

Abstract
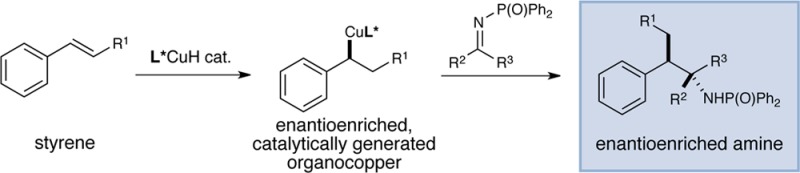
The copper-catalyzed intermolecular enantioselective addition of styrenes to imines has been achieved under mild conditions at ambient temperature. This process features the use of styrenes as latent carbanion equivalents via the intermediacy of catalytically generated benzylcopper derivatives, providing an effective means for accessing highly enantiomerically enriched amines bearing contiguous stereocenters. Mechanistic studies shed light on the origin of the preferential styrene hydrocupration in the presence of an imine with the Ph-BPE-derived copper catalyst.
Enantiomerically enriched amines constitute essential structural elements in a variety of pharmaceutically important small molecule therapeutics.1 Consequently, general methods for the synthesis of amines in optically pure form have long been regarded as an important objective for synthetic organic chemists.1 Among the numerous techniques available, asymmetric hydrogenation1,2 and biocatalysis1,3 are often employed. However, the addition of carbon nucleophiles to prochiral imines,4 particularly Ellman’s chiral tert-butylsulfinimines,4a often represents the method of choice for the preparation of free or protected chiral primary amines.
The need to utilize stoichiometric quantities of an organometallic reagent and the chiral auxiliary is a limitation of these processes. Additionally, the use of highly basic and nucleophilic organometallic compounds such as organomagnesium and organozinc reagents decreases the functional group compatibility of these methods. Moreover, one or multiple additional synthetic operations are usually required to prepare an organometallic reagent from an organic halide or an unsaturated hydrocarbon. In this context, the use of abundant or easily accessible olefins as latent carbanion equivalents for asymmetric nucleophilic addition to imines could obviate these problems. In recent years, Krische has pioneered the use of polyunsaturated olefins as latent carbon nucleophiles for carbonyl additions using noble metal catalysts (Rh, Ru, and Ir).5 While these processes have been exceptionally successful for the enantioselective addition to aldehydes,6 only a few examples of asymmetric addition to imines have been reported.7 Furthermore, the intermolecular, enantioselective coupling of less active olefins such as styrenes to imines is rare.8
In 2013, our group initiated a research program aimed at developing copper(I) hydride catalysis9,10 as a general platform for accessing enantiomerically enriched organocopper intermediates from easily available olefin precursors. By capturing the catalytically generated organocopper species with electrophilic aminating reagents, our laboratory10 and Hirano and Miura11 have independently developed a new approach for the enantioselective hydroamination of olefins. More recently, we have also demonstrated the feasibility of engaging organocopper intermediates in carbon–carbon bond-forming processes.12,13 In particular, we reported a CuH-catalyzed intramolecular addition of styrene-derived organocopper species to N-arylimines for the enantioselective synthesis of indolines (Figure 1A),12a as well as an enantioselective intermolecular coupling of (poly)unsaturated olefins and ketones.12e We note that Shibasaki and Lam have each previously devised elegant copper-catalyzed protocols for the enantioselective reductive Mannich reaction.14 We recently wondered whether our previously reported intramolecular cyclization of iminostyrenes could be extended to a more broadly applicable method for the coupling of readily available styrenes and N-protected imines. As illustrated in Figure 1B, the enantioselective addition of a phosphine-ligated copper catalyst (I) across the double bond of styrene II would furnish a transient benzylcopper intermediate (III).15 Nucleophilic addition of benzylcopper III to an N-protected imine (IV) would provide a copper amide (V), which is in turn protonated by t-BuOH to furnish addition product VII and release the copper tert-butoxide (VIII). Subsequent σ-bond metathesis of VIII with a hydridic silane (IX) would regenerate the copper hydride catalyst and complete the catalytic cycle.16 The strategy described herein provides rapid access to the enantioselective assembly of highly substituted amines bearing adjacent stereocenters from easily available olefin and imine building blocks. Furthermore, this mechanistic framework does not require the use of exogenous acidic or basic additives, thereby potentially allowing for sensitive functional groups to be tolerated.
Figure 1.
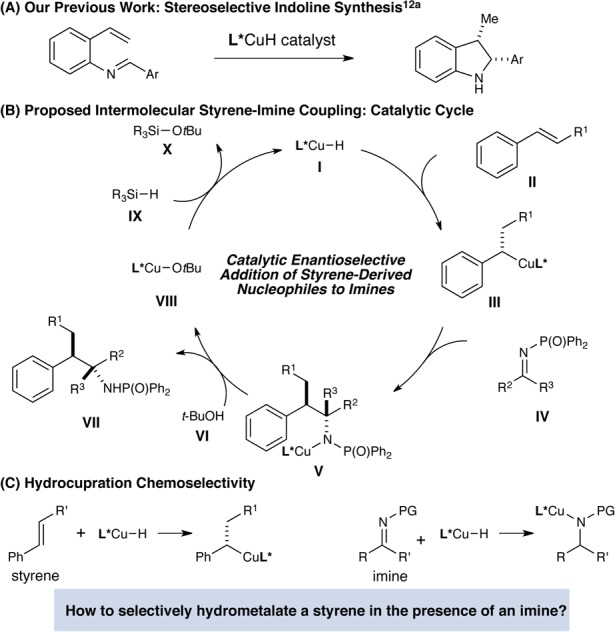
Copper-catalyzed enantioselective addition of styrene-derived nucleophiles to imines.
While successful in the intramolecular case, the development of an intermolecular transformation would require the chemoselective hydrometalation of a styrene in the presence of a suitably protected imine. The identification of reaction conditions with the desired selectivity was expected to be challenging, since several of the commonly used imine protecting groups are strongly electron-withdrawing, and hydrocupration of the imine was expected to be facile (Figure 1B). Indeed, important work by Lipshutz showed that phosphine-ligated copper hydride species can be highly efficient catalysts for the enantioselective reduction of N-phosphinoyl ketimines.17 The competitive hydrocupration of the imine component would lead to its unproductive reduction and result in diminished yields of the desired styrene-derived adduct.
Following a survey of imine protecting groups and reaction conditions, N-phosphinoylimines were identified as suitable substrates for the desired asymmetric addition reaction. Phosphinoylimines could be conveniently prepared from the parent carbonyl compounds, and the deprotection of the resulting N-phosphinoylamines could be effected under acidic conditions (see SI). The copper catalyst based on Ph-BPE (L6) was found to be highly effective for the asymmetric nucleophilic addition, providing the coupled product 3 in excellent yield with outstanding enantiocontrol (Table 1, entry 1). The effectiveness of this ligand is consistent with the ligand effects observed in our previously reported indoline synthesis, for which Ph-BPE was also found to be the optimal ligand.12a In contrast, the use of other ligands led to the formation of a significant amount of reduction product 4. For example, our optimal catalyst for hydroamination, based on DTBM-SEGPHOS (L1),10 provided only 9% product 3 and 85% reduced amine 4. In addition, bulky phenyl substituents on the phospholane moiety of the BPE ligand proved essential not only for achieving high levels of enantioselectivity but also for enabling the desired chemoselectivity (entries 4–6). Omitting tert-butanol resulted in a dramatic loss in yield (entry 7). We reasoned that the protonation of copper amide V (L*Cu-N(P(O)Ph2)R*, Figure 1) with t-BuOH bypassed the slow transmetalation of V with a hydrosilane and thus greatly facilitated the catalyst turnover.12a,18 The choice of the N-protecting group of the imine was crucial for the successful implementation of this transformation, with sulfonyl, aryl, and alkyl protecting groups providing much lower yields (entries 8 and 9, see SI for full details).
Table 1. Copper-Catalyzed Asymmetric Addition of Styrene-Derived Nucleophiles to Imines: Effect of Reaction Parametersa.

| entry | variation from the “standard conditions” | yield of 3 (dr) | ee of 3 | yield of 4 |
|---|---|---|---|---|
| 1 | none | 95% (3:1) | 99% ee (99% ee)b | 1.5% |
| 2 | L1 instead of L6 | 9% (1:1) | 95% ee (99% ee) | 85% |
| 3 | L2 instead of L6 | <5% | n.d. | 65% |
| 4 | L3 instead of L6 | <5%c | n.d. | 20% |
| 5 | L4 instead of L6 | 56% (1:1) | 86% ee (83% ee) | 35% |
| 6 | L5 instead of L6 | 46% (1:1) | 78% ee (70% ee) | 54% |
| 7 | no t-BuOH | 41% (3:1) | 99% ee (99% ee) | 31% |
| 8 | 2b instead of 2a | 5%d | n.d. | 36% |
| 9 | 2c instead of 2a | 6%e | n.d. | <10% |
Reaction conditions: 1 (0.2 mmol), 2 (0.1 mmol), Cu(OAc)2 (5 mol %), L (6 mol %), t-BuOH (2 equiv), (MeO)2MeSiH (5 equiv), THF (0.5 M), rt, 12 h. Yields were determined by 1H NMR spectroscopic analysis using 1,3,5-trimethoxybenzene as the internal standard. Enantiomeric excess values were determined by chiral HPLC analysis.
The ee of the minor diastereomer is shown in parentheses.
30% conv. of 2a.
41% conv. of 2b.
12% conv. of 2c.
We next set out to investigate the substrate scope of this reaction (Table 2). A number of styrenes with various electronic properties (5a–5f) could be successfully converted into the corresponding amine product with excellent levels of enantioselectivity and moderate levels of diastereoselectivity. Chlorinated (5e) and brominated (5f) styrenes were tolerated, thus opening up possibilities for further derivatization using cross-coupling technologies. Interestingly, the use of an ortho-substituted styrene furnished the coupled product with good diastereocontrol (5g), although slight decrease of enantioselectivity was also observed. Furthermore, nitrogen heterocycles (5h and 5i) and a β-substituted styrene (5j) were also suitable for this asymmetric addition. The lower yield of 5j is presumably due to the slower hydrocupration of these β-substituted styrenes.
Table 2. Substrate Scope of Styrenesa.


Yields are of isolated product as a mixture of two diastereomers on a 0.5 mmol scale. The relative and absolute stereochemistry of both diastereomers was determined by X-ray crystallography or further derivatization of the product, see SI for details. The ee of the minor diastereomer is shown in parentheses.
We next examined the scope of the imine coupling partner (Table 3). Our protocol was found to readily accommodate a variety of electron-rich (6a and 6b) and electron-poor (6c and 6d) imines. Furthermore, a broad spectrum of imines bearing a pendant heterocycle, including an indole (6e), a thiophene (6f), a pyridine (6g), an azaindole (6h), and a thiazole (6i), could be effectively transformed into the corresponding amine products in a highly enantioselective fashion. These results are notable because heterocycles are prevalent structural elements in pharmaceuticals and the metal-catalyzed enantioselective transformation of these substrates is generally challenging. Additionally, an α,β-unsaturated imine also underwent the current nucleophilic addition with excellent enantiocontrol (6j).19 Moreover, substantially less reactive ketimines (6k–6m) also represented suitable substrates, further highlighting the generality of the current enantioselective addition process. Finally, the absolute stereochemistry at the benzylic position is found to be opposite to that of the previously reported hydrofunctionalizations using the same ligand.11a,12a,12c Combined with results of our computational studies on the hydrocupration step, this observation suggests that the C–C bond formation (III + IV → V, Figure 1) is a stereoinvertive process (see SI for details).
Table 3. Substrate Scope of Iminesa.


Yields are of isolated product on a 0.5 mmol scale, see SI for details.
This copper-catalyzed enantioselective imine addition was scalable and required very low catalyst loadings. For example, we were able to couple 3-chlorostyrene (7) to imine 2a on a gram scale using only 0.5 mol % catalyst (Figure 2, eq 1). Furthermore, the chiral sulfinimines developed by Ellman4a could be used in lieu of N-phosphinoylimines, delivering enantiomerically enriched amine 9 with excellent diastereocontrol (eq 2).20 The relative and absolute configuration of 9 were ascertained by X-ray crystallographic analysis.
Figure 2.
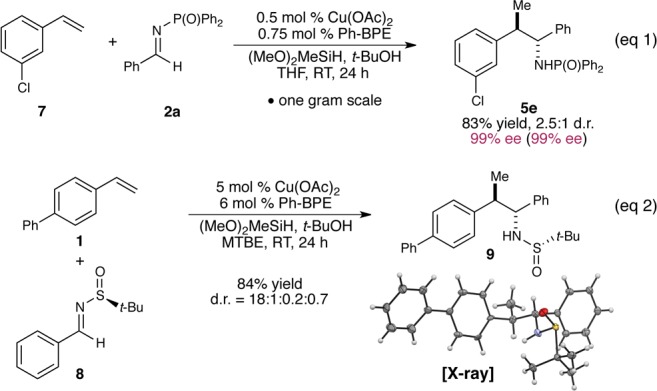
Gram-scale synthesis with low catalyst loadings and the use of chiral sulfinimines.
To gain further insight into the unique chemoselectivity during the hydrocupration event, we studied the rates of the CuH-catalyzed reduction of imine 2a with different supporting ligands by monitoring the reaction progress using calorimetric analysis established by Blackmond (Figure 3A).21 Our kinetic data suggested that the undesired imine reduction with Ph-BPE is considerably slower than that using DTBM-SEGPHOS and SEGPHOS. Furthermore, we performed density functional theory calculations for the styrene hydrocupration with the same set of CuH catalysts (Figure 3B).22 Our computational investigations revealed that while the activation barrier for styrene hydrocupration using Ph-BPE is comparable to that for DTBM-SEGPHOS, this Ph-BPE-based hydrocupration is ∼1.8 × 102 times faster than that using SEGPHOS. Taken together, these findings provide evidence for the unusual ability of the Ph-BPE-based CuH catalyst to suppress the undesired imine reduction while simultaneously facilitating the hydrocupration of weakly polarized styrene substrates.
Figure 3.
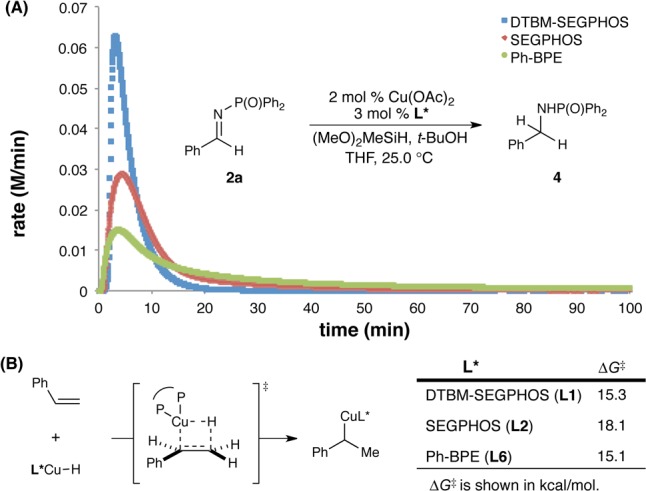
Mechanistic insight into the unusual chemoselectivity of hydrocupration.
In summary, we have developed a mild and general copper-catalyzed process for the enantioselective addition of styrene-derived nucleophiles to imines. This process tolerates a broad range of functionalized and heterocyclic substrates, providing rapid access to amines bearing vicinal stereogenic centers with excellent enantiocontrol. Utilizing a diverse range of other (poly)unsaturated hydrocarbons as latent nucleophiles in the asymmetric addition to imines is currently underway and will be reported in due course.
Acknowledgments
Financial support was provided by the National Institutes of Health (GM46059) and the MIT Undergraduate Research Opportunity Program (I.B.P). We acknowledge Dr. Gang Lu and Prof. Peng Liu (University of Pittsburgh) for helpful discussion on the computational study. We thank Dr. Yiming Wang (MIT) for his advice on this manuscript and Dr. Peter Müller (MIT) for X-ray crystallographic analysis. Calculations were performed at the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06299.
The authors declare no competing financial interest.
Supplementary Material
References
- Chiral Amine Synthesis; Nugent T. C., Ed.; Wiley-VCH: Weinheim, 2010. [Google Scholar]
- For recent reviews:; a Xie J.-H.; Zhu S.-F.; Zhou Q.-L. Chem. Rev. 2011, 111, 1713. 10.1021/cr100218m. [DOI] [PubMed] [Google Scholar]; b Tang W.; Zhang X. Chem. Rev. 2003, 103, 3029. 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- Hydrolases in Organic Synthesis; Bornscheuer U. T., Kazlauskas R. J., Ed.; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- For a review on the use of tert-butylsulfinamides:; a Robak M. T.; Herbage M. A.; Ellman J. A. Chem. Rev. 2010, 110, 3600. 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]; For reviews on catalytic asymmetric addition reactions to imines:; b Kobayashi S.; Mori Y.; Fossey J. S.; Salter M. M. Chem. Rev. 2011, 111, 2626. 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]; c Kobayashi S.; Ishitani Chem. Rev. 1999, 99, 1069. 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]
- Selected reviews:; a Jang H.-Y.; Krische M. J. Acc. Chem. Res. 2004, 37, 653. 10.1021/ar020108e. [DOI] [PubMed] [Google Scholar]; b Skucas E.; Ngai M.-Y.; Komanduri V.; Krische M. J. Acc. Chem. Res. 2007, 40, 1394. 10.1021/ar7001123. [DOI] [PubMed] [Google Scholar]
- Selected recent examples:; a Liang T.; Zhang W.; Krische M. J. J. Am. Chem. Soc. 2015, 137, 16024. 10.1021/jacs.5b12131. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zbieg J. R.; Yamaguchi E.; McInturff E. L.; Krische M. J. Science 2012, 336, 324. 10.1126/science.1219274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kong J.-R.; Cho C.-W.; Krische M. J. J. Am. Chem. Soc. 2005, 127, 11269. 10.1021/ja051104i. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Skucas E.; Kong J.-R.; Krische M. J. J. Am. Chem. Soc. 2007, 129, 7242. 10.1021/ja0715896. [DOI] [PubMed] [Google Scholar]; For a Ni-catalyzed asymmetric reductive coupling of alkynes and imines:; c Zhou C.-Y.; Zhu S.-F.; Wang L.-X.; Zhou Q.-L. J. Am. Chem. Soc. 2010, 132, 10955. 10.1021/ja104505t. [DOI] [PubMed] [Google Scholar]; For nonenantioselective examples:; d Chen T.-Y.; Tsutsumi R.; Montgomery T. P.; Volchkov I.; Krische M. J. J. Am. Chem. Soc. 2015, 137, 1798. 10.1021/ja5130258. [DOI] [PubMed] [Google Scholar]; e Oda S.; Franke J.; Krische M. J. Chem. Sci. 2016, 7, 136. 10.1039/C5SC03854E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a non-enantioselective example of reductive coupling between electron-deficient vinylazaarenes and imines:Komanduri V.; Grant C. D.; Krische M. J. J. Am. Chem. Soc. 2008, 130, 12592. 10.1021/ja805056g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on CuH catalysis:; a Deutsch C.; Krause N.; Lipshutz B. H. Chem. Rev. 2008, 108, 2916. 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]; b Rendler S.; Oestreich M. Angew. Chem., Int. Ed. 2007, 46, 498. 10.1002/anie.200602668. [DOI] [PubMed] [Google Scholar]
- For a review:Wang Y.-M.; Pirnot M. T. Angew. Chem., Int. Ed. 2016, 55, 48. 10.1002/anie.201507594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Miki Y.; Hirano K.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2013, 52, 10830. 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]; b Miki Y.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2014, 16, 1498. 10.1021/ol5003219. [DOI] [PubMed] [Google Scholar]; c Nishikawa D.; Hirano K.; Miura M. J. Am. Chem. Soc. 2015, 137, 15620. 10.1021/jacs.5b09773. [DOI] [PubMed] [Google Scholar]
- a Ascic E.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 4666. 10.1021/jacs.5b02316. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang Y.-M.; Bruno N. C.; Placeres Á. L.; Zhu S.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 10524. 10.1021/jacs.5b07061. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang Y.-M.; Buchwald S. L. J. Am. Chem. Soc. 2016, 138, 5024. 10.1021/jacs.6b02527. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bandar J. S.; Ascic E.; Buchwald S. L. J. Am. Chem. Soc. 2016, 138, 5821. 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yang Y.; Perry I. B.; Lu G.; Liu P.; Buchwald S. L. Science 2016, 353, 144. 10.1126/science.aaf7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related CuH-catalyzed C–C bond formation involving vinyl- or allylcopper intermediates:; c Uehling M. R.; Suess A. M.; Lalic G. J. Am. Chem. Soc. 2015, 137, 1424. 10.1021/ja5124368. [DOI] [PubMed] [Google Scholar]; d Suess A. M.; Uehling M. R.; Kaminsky W.; Lalic G. J. Am. Chem. Soc. 2015, 137, 7747. 10.1021/jacs.5b03086. [DOI] [PubMed] [Google Scholar]; e Iwasaki T.; Shimizu R.; Imanishi R.; Kuniyasu H.; Kambe N. Angew. Chem., Int. Ed. 2015, 54, 9347. 10.1002/anie.201503288. [DOI] [PubMed] [Google Scholar]
- a Du Y.; Xu L.-W.; Shimizu Y.; Oisaki K.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2008, 130, 16146. 10.1021/ja8069727. [DOI] [PubMed] [Google Scholar]; b Choi B.; Saxena A.; Smith J. J.; Churchill G. H.; Lam H. W. Synlett 2015, 26, 350. 10.1055/s-0034-1379548. [DOI] [Google Scholar]
- Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062. 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]
- For recent reviews on copper-catalyzed asymmetric nucleophilic addition to imines:; a Shibasaki M.; Kanai M. Chem. Rev. 2008, 108, 2853. 10.1021/cr078340r. [DOI] [PubMed] [Google Scholar]; b Yamada K.; Tomioka K. Chem. Rev. 2008, 108, 2874. 10.1021/cr078370u. [DOI] [PubMed] [Google Scholar]; Selected examples of copper-catalyzed enantioselective addition to imines using preformed organometallic reagents:; c Wada R.; Shibuguchi T.; Makino S.; Oisaki K.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2006, 128, 7687. 10.1021/ja061510h. [DOI] [PubMed] [Google Scholar]; d Vieira E. M.; Snapper M. L.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 3332. 10.1021/ja200311n. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Vieira E. M.; Haeffner F.; Snapper M. L.; Hoveyda A. H. Angew. Chem., Int. Ed. 2012, 51, 6618. 10.1002/anie.201202694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshutz B. H.; Shimizu H. Angew. Chem., Int. Ed. 2004, 43, 2228. 10.1002/anie.200353294. [DOI] [PubMed] [Google Scholar]
- For the use of t-BuOH in CuH-catalyzed reactions:; a Chen J.-X.; Daeuble J. F.; Stryker J. M. Tetrahedron 2000, 56, 2789. 10.1016/S0040-4020(00)00133-2. [DOI] [Google Scholar]; b Hughes G.; Kimura M.; Buchwald S. L. J. Am. Chem. Soc. 2003, 125, 11253. 10.1021/ja0351692. [DOI] [PubMed] [Google Scholar]; c Lipshutz B. H.; Servesko J. M.; Taft B. R. J. Am. Chem. Soc. 2004, 126, 8352. 10.1021/ja049135l. [DOI] [PubMed] [Google Scholar]; d Rainka M. P.; Aye Y.; Buchwald S. L. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5821. 10.1073/pnas.0307764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The use of aliphatic aldimines provided low yield.
- The use of (R,R)-Ph-BPE instead of (S,S)-Ph-BPE afforded the amine product in 80% yield and 1:1 dr.
- Blackmond D. G. Angew. Chem., Int. Ed. 2005, 44, 4302. 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- Calculations were performed at the M06/SDD(Cu)-6-311+G(d,p)/SMD(THF) level of theory with geometries optimized at the B3LYP/SDD(Cu)-6-31G(d) level. See SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.