

Abstract
Protein folding occurs on a time scale similar to peptide bond formation by the ribosome, which has long sparked speculation that altering translation rate could alter the folding mechanism or even the final folded structure of a protein in vivo. Recent results have provided strong support for this model: synonymous substitutions to codons with different usage frequency, which are often translated at different rates, have been shown to significantly alter the co-translational folding mechanism of some proteins, leading to altered cell function. Here we review recent progress towards understanding the connections between synonymous codon usage, translation rate and co-translational protein folding mechanisms.
Keywords: translation rate, ribosome, rare codons, aggregation, kinetic stability, non-native folding intermediate
Graphical Abstract
Introduction
Most amino acids are encoded by more than one codon. However, these synonymous codons are not used with equal frequency. In general, common codons are translated by the ribosome more quickly than their synonymous rare counterparts. For this reason, synonymous common codons were historically considered “optimal” for gene expression, as faster translation will facilitate rapid accumulation of a protein in the cell. Consistent with this hypothesis, many highly expressed genes are enriched in common codons [1]. For genes encoding less abundant proteins, synonymous codon usage was presumed to be essentially “silent”, representing an evolutionarily neutral mutation of one synonymous codon for another. Indeed, the presumption that synonymous mutations represent merely genomic “background noise” is the basis of the widely used dN/dS calculation (the ratio of non-synonymous versus synonymous substitutions in a coding sequence; also referred to as Ka/Ks) for functional selection [2].
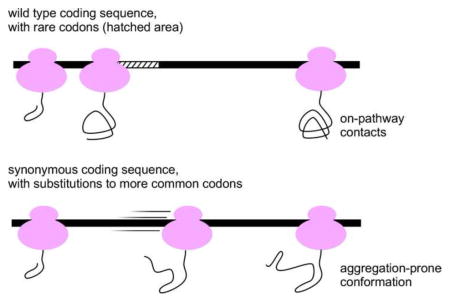
However, the longstanding focus of the effects of synonymous codon usage on protein level disregards another effect, on protein folding. Recent results have now provided irrefutable evidence that synonymous codon usage is non-random, and that synonymous substitutions can significantly perturb the folding efficiency of the encoded protein, in some cases leading to adverse effects on cell function (Figure 1). Clearly, for codons in these coding sequences, “common” and “optimal” cannot be used interchangeably [3]. These results are shifting attention to the specific effects on co-translational protein folding that can be achieved by modulating local translation rate. Questions include: What codon usage is “optimal” for each gene? Can codon usage affect the folding mechanism and/or native topology of the encoded protein? If so, how? More broadly, of the enormous numbers of rare codons found in naturally occurring coding sequences, which ones are most likely to impact co-translational folding or another aspect of protein production, versus have no effect (be truly “silent”)? Below we discuss the effects of synonymous codon substitutions on translation rate, several recent exciting studies reporting the effects of synonymous codon substitutions on the co-translational folding of specific proteins, and highlight other mechanisms available to modulate the local rate of protein synthesis in vivo.
Figure 1.

Effects of local translation rate on co-translational folding of the nascent chain. When translation is slow due to synonymous rare codons (shown here; hatched area within black mRNA), the partially synthesized nascent chain can achieve an equilibrium or near-equilibrium conformation that may not be kinetically accessible when C-terminal portions of the nascent chain are translated more quickly via synonymous common codons. See Figure 3 for an energy landscape perspective of these processes.
Ribosome Structure and the Logistics of Co-translational Protein Folding
In the cell, every protein is synthesized from N- to C-terminus by a ribosome. Peptide bond formation occurs deep within the ribosome, and the nascent polypeptide chain first passes through the ribosome exit tunnel (Figure 2) [4,5]. The narrowness and length of the exit tunnel (~20x100 Å) significantly constrains the most C-terminal residues of the nascent polypeptide chain to a small range of mostly extended or α-helical conformations [6,7]. The formation of bulky tertiary structure does not begin until the nascent chain is long enough to emerge from the exit tunnel (>35 aa), although there is evidence that the broader ‘vestibule’ near the end of the tunnel is wide enough to enable the nascent chain to fold back on itself to make some local tertiary structure contacts [8,9]. Synonymous codon substitutions near the 5′ end of a coding sequence are therefore unlikely to affect co-translational protein folding, as very little of the nascent chain has been translated and the portion that has been synthesized is constrained within the tunnel. Indeed, 5′ synonymous codon substitutions instead tend to alter protein abundance, due to altered translation initiation efficiency [10,11]. Synonymous substitutions farther within the coding sequence are more likely to affect co-translational folding mechanisms and are the focus of this article.
Figure 2.

Cross-section view of the E. coli ribosome (yellow and grey represent space-filling structures of the 30S and 50S ribosomal subunits, respectively). Shown are the position of the P-site tRNA (blue) and the path of the bound nascent chain (green, 36 aa polyalanine sequence) modeled through the exit tunnel to the ribosome surface. Image adapted from [4,5].
Synonymous Codon Calculations and Effects on Translation Rate
The effect of a single synonymous codon substitution on absolute in vivo translation rate has proven difficult to measure directly, or predict accurately. In general, there is an inverse correlation between codon rarity and translation rate [12,13], and codon usage frequencies have proven useful for predicting total translation time over an entire sequence [1]. However, there are clearly additional factors (including nutrient levels and codon context [14–17]) that affect the rate of translation of a single specific codon, although the relative importance and interplay between these factors is still poorly understood. For these reasons, codon usage frequency is less predictive of translation rate for an individual codon or short region than for an entire coding sequence. Nevertheless, the current paucity of absolute translation rate measurements and incomplete understanding of the specific mechanisms that regulate local translation rate has lead to the widespread use of codon usage frequency as a very convenient (albeit limited) proxy for local translation rate.
Codon usage has been quantified in many different ways, with the underlying mathematics designed to highlight a particular feature of interest. One of the first and most widely used calculators was codon adaptive index (CAI), which was developed to test connections between codon usage and expression level [1]. However, because CAI normalizes codon usage to very common synonymous codons, it is less suited to highlight regions encoded using less common codons. Likewise, calculators that compare absolute codon usage (comparing the usage frequency of one codon to all other 60 amino acid-encoding codons) are ill suited to identify synonymous codon usage differences that could affect folding mechanisms for identical protein sequences, because rare amino acids like Cys are only encoded by codons that are rare in an absolute sense, but one Cys codon is used more often than the other and hence is more common in a relative sense. Other calculators, like %MinMax [18], compare relative codon usage frequencies – the usage frequency of one codon that encodes an amino acid versus its synonyms – and were designed specifically to identify patterns in synonymous codon usage. %MinMax has accurately predicted the effects of synonymous codon substitutions on co-translational folding [12]. When evaluating codon usage calculators, it is important to keep in mind that individual codon usage signals are extremely weak: in E. coli, even the rarest codon encoding an amino acid (AGG; codes for arginine) still encodes >3% of all arginine residues. For this reason, in order to highlight regions of more statistically significant clusters of synonymous rare codons the %MinMax output is averaged over a sliding window of 17 codons.
An additional strength of calculators based only on codon usage is that this information is readily available for all fully sequenced genomes. Although more complicated calculators have also been developed to capture specific mechanisms hypothesized to regulate translation rate (including tRNA concentration and demand [19–21] and the effects of mismatch at the wobble position [22]), these calculators require additional information such as tRNA concentrations that is currently available for only a few organisms (e.g., Escherichia coli and Saccharomyces cerevisiae), and still suffer from limited predictive power. Initially, it was hoped that ribosome profiling [23] would reveal codon-specific effects on translation. However, the majority of profiling studies have detected only more dramatic effects on translation rate, for example those due to amino acid starvation or nascent chain features [24,25].
Effects of Synonymous Codon Substitutions on Protein Folding Mechanism and Structure
One of the earliest studies of the effects of synonymous codon substitutions on protein folding and structure was of chloramphenicol acetyltransferase (CAT) [26]. Substituting a large cluster of rare codons within the CAT coding sequence with synonymous common codons led to faster translation in vitro and increased protein production compared to the wild type coding sequence. Yet the products of this “optimized” CAT coding sequence had lower specific activity, suggesting that when translated faster CAT adopts an alternative conformation that is still soluble but less active [26]. More recently, several independent studies in a range of diverse cell types have expanded upon this finding, demonstrating that synonymous codon selection can affect the production of functional protein. For example, “optimizing” the codon usage in the Neurospora Frequency (FRQ) coding sequence was sufficient to eliminate circadian rhythms in vivo. These rare-to-common synonymous codon substitutions increased amount of FRQ produced [27], but also altered FRQ phosphorylation and its trypsin susceptibility. Taken together, these results indicate that when synthesized more quickly, FRQ folds to a different, less functional structure [28]. Synonymous codon substitutions that impact human cell function have also been identified: ‘silent’ single nucleotide polymorphisms (SNPs) in the human multidrug resistance 1 (MDR1) coding sequence correlate with altered effectiveness of chemotherapeutic drugs. These substitutions result in a gene product, P-glycoprotein (P-gp1), with altered drug transport properties and trypsin digestion pattern, possibly due to a different folded structure [29]. These results suggest that synonymous codon usage may play a role in the production of functional proteins in a wide array of (possibly all) organisms, and may represent a broader underlying mechanism for the effects of synonymous codon substitutions associated with human diseases [30]. Of note, in each of the studies described above, altered codon usage led to an alternative folded structure – an alternative energy minimum conformation. The ability of these proteins to adopt distinct, stably folded structures via manipulation of their folding pathway indicates that these proteins fold under kinetic control, rather than to a single global energy minimum structure [31] (see Figure 3). Although proteins that fold under kinetic control are rarely used as models of protein refolding in vitro, a significant fraction of the proteome exhibits this behavior [32,33], which may be a designable feature [34].
Figure 3.

A hypothetical folding energy landscape for a protein prone to misfolding to an enzymatically inactive, kinetically trapped conformation in vitro (I1). In addition to the energy landscape for the full-length, free protein, also shown are energy landscapes for short nascent chain lengths (red to blue) when tethered to the ribosome (tan). Two specific residues in contact in the native structure are shown as magenta filled circles; a third residue, which forms stabilizing non-native interactions at short nascent chain lengths, is shown as an open circle. These non-native interactions stabilize a non-native intermediate (I2) that is not stable enough to be populated during refolding in vitro yet represents the global energy minimum conformation for short nascent chain lengths. This intermediate may be stabilized solely via interactions within the nascent chain, or by intermolecular interactions with the ribosome surface or ribosome-bound molecular chaperones. Locally reducing the rate of translation during the synthesis of any portion of the N-terminus of the chain will provide additional time to populate productive, non-native folding intermediates such as I2, and avoid self-association. Conversely, accelerating the synthesis of C-terminal portions of the chain by substituting rare codons with common synonymous codons increases the probability of populating I1 instead of I2;; i.e., the longer nascent chain length (dark blue) will appear faster, enabling access to the I1 conformation.
While the studies above identified rare-to-common codon substitutions that led to an alternative, less-functional – but still soluble – structure, other synonymous codon changes have been found that increase aggregation of the encoded protein. For example, common synonymous codon substitutions at 57 positions in the first nucleotide binding domain (NBD1) of cystic fibrosis transmembrane conductance regulator (CFTR) increased the likelihood of NBD1 aggregation, and also led to increased aggregation of full-length CFTR [35]. Similarly, engineering the coding sequence of firefly luciferase to contain codons predicted to have fast translation rates led to a protein that was indeed translated faster – as demonstrated by pulse-chase analysis – but was more prone to aggregation and had lower specific activity [22]. More broadly, increasing the expression of low abundance tRNAs (which often correlate with rare codons) increased the aggregation of many E. coli cytoplasmic proteins [36], suggesting that slower translation might lead to higher folding yields for a variety of proteins. Consistent with this hypothesis, a correlation has been observed between translational pauses in E. coli coding sequences and protein solubility [37]
Although the genetic code is largely universal, codon usage frequencies are not. This means that expressing a heterologous coding sequence in E. coli can lead to major changes in local codon usage patterns and corresponding translation rates. In this scenario, the conventional wisdom is to ‘optimize’ codon usage by converting codons that are rare in E. coli to their more common counterparts, assuming that there is everything to be gained and nothing to be lost by translating a protein as quickly as possible. But if the goal is to optimize folding yield rather than the number of copies synthesized, a more appropriate approach might be to ‘harmonize’ codon usage, to match the codon usage frequency patterns in the host organism [16,38,39]. Because the translation rate in E. coli is approximately five times faster than in eukaryotic organisms, ‘harmonizing’ may still lead to the too-rapid appearance of the nascent chain. Consistent with this, the folding yield of some eukaryotic proteins was enhanced by globally reducing the rate of translation in E. coli [40]. However, synthesizing a protein too slowly can also lead to negative effects on folding, for example by allowing more time for the N-terminal portion of a nascent chain to form non-native interactions that lead to misfolding and aggregation [39].
Do we now understand enough about the connections between synonymous codon usage and co-translational folding to include synonymous codon selection as a feature of protein design, to regulate protein folding in vivo? This is a new area with much still to explore, but a recent study demonstrated that selection between two alternative folded structures can be steered by synonymous codon selection. In this case, the designed protein (YKB) consisted of three half-domains where the N- and C-terminal half-domains compete with each other to interact with the central half-domain [12]. YKB folding leads to one of two mutually exclusive folded structures with distinct fluorescent properties: yellow fluorescence, if the N-terminus wins the competition (to make YK-B), or cyan fluorescence, if the C-terminus wins (Y-KB). Synonymous codon substitutions in the sequence encoding the C-terminal half-domain altered the ratio of yellow to cyan fluorescence in vivo: slow translation with rare codons led to more N-terminal folding and hence more yellow fluorescence, whereas faster translation with common codons leads to a more even distribution of yellow and cyan fluorescence, closer to the 50:50 refolding yield observed in vitro [12].
Co-translational Folding: Domain-by-Domain?
Once emerged from the ribosome exit tunnel, the nascent chain can undergo significant folding even while its C-terminus is still tethered to the ribosome. Indeed, addition of a C-terminal extension, to enable the natural C-terminal residues of a domain to emerge from the ribosome exit tunnel, can enable formation of native structure, spectral properties and enzymatic activity for the ribosome-bound nascent chain [41–43]. These results have sparked many to wonder whether synonymous rare codons are more likely to appear 30–40 codons after an mRNA segment encoding a protein structural domain. Such a positioning could facilitate a domain-by-domain, beads-on-a-string folding mechanism: each domain would be granted additional time to fold just after it emerges from the exit tunnel, before the appearance of a C-terminal domain that might lead to misfolding and aggregation. The YKB study described above demonstrates that such positioning of synonymous rare codons can in principle increase N-terminal folding efficiency [12]. However, for naturally occurring coding sequences where synonymous codon substitutions have been shown to affect protein folding (see above), no correlation has been observed between the placement of rare codons and domain boundaries. Moreover, attempts to correlate codon usage broadly with protein domain boundaries have not identified a clear pattern [44], perhaps highlighting an important distinction between a possible function versus one selected through evolution. Alternatively, these results may reflect that proximity to the ribosome surface can alter folding of the nascent chain in unexpected ways [45]. Hence, if correlations between codon usage patterns and protein structure do exist in naturally occurring coding sequences, these patterns are likely much more subtle than a striking cluster of rare codons just after an obvious structural domain. If there is not a clear domain-by-domain pattern for rare codon usage, do we understand enough about co-translational folding mechanisms to predict in what other positions rare codons would be most likely to affect folding mechanisms? Below we focus on what is currently known regarding co-translational folding intermediates, and the extent to which synonymous codon substitutions and other mechanisms could perturb protein folding pathways.
Co-translational Folding Intermediates: Resemblance to In Vitro Refolding
What are the conformations of nascent chains once they emerge from the exit tunnel, but before an entire structural domain has appeared? Hypothetically, if translation rate was infinitely fast, the C-terminus of the nascent chain would be available for folding at the same time as the N-terminus, and the energy landscape for folding would resemble the landscape for refolding in vitro after dilution from a chemical denaturant. In reality, the rate of translation is much slower: in vivo, formation of each peptide bond takes 0.05 sec for bacterial ribosomes and 0.2 sec for eukaryotic ribosomes. This means that translation of a protein of average length (300–400 aa) takes 18–70 s, a generous time period within which co-translational folding can occur, particularly given that formation of stable secondary and tertiary structure often occurs on the millisecond to seconds time scale [33,46]. Indeed, recent FRET measurements of co-translational folding for a small five-helix bundle domain in an efficient reconstituted in vitro translation mixture (kelongation = 3.6 aa/sec) showed that formation of both α-helix secondary structure and the native tertiary structure were limited by the rate of translation [47]. This means that altering the rate of translation can alter the conformations populated during protein synthesis.
Intriguingly, the conformations adopted during co-translational folding need not resemble the conformations of intermediates populated during refolding in vitro [48–51]. Whether a ribosome-bound nascent polypeptide chain will adopt conformations not populated during in vitro refolding will be affected by the topology of the native protein structure and the kinetics of translation, which could alter access to the energy landscape for folding. For example, in some native protein topologies the N- and C-termini are in close proximity (Figure 3). During in vitro refolding of full length polypeptide chains, docking of the N- and C-terminal portions of these chains may stabilize an early folding intermediate, but this intermediate might represent a stable kinetic trap (Figure 3, I1) and/or be prone to self-association. In contrast, during protein synthesis it is unlikely that folding will begin from this intermediate, as initially the N-terminus of the chain will be available for folding before the C-terminus. The minimum energy structure for these partially synthesized chains may instead be stabilized by non-native interactions with other N-terminal portions of the nascent polypeptide chain, leading to the population of a folding intermediate distinct from those populated during refolding in vitro (Figure 3, I2). Whether a co-translational folding intermediate will or will not resemble an in vitro refolding intermediate will depend on the relative stabilities of a native-like N-terminal sub-structure versus a potential non-native folding intermediate like I2. Moreover, these partially synthesized nascent chain conformations might be stabilized by interactions with the ribosome surface and/or ribosome-bound molecular chaperones. Regardless of native state topology, it is thermodynamically unlikely that N-terminal portions of a nascent chain remain in a largely extended conformation until the C-terminus appears [46]. Note that although non-native folding intermediates are unusual features of protein refolding in vitro, the proteins with folding pathways now known to be affected by changes in local translation rate (e.g., CAT; see above) have folding mechanisms that are much more complex than typical proteins used to study in vitro refolding (CAT, for example, is a homotrimer, while MDR1 is a polytopic transmembrane protein). Going forward, it will be essential to develop experimental methods and theory to enable detailed investigations of these more complex folding pathways,
In general, if the intermediates populated during refolding in vitro define the only path to the native structure, slowing down the rate of translation would only slow the rate of formation of these folding intermediates (adopted co-translationally if energetically accessible; post-translationally if not). If instead there are multiple routes to the native structure, then minor shifts to the energy landscape for folding – such as increasing the time before the appearance of the nascent polypeptide C-terminus – could alter the flux of polypeptide chains through alternative conformations not populated during refolding in vitro. In this way, a reduction in local translation rate and its subsequent effects on co-translational folding can provide a mechanism to avoid populating certain partially folded conformations and instead populate others. The recent studies described above demonstrate that synonymous codon substitutions can achieve such fine-tuning of folding mechanisms. As mentioned above, a much more detailed understanding of the conformations of co-translational folding intermediates and the interactions that stabilize them is required before we will understand these effects in detail.
Caution Ahead: Other Effects of Synonymous Codon Substitutions
It is important to note that, in addition to altered co-translational folding, synonymous codon substitutions can affect a wide variety of other aspects of protein production [44], including altered splicing efficiency [52], translational fidelity [53], secretion efficiency [54–56] and mRNA half life [57]. Moreover, reducing translation rate simply to reduce expression level of some genes has been shown to provide a functional advantage [58,59]. Although these effects of synonymous codon substitutions are distinct from altered co-translational folding, many of these mechanisms also alter the amount of natively folded protein. As interest in the effects of synonymous codon substitutions continues to expand, keeping these alternative mechanisms in mind will be essential in order to design experiments that are capable of distinguishing between them. Moreover, it is important to remember that many synonymous substitutions will yield no discernable effect on the production of the encoded protein [18], akin to positions in the protein sequence that are widely tolerant to amino acid substitutions without significantly affecting the folding mechanism or stability of the native protein structure.
Other Mechanisms to Control Translation Rate
Beyond synonymous codon usage, a variety of other mRNA and nascent chain sequence features can affect local translation rate [17]. In most cases, however, these mechanisms constrain the amino acid sequence of the encoded nascent chain and as a result represent less versatile tools for tuning the co-translational folding of a desired protein sequence. Currently, it remains to be determined to what extent these sequence features alter co-translational protein folding. However, given that several of these mechanisms were discovered only recently, it would not be surprising if additional mechanisms and effects still remain to be discovered.
Amino acid sequences that affect translation rate
Specific amino acid sequences in the nascent protein can affect elongation rate. Ribosome arrest peptides (RAPs), such as the SecM stall sequence, interact with specific features of the ribosome exit tunnel during their biogenesis that transiently arrest elongation [60]. Similarly, stretches of positively charged amino acids in the nascent chain can slow translation by interacting with the overall negative electrostatic potential within the ribosome exit tunnel [61,62]. In addition, proline is translated more slowly than all other standard amino acids, due to its unusual N-alkyl group that both constrains its geometry and reduces the strength of the nitrogen nucleophile [63]. Translation of specific proline-rich motifs has been shown to inhibit elongation [64].
mRNA secondary structure
Despite the inherent helicase activity of the ribosome [65], stable mRNA secondary structure can reduce elongation rate [66,67]. mRNA secondary structure might therefore be used to regulate the rhythm of co-translational protein folding, for example in viral RNA [68]. It is important to note that mRNA structure can also trigger more drastic changes in translation, including reduced protein expression [57,69] and increased frameshifting [70].
Shine-Dalgarno (SD) sequences
Typically located ~8 nucleotide upstream of the AUG start codon in prokaryotic mRNAs, SD sequences base pair with anti-SD sequences in 16S rRNA to align the ribosome and initiate translation. SD sequences can also occur within coding regions. Although their impact on translation rate in vivo now appears smaller than previously thought [25,71], internal SD sequences are disfavored in E. coli coding sequences [24], possibly due to their negative effect on translation elongation rate. Consistent with this, introducing SD sequences within coding sequences has been shown to negatively impact protein accumulation [72].
Future Directions
The flurry of new findings described above have led to the inescapable conclusion that synonymous codon substitutions can alter not only the folding mechanism of an encoded protein but also its final folded structure. These results highlight a previously unrecognized importance of synonymous codon usage for production of functional proteins. It is still unknown what fraction of coding sequences will be affected by synonymous codon usage. However, it is striking that a significant fraction of both prokaryotic and eukaryotic coding sequences include statistically significant clusters of synonymous rare codons [18]. While it is important to keep in mind that these clusters could affect an aspect of protein production unrelated to co-translational folding, their sheer number suggests we have only just begun to understand the contributions of synonymous codon usage to efficient protein production.
Currently, we lack molecular-level details on precisely what, specifically, synonymous codon substitutions will change with regard to local translation rate and co-translational folding mechanism. As a result, we still lack a predictive understanding of the connections between synonymous codon choice and protein folding. Developing such an understanding will require addressing significant knowledge gaps with regard to the fundamental processes that underlie this connection, including the quantitative effect of synonymous codon substitutions on local translation rate, the effect(s) of the ribosome on co-translational folding, and what nascent chain sequences and native structure topologies are ideally suited for co-translational folding, to either a native-like or non-native conformation.
In light of these results, if the goal for a designed coding sequence is the highest possible folding efficiency, “optimizing” it by loading it up with very common synonymous codons should be regarded with skepticism. Techniques such as ‘harmonizing’ codon usage and/or adjusting bulk translation rate to more closely match the host organism might be more effective for producing the highest yield of functional protein. Such a shift in mindset will be needed in order to develop the new computational and experimental tools necessary to develop a truly predictive understanding of the effects of synonymous codon usage on co-translational folding, as well as other aspects of protein biogenesis.
Highlights.
Synonymous codon substitutions were long thought to be ‘silent’ mutations
In general, common codons are translated faster than rare synonyms
Recent results show these substitutions can affect folding of the encoded protein
These alterations can lead to an alternative folded structure, or increased aggregation
Matching codon usage to the host organism can improve folding during heterologous expression
Acknowledgments
Research in the Clark lab is supported by grants from the National Institutes of Health (R01 GM074807, R01 GM097573, U54 GM105816). G.J. is supported by a Notre Dame CBBI Graduate Fellowship (T32 GM075762) and the Protein Translation Research Network (U54 GM105816). We thank Daniel Wilson and Stefan Arenz for the preparation of Figure 2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Recent papers of particular interest (*) and exceptional interest (**) are highlighted.
- 1.Sharp PM, Li WH. The Codon Adaptation Index: A measure of directional synonymous codon usage bias, and its potential applications. Nucl Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimura M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 1977;267:275–276. doi: 10.1038/267275a0. [DOI] [PubMed] [Google Scholar]
- 3.Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med. 2014;20:604–613. doi: 10.1016/j.molmed.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bischoff L, Berninghausen O, Beckmann R. Molecular basis for the ribosome functioning as an L-tryptophan sensor. Cell Rep. 2014;9:469–475. doi: 10.1016/j.celrep.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Arenz S, Meydan S, Starosta AL, Berninghausen O, Beckmann R, Vazquez-Laslop N, Wilson DN. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol Cell. 2014;56:446–452. doi: 10.1016/j.molcel.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woolhead CA, McCormick PJ, Johnson AE. Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. Cell. 2004;116:725–736. doi: 10.1016/s0092-8674(04)00169-2. [DOI] [PubMed] [Google Scholar]
- 7.Lu J, Deutsch C. Folding zones inside the ribosomal exit tunnel. Nat Struct Mol Biol. 2005;12:1123–1129. doi: 10.1038/nsmb1021. [DOI] [PubMed] [Google Scholar]
- 8.Marino J, von Heijne G, Beckmann R. Small protein domains fold inside the ribosome exit tunnel. FEBS Lett. 2016;590:655–660. doi: 10.1002/1873-3468.12098. [DOI] [PubMed] [Google Scholar]
- 9.Tu L, Khanna P, Deutsch C. Transmembrane segments form tertiary hairpins in the folding vestibule of the ribosome. J Mol Biol. 2014;426:185–198. doi: 10.1016/j.jmb.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodman DB, Church GM, Kosuri S. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013;342:475–479. doi: 10.1126/science.1241934. [DOI] [PubMed] [Google Scholar]
- 11.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **12.Sander IM, Chaney JL, Clark PL. Expanding Anfinsen’s principle: contributions of synonymous codon selection to rational protein design. J Am Chem Soc. 2014;136:858–861. doi: 10.1021/ja411302m. YKB, an engineered protein that can adopt two alternative folded structures with different fluorescence properties, demonstrates that synonymous codon substitutions can alter the final structure of a protein in addition to the mechanism of co-translational folding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorensen MA, Kurland CG, Pedersen S. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 1989;207:365–377. doi: 10.1016/0022-2836(89)90260-x. [DOI] [PubMed] [Google Scholar]
- 14.Chevance FF, Le Guyon S, Hughes KT. The effects of codon context on in vivo translation speed. PLoS Genet. 2014;10:e1004392. doi: 10.1371/journal.pgen.1004392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramaniam AR, Zid BM, O’Shea EK. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell. 2014;159:1200–1211. doi: 10.1016/j.cell.2014.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS ONE. 2009;4:e7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Rodnina MV. The ribosome in action: Tuning of translational efficiency and protein folding. 2016 doi: 10.1002/pro.2950. A comprehensive review of our current understanding of nascent chain and mRNA sequence factors that can affect translation initiation, as well as elongation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke TF, Clark PL. Rare codons cluster. PLoS ONE. 2008;3:e3412. doi: 10.1371/journal.pone.0003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucl Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang G, Ignatova Z. Generic algorithm to predict the speed of translational elongation: implications for protein biogenesis. PLoS ONE. 2009;4:e5036. doi: 10.1371/journal.pone.0005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pechmann S, Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol. 2013;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spencer PS, Siller E, Anderson JF, Barral JM. Silent substitutions predictably alter translation elongation rates and protein folding efficiencies. J Mol Biol. 2012;422:328–35. doi: 10.1016/j.jmb.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammad F, Woolstenhulme CJ, Green R, Buskirk AR. Clarifying the Translational Pausing Landscape in Bacteria by Ribosome Profiling. Cell Rep. 2016;14:686–694. doi: 10.1016/j.celrep.2015.12.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komar AA, Lesnik T, Reiss C. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 1999;462:387–391. doi: 10.1016/s0014-5793(99)01566-5. [DOI] [PubMed] [Google Scholar]
- **27.Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol Cell. 2015;59:744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **28.Zhou M, Guo J, Cha J, Chae M, Chen S, Barral JM, Sachs MS, Liu Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013;495:111–115. doi: 10.1038/nature11833. This pair of studies (references 25 and 26) elegantly demonstrate that rare-to-common synonymous codon substitutions in the coding sequence of the circadian clock protein FRQ increase FRQ translation rate but result in altered FRQ structure and phosphorylation, and abolish circadian rhythms in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 30.Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30:308–321. doi: 10.1016/j.tig.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Baker D, Agard DA. Kinetics versus thermodynamics in protein folding. Biochemistry. 1994;33:7505–7509. doi: 10.1021/bi00190a002. [DOI] [PubMed] [Google Scholar]
- 32.Xia K, Manning M, Hesham H, Lin Q, Bystroff C, Colon W. Identifying the subproteome of kinetically stable proteins via diagonal 2D SDS/PAGE. Proc Natl Acad Sci USA. 2007;104:17329–17334. doi: 10.1073/pnas.0705417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braselmann E, Chaney JL, Clark PL. Folding the proteome. Trends Biochem Sci. 2013;38:337–344. doi: 10.1016/j.tibs.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broom A, Ma SM, Xia K, Rafalia H, Trainor K, Colon W, Gosavi S, Meiering EM. Designed protein reveals structural determinants of extreme kinetic stability. Proc Natl Acad Sci USA. 2015;112:14605–14610. doi: 10.1073/pnas.1510748112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *35.Kim SJ, Yoon JS, Shishido H, Yang Z, Rooney LA, Barral JM, Skach WR. Protein folding. Translational tuning optimizes nascent protein folding in cells. 2015;348:444–448. doi: 10.1126/science.aaa3974. Mutating rare, more slowly-translated codons to synonymous common codons in the cystic fibrosis transmembrane receptor (CFTR) led to increased CFTR aggregation. [DOI] [PubMed] [Google Scholar]
- 36.Fedyunin I, Lehnhardt L, Bohmer N, Kaufmann P, Zhang G, Ignatova Z. tRNA concentration fine tunes protein solubility. FEBS Lett. 2012;586:3336–3340. doi: 10.1016/j.febslet.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 37.Chadani Y, Niwa T, Chiba S, Taguchi H, Ito K. Integrated in vivo and in vitro nascent chain profiling reveals widespread translational pausing. Proc Natl Acad Sci USA. 2016;113:E829–E838. doi: 10.1073/pnas.1520560113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE. 2008;3:e2189. doi: 10.1371/journal.pone.0002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *39.Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, Komar AA. Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol Cell. 2016;61:341–351. doi: 10.1016/j.molcel.2016.01.008. Mutating rare, more slowly-translated codons to synonymous common codons in the cystic fibrosis transmembrane receptor (CFTR) led to increased CFTR aggregation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siller E, DeZwaan DC, Anderson JF, Freeman BC, Barral JM. Slowing bacterial translation speed enhances eukaryotic protein folding efficiency. J Mol Biol. 2010;396:1310–1318. doi: 10.1016/j.jmb.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 41.Ugrinov KG, Clark PL. Cotranslational folding increases GFP folding yield. Biophys J. 2010;98:1312–1320. doi: 10.1016/j.bpj.2009.12.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelkar DA, Khushoo A, Yang Z, Skach WR. Kinetic analysis of ribosome-bound fluorescent proteins reveals an early, stable, cotranslational folding intermediate. Journal of Biological Chemistry. 2012;287:2568–2578. doi: 10.1074/jbc.M111.318766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicola AV, Chen W, Helenius A. Co-translational folding of an alphavirus capsid protein in the cytosol of living cells. Nat Cell Biol. 1999;1:341–345. doi: 10.1038/14032. [DOI] [PubMed] [Google Scholar]
- *44.Chaney JL, Clark PL. Roles for synonymous codon usage in protein biogenesis. Ann Rev Biophys. 2015;44:143–166. doi: 10.1146/annurev-biophys-060414-034333. A comprehensive review of the diverse functions of synonymous codons and various ways to quantify codon usage. [DOI] [PubMed] [Google Scholar]
- *45.Cabrita LD, Cassaignau AM, Launay HM, Waudby CA, Wlodarski T, Camilloni C, Karyadi ME, Robertson AL, Wang X, Wentink AS, Goodsell LS, Woolhead CA, Vendruscolo M, Dobson CM, Christodoulou J. A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat Struct Mol Biol. 2016;23:278–285. doi: 10.1038/nsmb.3182. The authors use NMR to reveal that the proximity of the ribosome surface affects the structure of a nascent protein domain even after the entire domain has emerged from the exit tunnel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark PL. Protein folding in the cell: Reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- *47.Holtkamp W, Kokic G, Jager M, Mittelstaet J, Komar AA, Rodnina MV. Cotranslational protein folding on the ribosome monitored in real time. Science. 2015;350:1104–1107. doi: 10.1126/science.aad0344. Co-translational incorporation of fluorescent-labeled amino acids is used to monitor (via FRET) co-translational folding in real time in a highly efficient in vitro translation mixture. [DOI] [PubMed] [Google Scholar]
- 48.Clark PL, King J. A newly synthesized, ribosome-bound polypeptide chain adopts conformations dissimilar from early in vitro refolding intermediates. J Biol Chem. 2001;276:25411–25420. doi: 10.1074/jbc.M008490200. [DOI] [PubMed] [Google Scholar]
- 49.Evans MS, Sander IM, Clark PL. Cotranslational folding promotes β-helix formation and avoids aggregation in vivo. J Mol Biol. 2008;383:683–692. doi: 10.1016/j.jmb.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fedorov AN, Baldwin TO. Process of biosynthetic protein folding determines the rapid formation of native structure. J Mol Biol. 1999;294:579–586. doi: 10.1006/jmbi.1999.3281. [DOI] [PubMed] [Google Scholar]
- 51.Frydman J, Erdjument-Bromage H, Tempst P, Hartl FU. Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat Struct Biol. 1999;6:697–705. doi: 10.1038/10754. [DOI] [PubMed] [Google Scholar]
- 52.Gonzalez-Paredes FJ, Ramos-Trujillo E, Claverie-Martin F. Defective pre-mRNA splicing in PKD1 due to presumed missense and synonymous mutations causing autosomal dominant polycystic disease. Gene. 2014;546:243–249. doi: 10.1016/j.gene.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 53.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang G, Hubalewska M, Ignatova Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat Struct Mol Biol. 2009;16:274–280. doi: 10.1038/nsmb.1554. [DOI] [PubMed] [Google Scholar]
- 55.Pechmann S, Chartron JW, Frydman J. Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat Struct Mol Biol. 2014;21:1100–1105. doi: 10.1038/nsmb.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke TF, Clark PL. Increased incidence of rare codon clusters at 5′ and 3′ gene termini: Implications for function. BMC Genomics. 2010;11:118. doi: 10.1186/1471-2164-11-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12:205–216. doi: 10.1093/hmg/ddg055. [DOI] [PubMed] [Google Scholar]
- 58.Subramaniam AR, Deloughery A, Bradshaw N, Chen Y, O’Shea E, Losick R, Chai Y. A serine sensor for multicellularity in a bacterium. eLife. 2013;2:e01501. doi: 10.7554/eLife.01501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y, Ma P, Shah P, Rokas A, Liu Y, Johnson CH. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature. 2013;495:116–120. doi: 10.1038/nature11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ito K, Chiba S. Arrest peptides: cis-acting modulators of translation. Ann Rev Biochem. 2013;82:171–202. doi: 10.1146/annurev-biochem-080211-105026. [DOI] [PubMed] [Google Scholar]
- 61.Lu J, Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu J, Kobertz WR, Deutsch C. Mapping the electrostatic potential within the ribosomal exit tunnel. J Mol Biol. 2007;371:1378–1391. doi: 10.1016/j.jmb.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 63.Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, Forster AC. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc Natl Acad Sci USA. 2009;106:50–54. doi: 10.1073/pnas.0809211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *64.Woolstenhulme CJ, Parajuli S, Healey DW, Valverde DP, Petersen EN, Starosta AL, Guydosh NR, Johnson WE, Wilson DN, Buskirk AR. Nascent peptides that block protein synthesis in bacteria. Proc Natl Acad Sci USA. 2013;110:E878–E887. doi: 10.1073/pnas.1219536110. This article, together with the review article above (reference 60), provide an excellent introduction to the types of nascent chain sequences that can reduce translation rate in E. coli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takyar S, Hickerson RP, Noller HF. mRNA helicase activity of the ribosome. Cell. 2005;120:49–58. doi: 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 66.Qu X, Wen JD, Lancaster L, Noller HF, Bustamante C, Tinoco I., Jr The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature. 2011;475:118–121. doi: 10.1038/nature10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen C, Zhang H, Broitman SL, Reiche M, Farrell I, Cooperman BS, Goldman YE. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat Struct Mol Biol. 2013;20:582–588. doi: 10.1038/nsmb.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–1933. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 70.Giedroc DP, Cornish PV. Frameshifting RNA pseudoknots: structure and mechanism. Virus Res. 2009;139:193–208. doi: 10.1016/j.virusres.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Borg A, Ehrenberg M. Determinants of the rate of mRNA translocation in bacterial protein synthesis. J Mol Biol. 2015;427:1835–1847. doi: 10.1016/j.jmb.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 72.Vasquez KA, Hatridge TA, Curtis NC, Contreras LM. Slowing Translation between Protein Domains by Increasing Affinity between mRNAs and the Ribosomal Anti-Shine-Dalgarno Sequence Improves Solubility. ACS Synth Biol. 2016;5:133–145. doi: 10.1021/acssynbio.5b00193. [DOI] [PubMed] [Google Scholar]