

Abstract
Nucleotide excision repair is an important and highly conserved DNA repair mechanism with an exceptionally large range of chemically and structurally unrelated targets. Lesion verification is believed to be achieved by the helicases UvrB and XPD in the prokaryotic and eukaryotic processes, respectively. Using single molecule atomic force microscopy analyses, we demonstrate that UvrB and XPD are able to load onto DNA and pursue lesion verification in the absence of the initial lesion detection proteins. Interestingly, our studies show different lesion recognition strategies for the two functionally homologous helicases, as apparent from their distinct DNA strand preferences, which can be rationalized from the different structural features and interactions with other nucleotide excision repair protein factors of the two enzymes.
Keywords: atomic force microscopy (AFM), DNA helicase, DNA repair, nucleotide excision repair, protein-DNA interaction, single-molecule biophysics, DNA lesion recognition, UvrB, XPD
Introduction
Nucleotide excision repair (NER)4 is an important DNA repair mechanism with a large range of chemically and structurally unrelated targets. Examples range from bulky DNA adducts to interstrand DNA cross-links caused by antitumor drugs (1–3). In humans, NER is the only repair system for the removal of UV irradiation-induced photoproducts such as the intrastrand cross-linking cyclobutane-pyrimidine dimers (CPDs), and dysfunctional NER is responsible for severe diseases, including xeroderma pigmentosum (XP) (1–3).
The mechanism of NER is highly conserved between organisms. In bacteria, NER involves the proteins UvrA, UvrB, and UvrC. In the current model of prokaryotic NER, a hetero-tetrameric complex of UvrA and UvrB (UvrA2B2) scans the DNA for lesions. When a lesion is encountered, initial lesion sensing by a dimer of UvrA is based on detection of DNA distortion. Conformational changes in the UvrA2B2 complex result in an unwinding and opening of dsDNA around the lesion, providing an unpaired (bubble) region likely required by UvrB to thread onto one of the ssDNA strands. The helicase UvrB is believed to verify the presence of a lesion by insertion of a β-hairpin via interactions with residues at the base of the hairpin (2). Upon lesion verification by UvrB, UvrA dissociates from the complex, and ATP re-binding by UvrB results in the formation of the lesion-specific UvrB-DNA complex, which recruits the NER endonuclease UvrC (UvrBC complex). UvrC carries out two incisions on either side of the lesion. The 12–13-nucleotide (nt)-long ssDNA stretch (2) containing the lesion can then be removed together with the endonuclease by the helicase UvrD, and the resulting gap is filled and sealed by DNA polymerase I and ligase.
Eukaryotic NER encompasses a total of ∼30 proteins, including the xeroderma pigmentosum group proteins (XPA–XPG). Repair can either be initiated by a stalled RNA polymerase in transcription-coupled NER or via global genome NER. In global genome NER, upon initial detection of short destabilized DNA structures by the CEN2-XPC-HR23B complex, the ATPase/helicase XPB, which is part of the 10-subunit transcription factor IIH (TFIIH) complex, then directly interacts with XPC (4) and likely further enhances the size of the nascent DNA bubble via conformational rearrangements (5). XPD, the second helicase within TFIIH, can then load onto the DNA, where its activity is being regulated by interactions with the TFIIH component p44. Its 5′- to 3′-helicase activity is exploited to further increase the size of the unpaired region (6) to permit the binding of additional NER factors, which serve stabilizing and regulating roles in TFIIH. Importantly, XPD was proposed to assume a central role in damage verification (7–11).
Using atomic force microscopy (AFM) imaging, we could recently show that archaeal XPD exploits its helicase activity using different approaches for damage recognition depending on the type of lesion (12). Specifically, we observed distinct DNA strand preferences indicating different strategies for verification, as may serve to support the vast spectrum of NER targets. Our studies further revealed conformational changes in the specific XPD-lesion complexes (12). Such structural rearrangements may serve to trigger the recruitment of additional proteins, including the two endonucleases (XPG and XPF) in eukaryotic NER, resulting in the excision of a 24–32-nt stretch containing the lesion, and finally DNA re-synthesis and ligation by DNA polymerase and ligase, respectively (2). Although the high degree of similarity between archaeal and eukaryotic XPD allowed important deductions on the mechanistic details of eukaryotic NER from the archaeal enzyme, here we show for the first time lesion interactions by eukaryotic XPD using single molecule AFM analyses.
Interestingly, despite only low structural or sequence homologies in the involved proteins, recognition and repair efficiencies for different types of lesions are overall comparable between prokaryotic and eukaryotic NER (1, 13). Many of the mechanistic steps also show strong parallels such as the initial ATP-independent sensing of DNA helix distortions by UvrA (14) and XPC (15), or the ATP re-binding induced conformational changes necessary for the formation of a stable lesion-specific complex by UvrB (16, 17) and (archaeal) XPD (12).
In this study, we investigate the mechanistic conservation of lesion recognition in the prokaryotic and eukaryotic NER systems. Specifically, we focus on UvrB(C) from thermophilic bacteria (Bacillus caldotenax and Thermotoga maritima) and XPD/p44 from the thermostable fungus Chaetomium thermophilum (with comparable enzymatic properties as the human enzyme and 74% sequence homology (18)), in isolation from other NER factors. Potential reasons for the evolvement of differences in NER lesion recognition between the different kingdoms of life are discussed.
Results
DNA Substrates for Studying UvrB(C) and XPD(/p44) in the Absence of Other Proteins
In the general NER model, DNA interactions by UvrB and XPD are preceded by other proteins (UvrA or XPC and XPB in prokaryotic or eukaryotic NER, respectively) that bend and open dsDNA forming a DNA bubble on which UvrB and XPD can load. However, UvrBC as well as XPD have been shown to also be able to bind to DNA in the absence of UvrA or XPC and XPB, respectively (12, 18–20). To examine the damage verification activities of UvrB (±UvrC) and XPD (±p44) in the absence of other proteins, we provided a DNA bubble substrate for UvrB(C) and XPD(/p44) loading. UvrB(C) and XPD(/p44) bind to these substrates as shown in electrophoretic mobility shift assays (EMSAs, Figs. 1A and 3A).
FIGURE 1.

UvrB(C)-DNA interactions. A, UvrB-DNA (gray arrows, monomer and dimer) and UvrBC-DNA (black arrow) complexes and UvrC-DNA aggregates (white arrow) on DNA containing a lesion in the context of a DNA bubble. B, AUC sedimentation coefficient spectra of UvrB-DNA (gray arrows), UvrBC-DNA (black arrow), UvrB and UvrBC (* in left and right plot, respectively, gray solid lines), and DNA alone (*, gray dashed line). Solid black lines indicate lesion containing DNA, dashed lines non-damaged DNA. C, AFM images of UvrB-DNA (left) and UvrBC-DNA (middle) complexes, indicated by arrows. Right representative complexes consistent with UvrB (top) and UvrB2 or UvrBC (bottom) size. D, DNA incision by Uvr(A)BC for fluorescein lesion containing DNA with a bubble as loading site either directly around (Fcentral) or at a distance 5′ from the lesion (F5), in the presence of ATP or ATPγS (gray boxes). E, Uvr(A)BC incisions of DNA containing either F (left) or CPD (right) in the context of completely duplexed DNA (F−, CPD−), directly within a DNA bubble (Fcentral, CPDcentral), or either 5′ or 3′ from a DNA bubble (F3, CPD3 or F5 and CPD5, respectively). As a control for nonspecific DNA incisions by the endonuclease, UvrC was also incubated alone with lesion DNA (shown only for the Fcentral substrate) but displayed no incision activity. Schematics of the DNA substrates are shown (F, green circle; CPD, blue square). Black arrows indicate the uncut DNA substrates and white arrows the incision products. F, statistical analyses of DNA incisions by Uvr(A)BC on different DNA substrates (as in E). Significance levels are given as follows: *, p < 0.05; **, p < 0.01, and ***, p < 0.005, n.s., not significant. Capital letters in D--F indicate high (2 μm) [UvrB]; small letters indicate low protein concentrations (20 nm UvrA, 200 nm UvrB, 50 nm UvrC).
FIGURE 3.

XPD(/p44)-DNA interactions. A, EMSA of XPD and XPD/p44 binding to lesion containing and non-damaged DNA, as indicated (+, 1 μm; ++, 2 μm). Gray arrows indicate XPD-DNA, and the black arrow indicates XPD/p44-DNA complexes. B, AFM image of XPD/p44-DNA (black arrow) and XPD-DNA (gray arrow) complexes. The white arrow indicates a potential p44 complex or mere DNA superstructure. C, representative examples of the three different types of DNA bound complexes observed in the images, interpreted as p44, XPD, and XPD/p44 complexes based on their AFM volumes.
DNA Binding and Lesion Processing by UvrB(C)
Once loaded onto DNA, UvrB forms a specific UvrBC-DNA complex at a DNA lesion in the presence of UvrC (Fig. 1A, black arrow). AUC sedimentation coefficients indicate a major DNA lesion-bound species with a molecular weight consistent with a heterodimeric UvrBC complex (Fig. 1B). AFM imaging (Fig. 1C) also reveals distinct protein complexes formed on DNA in the presence of UvrB ± UvrC (molecular mass ∼75 and 68 kDa, respectively (supplemental Fig. S1)). DNA-bound UvrB in the absence of UvrC displayed two species with approximate average volumes of (70 ± 25) nm3 and (140 ± 30) nm3 (Fig. 1C, left, and supplemental Fig. S2), corresponding to 60 ± 26 and 122 ± 30 kDa (based on prior AFM volume calibration, see under “Materials and Methods” (21)), consistent with monomeric and dimeric UvrB, respectively (22). Volume distributions of DNA-bound complexes for incubations containing both UvrB and UvrC also displayed (at least) two species (Fig. 1C, middle, and supplemental Fig. S2), which were classed as monomeric UvrB-DNA or UvrC-DNA complexes (50–110 nm3, class 1) and dimeric UvrBC or UvrB2 (110–250 nm3, class 2). In incubations containing UvrC, larger clusters on DNA were also apparent (with volumes > 300 nm3) that likely represent UvrC-DNA aggregates (supplemental Fig. S3), as also observed in EMSAs (white arrow in Fig. 1A). Although we cannot unambiguously distinguish between UvrBC and UvrB2 based on their AFM volumes, the volume distributions for mixed incubations (UvrB and UvrC) showed an increased frequency of dimeric volumes (UvrBC or UvrB2, ∼45% versus ∼30% of all complexes) as well as a slight shift to smaller volumes of the larger complex species compared with incubations containing only UvrB (supplemental Fig. S2).
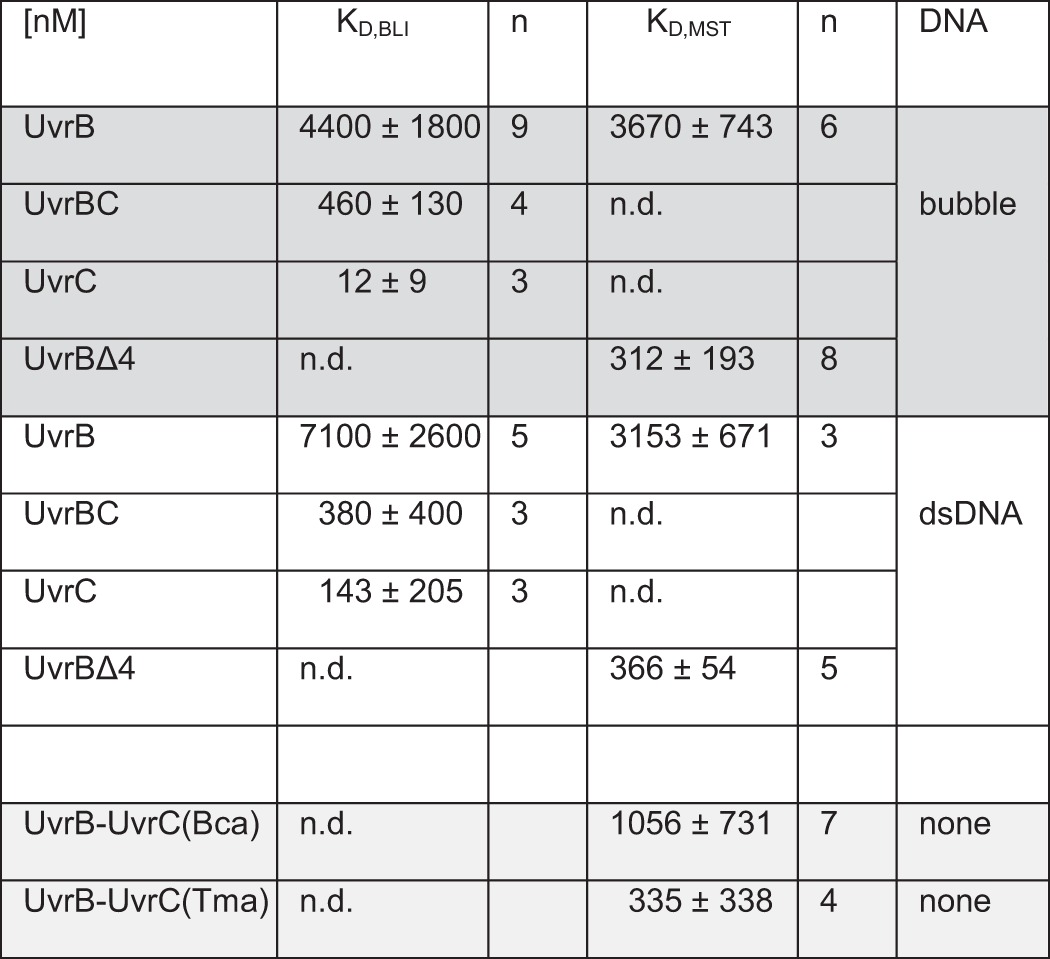
UvrB and UvrC interact with medium affinity (KD ∼500 nm) as measured by microscale thermophoresis (MST) in the absence of DNA (supplemental Fig. S4). Using MST and biolayer interferometry (BLI), we measured enhanced DNA binding affinities for UvrBC compared with UvrB alone (KD of ∼4 μm for UvrB versus ∼500 nm for UvrBC, see Table 1 and supplemental Fig. S4). This ∼10-fold increased affinity is consistent with DNA binding observed only for UvrBC and not for UvrB alone by Kad and co-workers (20) and is suggestive of a role of the UvrBC interaction in enhancing DNA binding by UvrB, potentially through repositioning of the autoinhibitory domain 4 of UvrB away from the protein core. Consistent with this interpretation, a UvrB variant that does not contain domain 4 (UvrBΔ4) showed similar DNA binding affinity (KD ∼400 nm) as UvrBC (Table 1).
TABLE 1.
Dissociation constants (KD) obtained from BLI and/or MST measurements
KD were determined for UvrB, preincubated UvrBC, UvrC, and UvrBΔ4 complexes binding to 48-bp fragments of homoduplex DNA (dsDNA, white background), and DNA containing an 8-nt unpaired (bubble, dark gray background) region. The interaction between UvrB and UvrC (in the absence of DNA, light grey background) was probed for UvrC both (from B. caldotenax) and from T. maritima by fluorescently labeling UvrB (from B.caldotenax) in MST experiments. Representative data are shown in Supplemental Fig. S3. The errors signify the standard deviations from n contributing experiments. ND means not determined.
To further investigate lesion identification by UvrB, we measured specific lesion excision by the UvrBC complex in biochemical experiments (Fig. 1, D and E). Either UvrABC or UvrBC was incubated with DNA containing a lesion (a bulky fluorescein adduct (F) placed centrally within the context of a DNA bubble). When UvrBC was thus loaded directly at the lesion (in the absence of UvrA), only ATP binding, not hydrolysis, was required for lesion excision (compare Fcentral + UvrBC in the presence of ATP in Fig. 1E and in D, gray box, in the presence of the non- or only poorly hydrolysable ATP analog ATPγS). In contrast, in the presence of UvrA, DNA incisions by UvrC were inhibited in the presence of ATPγS (Fig. 1D, gray boxes), presumably due to impaired UvrA dissociation from the complex and subsequently blocked conformational changes in UvrB. Importantly, lesion excision products are the same for UvrBC and UvrABC and consistent with prokaryotic NER incisions at positions 4–5 nts 3′ and 8 nts 5′ from the lesion (Fig. 1D, white arrow) (23). UvrB is hence able to initiate NER in the absence of UvrA.
DNA Damage Recognition by UvrB(C)
In the next step, the separation of the loading (DNA bubble) and lesion site allowed us to disentangle effects of lesion recognition and DNA translocation by UvrB(C). Upon loading of UvrB(C) at a distance 5′ from the lesion (Fig. 1D, F5 DNA substrate), subsequent translocation on DNA toward the lesion clearly requires ATP hydrolysis, as demonstrated by the absence of any incision product when incubations were conducted in the presence of ATPγS (box for F5 in Fig. 1D).
To investigate DNA strand preferences for lesion detection, we placed the DNA bubble either centrally around (Fcentral) or at a distance either 5′ (F5) or 3′ (F3) from a fluorescein adduct (Fig. 1, E and F, left). A fourth DNA substrate did not contain any DNA bubble (F−). We also analyzed detection of the UV irradiation product CPD, a prominent NER target lesion (CPDcentral, CPD5, CPD3, and CPD-DNA substrates, analogous to F substrates, Fig. 1, E and F, right). In our DNA incision assays, lesion recognition resulted in subsequent excision of the two different types of lesions, F and CPD, when UvrBC was loaded onto the DNA either directly at the lesion (Fcentral and CPDcentral) or 5′ from the lesion (F5, CPD5). In contrast, loading of the protein complex 3′ to the lesion (F3, CPD3) did not result in DNA incision by UvrBC. This finding is consistent with 5′-to-3′-helicase directionality and lesion recognition by the protein complex on the DNA strand on which it translocates, as further discussed below. Furthermore, the presence of a DNA bubble in the substrates was a necessary prerequisite for DNA incisions by UvrBC around the lesion (no incision for lesion substrates without a bubble, F− and CPD−, Fig. 1, E and F). In the absence of UvrA, the UvrBC complex clearly requires a short unpaired DNA region as a loading site onto the DNA, presumably for UvrB to thread onto one of the ssDNA.
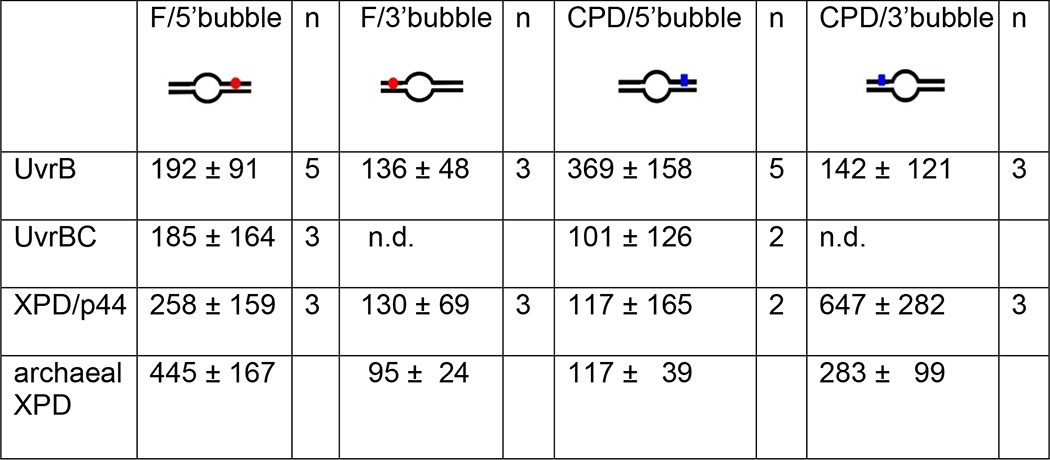
To directly probe lesion recognition by UvrB versus UvrBC complexes, we employed AFM imaging. Measuring the volumes of DNA bound protein complexes allowed us to distinguish between monomeric (UvrB or UvrC) and dimeric (UvrBC or UvrB2) complexes (see above). In these studies, we used long (916 bp) DNA substrates containing a lesion (F or CPD) at ∼30% of the DNA length. Control over the exact position of the lesion within the DNA substrates (24) allows us to distinguish between specific (lesion-bound, at ∼30% of the DNA length) and nonspecific (lesion search) complexes dependent on their positions on the DNA. For a DNA repair helicase such as UvrB(C), the fraction of specific complexes represents the amount of molecules stalled at the lesion, indicating lesion recognition, as described previously (12). In addition to the lesion site, the DNA contained an 8-nt DNA bubble that served as loading site for UvrB(C). The bubble was located at a distance of 26 nt (F5 and F3) or 23 nt (CPD5 and CPD3) either 5′ or 3′ from the lesion. Control over the exact position of the bubble allows us to distinguish between lesion recognition on the translocated ssDNA strand (for loading 5′ from the lesion, F5 and CPD5) and on the opposite, non-translocated strand (for loading 3′ from the lesion, F3 and CPD3), analogous to the DNA incision assays above. In this context it is worth noting that the lesion and bubble positions cannot be distinguished within the resolution limits of the AFM images (< 10 nm distance, see under “Materials and Methods”). However, we can calculate the preference or specificity, S, of a protein system for lesion recognition over nonspecific DNA dependent on whether the lesion is placed 3′ or 5′ from the bubble, from the ratio of specific to nonspecific complexes (25). The fraction of proteins bound at a lesion site (here at ∼30% of DNA length) increases significantly with increasing S. Surprisingly, analyses of UvrB (monomeric and dimeric species, 50–250 nm3) and UvrBC (110–250 nm3 in mixed incubations, likely also containing UvrB2) complex positions on DNA revealed no enhancement in lesion recognition specificity in the UvrBC complex compared with UvrB alone (compare Fig. 2, A, C, E, and F, SB, F5 = 192 ± 91 and SBC,F5 = 185 ± 164, SB, CPD5′ = 369 ± 158 and SBC,CPD5 =101 ± 126, see Table 2). Dimeric UvrB species (with volumes 110–250 nm3) in incubations containing no UvrC also showed comparable specificities (SB2,F5 = 192 ± 78 and SB2,CPD5 = 306 ± 209, data not shown). Importantly, UvrB was clearly able to recognize both fluorescein and CPD also in the absence of UvrC. Consistent with our DNA incision data, we detect a clear preference for recognition of lesions (either fluorescein or CPD) by UvrB when loaded onto the lesion containing strand 5′ from the lesion as compared to loaded 3′ from the lesion. Specifically, SB, F5 ∼200 and SB, CPD5 ∼400 for loading at a DNA bubble 5′ from the lesion site, for fluorescein and CPD, respectively, versus SB, F3 and SB, CPD3 ∼100 for loading 3′ from the lesion (Fig. 2, A–D, and Table 2). Because UvrB preferentially binds to ssDNA over dsDNA, residual binding preference of UvrB at ∼30% of the DNA fragment length for DNA substrates with bubbles located 3′ from the lesion sites is likely due to enhanced loading of the protein complexes at the DNA bubble rather than stalling at the lesion.
FIGURE 2.

AFM position distributions of UvrB and UvrBC on DNA lesion substrates. A–D, UvrB with F5 DNA (A), F3 DNA (B), CPD5 DNA (C), and CPD3 DNA (D). E and F, UvrBC (class 2, dimers only) with (E) F5 and F, CPD5 DNA. Position distributions are shown from 2.5% of DNA fragment length (excluding fragment ends) to 50% (center of the DNA substrate).
TABLE 2.
Lesion specificities of UvrB(C) and XPD(/p44) obtained from AFM imaging
Specificities were quantified from statistical analyses of protein position distributions on DNA containing a fluorescein adduct (F) or a CPD lesion in the context of a DNA bubble located either 5′ or 3′ from the lesion (see schematics). The specificities shown in the table are averages and standard deviations from n experiments. For comparison, specificities for archaeal XPD (without p44) are quoted from previously reported work (12). Position distributions of protein complexes on the different DNA substrates are shown in Figs. 2 and 4. ND means not determined.
DNA Binding by Eukaryotic XPD(/p44)
Eukaryotic XPD requires interaction with the TFIIH subunit p44 to activate its helicase activity (supplemental Fig. S5). However, no protein interactions are required for DNA binding by XPD. DNA binding affinity with a KD of ∼100 nm has been reported for XPD ± p44 (18). Our EMSA studies show comparable binding of XPD to lesion-containing and nondamaged DNA in the absence or presence of p44 (Fig. 3A). In AFM images, we measured the volumes of DNA-bound peaks and interpreted them based on prior AFM volume calibration (“Materials and Methods” and see Ref. 21) and the monomeric molecular masses of ctp44 (residues 1–285, ∼40 kDa) and ctXPD (∼95 kDa (supplemental Fig. S1)). For incubations containing only XPD, we observed volumes of 110 ± 30 nm3, corresponding to ∼95 kDa, consistent with monomeric XPD (supplemental Fig. S6B). The volume distribution also shows a smaller species with an AFM volume of ∼50 nm3, which we interpret as due to DNA superstructures. In the presence of XPD and p44 (supplemental Fig. S6 D), these species are also apparent in the volume distribution (maxima at ∼50 nm3 and ∼105 nm3). In addition, we observed two distinct types of DNA-bound species with AFM volumes of ∼70 and ∼150 nm3. We attribute these volumes (corresponding to molecular masses of ∼60 and ∼130 kDa) to p44- and XPD/p44-DNA complexes, respectively. DNA-bound peaks were hence classified as p44 or DNA structures (50–80 nm3, class 1), XPD (80–120 nm3, class 2), and XPD/p44 (140–250 nm3, class 3). Importantly, XPD (± p44) is able to bind to unpaired DNA (DNA bubble) in the absence of other NER proteins, similar to UvrB.
Lesion Recognition by Eukaryotic XPD
Analogous to UvrB(C), we performed AFM imaging studies with eukaryotic XPD/p44. We have previously shown for an archaeal homolog of eukaryotic XPD that a DNA bubble appears to be absolutely required as a loading site to achieve lesion recognition-competent complexes on the DNA (12). Furthermore, the presence (and hydrolysis) of ATP was necessary for lesion processing (and DNA translocation) by archaeal XPD when loaded at a DNA bubble distant from the DNA lesion (12). Here, we included ATP in all incubations of XPD or XPD/p44 with DNA substrates containing either a fluorescein adduct or a CPD lesion and a DNA bubble either 5′ (F5, CPD5) or 3′ (F3, CPD3) from the lesion. As for the experiments described above for UvrBC, relative enhancement of protein localization at the lesion position (∼30% from DNA fragment ends) allows us to deduce important information on lesion-induced stalling of XPD/p44 complexes. Representative AFM images of the different types of complexes observed in these experiments are shown in Fig. 3, B and C. Based on their measured volumes (see above) the complexes were classified as p44 or non-protein peaks (class 1), XPD (class 2), or XPD/p44 (class 3), and their binding positions on the DNA substrates were determined separately. Class 2 protein peaks (XPD) did not preferentially localize at the lesion site for either of the DNA substrates (supplemental Fig. S6, E and F). In contrast, class 3 complexes (XPD/p44) showed enhanced binding (indicating stalling and lesion recognition) at the lesion site for the F5 and CPD3 compared with the F3 and CPD5 substrates (Fig. 4). Specifically, we calculated specificities of SXPD/p44, F5 ∼300 and SXPD/p44, CPD3 ∼600 for F5 and CPD versus SXPD/p44, F3 and SXPD/p44, CPD5 ∼100 for F3 and CPD5 substrates (Table 2). These results are consistent with the requirement for a loading site (DNA bubble) for XPD to correctly thread onto the DNA as well as helicase activity (induced by interaction with p44 (supplemental Fig. S5) (18)) for XPD scanning the DNA in a lesion recognition-competent mode. Interestingly, because XPD is known to be a 5′- to 3′-helicase, specific recognition hence occurred predominantly on the translocated strand for bulky fluorescein but on the opposite non-translocated strand for the CPD lesion.
FIGURE 4.

AFM position distributions of XPD(/p44) on DNA lesion substrates. A, F5; B, F3; C, CPD5; D, CPD3. Position distributions are shown from 3.5% of DNA fragment length (excluding fragment ends) to 50% (center of the DNA substrate).
Discussion
The mechanism of NER is highly conserved between organisms with largely comparable target spectra and repair efficiencies. Target site identification in NER is achieved via a bi- or even tripartite substrate discrimination approach (26–28) in which initial detection is carried out by UvrA or XPC (requiring lesion marking by additional proteins for some more inconspicuous targets such as UV lesions). Lesion verification constitutes the next step of the intricate proofreading mechanism and is believed to involve the NER helicases UvrB and XPD (in prokaryotic and eukaryotic NER, respectively). In addition, at least in the eukaryotic system, lesion recognition is possibly aided by further protein interactions, e.g. by XPA (28). To disentangle the complex multiprotein, multistep mechanism of NER lesion identification, we investigated the isolated lesion recognition approaches employed by UvrB and XPD. Similarities and differences in their mechanisms will be discussed below.
UvrB and XPD Helicase Activities Are Activated by Protein Interactions
UvrC interaction with UvrB is absolutely required for the first, specific 3′-incision by UvrC (29, 30). Our data suggest that this interaction may serve the further purpose of enhancing DNA binding by UvrB and/or stability of the UvrB-DNA complex. The affinity of UvrB for dsDNA and also for pre-formed DNA bubbles is low in the absence of UvrA, and it has previously been reported as below the detection limit using different experimental approaches (19, 20). Our MST as well as BLI and AFM data clearly demonstrate DNA binding by UvrB with affinities in the low micromolar range (Table 1). Importantly, we observed significantly enhanced DNA binding affinity for a pre-formed UvrBC complex with a KD of ∼500 nm compared with UvrB alone (KD of ∼4 μm). These findings are consistent with observations by Kad and co-workers (20), who found the UvrBC complex to dissociate considerably less easily from undamaged DNA than UvrB alone. A possible mechanism for this enhancement may be via interactions of UvrC with domain 4 of UvrB (31) because the DNA binding affinities were comparable for UvrBC and the UvrBΔ4 variant that lacks this domain (Table 1). Domain 4 of UvrB has been found to play an autoinhibitory role for UvrB's ATPase activity (22), and interactions with UvrC may free UvrB for DNA interactions.
DNA binding enhances the ATPase activity of UvrB (22, 32), which concomitantly activates UvrB's helicase activity. In contrast, DNA binding affinity of XPD is independent of protein interactions (18). However, interactions of eukaryotic XPD with the TFIIH component p44 are required to activate XPD's helicase activity (supplemental Fig. S5) (18). XPD in the absence of p44 would hence not translocate to a lesion from a separate loading site, would subsequently not be able to form the lesion-specific stable complex, and would likely dissociate from the DNA (consistent with our data). In this context, it is interesting to compare the results on eukaryotic XPD with our previous findings on the archaeal enzyme (12). Archaeal species do not contain the full set of TFIIH subunits (including p44), and archaeal XPD does not require protein interactions to activate its helicase activity. Our experiments show comparable target site recognition by archaeal XPD and eukaryotic XPD/p44, indicating that the p44 interaction is likely required exclusively for helicase activity and not for target site interactions by XPD.
Isolated UvrB(C) and XPD/p44 Are Functional in NER Lesion Detection
Our DNA incision assays (Fig. 1, D–F) show that UvrBC is competent in NER lesion excision in the absence of UvrA. These assays also demonstrate that an access site (such as a DNA bubble) is required for proper threading of UvrB onto one of the ssDNA strands as a prerequisite for specific DNA incisions by UvrC, as reported previously (19, 33). There are two distinct conceivable scenarios as follow: 1) a preassembled complex of UvrBC loads and carries out lesion search along the DNA, or 2) UvrB alone binds to and translocates along the DNA in search of lesions and only recruits the endonuclease UvrC upon identifying a target site. Binding on and translocation along dsDNA have been observed for preassembled UvrBC complexes (20). UvrB alone did not bind to dsDNA under the conditions of these experiments (low nanomolar protein concentrations). Importantly, in these studies, the DNA substrates did not contain lesions or loading sites for UvrBC (such as DNA bubbles). We also observe UvrB(C) binding to fully intact dsDNA by AFM imaging (nonspecific strand-internal complexes). A role of UvrBC complexes in speeding up the repair reaction by transferring the endonuclease (which exists only in low copy numbers in the cell) when it reaches a lesion-bound UvrB loaded by UvrA has been proposed (20). Such complexes bound to intact dsDNA may involve a different binding mode, resulting in complexes that are competent in translocating along the DNA albeit with a different lesion recognition (and hence incision)-incompetent translocation mode, as also proposed for archaeal XPD (12).
Although UvrA and UvrB are up-regulated by the bacterial SOS response (to ∼0.2 and 1 μm, respectively (34, 35)), UvrC is not (∼10 nm). Our MST and BLI measurements indicated a KD of ∼500 nm for the formation of the UvrBC complex in the absence of DNA (supplemental Fig. S4 and Table 1). Furthermore, UvrBC complex formation has also been reported for incubations at much lower protein concentrations (20, 36). Up-regulated UvrB would hence be able to form such complexes and thus achieve higher affinity for destabilized DNA regions to initiate NER lesion excision independent of UvrA. Destabilized DNA regions as represented by a DNA bubble in our studies are in fact available in the negatively supercoiled genome of bacterial organisms such as B. caldotenax. An example of an AFM image showing UvrBC complexes bound to non-damaged negatively supercoiled circular plasmid DNA is depicted in supplemental Fig. S7. However, UvrB and UvrA deletion mutants show comparable sensitivity to UV irradiation,5 arguing against a role of additional UvrA-independent NER initiations under SOS conditions.
Although XPD(/p44) possesses roles that are independent of the other TFIIH complex components (37), a potential function of isolated XPD/p44 in eukaryotic NER is unlikely. XPD/p44 is part of the multisubunit TFIIH complex, and DNA lesion search, recognition, and verification as well as removal are all tightly regulated via protein-protein interactions (38). Recent studies by Yang and co-workers (28) have demonstrated stalling of XPD helicase activity by bulky DNA lesions (indicating target recognition) in the context of the (seven subunits) TFIIH core complex. Furthermore, data from Sugasawa et al. (26) showed NER-specific incisions of CPD containing DNA substrates for cell-free extracts of TFIIH in complex with XPC. Our studies show that mechanistically isolated XPD activated by p44 (like its prokaryotic counterpart UvrB) is also able to specifically recognize target lesions in the absence of other NER proteins.
UvrBC and XPD/p44 Have Different Strand Preferences in Lesion Verification
In our studies, we separated the locations of loading site (DNA bubble) and DNA lesion. For UvrB(C), lesion recognition and subsequent excision from these DNA substrates required ATP hydrolysis and occurred only when UvrB(C) was loaded 5′ from the lesion (either F or CPD) and not when the loading site was placed 3′ from the lesion in our biochemical assays. Enhanced binding at DNA lesion sites in the presence of ATP in our AFM experiments also indicated that DNA translocation is stalled when UvrB(C) encounters a lesion (F or CPD) in the DNA 3′ from the loading site and not when loading occurred 3′ from the lesion. These findings suggest ATP hydrolysis-dependent translocation by UvrB(C) on ssDNA with 5′-to-3′ directionality and recognition of lesions on the translocated DNA strand (Figs. 1, D–F, and 2).
An alternative theoretical possibility would be recognition of the two tested lesion types (F and CPD) on the non-translocated strand with 3′-to-5′ directionality of UvrB. The 3′-to-5′-helicase directionality had previously been proposed for UvrB based on the orientation of the protein in a crystal structure of UvrB bound to a short 3′ ssDNA overhang in the absence of ATP (39). Preferential detection of its diverse target lesions by UvrB on the non-translocated strand is, however, an unlikely scenario. In support of our interpretation of the DNA incision and AFM data, 5′-to-3′ directionality had also previously been demonstrated for the UvrAB complex (40). Our findings are also consistent with previous reports that UvrBC could excise lesions located close to the 5′ DNA end in the absence of UvrA only under conditions of ATP hydrolysis by UvrB (41). Interestingly, our AFM data suggest that the presence of UvrC in the complex is not necessary for lesion search and detection by UvrB. In fact, UvrBC complexes even showed reduced lesion recognition compared with UvrB alone, likely due to enhanced background binding to nonspecific DNA (Fig. 2 and Tables 1 and 2). Kad and co-workers (20) showed that in their nonspecific (lesion search) UvrBC-DNA complexes, the DNA directly interacts with the UvrB molecule and not with UvrC. The dominant role of UvrB in the DNA translocation and lesion recognition processes supports a model in which the main contribution of UvrC to lesion search and detection is to enhance DNA binding by UvrB.
Interestingly, the preference for recognition of different target lesions (bulky adducts as well as CPD lesions) by UvrB on the same (translocated) strand is in contrast to the strand preferences that we observed for its eukaryotic functional homolog XPD. Our AFM studies clearly show that the XPD/p44 complex preferentially identifies a bulky adduct (fluorescein) on the translocated ssDNA strand but the non-bulky UV lesion CPD on the opposite non-translocated strand. These findings are consistent with the model that we previously suggested based on our data on archaeal XPD, in which XPD employs (simultaneously) a combination of different lesion-sensing approaches (concomitant with different strand preferences) to enhance its target spectrum (12). In support of our model, recent studies on the entire reconstituted human TFIIH complex also suggested recognition of strongly helix-distorting cisplatin as well as Cy3 adducts on the translocated strand (28). Based on crystallographic and biochemical data in combination with mutational analyses, a structural model has been suggested of how XPD interacts with DNA to recognize lesions (10, 11, 42). In this model, DNA interactions with amino acid residues along a channel in XPD direct the translocated ssDNA through a pore in the enzyme (see also below). Furthermore, the model proposes DNA interactions in close proximity to the FeS cluster in the enzyme, at the 3′ end of the translocated strand where the fork between single- and double-stranded DNA would likely be located in the DNA-bound protein. This may position a DNA lesion in the opposite non-translocated ssDNA strand also close to the FeS cluster-containing domain (11, 42), allowing direct interactions for lesion sensing (Fig. 5). Although lesion recognition on the non-translocated strand can hence be directly pictured, the model does not provide an explanation for the absence of lesion recognition on the translocated strand, as observed in our AFM studies for CPD lesions (Fig. 4). Conflicting observations have been reported for recognition of these UV lesions by XPD from different archaeal organisms on the translocated or on the opposite non-translocated strand (9, 11, 12, 43). XPD from Sulfolobus acidocaldarius showed no stalling at fluorescein adducts or CPD lesions either in the translocated or in the displaced strand for short dsDNA substrates (19–45 bp) with 5′ overhangs at 30 or 45 °C (43). In contrast, DNA unwinding of forked DNA substrates with a longer paired region of 81 bp was stalled at a CPD lesion in the translocated DNA strand and also to a lesser degree in the displaced strand for the XPD homolog from Ferroplasma acidarmanus at a lower temperature (25 °C) (9, 11). Finally, we have previously reported stalling of XPD from Thermoplasma acidophilum (incubated at 37 °C) at fluorescein adducts in the translocated DNA strand and CPD lesions in the non-translocated strand of long (916 bp) fragments after loading of the protein at a DNA bubble (12). These differences may be due to different experimental approaches, e.g. more physiological DNA substrate length in our studies, and/or different organisms. Importantly, our AFM data clearly show different strand preferences for F and CPD recognition by eukaryotic XPD. Despite high structural similarity of archaeal and eukaryotic XPD, there are important differences between these two kingdoms. In archaea, XPD helicase activity is not regulated via protein-protein interactions, most of the eukaryotic proteins forming the TFIIH complex do not exist in archaea, and a function of archaeal XPD (or the other XP group proteins) in NER has not yet been demonstrated in vivo. Importantly, here we describe for the first time lesion recognition by the isolated eukaryotic XPD/p44 component of the TFIIH complex. A closer understanding of the DNA investigation mechanism employed by eukaryotic XPD will have to await higher resolution information from crystal structures of XPD-lesion complexes.
FIGURE 5.
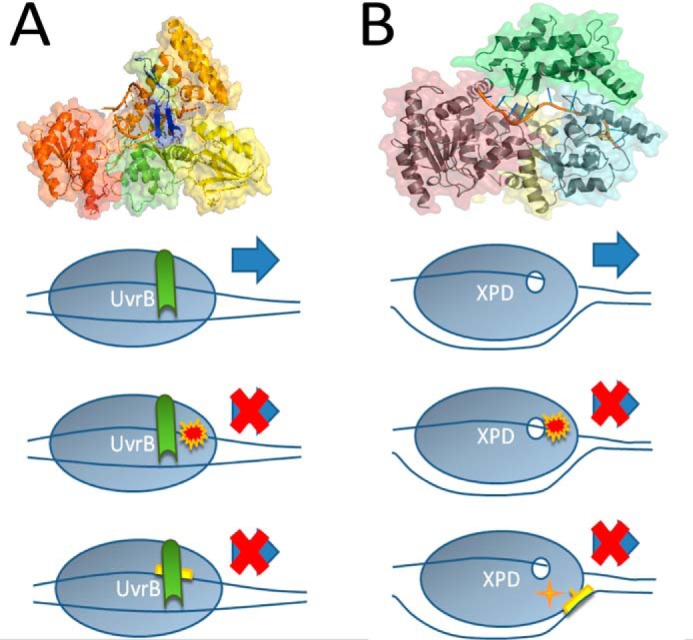
Lesion sensing by UvrB and XPD. A, crystal structure of UvrB from Bacillus caldotenax in complex with ssDNA (2FDC.pdb). The two RecA-like domains (domains 1a and 3, green and red, respectively) enclose the ATP binding site. Domains 1b and 2 are shown in orange and yellow, respectively. The C-terminal domain 4, which hosts the interface for UvrC interaction, is not resolved in the structure due to a flexible linker connection to domain 3. The DNA strand (in orange) can be seen threaded behind a β-hairpin (blue), which is involved in DNA lesion verification. In our model, UvrB threads the translocated DNA strand behind its β-hairpin probe and verifies lesions on this strand via interactions with residues on the inside of the hairpin. B, crystal structure of XPD from Thermoplasma acidophilum (pdb coordinates 4A15). Only the first four DNA nucleotides from the 5′ end of the ssDNA substrate are visible in the original data (not shown), the remaining DNA is modeled into the structure (shown in “orange”) threading through a pore in the protein. Different domains of XPD are shown in yellow (RecA-like helicase domain 1), purple (RecA-like helicase domain 2), green (arch domain), and cyan (iron sulfur, FeS, cluster containing domain). The FeS cluster, shown in stick representation, is located close to the ss/dsDNA junction at the “plow” of the 5′-to-3′ helicase. In the model below, this places the FeS cluster in a suitable position for lesion sensing on the non-translocated DNA strand (as seen for CPD in our DNA substrates). Lesion verification is likely achieved by a combination of interactions with the translocated and with the non-translocated DNA strand. Verification of (bulky) DNA adducts (such as fluorescein in our substrates), for instance, occurs predominantly on the translocated strand.
The current model of NER also does not offer a clear understanding of why the lesion verification step by XPD may need to investigate both the translocated and the opposite non-translocated DNA strand for lesions. In GG-NER, TFIIH is recruited by XPC, which is believed to load the complex on the ssDNA strand that contains the lesion (26). Although XPC nonspecifically recognizes DNA distortion by lesions and hence requires an additional verification step of NER target sites, this process could, in principle, orient XPD on the lesion-containing strand and eliminate the need for investigation of the non-translocated strand. However, multiple protein interactions within TFIIH (e.g. XPD) as well as with additional NER factors (e.g. XPA (44)) subsequently contribute to fine-tune verification of the diverse target lesions. The potential of XPD(/p44) to interact with lesions on the non-translocated strand as observed in our studies adds important information on mechanistic details. In future studies, it will be interesting to resolve this mechanistic step with successively added NER factors, ultimately reconstituting the entire NER system, at the molecular level.
Structural Conservation and Differences between UvrB and XPD
UvrB and XPD are both members of the same helicase superfamily 2 (SF2). Crystal structures for UvrB (PDB 1D9X for apo- and 2FDC for protein-DNA complex) and archaeal XPD (2VSF for apo- and 4A15 for protein-DNA complex) show that both proteins contain two RecA-like helicase domains (green and red for UvrB and purple and yellow for XPD in Fig. 5) that can mediate ATP hydrolysis-dependent movement along the DNA. XPD is a 5′- to 3′-helicase (10, 42, 45). It has been controversial whether DNA translocation by Uvr(A)B involves helicase activity or merely ATP-dependent conformational rearrangements of the DNA around the lesion (46–48). In prokaryotic NER, the single helicase/ATPase UvrB may have to fulfill comparable tasks as the two enzymes, XPB and XPD, in the eukaryotic system. UvrB may hence harbor both DNA rearrangement and helicase activities (as likely achieved by XPB and XPD, respectively). Our data show that an unpaired region is clearly required by UvrB (in the absence of UvrA) to load onto DNA in a lesion recognition-competent mode and that UvrB hydrolyzes ATP to translocate from the loading site to the DNA lesion site (analogous to XPD (12)). In contrast to previous speculations based on crystallographic data (39), our AFM and DNA incision data further indicate that UvrB, like XPD, possesses 5′ to 3′ directionality.
Despite high structural similarity of UvrB and XPD (7), their crystal structures, however, also reveal interesting differences (Fig. 5). UvrB harbors a β-hairpin, which is believed to be inserted into the DNA double helix to probe for lesions as it translocates along one of the ssDNA strands (39). Interactions with the lesion occur with hydrophobic residues in the hairpin (39, 49–51). Our data clearly support a lesion recognition approach, which requires the lesion to be located on the translocated strand. The current model of prokaryotic NER envisions two molecules of UvrB probing for DNA lesions (each bound to one UvrA monomer in a UvrA2B2 complex) (17). Each UvrB monomer can therefore focus exclusively on one of the DNA strands. In contrast, in eukaryotic NER, XPD is likely present as a single copy in the multisubunit TFIIH complex, as visualized in several electron microscopy studies (52, 53). The crystal structure of XPD shows a small (∼1 nm diameter) pore in the protein formed by domain 1 together with the arch and iron sulfur (FeS) cluster containing domains through which the ssDNA is believed to thread (10, 54). As XPD translocates along the DNA, the arch and FeS cluster domains are believed to act as a “ploughshare” that separates the two ssDNA strands. This brings a lesion on the non-translocated strand at the ss/dsDNA junction into close proximity of the redox active FeS cluster in the protein, which may assume a function in lesion verification (10, 12, 55). In addition, bulky lesions (such as fluorescein, Cy3, or cisplatin adducts) may serve as mechanical roadblocks. Such lesions would hence directly hinder DNA translocation of the helicase leading to its stalling at the lesion site on the translocated DNA strand, as suggested previously for archaeal and human XPD (10, 12, 28). It is worth noting, however, that the diameter of the pore is too large to be blocked by typical bulky DNA lesions so that further interactions between the lesion and protein residues in the proximity of the pore are likely involved in specific lesion interactions. Deeper insight into lesion recognition approaches by XPD will have to await the resolution of a crystal structure of XPD complexed with a DNA lesion site.
Conclusions
Our studies focus on the lesion recognition and verification step carried out by the NER helicases, UvrB in prokaryotes and XPD in eukaryotes, as part of the complex, multistep and multimolecular mechanism of NER. We have shown that both UvrB(C) and XPD/p44 are able to recognize NER target lesions in the absence of additional NER factors. In prokaryotic NER, interactions with the endonuclease UvrC appear to be required only for lesion recognition-competent DNA loading of the helicase UvrB and not for lesion verification. In the eukaryotic system, interaction of XPD with p44 is a necessary requirement for helicase activity and hence lesion recognition-competent loading of the complex. Interactions on the inside of a β-hairpin of UvrB lead to NER target recognition exclusively on the translocated DNA strand, supporting a model of two UvrB molecules in the heterotetrameric UvrA2B2 lesion search complex that can each focus on one of the ssDNA strands. In eukaryotic NER, diverse interactions of lesions in the translocated and the opposite ssDNA strand with different parts of XPD may serve to support the large target spectrum of NER by only one copy of XPD in the multimeric TFIIH complex.
Materials and Methods
Protein Expression and Purification
UvrB wild type (WT) and domain 4 deletion mutant (UvrBΔ4) from B. caldotenax were expressed and purified by affinity and size exclusion chromatography as described (56–58). WT UvrC from B. caldotenax and T. maritima were expressed and purified analogously with the exception of a pH 9 Tris-HCl buffer containing additionally 100 μm EDTA and the detergents Triton X (0.2%) and Tween 20 (0.2%) in the elution buffer for affinity chromatography for T. maritima UvrC. For protein-DNA complex analysis via native PAGE, UvrB and UvrC from B. caldotenax were used. BcaUvrC and UvrC from T. maritima showed comparable DNA binding in the presence of BcaUvrB (data not shown). However, TmaUvrC has been shown previously to form a productive complex with BcaUvrB (57, 59) and is more active in DNA incisions than BcaUvrC. BcaUvrC was used in the AUC and MST studies, and TmaUvrC was used in the DNA incision, AFM, MST, and BLI analyses.
XPD and p44 (residues 1–285) from C. thermophilum were expressed and purified via affinity and size exclusion followed by anion exchange chromatography, as described (18). All proteins were purified to at least 95% homogeneity as judged from Coomassie-stained SDS gels (supplemental Fig. S1).
DNA Substrates
For native PAGE and analytical ultracentrifugation (AUC), 24-bp DNA fragments were used. The sequences of the employed DNA oligomers are shown in (supplemental Table S1). Oligonucleotides 1, 1′ (containing a fluorescein (F) modification), and 2 were obtained from Sigma and stored as well as annealed at equimolar concentrations in 10 mm Tris-HCl, pH 7.5, 1 mm EDTA. The annealed 24-bp DNA substrates contain an unpaired region of six bases (6-base DNA bubble, underlined in (supplemental Table S1)) and either no damage (oligonucleotides 1 and 2) or a fluorescein-modified thymine at the central position within the unpaired region (oligonucleotides 1′ and 2). Fluorescein has been shown to be recognized by the NER mechanism (34) and served as a mimic for bulky DNA damage in our experiments.
For DNA incision assays with UvrBC, we used longer DNA oligonucleotides of 100- and 59-bp lengths for the fluorescein (F) and CPD lesion containing substrates, respectively. Sequences are shown in supplemental Table S1 (oligonucleotides 3–12). For substrate annealing, we used equimolar amounts of the top strand (substrates 3 and 8 in supplemental Table S1) containing the lesion and the bottom strand coding for a 6-nt unpaired DNA region (DNA bubble, underlined in substrates 5–7 and 10–12 in supplemental Table S1) if present. The fluorescein adduct on a thymine base (substrate 3 in supplemental Table S1) served both as the lesion and fluorescence label, although for the CPD substrates the lesion containing the top strand (substrate 8 in supplemental Table S1) was obtained with a fluorescein fluorophore attached at the 5′ end to provide the fluorescence signal. The resulting substrates were either fully double-stranded (F- and CPD-, substrates 3/4 and 8/9 in supplemental Table S1, respectively) or contained a DNA bubble either centrally located around the F or CPD lesion (Fcentral and CPDcentral, 3/5 and 8/10 in supplemental Table S1) or 27 nt 5′ (F5 and CPD5, 3/6 and 8/11 in supplemental Table S1) or 3′ 3/7 (F3 and CPD3, 3/7 and 8/12 in supplemental Table S1 from the lesion. Oligonucleotides for fluorescein-containing DNA substrates were purchased from Integrated DNA Technologies (IDT), CPD-containing oligonucleotides from TriLink Biotechnologies, and complementary oligonucleotides (9–12 in supplemental Table S1) from Sigma.
For AFM experiments, long (several hundred bp) DNA substrates optionally containing a lesion (either fluorescein or CPD) and/or a region of eight unpaired bases were prepared as described previously (12, 24). Briefly, we used a modified plasmid (puc19N) containing several N.BstNBI nickase sites, which has been kindly provided by the Wilson laboratory (NIEHS, National Institutes of Health) (60). After application of N.BstNBI (New England Biolabs), an ssDNA stretch framed by two nickase sites separated by 48 nt can be removed by incubation in the presence of an excess of complementary DNA (oligonucleotide 13 in supplemental Table S1) at ∼70 °C for 30 min immediately followed by centrifugation through a 125-nt cutoff filter (Amicon Ultra, Millipore). Short ssDNA and annealed dsDNA fragments were removed by repeating this step at least three times. The ssDNA stretch can then be replaced by a 5′-phosphorylated oligonucleotide of matching sequence containing any choice of unpaired regions (DNA bubble) and/or damages, which is ligated into the gap overnight at ambient temperature by T4 DNA ligase (New England Biolabs). For our experiments, we introduced a lesion-containing oligonucleotide with an altered sequence producing an 8-nt unpaired region 5′ or 3′ from the lesion (supplemental Table S1) as follows: oligonucleotides 14 and 15 containing a fluorescein adduct and oligonucleotides 16 and 17 containing a CPD lesion, kindly provided by the Carrell laboratory. The separation between DNA bubble and lesion in the resulting DNA substrates is 27 and 26 nts for the F/5′ bubble (F5) and F/3′ bubble (F3), respectively, and 23 nts for the CPD/5′ bubble (CPD5) and CPD/3′ bubble (CPD3) substrates. Finally, fragments of the desired length were excised from the plasmid using appropriate restriction enzymes. For our experiments, we applied SSpI and BspQI (New England Biolabs) to produce 916-bp fragments with a fluorescein or a CPD lesion at 30% of the DNA fragment length. Fragments are separated and purified using agarose gel extraction. To avoid accidental UV damage (which is a target of NER), the gel extracts used for DNA substrate purification are at no time exposed to UV light. Instead, they are excised from the gel analogous to the position in the separately exposed, first lane.
DNA substrates for BLI and MST measurements (see below) were obtained through annealing of the same 48-nt sequence as used in DNA substrate preparation for AFM (oligonucleotide 13 in supplemental Table S1) with complementary strands to provide either a fully base-paired DNA substrate (sequence 18 in supplemental Table S1) or a DNA substrate containing an 8-nt DNA bubble with or without a fluorescein or CPD lesion in the center of the unpaired region (sequences 19, 19′, and 19″ in supplemental Table S1). In addition, the 48-nt bottom strand (sequence 13 in supplemental Table S1) was obtained biotinylated or Alexa Fluor 647-labeled at its 3′ end (from IDT) to allow for attachment to the BLI probe via biotin-streptavidin interaction or to provide the fluorescence signal for detection, for BLI and MST experiments, respectively. For the helicase assay, DNA oligonucleotides 20 and 21 (supplemental Table S1) were incubated at equimolar concentrations, resulting in a DNA substrate with a Dabcyl-quenched Cy3 label at one end and a 22-nt 5′ ssDNA overhang for protein loading at the other end.
Native PAGE
UvrABC proteins were preincubated at 65 °C for 10 min at concentrations of 0.5 or 1 μm. In subsequent incubations with DNA, protein concentrations varied between 0.2 and 10 μm, as indicated. XPD/p44 were incubated at 1 and 2 μm (equimolar amounts of both proteins if present) with 20 nm DNA. All incubations were carried out for 30–60 min at 37 °C in UvrABC incubation buffer (20 mm Tris-HCl pH 7.5, 50 mm KCl, 10 mm MgCl2, 5 mm DTT) for the UvrABC system or in XPD incubation buffer (20 mm Tris-HCl pH 7.5, 10 mm KCl, 5 mm MgCl2, 1 mm tris(2-carboxyethyl)phosphine, 2 mm ATP) for XPD/p44. The DNA substrates contained a fluorescein lesion in the context of a DNA bubble (supplemental Table S1, oligonucleotides 1′/2 for UvrABC, and terminally Alexa Fluor 647-labeled oligonucleotide 18 annealed with 19′ for XPD/p44) or only a DNA bubble (supplemental Table S1, terminally Alexa Fluor 647-labeled oligonucleotide 18 annealed with 19 for XPD/p44). Samples were loaded on an 8% (for UvrABC) or 10% (for XPD/p44) polyacrylamide gel containing 0.5× Tris-Borate EDTA buffer (TBE, 45 mm Tris borate, pH 8.3, 1 mm EDTA) and 10 mm MgCl2 and run at 100–140 V at ambient temperature.
AUC
Samples of 2.5 μm UvrB ± 500 nm UvrC (both from B. caldotenax) and ±500 nm DNA substrates (substrates 1/2 and 1′/2 containing a fluorescein lesion, supplemental Table S1) in incubation buffer (see under “Native PAGE” above) were subjected to sedimentation velocity analytical ultracentrifugation. Sedimentation velocity experiments were conducted in a Beckman Optima XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) using an eight-hole An-50 Ti rotor at 40,000 rpm and at 20 °C, with 400-μl samples in standard double-sector charcoal-filled Epon centerpieces equipped with sapphire windows. Data were collected in continuous mode at a step-size of 0.003 cm without averaging using absorption optical detection at wavelengths of 280 nm and analyzed using the software SEDFIT to determine continuous distributions for solutions to the Lamm equation c(s), as described previously (61). Analysis was performed with regularization at confidence levels of 0.68 and floating frictional ratio (f/f0), time-independent noise, baseline, and meniscus position to root mean square values of <0.016.
Uvr(A)BC DNA Incision Assays
For DNA incision assays, DNA substrates 3–7 (3/4, 3/5, 3/6, and 3/7) and 8–12 (8/9, 8/10, 8/11, and 8/12) were used as given in supplemental Table S1. Because B. caldotenax UvrC has been described to carry out only the 5′ incision (59), for the incision assays we used UvrC from T. maritima, which has been previously shown to interact with UvrB from B. caldotenax (57). Experiments were carried out using at least three different protein batches. Proteins were heated to 65 °C for 10 min prior to incubations at 20 nm DNA and 20 nm UvrA, 200 nm UvrB, 200 nm UvrC (for UvrABC), or 4 μm UvrB and 200 nm UvrC (for UvrBC) in volumes of 20 μl at 55 °C in incubation buffer (see above, native PAGE) in the presence of 1 mm ATP or ATPγS as indicated. To achieve sufficient amounts of product formation for only the very slowly processed CPD lesion, samples were incubated for 5 h. DNA substrates were then heated for 10 min to 95 °C in urea sample buffer (5 m urea 10 mm Tris-HCl, pH 7.5, 0.3% (w/v) OrangeG), loaded on 4 m urea 15% polyacrylamide gels, and run for 30 min at 300 V at ambient temperature. Signal detection was based on the fluorescence from either the fluorescein adduct that simultaneously served as bulky lesion mimic in the assays or from a fluorescent end-label for the CPD lesion containing DNA substrate (see above and supplemental Table S1). Incision activity was quantified based on relative fluorescence intensities of non-incised DNA and DNA incision products as determined using the software ImageJ and plotted using Origin. Significance levels were calculated using the one-sided Student's t test.
AFM
UvrBC complex formation was supported by a 10-min preincubation of 2 μm UvrB and 400 nm to μm UvrC at 37 °C prior to incubation with DNA. Protein-DNA complexes were then formed by incubating 500 nm UvrB from B. caldotenax (WT or δ4 variant) and 50–100 nm DNA substrate in the absence or presence of UvrC from T. maritima (100–250 nm) for 15 min at 37 °C in incubation buffer (see above, native PAGE). XPD and p44 were preincubated in XPD incubation buffer (see above, native PAGE) containing 2 mm ATP at concentrations of 10 μm each for 10 min at 37 °C. 1 μm XPD ± 1 μm p44 was then incubated with 100 nm DNA substrate for 30 min at 37 °C in XPD incubation buffer containing 2 mm ATP. Reactions were diluted 50–100-fold in AFM deposition buffer (25 mm HEPES, pH 7.5, 25 mm sodium acetate, 10 mm magnesium acetate) and deposited immediately onto freshly cleaved mica (SPI Supplies) at room temperature. The mica surface was rinsed with HPLC grade water, blotted, and then dried under a stream of nitrogen, leaving the protein and DNA fixed to the substrate surface but in a hydrated state due to the extremely hygroscopic character of mica. The images were captured in air with a Molecular Force Probe 3D (MFP-3D) atomic force microscope (Asylum Research, Santa Barbara, CA) in tapping mode. OMCL-AC240TS reflection-coated tapping mode silicon probes (Olympus) with spring constants of ∼2 n·m−1 and resonance frequencies of ∼70 kHz were used for all imaging. Images were collected at a scan speed of 2.5 μm/s, image sizes of 4 × 4 μm to 8 × 8 μm, and pixel resolutions of 2048–4096.
Volume analysis of the imaged protein complexes was carried out using the section tool of the MFP software and a spherical cap model to estimate their AFM volume as shown in Equation 1,
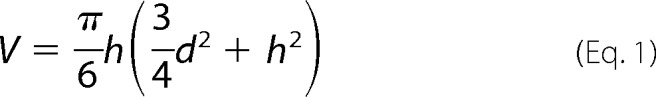 |
where h is the height of the complex and d its diameter as measured. The resulting volume distributions were plotted using Origin. Gaussian fits to the protein volume distributions provide the average AFM volume of protein complexes. Different types of complexes (UvrB, UvrBC, XPD, and XPD/44) were classed according to their measured AFM volumes.
Position distributions of protein complexes on DNA were determined by measuring the distances of protein peaks from DNA fragment ends using the ImageJ software, as described previously (12). Many DNA repair proteins have a tendency to bind to destabilized DNA fragment ends, as discussed previously (62). For UvrB(C) and XPD(/p44), end binding preference varied strongly between experiments from 0 to 54% (with averages of 9% for UvrB and XPD alone, 4% for UvrBC, and 22% for XPD/p44). As our focus is on DNA strand internal binding, the position distributions were plotted excluding DNA ends (starting from 4% of the DNA fragment length for fits to individual experiments, 3.5% of DNA length for pooled XPD/p44 data, and 2.5% of DNA length for pooled UvrB(C) data, Figs. 2 and 4). The program Origin (Microcal Inc.) was used for statistical Gaussian fits to the data to determine the ratio of protein complexes bound at the specific (lesion) site (Aspec) and those bound at nonspecific sites (Ansp) from n repeated independent experiments. Specificities S of UvrB(C) and XPD/p44 for the lesions were then calculated as described (63); S = (Aspec/Ansp)·N + 1, where N is the number of possible binding sites along the DNA (n = 916 to 73 = 843, excluding 4% of DNA length at each fragment end). For XPD, UvrB, and UvrC, at least two (for UvrB and UvrC three) different batches of independent protein purifications were analyzed, compared, and pooled. Lesion specificities are given as averages from n experiments with standard deviations between experiments (Table 2). Plots shown in Figs. 2 and 4 were produced by pooling the data from all experiments for each DNA substrate type normalized to DNA.
Helicase Activity Assay
The (undamaged) DNA substrate in these experiments contained a 22-nt 5′ ssDNA overhang for loading of XPD(/44) and a Cy3 fluorophore attached at the other DNA end. Cy3 fluorescence was quenched in the dsDNA substrate by a Dabcyl label on the complementary DNA strand end. Helicase activity was detected as the increase in Cy3 fluorescence upon DNA substrate unwinding the protein complexes. Fluorescence was excited at 550 nm and detected at 570 nm using a FluoroMax-4 spectrophotometer (Horiba Jobin Yvone). 1 μm XPD ± 2 μm p44 were mixed with 100 nm DNA substrate solution in 20 mm Tris-HCl, pH 7.5, 10 mm KCl, 5 mm MgCl2, 1 mm tris(2-carboxyethyl)phosphine directly in the measurement cuvette. Cy3 fluorescence increase upon addition of 5 mm ATP was measured for 30–60 s.
MST
MST was performed to determine binding of UvrB (from B. caldotenax) to various DNA substrates as well as to quantify the affinity of the UvrB-UvrC interaction. To measure DNA binding, UvrB was diluted in 16 steps in a 1:2 dilution series starting at 150 μm, prior to adding DNA to 50 nm to each dilution. For fluorescence detection, the DNA substrates used in these experiments were obtained with a terminal Alexa Fluor 647 label from IDT (see under “DNA Substrates” above and supplemental Table S1 oligonucleotides 18–21). The UvrB-UvrC interaction was probed by labeling UvrB amines with NT495 following the protocol of a labeling kit based on succinimidyl ester conjugation (NanoTemper Technologies). UvrC from B. caldotenax was then diluted in a 1:2 series starting from 50 or 100 μm, and NT495-UvrB conjugate was added to each dilution to a concentration of 50 nm. To remove aggregates, samples were centrifuged for 5 min at 10,000 × g before they were filled into standard treated capillaries (NanoTemper Technologies). MST measurements were performed with a MonolithTM NT.115 instrument (NanoTemper Technologies) at 20 and 40% MST Power and 80 and 100% LED Power. Experiments were performed in UvrABC incubation buffer (without ATP) in at least triplicate and with two different protein batches. Dissociation constants (KD, MST) were derived based on the different thermophoretic properties of the individual protein molecules and their complexes (UvrBC or UvrB-DNA) from fits of the Hill equation to the resulting concentration-dependent fluorescence signals using Origin (Equation 2),
 |
with x being ligand (protein) concentration and n = 1.15 ± 0.05. Average KD values were then calculated as the average from a total of five experimental series, each involving multiple measurements. The error in the average KD value of the UvrB-UvrC interaction was dominated by the variation between different experiments (standard deviation) rather than within each experimental series (error propagation).
BLI
DNA binding affinities were also measured by BLI on an Octet RED system (ForteBio, Menlo Park, CA) similarly as described previously (12). Experiments were performed using 100 nm DNA and protein concentrations of 4–10 μm B. caldotenax UvrB and 10–450 nm T. maritima UvrC in incubation buffer (20 mm Tris-HCl, pH 7.5, 50 mm KCl, 10 mm MgCl2, 5 mm DTT) supplemented with 0.1 mg/ml BSA, in at least triplicate and with at least two different protein batches of UvrB and UvrC. DNA oligonucleotides (supplemental Table S1 oligonucleotides 18–21) were purchased from IDT with a biotin group attached to the 3′ end of the bottom strand (see also “DNA Substrates” above), annealed, and coupled to the sensor surface via streptavidin-biotin interaction. Dissociation (kd) and association rate constants (ka) were determined with the Octet analysis software from protein layer thicknesses for the minimum protein concentration required to achieve DNA binding for each experimental series. BLI dissociation constants (KD, BLI) were then calculated for this protein concentration c as shown in Equation 3.
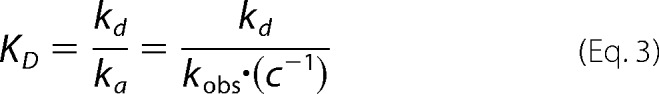 |
Author Contributions
Experiments and analyses were performed by N. W., J. G., H. M. R., C. N. B., and I. T.; AFM was done by N. W., J. G., and I. T.; biochemistry was by H. M. R., N. W., J. G., and I. T.; BLI and MST was done by C. N. B., N. W., H. M. R., and I. T. The experiments were conceived by I. T. Conceptual and financial support was provided by C. K. The manuscript was written by I. T. and edited by all authors.
Supplementary Material
Acknowledgments
We thank Gudrun Michels for providing p44 and help with XPD purification; Jochen Kuper for the XPD-DNA model and helpful discussions; Korbinian Heil and Thomas Carrell for providing CPD lesion DNA; Samuel Wilson for plasmid DNA; and Bennett van Houten for invaluable discussions.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grants KI-562/7-1, Forschungszentrum FZ82, and TE-671/4 (to C. K. and I. T.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1 and Figs. S1–S7.
N. Goosen, personal communication.
- NER
- nucleotide excision repair
- AFM
- atomic force microscopy
- CPD
- cyclobutane-pyrimidine dimer
- ssDNA
- single strand DNA
- BLI
- biolayer interferometry
- MST
- microscale thermophoresis
- XP
- xeroderma pigmentosum
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- nt
- nucleotide
- AUC
- analytical ultracentrifugation
- PDB
- Protein Data Bank.
References
- 1. Batty D. P., and Wood R. D. (2000) Damage recognition in nucleotide excision repair of DNA. Gene 241, 193–204 [DOI] [PubMed] [Google Scholar]
- 2. Kuper J., and Kisker C. (2012) Damage recognition in nucleotide excision DNA repair. Curr. Opin. Struct. Biol. 22, 88–93 [DOI] [PubMed] [Google Scholar]
- 3. de Boer J., and Hoeijmakers J. H. (2000) Nucleotide excision repair and human syndromes. Carcinogenesis 21, 453–460 [DOI] [PubMed] [Google Scholar]
- 4. Bernardes de Jesus B. M., Bjørås M., Coin F., and Egly J. M. (2008) Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Mol. Cell. Biol. 28, 7225–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oksenych V., Bernardes de Jesus B., Zhovmer A., Egly J. M., and Coin F. (2009) Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 28, 2971–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coin F., Oksenych V., and Egly J. M. (2007) Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell 26, 245–256 [DOI] [PubMed] [Google Scholar]
- 7. Wolski S. C., Kuper J., Hänzelmann P., Truglio J. J., Croteau D. L., Van Houten B., and Kisker C. (2008) Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 6, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fan L., Fuss J. O., Cheng Q. J., Arvai A. S., Hammel M., Roberts V. A., Cooper P. K., and Tainer J. A. (2008) XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133, 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathieu N., Kaczmarek N., and Naegeli H. (2010) Strand- and site-specific DNA lesion demarcation by the xeroderma pigmentosum group D helicase. Proc. Natl. Acad. Sci. U.S.A. 107, 17545–17550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuper J., Wolski S. C., Michels G., and Kisker C. (2012) Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 31, 494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mathieu N., Kaczmarek N., Rüthemann P., Luch A., and Naegeli H. (2013) DNA quality control by a lesion sensor pocket of the xeroderma pigmentosum group D helicase subunit of TFIIH. Curr. Biol. 23, 204–212 [DOI] [PubMed] [Google Scholar]
- 12. Buechner C. N., Heil K., Michels G., Carell T., Kisker C., and Tessmer I. (2014) Strand-specific recognition of DNA damages by XPD provides insights into nucleotide excision repair substrate versatility. J. Biol. Chem. 289, 3613–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y., Reeves D., Kropachev K., Cai Y., Ding S., Kolbanovskiy M., Kolbanovskiy A., Bolton J. L., Broyde S., Van Houten B., and Geacintov N. E. (2011) Probing for DNA damage with β-hairpins: similarities in incision efficiencies of bulky DNA adducts by prokaryotic and human nucleotide excision repair systems in vitro. DNA Repair 10, 684–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaciuk M., Nowak E., Skowronek K., Tańska A., and Nowotny M. (2011) Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat. Struct. Mol. Biol. 18, 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugasawa K., Okamoto T., Shimizu Y., Masutani C., Iwai S., and Hanaoka F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 15, 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verhoeven E. E., Wyman C., Moolenaar G. F., Hoeijmakers J. H., and Goosen N. (2001) Architecture of nucleotide excision repair complexes: DNA is wrapped by UvrB before and after damage recognition. EMBO J. 20, 601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verhoeven E. E., Wyman C., Moolenaar G. F., and Goosen N. (2002) The presence of two UvrB subunits in the UvrAB complex ensures damage detection in both DNA strands. EMBO J. 21, 4196–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuper J., Braun C., Elias A., Michels G., Sauer F., Schmitt D. R., Poterszman A., Egly J. M., and Kisker C. (2014) In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol. 12, e1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zou Y., and Van Houten B. (1999) Strand opening by the UvrA(2)B complex allows dynamic recognition of DNA damage. EMBO J. 18, 4889–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hughes C. D., Wang H., Ghodke H., Simons M., Towheed A., Peng Y., Van Houten B., and Kad N. M. (2013) Real-time single-molecule imaging reveals a direct interaction between UvrC and UvrB on DNA tightropes. Nucleic Acids Res. 41, 4901–4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roth H. M., Römer J., Grundler V., Van Houten B., Kisker C., and Tessmer I. (2012) XPB helicase regulates DNA incision by the Thermoplasma acidophilum endonuclease Bax1. DNA Repair 11, 286–293 [DOI] [PubMed] [Google Scholar]
- 22. Wang H., DellaVecchia M. J., Skorvaga M., Croteau D. L., Erie D. A., and Van Houten B. (2006) UvrB domain 4, an autoinhibitory gate for regulation of DNA binding and ATPase activity. J. Biol. Chem. 281, 15227–15237 [DOI] [PubMed] [Google Scholar]
- 23. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 [DOI] [PubMed] [Google Scholar]
- 24. Buechner C. N., and Tessmer I. (2013) DNA substrate preparation for atomic force microscopy studies of protein-DNA interactions. J. Mol. Recognit. 26, 605–617 [DOI] [PubMed] [Google Scholar]
- 25. Yang Y., Wang H., and Erie D. A. (2003) Quantitative characterization of biomolecular assemblies and interactions using atomic force microscopy. Methods 29, 175–187 [DOI] [PubMed] [Google Scholar]
- 26. Sugasawa K., Akagi J., Nishi R., Iwai S., and Hanaoka F. (2009) Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol. Cell 36, 642–653 [DOI] [PubMed] [Google Scholar]
- 27. Maillard O., Camenisch U., Blagoev K. B., and Naegeli H. (2008) Versatile protection from mutagenic DNA lesions conferred by bipartite recognition in nucleotide excision repair. Mutat. Res. 658, 271–286 [DOI] [PubMed] [Google Scholar]
- 28. Li C. L., Golebiowski F. M., Onishi Y., Samara N. L., Sugasawa K., and Yang W. (2015) Tripartite DNA lesion recognition and verification by XPC, TFIIH, and XPA in nucleotide excision repair. Mol. Cell 59, 1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moolenaar G. F., Franken K. L., Dijkstra D. M., Thomas-Oates J. E., Visse R., van de Putte P., and Goosen N. (1995) The C-terminal region of the UvrB protein of Escherichia coli contains an important determinant for UvrC binding to the preincision complex but not the catalytic site for 3′-incision. J. Biol. Chem. 270, 30508–30515 [DOI] [PubMed] [Google Scholar]
- 30. Moolenaar G. F., Franken K. L., van de Putte P., and Goosen N. (1997) Function of the homologous regions of the Escherichia coli DNA excision repair proteins UvrB and UvrC in stabilization of the UvrBC-DNA complex and in 3′-incision. Mutat. Res. 385, 195–203 [DOI] [PubMed] [Google Scholar]
- 31. Sohi M., Alexandrovich A., Moolenaar G., Visse R., Goosen N., Vernede X., Fontecilla-Camps J. C., Champness J., and Sanderson M. R. (2000) Crystal structure of Escherichia coli UvrB C-terminal domain, and a model for UvrB-uvrC interaction. FEBS Lett. 465, 161–164 [DOI] [PubMed] [Google Scholar]
- 32. Caron P. R., and Grossman L. (1988) Involvement of a cryptic ATPase activity of UvrB and its proteolysis product, UvrB* in DNA repair. Nucleic Acids Res. 16, 10891–10902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zou Y., Walker R., Bassett H., Geacintov N. E., and Van Houten B. (1997) Formation of DNA repair intermediates and incision by the ATP-dependent UvrB-UvrC endonuclease. J. Biol. Chem. 272, 4820–4827 [DOI] [PubMed] [Google Scholar]
- 34. Truglio J. J., Croteau D. L., Van Houten B., and Kisker C. (2006) Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 106, 233–252 [DOI] [PubMed] [Google Scholar]
- 35. Schendel P. F., Fogliano M., and Strausbaugh L. D. (1982) Regulation of the Escherichia coli K-12 uvrB operon. J. Bacteriol. 150, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moolenaar G. F., Bazuine M., van Knippenberg I. C., Visse R., and Goosen N. (1998) Characterization of the Escherichia coli damage-independent UvrBC endonuclease activity. J. Biol. Chem. 273, 34896–34903 [DOI] [PubMed] [Google Scholar]
- 37. Li X., Urwyler O., and Suter B. (2010) Drosophila Xpd regulates Cdk7 localization, mitotic kinase activity, spindle dynamics, and chromosome segregation. PLoS Genet. 6, e1000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo J., Cimermancic P., Viswanath S., Ebmeier C. C., Kim B., Dehecq M., Raman V., Greenberg C. H., Pellarin R., Sali A., Taatjes D. J., Hahn S., and Ranish J. (2015) Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol. Cell 59, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Truglio J. J., Karakas E., Rhau B., Wang H., DellaVecchia M. J., Van Houten B., and Kisker C. (2006) Structural basis for DNA recognition and processing by UvrB. Nat. Struct. Mol. Biol. 13, 360–364 [DOI] [PubMed] [Google Scholar]
- 40. Oh E. Y., and Grossman L. (1989) Characterization of the helicase activity of the Escherichia coli UvrAB protein complex. J. Biol. Chem. 264, 1336–1343 [PubMed] [Google Scholar]
- 41. Moolenaar G. F., Monaco V., van der Marel G. A., van Boom J. H., Visse R., and Goosen N. (2000) The effect of the DNA flanking the lesion on formation of the UvrB-DNA preincision complex. Mechanism for the UvrA-mediated loading of UvrB onto a DNA damaged site. J. Biol. Chem. 275, 8038–8043 [DOI] [PubMed] [Google Scholar]
- 42. Pugh R. A., Wu C. G., and Spies M. (2012) Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 31, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rudolf J., Rouillon C., Schwarz-Linek U., and White M. F. (2010) The helicase XPD unwinds bubble structures and is not stalled by DNA lesions removed by the nucleotide excision repair pathway. Nucleic Acids Res. 38, 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koch S. C., Kuper J., Gasteiger K. L., Simon N., Strasser R., Eisen D., Geiger S., Schneider S., Kisker C., and Carell T. (2015) Structural insights into the recognition of cisplatin and AAF-dG lesion by Rad14 (XPA). Proc. Natl. Acad. Sci. U.S.A. 112, 8272–8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coin F., Marinoni J. C., Rodolfo C., Fribourg S., Pedrini A. M., and Egly J. M. (1998) Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat. Genet. 20, 184–188 [DOI] [PubMed] [Google Scholar]
- 46. Gordienko I., and Rupp W. D. (1997) The limited strand-separating activity of the UvrAB protein complex and its role in the recognition of DNA damage. EMBO J. 16, 889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bessho T., Mu D., and Sancar A. (1997) Initiation of DNA interstrand cross-link repair in humans: the nucleotide excision repair system makes dual incisions 5′ to the cross-linked base and removes a 22- to 28-nucleotide-long damage-free strand. Mol. Cell. Biol. 17, 6822–6830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Theis K., Skorvaga M., Machius M., Nakagawa N., Van Houten B., and Kisker C. (2000) The nucleotide excision repair protein UvrB, a helicase-like enzyme with a catch. Mutat. Res. 460, 277–300 [DOI] [PubMed] [Google Scholar]
- 49. Moolenaar G. F., Höglund L., and Goosen N. (2001) Clue to damage recognition by UvrB: residues in the β-hairpin structure prevent binding to non-damaged DNA. EMBO J. 20, 6140–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zou Y., Ma H., Minko I. G., Shell S. M., Yang Z., Qu Y., Xu Y., Geacintov N. E., and Lloyd R. S. (2004) DNA damage recognition of mutated forms of UvrB proteins in nucleotide excision repair. Biochemistry 43, 4196–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jia L., Kropachev K., Ding S., Van Houten B., Geacintov N. E., and Broyde S. (2009) Exploring damage recognition models in prokaryotic nucleotide excision repair with a benzo[a]pyrene-derived lesion in UvrB. Biochemistry 48, 8948–8957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. He Y., Fang J., Taatjes D. J., and Nogales E. (2013) Structural visualization of key steps in human transcription initiation. Nature 495, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gibbons B. J., Brignole E. J., Azubel M., Murakami K., Voss N. R., Bushnell D. A., Asturias F. J., and Kornberg R. D. (2012) Subunit architecture of general transcription factor TFIIH. Proc. Natl. Acad. Sci. U.S.A. 109, 1949–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Constantinescu-Aruxandei D., Petrovic-Stojanovska B., Penedo J.C., White M. F., and Naismith J. H. (2016) Mechanism of DNA loading by the DNA repair helicase XPD. Nucleic Acids Res. 44, 2806–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pugh R. A., Honda M., Leesley H., Thomas A., Lin Y., Nilges M. J., Cann I. K., and Spies M. (2008) The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J. Biol. Chem. 283, 1732–1743 [DOI] [PubMed] [Google Scholar]
- 56. Theis K., Chen P. J., Skorvaga M., Van Houten B., and Kisker C. (1999) Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 18, 6899–6907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Truglio J. J., Rhau B., Croteau D. L., Wang L., Skorvaga M., Karakas E., DellaVecchia M. J., Wang H., Van Houten B., and Kisker C. (2005) Structural insights into the first incision reaction during nucleotide excision repair. EMBO J. 24, 885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang H., Tessmer I., Croteau D. L., Erie D. A., and Van Houten B. (2008) Functional characterization and atomic force microscopy of a DNA repair protein conjugated to a quantum dot. Nano Lett. 8, 1631–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiang G. H., Skorvaga M., Croteau D. L., Van Houten B., and States J. C. (2006) Robust incision of benzo[a]pyrene-7,8-dihyrodiol-9,10-epoxide-DNA adducts by a recombinant thermoresistant interspecies combination UvrABC endonuclease system. Biochemistry 45, 7834–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hou E. W., Prasad R., Asagoshi K., Masaoka A., and Wilson S. H. (2007) Comparative assessment of plasmid and oligonucleotide DNA substrates in measurement of in vitro base excision repair activity. Nucleic Acids Res. 35, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tessmer I., Melikishvili M., and Fried M. G. (2012) Cooperative cluster formation, DNA bending and base-flipping by the O6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 40, 8296–8308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang Y., Sass L. E., Du C., Hsieh P., and Erie D. A. (2005) Determination of protein-DNA binding constants and specificities from statistical analyses of single molecules: MutS-DNA interactions. Nucleic Acids Res. 33, 4322–4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.