barcode values were calculated with the frma [20] and frma-associated hgu133abarcodevecs package, using the method described by McCall et al. [22, 23] (http://rafalab.jhsph.edu/barcode/). A probe set was defined as expressed (= 1) or non-expressed (= 0) in a given sample according to fRMA cutoffs. BE barcodes were filtered per dataset using very stringent criteria. A probe set was integrated in the dataset barcode if expressed in 100% or ≥ 75% in P-BE or nonP-BE samples, respectively. Group-specific barcodes were calculated by intersecting probe set IDs expressed in each dataset. EA-specific dataset barcodes were estimated as for nonP-BE i.e. probe sets were expressed in ≥ 75% samples to be integrated in the EA barcode.
Gene set enrichment analysis
To find over-represented gene ontology biological processes (GO-BP) among specific sets of genes we used the gene set enrichment analysis (GSEA) tool from InnateDB (http://www.innatedb.ca/) using Entrez ID as gene identifier.
GeneMANIA network analysis
The guilt-by-association GeneMANIA Cytoscape plugin algorithm [24] (http://genemania.org/) was used to identify genes functionally-related to our query genes, with gene symbols as identifiers.
Samples and clinical data
For validation we used 19 formalin fixed paraffin-embedded (FFPE) BE samples from P-BE and nonP-BE (clinical data in S1 Table) of a cohort of 331 non-dysplastic Barrett’s esophagus enrolled in an endoscopic surveillance program (mean surveillance of 6.2 years ranging from 1 to 25) at the Instituto Português de Oncologia de Lisboa Francisco Gentil (IPOLFG), with an observed incidence of high-grade dysplasia or adenocarcinoma of 3,4/1000 patient-years during about the 30 years of existence of the surveillance program. All cases, at diagnosis, were analysed as per normal routine by two experienced GI pathologists. Patients select for this study were part of a cohort of patients diagnosed with Barrett’s Esophagus under surveillance at the IPO. This included a group of nine patients that progressed to high grade dysplasia or adenocarcinoma during surveillance (Progressed, P-BE), being the diagnostic confirmed by two independent pathologists, as standard practice and international recommendations. It included another group, as control, of 10 patients that did not display any dysplasia or carcinoma at any time during surveillance (termed non-Progressed, nonP-BE). In our cohort, progressor patients were defined as those with no dysplastic Barrett´s esophagus in the index endoscopy who progress during follow up to HGD or ADC. Non-progressors were defined as patients with no dysplastic Barret’s esophagus in the index endoscopy who remind free of dysplasia or ADC during a mean follow-up similar to that of progressors. The non-Progressed patients were randomly selected from our database. In the P-BE patients we analyzed samples from two time points, before and after malignant progression, named as t0 and t1. In the first time point (t0) we studied the initial biopsy diagnosed BE negative for dysplasia. In the second time point (t1) we examined two areas, the BE and the adjacent HGD/EA on mucosectomies or surgical pieces from these same patients. In the control group of nonP-BE patients we also studied two time points: t0 and t1 for the index and for most recent follow-up biopsies, respectively. In this set of patients all samples from t0, t1 or in any other follow-up archived sample (between t0 and t1) displayed any signs of malignancy. We chose to use a balanced number of non-progressors to avoid artificially inflating p-values in the comparisons. Histopathological characterization and area selection was carried out on hematoxylin- and eosin-stained sections under the supervision of an experienced pathologist. The study was approved by the institutional review board and a waiver of consent was obtained prior to initiating this retrospective study (project GIC/721 IPOLFG, EPE).
RNA extraction and cDNA synthesis
Archived BE FFPE tissue sections (5 μm) were deparaffinized and counterstained with Mayer’s hematoxylin and eosin. BE-enriched areas were needle microdissected under the pathologist guidance. Total RNA was extracted with the RNeasy FFPE kit (Qiagen), according to manufacturer’s instructions with a slight modification: proteinase K cell-lysis at 56°C was performed overnight. The RNase-Free DNase Set (Qiagen) “on column” DNA digestion procedure was included. Each extracted RNA was reverse-transcribed with the First-Strand cDNA Synthesis kit (GE Healthcare), using a 1:1 mixture of random primers (pd(N)6) and oligo-dT primers (NotI-d(T)18. High quality total RNA (3 μg) from two control cell lines (HCT116 and a primary skin fibroblasts) was used to synthesize cDNA to be used as dilution standards in qRT-PCR.
Quantitative real-time PCR
RNA concentration and integrity could not be assessed using standard methods due to known FFPE degradation issues and to the small amounts of extracted samples. Thus, to indirectly check the amount of each isolated total RNA FFPE sample and its quantitative real-time PCR (qRT-PCR) downstream performance, we prepared two standards dilution series using cDNA from the two control cell lines, corresponding to 100, 10 1, 0.1, 0.01, 0.001 and 0.0001 ng of the original total RNA. These series were subsequently used to calculate a qRT-PCR standard curve for the non-differentially expressed gene MAPKAPK2 (Lods = -2.7). Primer sets were designed with the NCBI Primer-BLAST tool [25], to work at 59°C and with an amplicon length of 70–100bp (S2 Table). Duplicates of each BE sample were analyzed by qRT-PCR using SsoFast™ EvaGreen® Supermix (Bio-Rad, Hercules CA, USA) reagent in 10μL of reaction mixture containing template (2μL, ~200pg/μL) and primers (0.5μM each). Samples were processed in a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules CA, USA) according to the cycling program: 95°C for 60 s, 50 cycles of 95°C for 10s and 59°C for 15s. Fluorescence data collection occurred at 59°C. Relative differential expression analysis of target genes by qRT-PCR was based on the 2-ΔΔCt methodology from Livak et al. [26] using mean quantification cycle of duplicates as cycle threshold (Ct) compared to the Ct of the calibrator gene GAPDH.
Immunohistochemistry
Immunohistochemistry (IHC) of BE samples (3 μm thick tissue sections) was performed according to standard protocols. Primary antibodies were diluted in Bond Primary Antibody Diluent (Leica Microsystems) plus background-reducing components at the dilutions: CYR61 (1:600, mouse monoclonal [3H3], Abcam, ab80112), TAZ (1:300, mouse monoclonal, Abcam, ab118373), E-Cadherin (1:80, mouse monoclonal 4A2C7, Invitrogen, 33-4000). Antigen retrieval consisted of pressure-cooking for 6 minutes in pH6 sodium citrate 0.01M buffered solution for CYR61, of 20 minutes of microwave exposure at 750W in pH6 sodium citrate 0.01M buffer and of 25 minutes in Epitope Retrieval Solution 2 (ER2) Leica Bond III system for E-Cadherin. Signal detection of CYR61 and TAZ was obtained using the Rabbit/Mouse Peroxidase/DAB+ Dako REAL Envision Detection System while E-Cadherin visualization was performed in the Leica Bond III system with the detection system Bond Polymer Refine Detection plus Bond DAB Enhancer. Nuclei were counterstained with Mayer’s hematoxylin. Images were acquired on a Leica DM5500 microscope.
IHC staining specificity was evaluated with a three level score. IHC scores for CYR61 antibody (ab80112) were defined as “Low” when a weakly positive diffuse protein staining was observed (+), “Intermediate” when scored areas included few areas of weakly positive and most areas with positive diffuse staining (++) and “High” when no negative areas were observed and the majority of evaluated areas presented a strongly positive diffuse staining (+++). IHC scores for TAZ antibody (ab118373) were defined as “Low” when protein staining was negative to weakly positive cytoplasmatic diffuse staining in the majority of visualized areas (-), “Intermediate” when weakly positive cytplasmatic and low to strong nuclear staining was observed in some areas (+) and “High” when low to strong cytoplasmatic staining and very strong nuclear staining was observed in the majority of evaluated areas (++).
Western-blotting
The western-blotting procedure was performed according to standard protocols. Total protein extracts (20μg) from the two breast cancer cell lines MDA231 and MCF7 were resolved by SDS-PAGE on a 12% acrylamide gel (BIO-RAD) and transferred to a PVDF membrane (GE Healthcare) on a Mini-Protean system (BIO-RAD). The molecular weight marker used for electrophoresis was the Kaleidoscope (∫BIO-RAD) and transfer conditions were 250 mA, 100 min. CYR61 (ab80112) and TAZ (ab118373) antibodies were both diluted 1:200 in 0.2% fish skin gelatin, 1x TBST and the loading control β-Actin antibody (Sigma) was diluted 1:2000 in 1% milk, 1x PBS. All primary antibodies were incubated overnight at 4°C. Detection for all antibodies was performed by incubation with mouse Horseradish Peroxidase antibody (Jackson Immunoresearch) at 1:10.000 dilution and the western-blot signal developed with and ECL system (BIO-RAD) and detected on an x-ray Amersham Hyperfilm ECL (GE Healthcare).
Statistical analysis
Data analysis was performed with R language for Statistical Computing [14]. Expression differences between P-BE and nonP-BE microarray data was determined with a Bayesian T-test implemented in the R package limma [21]. We used hypergeometric testing to assess gene set enrichment. Statistical significance of qRT-PCR data was calculated with Wilcoxon Rank Sum test (confidence level = 0.95). IHC categorical data was analyzed with Pearson’s Chi-squared test.
Results
Hypotheses generation: mining public gene transcriptomics data
We implemented a bioinformatics pipeline for data mining that takes as input the small number of expression profiling data sets available for nonP-BE, P-BE and EA (Fig 1A), insures that data is comparable using fRMA normalization (Fig 1B) and identifies genes differentially expressed between P-BE and nonP-BE (Fig 1C). Subsequently cross checks these against a database of human gene expression patterns to binarize the genes with bimodal gene expression (Fig 1D) and implements a network analysis to cross check the selected list of candidate genes against other data types (such as protein interactions, gene co-expression, etc) for plausibility (Fig 1E). Finally, the output of this pipeline is submitted to one additional filtering step, based on manual literature curation of selected genes (Fig 1F, Table 1). From a list of 12749 unique starting genes, our approach predicts the two genes CYR61 and WWTR1 (alias TAZ) that in silico can distinguish P-BE from nonP-BE samples. Details of the methods are given in methods section, and a detailed step-by-step description of each step and resulting lists of genes is given as supplementary material (S1 Fig and S1 Results).
Fig 1. Bioinformatics analysis workflow of BE datasets for biomarker discovery.

A. Three publicly available microarray datasets of BE data containing 33 BE samples with progression information were used to interrogate the expression levels of 12719 unique genes. B. After data normalization with frozen robust multi-array (fRMA) we first performed C. differential gene expression analysis, which allowed the identification of 799 differentially expressed genes. Normalized fRMA data was subsequently submitted to D. gene expression barcode which further restricted the number of selected candidates to 19. The combined usage of a E. systems biology approach plus F. manual literature curation selected the two most promising candidates.
Table 1. Cancers where CYR61 and TAZ over-expression has been previously correlated with poor outcome.
| Cancer Type | CYR61 (references) | TAZ (references) |
|---|---|---|
| Breast | [27] | [28, 29] |
| Prostate | [30–32] | --- |
| Colorectal | [33] | [34] |
| Gastric | [35, 36] | --- |
| Esophageal Squamous Cell | [37, 38] | --- |
| Esophageal Adenocarcinoma | [39] | --- |
| Pancreas | [40, 41] | --- |
| Hepatocellular | [42] | --- |
| Non-Small Cell Lung | [43]--- | [43],[44] |
| Thyroid Carcinoma | [45] | [46]ª |
| Renal cell carcinoma | [47] | --- |
| Ovary | [48] | --- |
| Glioma | [49] | [50] |
| Osteosarcoma | [51] | --- |
| Epithelioid Hemangioendothelioma | --- | [52] |
| Oral (squamous cell) | [53, 54] | --- |
ªPapillary
mRNA levels of CYR61 and TAZ distinguish nonP-BE from P-BE in paraffin-embedded samples
Given the existing variability associated with microarray technology results (lab-, user- platform-associated, etc) and the technology limitations (probe sensitivity and specificity) it is essential to use an independent mean to verify and reproduce the results of candidate genes.
We evaluated CYR61 and TAZ as early biomarkers of BE progression in a validation set of FFPE samples from 19 BE patients (detailed characteristics of the patients used in the validation set of the present study is available in S1 Table). In total, 9 P-BE patients (t0, n = 9 and t1, n = 9 samples) and 10 nonP-BE patients (t0, n = 10 and t1, n = 10 samples) were included (see Materials and Methods)
Using qRT-PCR we compared CYR61 and TAZ mRNA levels from t1 P-BE tissue co-occurring with HGD/EA with nonP-BE samples without any histological signs of malignancy. The analysis revealed that the transcriptional levels of both genes were significantly increased (P value<0.005) in P-BE samples (Fig 2A) as predicted in silico. We have also detected a significant up-regulation (average fold change >2, P value <0.01) in the index samples (t0) of P-BE patients, years before the development of HGD/EA as compared to nonP-BE index samples (t0) from patients that never developed HGD/EA (Fig 2A, Table 2). This early up-regulation could be detected as early as 13 years (average: 4.6 years; range: 1-13 years) in the P-BE group. In the nonP-BE group, the maximum follow-up interval was of 17 years (average: 9.4 years; range: 3-17 years). In addition, using the microarray and qRT-PCR data we verified that CYR61 and TAZ expression levels are not correlated and thus P-BE and nonP-BE samples could be better segregated when using independent information from both markers (S4 Fig). Their combined usage may enhance sensitivity for the early detection of patients at risk of BE malignant progression in BE index samples.
Fig 2. CYR61 and TAZ mRNA and protein levels are significantly increased in early and late at risk BE biopsies.

A. Timeline analysis of CYR61 (left panel) and TAZ (right panel) expression levels by qRT-PCR of P-BE associated with EA (t1) and in the patient-matched BE index biopsies, free of dysplasia/EA (t0). Index BE biopsies were collected at t0 while, after several years of follow-up, EA-associated BE biopsies in the P-BE group and EA-free BE biopsies in the nonP-BE group were designated as collected at t1. The average years of follow-up between t0 and t1 was 4.6 and 9.4 years for P-BE and nonP-BE samples. B. and C. panels display respectively representative samples of CYR61 and TAZ protein levels in BE (t0 and t1) and in EA, evaluated by immunohistochemistry. Staining patterns used to score the protein levels (low, intermediate and high) are represented on the top right of each panel. The counts and statistical test (Pearson’s Chi-squared test) results are represented in the top left of the panels. (Magnification ×200).
Table 2. Per patient quantitative assessment of CYR61 and TAZ expression levels by qRT-PCR and IHC.
| Age at | t0 | t1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (biopsy order) | YearsFUp | qRT-PCR | IHC | qRT-PCR | IHC | |||||||
| Group | ID | 1st | last | CYR61 | TAZ | CYR61 | TAZ | CYR61 | TAZ | CYR61 | TAZ | |
| nonP-BE | 11 | 64 | 81 | 17 | Low | Low | ++ | + | Low | Low | + | + |
| nonP-BE | 13 | 62 | 65 | 3 | Low | Low | ++ | + | Low | Low | + | + |
| nonP-BE | 14 | 65 | 75 | 10 | Low | Low | ++ | ++ | Low | Low | + | + |
| nonP-BE | 21 | 66 | 77 | 11 | Low | Low | + | - | Low | Low | ++ | - |
| nonP-BE | 22 | 67 | 74 | 7 | Low | Low | + | + | Low | Low | + | + |
| nonP-BE | 25 | 32 | 42 | 10 | High | High | ++ | - | Low | Low | + | - |
| nonP-BE | 26 | 64 | 76 | 12 | Low | High | + | - | Low | Low | ++ | + |
| nonP-BE | 27 | 42 | 52 | 10 | Low | Low | ++ | - | Low | Low | ++ | + |
| nonP-BE | 28 | 52 | 63 | 11 | Low | Low | +++ | - | Low | Low | + | - |
| nonP-BE | 29 | 51 | 54 | 3 | Low | Low | + | + | Low | Low | + | - |
| P-BE | 2 | 71 | 72 | 1 | Low | High | +++ | + | Low | High | +++ | + |
| P-BE | 3 | 46 | 59 | 13 | High | High | ++ | ++ | Low | High | + | ++ |
| P-BE | 4 | 50 | 54 | 4 | Low | High | +++ | + | Low | Low | ++ | + |
| P-BE | 5 | 51 | 53 | 2 | Low | High | ++ | ++ | Low | Low | + | ++ |
| P-BE | 6 | 45 | 48 | 3 | High | High | +++ | + | High | High | ++ | - |
| P-BE | 17 | 62 | 67 | 5 | High | High | +++ | ++ | High | High | + | + |
| P-BE | 18 | 48 | 54 | 6 | High | High | + | - | High | High | +++ | + |
| P-BE | 19 | 68 | 69 | 1 | Low | Low | +++ | ++ | High | Low | + | ++ |
| P-BE | 20 | 51 | 57 | 6 | High | High | + | ++ | Low | Low | ++ | ++ |
Degree of immunostaining is indicated by the “+” and “-”signs: “+++” = very strong staining; “++” = strong staining; “+” = weak staining; “-”= absence of positive staining. YearsFUp = Years of follow-up; qRT-PCR = quantitative real time PCR; IHC = immunohistochemistry.
Protein levels of CYR61 and TAZ distinguish P-BE from nonP-BE in paraffin embedded samples
Validation of the in silico predictions by qRT-PCR is encouraging, but clinical use could be simpler if routine techniques such as immunohistochemistry were to be used. We have thus implemented an IHC assay. We have started by evaluating the specificity of both antibodies by Western-blot analysis (S5A Fig). IHC results of CYR61 and TAZ proteins were categorized into three different groups according to staining intensities (Fig 2B, Table 2): low, intermediate and high. CYR61 antibody presented a diffuse cytoplasmatic and/or nuclear staining as shown in the positive control (S5B Fig) and TAZ antibody mostly stained nuclei although it is also presented a diffuse pattern in the cytoplasm (S5C Fig). Blinded analysis showed that despite the somewhat heterogeneous cytoplasmatic staining of CYR61 protein, its levels were mainly intermediate to high in the P-BE group and mostly varied from low to intermediate in the nonP-BE group (Fig 2B). CYR61 protein over-expression differences were more pronounced in the early time point t0. Interestingly, samples displaying the highest amounts of CYR61 often exhibited strong nuclear accumulation. As for CYR61, despite some heterogeneity of TAZ IHC pattern (Fig 2C, Table 2), TAZ protein levels were increased in the P-BE as compared to nonP-BE group, with P-BE samples from t0 displaying a more distinct TAZ over-expression. Overall, protein levels validated the in silico transcriptional changes and correlated with qRT-PCR results (Table 2). Further, they highlighted once more the very early (at t0) differences of CYR61 and TAZ expression between progressors and non-progressors.
CYR61 and TAZ up-regulation is correlated to an epithelial-to-mesenchymal transition phenotype
In our network analysis for prioritizing genes (Fig 1E), we found that EMT and stemness-related genes are significantly over-represented in the in silico P-BE samples (p = 5.5×10-8). Out of the 19 barcoded genes, 52% (SPARC, CYR61, JUN, ACTN1, COL4A1, PPAP2B, DUSP1, CTSB and TAZ) have been previously detected in molecular signatures of EMT/stemness phenotypes [55–57]. Such phenotypes are clearly associated with an aggressive clinical behavior, poor outcome and resistance to treatment (reviewed in [58]). Given the involvement of CYR61, TAZ and other P-BE up-regulated genes in stemness/EMT-related cellular functions (e.g. cell adhesion/motility, inflammation, differentiation/wounding, extracellular matrix) (reviewed in [58]) we checked if core EMT markers such as TWIST1, ZEB1, SNAI1, SNAI2 and CDH1 (alias E-cadherin) (reviewed by [59]) were also differentially expressed. TWIST1 was the only significantly over-expressed gene (Lods = 5.84, fold change>2) in P-BE samples. As shown in Fig 3A, we validated TWIST1 up-regulation by qRT-PCR analysis in early P-BE samples (t0) before the emergence of any microscopic signs of malignancy. Furthermore, using routine pathology IHC for E-cadherin we also detected foci of lower E-cadherin expression in P-BE samples both in early (t0) and late (t1) P-BE samples (Fig 3B), an observation usually associated with invasive EA cells. The appearance of such foci is indicative of very early P-BE cellular adhesion and/or extracellular matrix changes absent in nonP-BE samples. This observation suggests that an aggressive mechanism, typical of advanced metastatic lesions, is active in non-malignant BE cells of at risk patients. These features of neoplastic progression occur in P-BE in a time point far earlier than we anticipated. The presence in P-BE samples of alterations typical of aggressive behavior in the context of cancer, suggest that at very early stages in BE there is already a proneness for later development of dysplasia and EA.
Fig 3. Changes in epithelial-to-mensenchymal biomarkers are visible in early and late BE backgrounds.
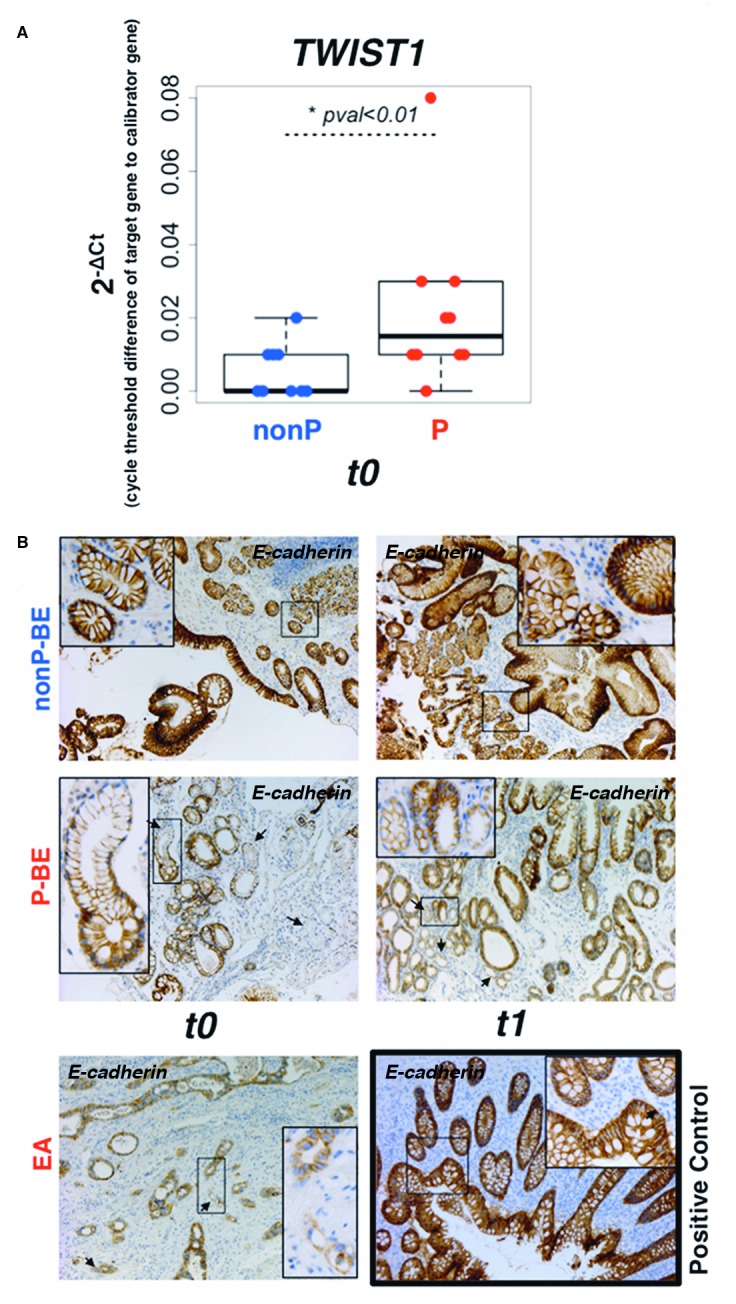
A. qRT-PCR validation of TWIST1 transcription factor in the patient-matched BE index biopsies, free of dysplasia/EA (t0). B. E-cadherin protein levels were evaluated by immunohistochemistry staining in P-BE associated with EA (t1) and in the patient-matched BE index biopsies, free of dysplasia/EA (t0). Arrows denote foci of lower E-cadherin expression. Normal appendix was used as E-cadherin immunostaining positive control. (Magnification: picture ×100, detail ×200).
Discussion
In the present work, we aimed at maximizing the identification of potentially translatable biomarkers of early BE progression to EA using an original bioinformatics pipeline applied to public BE expression profiling data. This discovery framework allowed the straightforward comparison of BE samples from distinct datasets and the trimming of promising over-expressed biomarkers to CYR61 and TAZ genes. Validation with qRT-PCR and IHC emphasized CYR61 and TAZ over-expression as early markers of at risk BE index samples, years before HGD/EA emergence. The access to Barrett’s patients that progressed during the surveillance program allowed the unique opportunity to validate biomarkers in Barrett’s samples from the same group of patients before and after malignant progression. This allowed us to overcome the limitations derived from inter-patient variability in studies where different sets of patients were used in each step of Barrett’s malignant progression. To our knowledge, this is the first time that risk stratification biomarkers were validated in BE samples coming from the same set of patients. Finally, changes in EMT biomarkers were detected in index samples of at risk BE. This observation fits into CYR61 and TAZ functional context and further suggests that in at risk BE, proteins associated with EMT can be operational from very early, before any visible signs of malignancy.
CYR61 and TAZ emerged from our pipeline as the most promising biomarkers of at risk BE and we experimentally validated their up-regulation in a different cohort. Several studies have implicated CYR61 and TAZ in the biology of major cancers (Table 1). BE is a metaplastic response of the esophageal surface to chronic injury caused by gastric reflux, possibly amplified by an inflammatory response [60]. BE progression to EA could plausibly be mediated by the known functions of these two genes in extracellular matrix, cell migration, angiogenesis and stemness/EMT. CYR61 is an important factor in acid-induced esophageal epithelial transformation [61] and its up-regulation is an early response of esophageal cells exposed to low pH [62]. In a non-esophageal context, CYR61 plays a role in inflammation, it is notably expressed at wounded tissues [63] and also up regulated upon mechanical stress (reviewed in [64]). CYR61 has also an independent prognostic value in esophageal squamous cell carcinoma [37, 38] and is a negative predictor in early onset sporadic colorectal [65, 66] and ovarian [48] cancers. Functionally, CYR61 is a ligand for several integrins which in turn can trigger cancer cells motility (reviewed in [67]) as it was recently demonstrated in pancreatic cancer [41]. TAZ is a key mediator of mechanotransduction, also implicated in human tumorigenesis: TAZ transducer activities are required to sustain self-renewal and tumor-initiation capacities [29], cell proliferation and EMT in breast cancer stem cells [28, 68] and to regulate mesenchymal differentiation in malignant gliomas [50].
Many putative biomarkers of BE malignant progression resulted from previous studies (reviewed in [11]) but no biomarker has been used in routine clinical practice [12]. Typically, most biomarkers were discovered in a context of a detectable high-grade dysplasia/EA, a time point where the neoplasia is already established and therefore where markers of tumor development are of little use and only cancer progression is at stake. This type of cancer-associated molecular alterations in non-cancer tissues (e.g. BE and NE) but adjacent to a tumor are often referred to as a cancer field effect and have already been described in BE [69]. We analyzed CYR61 and TAZ levels in non-dysplastic/EA-free BE index biopsies of P-BE and nonP-BE patients by qRT-PCR to distinguish whether CYR61 and TAZ up-regulation were a cancer field effect or an early property of P-BE samples. We found that, in fact, the up-regulation of these genes in BE years before the appearance of dysplasia (t0), reveals the establishment of a signaling pathway prone for progression to dysplasia/EA at very early stages through an intrinsic alteration of cell properties that directly, or by interaction with stromal tissue will facilitate tumor initiating features. Interestingly, we observed that CYR61 and to a lesser degree also TAZ expression levels, slightly decreased when progressing from index (t0) to advanced (t1) BE. Differences between P-BE and nonP-BE are thus more significant at t0. This phenomenon of increased expression on localized benign disease and a decreased expression upon progression to metastasis is known to occur in several contexts such as for CYR61 in prostate cancer (reviewed in [70]). While no mechanistic explanation justifies yet this observation in BE, it is possible that, as in prostate, CYR61 and TAZ are more important in the neoplastic initiation than progression.
Although CYR61 up-regulation was previously described in BE samples where dysplasia/EA is already present [39], our work is the first to describe that such up-regulation is in fact a very early event in Barrett’s tumorigenic process, being an indicator for a later establishment of dysplasia/EA, since it is detected in BE index biopsies. CYR61 belongs to the CCN family of six structurally related proteins, a multi-tasking group of secreted proteins which primarily function in adhesion, migration, proliferation, ECM synthesis, inflammation and mechanical stress regulation (reviewed in [67]). Furthermore, CYR61 has an already established role in cancer malignant progression and prognosis in major and diverse tumors (Table 1) and is a downstream target of TAZ [71]. TAZ is a major downstream effector and is regulated by the Hippo tumor suppressor pathway, a pathway relevant in organ size control, tissue regeneration, stem cell self-renewal (reviewed by [72]), cell polarity and cancer (reviewed by [73]). TAZ has also been implicated in the malignant phenotype of several tumors (Table 1) but differently from CYR61, TAZ over-expression has never been under scrutiny in human BE or EA. We anticipate that the biological functions of both genes, TAZ as a major regulator of the hippo pathway and its downstream effector CYR61, may contribute to BE progression to EA and we demonstrate for the fist time that they have predictive value.
The functional context of our validated targets and of other genes detected in our analysis suggested the early occurrence of mechanisms known to operate in EMT. As additional examples of such, we detected early TWIST1 up-regulation and lower E-cadherin expression foci very early in BE (i.e. in BE index samples) from patients that progressed later on to cancer. These are classical biomarkers associated to aggressive features of malignant progression such as EMT. The early occurrence of in vivo EMT in the absence of any histological signs of cancer was recently demonstrated in pancreatic cancer and can be partially facilitated by inflammation [74]. EMT occurs both in wound healing and tumors (reviewed in [75]) a scenario that fits into CYR61 and TAZ functional context. How EMT could contribute to early stages of BE malignant progression and/or if only some EMT-related pathways are activated is currently unknown. However, EMT occurrence was described in EA [76] and in an immortalized normal epithelial cell line. In the latter CYR61 up-regulation was critical and exacerbated acid-induced EMT phenotypes such as triggering E-cadherin loss [61]. Moreover, it was recently reported that TAZ and CYR61 were implicated in lung cancer progression and EMT via angiomotin [43].
Our observations indicate that in particular, being CYR61 an extracellular matrix secreted protein it harbors the potential to become a serum biomarker to stratify the risk of progression to malignancy in BE [32, 36] allowing for less invasive follow-up exams. This could supply an extra tool to define the risk of malignant progression and the possibility of reducing the number of biopsies needed, in particular for low risk patients.
Despite the very low BE progression frequency, we had access to a small, yet precious and very rare, cohort of FFPE follow-up samples for validation and more importantly to implement CYR61 and TAZ detection by IHC, a method directly translatable to all pathology labs. Although FFPE-based qRT-PCR CYR61 and TAZ measurements are a valid profiling option, already validated in oncologic diagnosis (reviewed in [77, 78]), assaying mRNA in the clinical routine may not be the most desirable because of technical and cost constrains. Since IHC is a method not limited by the quantity of material and can assess protein presence at single cell level, we tested commercially available antibodies against our two gene candidates and found two that, after optimization, the expected immunostains were obtained and the results could be easily translated to a routine diagnostic lab. Though the qRT-PCR and IHC data showed independently a significant difference between P-BE and nonP-BE samples in both timepoints, we have not observed clear correlation between the results of both methodologies within the same patient. The discrepant results observed in Table 2 may be justified do to inherent differences between qPCR and IHC, two techniques that are very different in nature. qPCR is quantitative, whether IHC is qualitative; furthermore, for the qPCR analysis we enriched the sample by microdissection, whereas no equivalent procedure was possible for IHC. In addition, IHC is prone to variation-dependent biases while qPCR is not observer dependent.
Given the very low BE progression frequency we will always be limited by the number of available archived collections of P-BE index samples (t0) and its follow-up samples until histological signs of malignancy are displayed (t1), which spans periods of many years. Additionally, our country has lower BE incidence rates when compared to other developed countries and we do not have a national BE register. Despite all this, our small cohort of patients that progressed during surveillance reproduces the actual expected risk progression in Barrett’s patients. Indeed, compared with the several international cohorts reviewed by the British Society of Gastroenterology guidelines [79] our cohort is quite representative. We are aware that larger numbers are needed in future tests with these two markers, which will certainly implicate several international multi-institutional collaborations.
Conclusions
Given the running debate on the costs/benefits of BE surveillance programs [80–82] and despite the diminished risk of progression [83] biomarkers to better stratify the patients who have real increased risk of neoplastic progression are required. Furthermore, it is important to stress that the risk may be low but still, BE is the precursor lesion of one of the fastest growing cancer types in developed countries over the past decades. Our results support the use of CYR61 and TAZ as early biomarkers to discriminate which BE patients have an increased risk to progress to dysplasia and cancer and further suggest that proteins/genes involved in EMT are critical to trigger the BE lesions that evolve to more aggressive lesions. Our findings also highlighted that these and other yet unknown markers should be supervised right from the non-dysplastic BE index biopsy. Such procedure has the potential to greatly improve BE management by sparing low risk patients from unnecessary invasive exams and to impact the overall costs (ethical and economical) of surveillance programs.
Supporting Information
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We are grateful to Leonor David from the Institute of Molecular Pathology and Immunology of the University of Porto (Portugal) for her helpful comments on our manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by funding from FCT grants HMSP-CTSAU-ICT-0075-2009 and SFRH-BPD-26487-2006. Ophiomics Precision Medicine provided support in the form of salaries for authors JC and JPL but these funders did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the 'author contributions' section.
References
- 1.Chandrasoma PT, Der R, Dalton P, Kobayashi G, Ma Y, Peters J, et al. Distribution and significance of epithelial types in columnar-lined esophagus. Am J Surg Pathol. 2001;25(9):1188–93. Epub 2001/11/02. . [DOI] [PubMed] [Google Scholar]
- 2.Cook MB, Wild CP, Everett SM, Hardie LJ, Bani-Hani KE, Martin IG, et al. Risk of mortality and cancer incidence in Barrett's esophagus. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2090–6. Epub 2007/09/25. 1055-9965.EPI-07-0432 [pii] 10.1158/1055-9965.EPI-07-0432 . [DOI] [PubMed] [Google Scholar]
- 3.Edgren G, Adami HO, Weiderpass Vainio E, Nyren O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut. 2012. Epub 2012/08/25. gutjnl-2012-302412 [pii] 10.1136/gutjnl-2012-302412 . [DOI] [PubMed] [Google Scholar]
- 4.Wang KK, Sampliner RE. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett's esophagus. Am J Gastroenterol. 2008;103(3):788–97. Epub 2008/03/18. AJG1835 [pii] 10.1111/j.1572-0241.2008.01835.x . [DOI] [PubMed] [Google Scholar]
- 5.Shaheen NJ, Falk GW, Iyer PG, Gerson LB. ACG Clinical Guideline: Diagnosis and Management of Barrett's Esophagus. Am J Gastroenterol. 2015;111(1):30–50. Epub 2015/11/04. ajg2015322 [pii] 10.1038/ajg.2015.322 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whiteman DC, Appleyard M, Bahin FF, Bobryshev YV, Bourke MJ, Brown I, et al. Australian clinical practice guidelines for the diagnosis and management of Barrett's esophagus and early esophageal adenocarcinoma. J Gastroenterol Hepatol. 2015;30(5):804–20. Epub 2015/01/23. 10.1111/jgh.12913 . [DOI] [PubMed] [Google Scholar]
- 7.Spechler SJ, Fitzgerald RC, Prasad GA, Wang KK. History, molecular mechanisms, and endoscopic treatment of Barrett's esophagus. Gastroenterology. 2010;138(3):854–69. Epub 2010/01/19. S0016-5085(10)00018-1 [pii] 10.1053/j.gastro.2010.01.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varghese S, Lao-Sirieix P, Fitzgerald RC. Identification and clinical implementation of biomarkers for Barrett's esophagus. Gastroenterology. 2012;142(3):435–41 e2. Epub 2012/01/24. S0016-5085(12)00081-9 [pii] 10.1053/j.gastro.2012.01.013 . [DOI] [PubMed] [Google Scholar]
- 9.Nancarrow DJ, Clouston AD, Smithers BM, Gotley DC, Drew PA, Watson DI, et al. Whole genome expression array profiling highlights differences in mucosal defense genes in Barrett's esophagus and esophageal adenocarcinoma. PLoS One. 2011;6(7):e22513 Epub 2011/08/11. 10.1371/journal.pone.0022513 PONE-D-11-05378 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Qin R, Ma Y, Wu H, Peters H, Tyska M, et al. Differential gene expression in normal esophagus and Barrett's esophagus. J Gastroenterol. 2009;44(9):897–911. Epub 2009/05/27. 10.1007/s00535-009-0082-2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ong CA, Lao-Sirieix P, Fitzgerald RC. Biomarkers in Barrett's esophagus and esophageal adenocarcinoma: predictors of progression and prognosis. World J Gastroenterol. 2010;16(45):5669–81. Epub 2010/12/04. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moyes LH, Going JJ. Still waiting for predictive biomarkers in Barrett's oesophagus. J Clin Pathol. 2011;64(9):742–50. Epub 2011/05/25. jclinpath-2011-200084 [pii] 10.1136/jclinpath-2011-200084 . [DOI] [PubMed] [Google Scholar]
- 13.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37(Database issue):D885–90. Epub 2008/10/23. gkn764 [pii] 10.1093/nar/gkn764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2009.
- 15.Kimchi ET, Posner MC, Park JO, Darga TE, Kocherginsky M, Karrison T, et al. Progression of Barrett's metaplasia to adenocarcinoma is associated with the suppression of the transcriptional programs of epidermal differentiation. Cancer Res. 2005;65(8):3146–54. Epub 2005/04/19. 65/8/3146 [pii] 10.1158/0008-5472.CAN-04-2490 . [DOI] [PubMed] [Google Scholar]
- 16.Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, et al. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett's esophagus. PLoS One. 2008;3(10):e3534 Epub 2008/10/28. 10.1371/journal.pone.0003534 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watts GS, Tran NL, Berens ME, Bhattacharyya AK, Nelson MA, Montgomery EA, et al. Identification of Fn14/TWEAK receptor as a potential therapeutic target in esophageal adenocarcinoma. Int J Cancer. 2007;121(10):2132–9. Epub 2007/06/28. 10.1002/ijc.22898 . [DOI] [PubMed] [Google Scholar]
- 18.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80 Epub 2004/10/06. gb-2004-5-10-r80 [pii] 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15. Epub 2004/02/13. 10.1093/bioinformatics/btg405 20/3/307 [pii]. . [DOI] [PubMed] [Google Scholar]
- 20.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA). Biostatistics. 2010;11(2):242–53. Epub 2010/01/26. kxp059 [pii] 10.1093/biostatistics/kxp059 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. Epub 2006/05/02. 10.2202/1544-6115.1027 . [DOI] [PubMed] [Google Scholar]
- 22.McCall MN, Uppal K, Jaffee HA, Zilliox MJ, Irizarry RA. The Gene Expression Barcode: leveraging public data repositories to begin cataloging the human and murine transcriptomes. Nucleic Acids Res. 2011;39(Database issue):D1011–5. Epub 2011/01/05. gkq1259 [pii] 10.1093/nar/gkq1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zilliox MJ, Irizarry RA. A gene expression bar code for microarray data. Nat Methods. 2007;4(11):911–3. Epub 2007/10/02. nmeth1102 [pii] 10.1038/nmeth1102 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26(22):2927–8. Epub 2010/10/12. btq562 [pii] 10.1093/bioinformatics/btq562 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134 Epub 2012/06/20. 1471-2105-13-134 [pii] 10.1186/1471-2105-13-134 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. Epub 2002/02/16. 10.1006/meth.2001.1262 S1046-2023(01)91262-9 [pii]. . [DOI] [PubMed] [Google Scholar]
- 27.Tsai MS, Bogart DF, Castaneda JM, Li P, Lupu R. Cyr61 promotes breast tumorigenesis and cancer progression. Oncogene. 2002;21(53):8178–85. Epub 2002/11/22. 10.1038/sj.onc.1205682 . [DOI] [PubMed] [Google Scholar]
- 28.Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68(8):2592–8. Epub 2008/04/17. 68/8/2592 [pii] 10.1158/0008-5472.CAN-07-2696 . [DOI] [PubMed] [Google Scholar]
- 29.Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147(4):759–72. Epub 2011/11/15. S0092-8674(11)01218-9 [pii] 10.1016/j.cell.2011.09.048 . [DOI] [PubMed] [Google Scholar]
- 30.D'Antonio KB, Toubaji A, Albadine R, Mondul AM, Platz EA, Netto GJ, et al. Extracellular matrix associated protein CYR61 is linked to prostate cancer development. J Urol. 2010;183(4):1604–10. Epub 2010/02/23. S0022-5347(09)03150-4 [pii] 10.1016/j.juro.2009.12.006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franzen CA, Chen CC, Todorovic V, Juric V, Monzon RI, Lau LF. Matrix protein CCN1 is critical for prostate carcinoma cell proliferation and TRAIL-induced apoptosis. Mol Cancer Res. 2009;7(7):1045–55. Epub 2009/07/09. 1541-7786.MCR-09-0017 [pii] 10.1158/1541-7786.MCR-09-0017 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terada N, Shiraishi T, Zeng Y, Mooney SM, Yeater DB, Mangold LA, et al. Cyr61 is regulated by cAMP-dependent protein kinase with serum levels correlating with prostate cancer aggressiveness. Prostate. 2011;72(9):966–76. Epub 2011/10/26. 10.1002/pros.21501 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ladwa R, Pringle H, Kumar R, West K. Expression of CTGF and Cyr61 in colorectal cancer. J Clin Pathol. 2011;64(1):58–64. Epub 2010/11/18. jcp.2010.082768 [pii] 10.1136/jcp.2010.082768 . [DOI] [PubMed] [Google Scholar]
- 34.Pan J, Li S, Chi P, Xu Z, Lu X, Huang Y. Lentivirus-mediated RNA interference targeting WWTR1 in human colorectal cancer cells inhibits cell proliferation in vitro and tumor growth in vivo. Oncol Rep. 2012;28(1):179–85. Epub 2012/04/04. 10.3892/or.2012.1751 . [DOI] [PubMed] [Google Scholar]
- 35.Lin MT, Chang CC, Lin BR, Yang HY, Chu CY, Wu MH, et al. Elevated expression of Cyr61 enhances peritoneal dissemination of gastric cancer cells through integrin alpha2beta1. J Biol Chem. 2007;282(47):34594–604. Epub 2007/10/02. M706600200 [pii] 10.1074/jbc.M706600200 . [DOI] [PubMed] [Google Scholar]
- 36.Zhao ZS, Li L, Wang HJ, Wang YY. Expression and prognostic significance of CEACAM6, ITGB1, and CYR61 in peripheral blood of patients with gastric cancer. J Surg Oncol. 2011;104(5):525–9. Epub 2011/05/28. 10.1002/jso.21984 . [DOI] [PubMed] [Google Scholar]
- 37.Xie JJ, Xu LY, Xie YM, Du ZP, Feng CH, Dong H, et al. Involvement of Cyr61 in the growth, invasiveness and adhesion of esophageal squamous cell carcinoma cells. Int J Mol Med. 2011;27(3):429–34. Epub 2011/01/21. 10.3892/ijmm.2011.603 [DOI] [PubMed] [Google Scholar]
- 38.Zhou ZQ, Cao WH, Xie JJ, Lin J, Shen ZY, Zhang QY, et al. Expression and prognostic significance of THBS1, Cyr61 and CTGF in esophageal squamous cell carcinoma. BMC Cancer. 2009;9:291 Epub 2009/08/25. 1471-2407-9-291 [pii] 10.1186/1471-2407-9-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Martino E, Wild CP, Rotimi O, Darnton JS, Olliver RJ, Hardie LJ. IGFBP-3 and IGFBP-10 (CYR61) up-regulation during the development of Barrett's oesophagus and associated oesophageal adenocarcinoma: potential biomarkers of disease risk. Biomarkers. 2006;11(6):547–61. Epub 2006/10/24. K16Q733086XL4HM8 [pii] doi: 10.1080/13547500600896791 . [DOI] [PubMed] [Google Scholar]
- 40.Haque I, Mehta S, Majumder M, Dhar K, De A, McGregor D, et al. Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol Cancer. 2011;10:8 Epub 2011/01/15. 1476-4598-10-8 [pii] 10.1186/1476-4598-10-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maity G, Mehta S, Haque I, Dhar K, Sarkar S, Banerjee SK, et al. Pancreatic tumor cell secreted CCN1/Cyr61 promotes endothelial cell migration and aberrant neovascularization. Sci Rep. 2014;4:4995 Epub 2014/05/17. srep04995 [pii] 10.1038/srep04995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li ZQ, Ding W, Sun SJ, Li J, Pan J, Zhao C, et al. Cyr61/CCN1 is regulated by Wnt/beta-catenin signaling and plays an important role in the progression of hepatocellular carcinoma. PLoS One. 2012;7(4):e35754 Epub 2012/04/28. [pii]. 10.1371/journal.pone.0035754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu YL, Hung JY, Chou SH, Huang MS, Tsai MJ, Lin YS, et al. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene. 2015;34(31):4056–68. Epub 2014/11/11. onc2014333 [pii] 10.1038/onc.2014.333 [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene. 2011;30(18):2181–6. Epub 2011/01/25. onc2010606 [pii] 10.1038/onc.2010.606 [DOI] [PubMed] [Google Scholar]
- 45.Chen G, Jin M, Lin S, Wang P, Liu J, Li W, et al. Increased expression cysteine-rich61 (Cyr61) in patients with thyroid carcinomas. African Journal of Pharmacy and Pharmacology. 2011;5(22):2517–21. Epub 15 December, 2011. 10.5897/AJPP11.708 [DOI] [Google Scholar]
- 46.de Cristofaro T, Di Palma T, Ferraro A, Corrado A, Lucci V, Franco R, et al. TAZ/WWTR1 is overexpressed in papillary thyroid carcinoma. Eur J Cancer. 2010;47(6):926–33. Epub 2010/12/07. S0959-8049(10)01076-2 [pii] 10.1016/j.ejca.2010.11.008 [DOI] [PubMed] [Google Scholar]
- 47.Chintalapudi MR, Markiewicz M, Kose N, Dammai V, Champion KJ, Hoda RS, et al. Cyr61/CCN1 and CTGF/CCN2 mediate the proangiogenic activity of VHL-mutant renal carcinoma cells. Carcinogenesis. 2008;29(4):696–703. Epub 2008/01/24. bgn019 [pii] 10.1093/carcin/bgn019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen H, Cai M, Zhao S, Wang H, Li M, Yao S, et al. CYR61 overexpression associated with the development and poor prognosis of ovarian carcinoma. Med Oncol. 2014;31(8):117 Epub 2014/07/23. 10.1007/s12032-014-0117-2 [DOI] [PubMed] [Google Scholar]
- 49.Xie D, Yin D, Wang HJ, Liu GT, Elashoff R, Black K, et al. Levels of expression of CYR61 and CTGF are prognostic for tumor progression and survival of individuals with gliomas. Clin Cancer Res. 2004;10(6):2072–81. Epub 2004/03/26. . [DOI] [PubMed] [Google Scholar]
- 50.Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011;25(24):2594–609. Epub 2011/12/23. 25/24/2594 [pii] 10.1101/gad.176800.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabile AA, Arlt MJ, Muff R, Bode B, Langsam B, Bertz J, et al. Cyr61 expression in Osteosarcoma indicates poor prognosis and promotes intratibial growth and lung metastasis in mice. J Bone Miner Res. 2012. Epub 2011/10/07. 10.1002/jbmr.535 . [DOI] [PubMed] [Google Scholar]
- 52.Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50(8):644–53. Epub 2011/05/18. 10.1002/gcc.20886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kok SH, Chang HH, Tsai JY, Hung HC, Lin CY, Chiang CP, et al. Expression of Cyr61 (CCN1) in human oral squamous cell carcinoma: An independent marker for poor prognosis. Head Neck. 2010;32(12):1665–73. Epub 2010/09/18. 10.1002/hed.21381 [DOI] [PubMed] [Google Scholar]
- 54.Tang QL, Fan S, Li HG, Chen WL, Shen XM, Yuan XP, et al. Expression of Cyr61 in primary salivary adenoid cystic carcinoma and its relation to Ki-67 and prognosis. Oral Oncol. 2011;47(5):365–70. Epub 2011/03/29. S1368-8375(11)00081-9 [pii] 10.1016/j.oraloncology.2011.02.022 . [DOI] [PubMed] [Google Scholar]
- 55.Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22(46):7155–69. Epub 2003/10/17. 1206887 [pii]. 10.1038/sj.onc.1206887 . [DOI] [PubMed] [Google Scholar]
- 56.Kim H, Watkinson J, Varadan V, Anastassiou D. Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC Med Genomics. 2010;3:51 Epub 2010/11/05. 1755-8794-3-51 [pii] 10.1186/1755-8794-3-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107(35):15449–54. Epub 2010/08/18. 1004900107 [pii] 10.1073/pnas.1004900107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. Epub 2009/12/01. S0092-8674(09)01419-6 [pii] 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 59.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–37. Epub 2009/06/03. 36183 [pii] 10.1172/JCI36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fitzgerald RC. Pre-Invasive Disease: Pathogenesis and Clinical Management In: Fitzgerald RC, editor. Pre-Invasive Disease: Pathogenesis and Clinical Management. New York: Springer Science+Business Media; 2011. p. 315–40. [Google Scholar]
- 61.Modak C, Mouazzen W, Narvaez R, Reavis KM, Chai J. CCN1 is critical for acid-induced esophageal epithelial cell transformation. Biochem Biophys Res Commun. 2010;392(4):533–7. Epub 2010/01/26. S0006-291X(10)00106-3 [pii] 10.1016/j.bbrc.2010.01.057 [DOI] [PubMed] [Google Scholar]
- 62.Duggan SP, Gallagher WM, Fox EJ, Abdel-Latif MM, Reynolds JV, Kelleher D. Low pH induces co-ordinate regulation of gene expression in oesophageal cells. Carcinogenesis. 2006;27(2):319–27. Epub 2005/08/23. bgi211 [pii] 10.1093/carcin/bgi211 . [DOI] [PubMed] [Google Scholar]
- 63.Chen CC, Mo FE, Lau LF. The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem. 2001;276(50):47329–37. Epub 2001/10/05. M107666200 [pii]. 10.1074/jbc.M107666200 . [DOI] [PubMed] [Google Scholar]
- 64.Chaqour B, Goppelt-Struebe M. Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J. 2006;273(16):3639–49. Epub 2006/07/22. EJB5360 [pii] 10.1111/j.1742-4658.2006.05360.x . [DOI] [PubMed] [Google Scholar]
- 65.Hong Y, Ho KS, Eu KW, Cheah PY. A susceptibility gene set for early onset colorectal cancer that integrates diverse signaling pathways: implication for tumorigenesis. Clin Cancer Res. 2007;13(4):1107–14. Epub 2007/02/24. 13/4/1107 [pii] 10.1158/1078-0432.CCR-06-1633 . [DOI] [PubMed] [Google Scholar]
- 66.Ladwa R, Pringle H, Kumar R, West K. Expression of CTGF and Cyr61 in colorectal cancer. J Clin Pathol. 2010;64(1):58–64. Epub 2010/11/18. jcp.2010.082768 [pii] 10.1136/jcp.2010.082768 [DOI] [PubMed] [Google Scholar]
- 67.Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41(4):771–83. Epub 2008/09/09. S1357-2725(08)00318-X [pii] 10.1016/j.biocel.2008.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28(7):2426–36. Epub 2008/01/30. MCB.01874-07 [pii] 10.1128/MCB.01874-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selaru FM, Wang S, Yin J, Schulmann K, Xu Y, Mori Y, et al. Beyond Field Effect: Analysis of Shrunken Centroids in Normal Esophageal Epithelia Detects Concomitant Esophageal Adenocarcinoma. Bioinform Biol Insights. 2007;1:127–36. Epub 2008/04/22. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terada N, Kulkarni P, Getzenberg RH. Cyr61 is a potential prognostic marker for prostate cancer. Asian J Androl. 2012;14(3):405–8. Epub 2012/02/22. aja2011149 [pii] 10.1038/aja.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. 2009;284(20):13355–62. Epub 2009/03/28. M900843200 [pii] 10.1074/jbc.M900843200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13(8):877–83. Epub 2011/08/03. ncb2303 [pii] 10.1038/ncb2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2011;12(1):23–38. Epub 2011/12/16. nrc3169 [pii] 10.1038/nrc3169 [DOI] [PubMed] [Google Scholar]
- 74.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–61. Epub 2012/01/24. S0092-8674(11)01369-9 [pii] 10.1016/j.cell.2011.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leopold PL, Vincent J, Wang H. A comparison of epithelial-to-mesenchymal transition and re-epithelialization. Semin Cancer Biol. 2012;22(5–6):471–83. Epub 2012/08/07. S1044-579X(12)00104-6 [pii] 10.1016/j.semcancer.2012.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomizawa Y, Wu TT, Wang KK. Epithelial mesenchymal transition and cancer stem cells in esophageal adenocarcinoma originating from Barrett's esophagus. Oncol Lett. 2012;3(5):1059–63. Epub 2012/07/12. [pii]. 10.3892/ol.2012.632ol-03-05-1059 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Farragher SM, Tanney A, Kennedy RD, Paul Harkin D. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem Cell Biol. 2008;130(3):435–45. Epub 2008/08/06. 10.1007/s00418-008-0479-7 [DOI] [PubMed] [Google Scholar]
- 78.Paik S, Kim CY, Song YK, Kim WS. Technology insight: Application of molecular techniques to formalin-fixed paraffin-embedded tissues from breast cancer. Nat Clin Pract Oncol. 2005;2(5):246–54. Epub 2005/11/03. ncponc0171 [pii] 10.1038/ncponc0171 . [DOI] [PubMed] [Google Scholar]
- 79.Fitzgerald RC, di Pietro M, Ragunath K, Ang Y, Kang JY, Watson P, et al. British Society of Gastroenterology guidelines on the diagnosis and management of Barrett's oesophagus. Gut. 2013;63(1):7–42. Epub 2013/10/30. gutjnl-2013-305372 [pii] 10.1136/gutjnl-2013-305372 [DOI] [PubMed] [Google Scholar]
- 80.Aldulaimi DM, Cox M, Nwokolo CU, Loft DE. Barrett's surveillance is worthwhile and detects curable cancers. A prospective cohort study addressing cancer incidence, treatment outcome and survival. Eur J Gastroenterol Hepatol. 2005;17(9):943–50. Epub 2005/08/12. doi: 00042737-200509000-00010 [pii]. . [DOI] [PubMed] [Google Scholar]
- 81.Hirst NG, Gordon LG, Whiteman DC, Watson DI, Barendregt JJ. Is endoscopic surveillance for non-dysplastic Barrett's esophagus cost-effective? Review of economic evaluations. J Gastroenterol Hepatol. 2011;26(2):247–54. Epub 2011/01/26. 10.1111/j.1440-1746.2010.06506.x [DOI] [PubMed] [Google Scholar]
- 82.Somerville M, Garside R, Pitt M, Stein K. Surveillance of Barrett's oesophagus: is it worthwhile? Eur J Cancer. 2008;44(4):588–99. Epub 2008/02/15. S0959-8049(08)00007-5 [pii] 10.1016/j.ejca.2008.01.015 [DOI] [PubMed] [Google Scholar]
- 83.Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett's esophagus. N Engl J Med. 2011;365(15):1375–83. Epub 2011/10/15. 10.1056/NEJMoa1103042 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.