

Abstract
The endometrial stromal fibroblast (ESF) is a cell type present in the uterine lining of therian mammals. In the stem lineage of eutherian mammals, ESF acquired the ability to differentiate into decidual cells in order to allow embryo implantation. We call the latter cell type “neo-ESF” in contrast to “paleo-ESF” which is homologous to eutherian ESF but is not able to decidualize. In this study, we compare the transcriptomes of ESF from six therian species: Opossum (Monodelphis domestica; paleo-ESF), mink, rat, rabbit, human (all neo-ESF), and cow (secondarily nondecidualizing neo-ESF). We find evidence for strong stabilizing selection on transcriptome composition suggesting that the expression of approximately 5,600 genes is maintained by natural selection. The evolution of neo-ESF from paleo-ESF involved the following gene expression changes: Loss of expression of genes related to inflammation and immune response, lower expression of genes opposing tissue invasion, increased markers for proliferation as well as the recruitment of FOXM1, a key gene transiently expressed during decidualization. Signaling pathways also evolve rapidly and continue to evolve within eutherian lineages. In the bovine lineage, where invasiveness and decidualization were secondarily lost, we see a re-expression of genes found in opossum, most prominently WISP2, and a loss of gene expression related to angiogenesis. The data from this and previous studies support a scenario, where the proinflammatory paleo-ESF was reprogrammed to express anti-inflammatory genes in response to the inflammatory stimulus coming from the implanting conceptus and thus paving the way for extended, trans-cyclic gestation.
Keywords: transcriptome evolution, cell type evolution, Eutheria, Marsupialia, endometrial stromal cells
Introduction
A major mode of the evolution of organismal complexity in animals is the origin of novel cell types (Valentine et al. 1994; Arendt 2008; Wagner 2014). Cell types are classes of cells that share a specific gene regulatory network state that enables the expression of cell type-specific “realizer” genes which in turn constitute the functional cell phenotype (Graf and Enver 2009; Hobert 2011). A likely scenario for the evolution of a novel cell type is the so-called sister cell type model, in which pairs of novel cell types differentiate from an ancestral cell type (Arendt 2008; Kin 2015). Although there is increasing evidence that the sister cell type process is underlying a major part of cell type diversity (Liang et al. 2015; Musser and Wagner 2015), the molecular mechanisms of sister cell type origination are not well understood. Here we investigate one case of cell type origination, the origin of the eutherian endometrial stromal fibroblasts (ESFs) and decidual cells, by comparing the transcriptomes of homologous cell populations from six therian species. The ESF is a cell type present in the uterus of eutherian mammals (Mossman 1987), and marsupials (Kin et al. 2014). In eutherian mammals with invasive placentation they undergo a characteristic cellular transformation, called decidualization, and become decidual stromal cells (DSCs), either spontaneously during the sexual cycle, as in humans (Emera et al. 2012; Gellersen and Brosens 2014), or upon pregnancy. In contrast, the ESFs of marsupials have not been found to decidualize and thus represent a different kind of cell than that of eutherians (Kin et al. 2014).
DSCs play various functional roles for the successful implantation of embryos as well as the maintenance of pregnancy, such as the regulation of trophoblast invasion, modulation of maternal immune and inflammatory reactions, control of tissue remodeling of the endometrium (Gellersen and Brosens 2014; Chavan et al. 2016). The DSC type is an evolutionary novelty in eutherian mammals (Mess and Carter 2006) and DSC is thought to have evolved in the stem lineage of eutherian mammals coincidentally with the evolution of invasive placenta (Kin et al. 2014; Chavan et al. 2016).
Although traditionally classified as “nonplacental” animals, marsupials do form a short-lived placenta (Freyer et al. 2003; Renfree 2010). It is generally considered an epitheliochorial type placenta where the invasion of trophoblast into the endometrium remains minimal and does not involve direct interaction between trophoblast and the endometrial stroma (Wagner et al. 2014). A major transition in the mode of placentation occurred in the stem lineage of eutherian mammals. Invasive placentation, where the trophoblast breaches the endometrial epithelium and actively interacts with maternal stromal cells, is the ancestral mode for eutherian mammals (Mess and Carter 2006; Wildman et al. 2006; Martin 2008; Elliot and Crespi 2009). During the evolution of eutherian mammals, the mode of placentation shifted multiple times. Most notably, the epitheliochorial type of placenta re-evolved in the hoofed animals, such as cows and horses and their relatives, as well as in basal primate lineages (lemurs). Although DSCs are essential for species with invasive placentation, eutherian mammals with noninvasive placenta are known to have lost decidual reactions and thus DSC (Mess and Carter 2006), even though some remnants of decidualization have been described in sheep (Johnson et al. 2003) and in the so-called “endometrial cups” of the horse (Ramsey 1982).
In figure 1, we set the stage for the comparative analysis of ESF transcriptomes in this study by presenting a hypothetical cell type tree for endometrial stromal cells. Based on a previous study we suggest that the sister cell type of the endometrial stromal cells in mammals is the follicular dendritic cell, FDC (Kin et al. 2015). The sister cell type split between FDC and ESF produces the ancestral form of the ESF, a cell type that we call here “paleo-ESF.”A paleo-ESF is defined as a cell type homologous to the ESF in eutherian mammals, but ancestrally not able to decidualize. A fibroblast-like cell type identified in the endometrial stroma of the opossum, Monodelphis domestica, is an example (Kin et al. 2014). The DSC then arises with a further sister cell type split leading also to the neo-ESF/DSC sister cell type pair. Here we identify neo-ESF as a novel cell type, because it acquired the ability to give rise to DSC. We also note that during ontogeny DSC arises from neo-ESF, as indicated by the arrow in figure 1. This ontogenetic transformation is different from the evolutionary cell-typogenetic event that gave rise to the neo-ESF/DSC sister cell types.
Fig. 1.—

A hypothetical cell type tree for ESF. The tree contains two kinds of inner nodes. One that represents cell type origination events, black ovals, and the other represents species lineage splits, white ovals. In this tree paleo-ESF originate from a cell type splitting event with FDC and the extant paleo ESF in this diagram is related to the neo-ESF/DSC pair by the species lineage splitting event that gave rise to the marsupial and the eutherian stem lineages. The neo-ESF/DSC sister cell type pair arose in the stem lineage of eutherian mammals. The arrow represents the ontogenetic transformation of ESF into DSC.
As we have seen above, it is known that paleo-ESF and neo-ESF differ in one crucial aspect, whether they decidualize or not. However, not much else is known about their similarities or differences associated with their presumably different roles in pregnancy. Another important question is whether eutherian neo-ESF which secondarily lost its ability to decidualize share any characteristics with marsupial ESF. In this study, we aim at identifying the gene expression changes that were associated with the origin and subsequent modifications of neo-ESF. Specifically, we collected RNA-Seq data of ESF in culture from six mammals, including five eutherians and one marsupial and analyzed the data in a phylogenetic framework. As we will discuss below, the data reveal various previously unknown aspects in the evolution of ESF, including the nature of paleo-ESF as a pro-inflammatory immune cell type and shared molecular characteristics between paleo-ESF and secondarily nondecidualizing neo-ESF.
Materials and Methods
A detailed description of experimental protocols and methods is provided in the supplementary methods, Supplementary Material online. Below we focus on methods of data analysis.
Transcriptomic Data Analysis
Orthologous genes among species were retrieved thorough Ensemble Biomart, which uses the EnsembleCompara algorithm for assigning orthologs (Vilella et al. 2009). The retrieved list contained 10,259 one-to-one orthologs. We normalized the data by calculating the transcripts per million (TPM) values (Wagner et al. 2012). The transcript length information was obtained from the Ensemble database with Biomart. When a gene has multiple transcripts, a median length of all transcripts for the gene was used. For the purpose of phylogenetic analyses, TPM values were transformed into binary values (1 or 0) representing presence or absence of gene expression, with TPM values above 3 were called present and those with TPM values less than 3 were called absent based (Wagner et al. 2013).
Phylogenetic Reconstruction Using the Threshold Model
The divergence times of the time tree for six species included in this study were taken from dos Reis et al. (2012). We used the ancThresh function implemented in the R package phytools (Revell 2014) for estimating the ancestral states in inner nodes. For each of the 64 (=26) possible states on the time tree, we ran ancThresh for 1,000,000 generations under the Brownian motion model for the liability evolution, sampling every 100 generations. We assigned the state with largest posterior probability to be the state of each node.
Gene Annotation and Overrepresentation Analysis
The lists of genes gained or lost on each branch of the tree were annotated based on gene ontology (GO) and associated pathways, and overrepresented GO terms and pathways in each list were detected using ConsensusPathDB (http://consensuspathdb.org/, last accessed on July 15) (Kamburov et al. 2011). The list of 10,259 genes that were present in all species was used as a background for overrepresentation analysis. GO terms were summarized and visualized in semantic space by REVIGO (http://revigo.irb.hr/, last accessed on July 15) (Supek et al. 2011).
Results
Isolation of Endometrial Stromal Cells from Monodelphis domestica
To understand the origin of eutherian pregnancy, the comparison of eutherian with noneutherian species is essential. Here we use the gray short-tailed opossum, Monodelphis domestica, as an outgroup species. This species belongs to the most basal lineage among marsupials (Nilsson et al. 2010) and is considered representative of the ancestral therian condition with respect to many of its female reproductive traits (Freyer et al. 2003). In Monodelphis, endometrial fibroblasts have been described as a sup-epithelial layer of mesenchymal cells expressing HoxA11, CEBPB, and PGR (Kin et al. 2014). Here, we report the isolation of these cells for primary culture using Percoll gradient centrifugation. To assess the homogeneity of isolated stromal fibroblasts, we performed immunohistochemistry on the cells using vimentin, a marker for mesenchymal cells, and cytokeratin, a molecular marker for epithelial cells. By double immunofluorescence, epithelial cells should appear CK+, VIM- and stromal fibroblasts should appear CK-, VIM+. In three independent experiments, the degree of epithelial contamination was determined to be 1.5% (5 epithelial cells of 323 total cells), 6.6% (16/241), and 3.3% (38/1,166), respectively (see also fig. 2B). These cells were then processed for RNA-Seq. High expression of PGR mRNA as observed in RNA-Seq data (not shown) also confirmed that the isolated cells are ESF rather than connective tissue fibroblasts.
Fig. 2.—

(A) Time tree of six species used in this study adopted from dos Reis et al. (2012). Events of appearance of five major mammalian clades are shown by black full circles on the tree. The time scale is shown at the bottom. Ma = million years ago. Branch colors represent the types of placentation associated with each branch. (B) Double-immunofluorescent images of cultured ESF used in this study. On column, from left to right, vimentin expression (red), cytokeratin expression (green), cell nuclei visualized with DAPI (blue), and merged image of the above three are shown for each species. In all species, almost all the cells express vimentin whereas few cells express cytokeratin, showing that the contamination of epithelial cells was kept minimum. Scale bar = 100 µm.
Overall Results of RNA-Seq and the Pattern of Transcriptome Divergence
In addition to opossum (Monodelphis domestica) ESF, we collected data from ESFs of five eutherian species: Cow (Bos taurus), mink (Neovision vision), rabbit (Oryctolagus cuniculus), rat (Rattus norvegicus), and human (Homo sapiens). This taxon sample spans the taxonomic range of the main eutherian lineages (Boreoeutheria) with the exception of the most basal eutherian lineages, Xenarthra (armadillos, sloth, anteaters, etc.) and Afrotheria (elephants, hyrax, tenrecs, etc.) (see fig. 2A for their phylogenetic relationships). The homogeneity of stromal cell populations from each species was confirmed by immunocytochemistry (fig. 2B). On average, 29.6 million mRNA-Seq reads (18.0M–46.5M reads; supplementary table S1, Supplementary Material online) were uniquely mapped to the genome of each species. The RNA-Seq reads from mink ESF were mapped to the ferret genome (Mustela putorius furo). The percentage of reads from mink ESF uniquely mapped to the ferret genome was comparable to those in other species (average for all samples = 65.1%, average for mink samples = 66.3%; supplementary table S1, Supplementary Material online).
For comparative analysis, a timed tree of mammals from a previous phylogenomic study (dos Reis et al. 2012) was adopted with the point estimates for the age of each node under the “Atlantogenata” topology (fig. 2A). We first explored the overall trends in our data by plotting the distances among ESF transcriptomes against phylogenetic divergence time. The plot shows that the degree of ESF transcriptome divergence as measured by Spearman’s correlation coefficient is generally within the range described for the transcriptome divergence of various organs as reported by Brawand et al. (2011) (fig. 3A). In contrast to the data by Brawand et al. we find that there is no difference in average divergence at 70–90 Myr compared with 175 Myr, that is, opossum. The same pattern is seen if divergence is measured by Pearson correlation of square-root TPM or Hamming distance on discretized data (expressed/nonexpressed) (fig. 3B). These data suggest that transcriptome divergence saturates at or before 70 Myr. We used this observation to estimate the degree of selective constraint on the gene expression profile.
Fig. 3.—

The relationship between phylogenetic divergence time and divergence in cell transcriptome. (A) The plot showing the relationship between divergence time and the distance among cell transcriptomes as measured by Spearman’s correlation coefficients. The data points from this study are shown by open circles. Triangles and squares represent the phylogenetic divergence versus transcriptome divergence plot for brain and testis, respectively, adopted from Brawand et al. (2011). (B) The plot showing the relationship between divergence time and the distance among cell transcriptomes as measured by relative Hamming distance of presence/absence matrix of gene expression. Both in (A) and (B), the data points from comparisons against the mink transcriptome are shown as blue x-mark.
Estimating Selective Constraints on Transcriptome
We noted that divergence compared with mink ESF is on average higher than any other compared transcriptomes (fig. 3). We think this is due to the fact that the mink cells are immortalized and thus may be subject to artifacts due to immortalization. In the analysis of transcriptome divergence below we will exclude the data from mink.
We use the following model to explain saturation of transcriptome divergence and to infer the number of genes under selective constraint. Let N be the number of genes recorded in the study, and Nc, the number of constrained genes, that is, genes that cannot be turned on or off without affecting the cell. Then Nf = N − Nc is the number of genes that can be changed without affecting the function of the cells. For technical reasons, this number also includes genes which have expression levels that fluctuate around the operational threshold. Saturation happens when the expression profile of the Nf genes is randomized, and further random change does not lead to higher overall divergence. The expected equilibrium distance between random 0/1 sequences is 〈Heq〉 = Nf/2. This simply follows from the fact that the probability of a site having different states is 0.5 if a 0/1 sequence is completely randomized, and the average distance then is Nf/2. Assuming this model we can use the measured equilibrium divergence among our transcriptomes to estimate the number of genes that are free to change, Nf = Heq*2. We further can estimate the number of genes that remain expressed across species due to natural selection. This model assumes that the number of genes that change due to directional natural selection is small. In our data, we recorded the expression of N = 10,259 genes (fig. 4). The average Hamming distance among all pairs of transcriptomes is Heq = 1,294.4. From this we estimate that the number of genes free to change is Nf = 2,589, and the number of constrained genes Nc ≃ 7,670. On average, there are 6,852 (±137 SD) genes classified as expressed in each species (fig. 4). This number incudes genes that are expressed and maintained by natural selection across species as well as genes that are expressed by chance among the Nf genes that are free to vary. The expected number of the latter class of genes is Nf/2. This implies, according to the model, that there are Nc+ 6,852 − 1,294 = 5,558 (±625 SD) genes that remain expressed due to functional constraints. This inference can be tested by determining the number of genes that are classified as expressed in all five species compared (fig. 4). This number is 5,507 which is only 1.5% off the predicted number based on the measurement of divergence. This confirms that the model inferences are biologically reasonable. There is a subset of approximately 2,600 genes that are either free to evolve or have spurious expression levels (close to the threshold) and may change randomly due to estimation noise. As expected, the average expression level of the genes expressed in all five species is higher (165 TPM) than those genes that are variable across species (32.5 TPM). Nevertheless, the expressed genes conserved across the five species also include moderately expressed genes (1,090 genes with expression levels <20 TPM). Overall our results suggest that the expression state of more than 50% genes measured here is maintained by natural selection.
Fig. 4.—

A graph showing the estimated numbers of constrained and unconstrained genes in the ESF transcriptome. The total number of genes analyzed in the present study is 10,259 as shown in the left column. It is estimated from the ESF RNA-Seq data that the gene expression states of 7,670 genes are constrained whereas those of 2,589 genes are free to change. The average number of genes expressed in a single species is 6,852 as shown in the center column. As explained in the text, we can estimate that 5,558 of them are constrained to be expressed. This number, N_all+, closely matches the number of genes expressed in all species shown in the right column as indicated by a dashed line.
Reconstruction of Ancestral Transcriptomes
We next reconstructed the ancestral transcriptomes using the threshold model of discrete character evolution (Felsenstein 2012) as implemented by Revell (2014). The overall result is shown in figure 5 and the list of genes gained or lost on each branch of the tree is available in supplementary table S2, Supplementary Material online.
Fig. 5.—

Changes in gene expression shown on the mammalian time tree. (A) The number of genes gained (+) or lost (−) on each branch reconstructed by ancThresh is shown. Branch colors represent the types of placentation associated with each branch. (B) The plot showing the relationship between the branch length and the total number of changes in gene expression (the sum of gene expression gain and loss). The numbers on the dots correspond to the branch numbers as appear on (A). The blue dotted line is the linear regression line through the origin for all the data points, whereas the red dashed line is the linear regression line through the origin only for data points from eutherian mammals (i.e., excluding the opossum branch, #10). The slope for the blue dotted line is 6.5 changes/Myr, and that for the red dashed line is 9.4 changes/Myr.
We performed GO and pathway overrepresentation analyses the lists of expressed genes reconstructed as gained or lost during certain periods in evolution. All the overrepresented GO terms and pathways are listed in supplementary table S3, Supplementary Material online. Because the numbers of gene expression changes mapped to the internal branches is small, they are excluded from further analysis, except the branch leading to eutherian mammals (branch #1), where a substantial number of changes were detected.
The Monodelphis ESF Transcriptome
The transcriptome of the opossum ESF was included in this study as a representative of a “paleo-ESF,” meaning a uterine ESF homologous to the ESF of eutherians but that ancestrally does not differentiate into decidual cells (Kin et al. 2014). Hence, genes expressed differently between opossum ESF and eutherian ESF are informative about the biological changes that occurred during the evolution of neo-ESF from paleo-ESF.
A formal phylogenetic reconstruction of changes along the most basal lineages in the phylogeny of our study species is not possible because we do not have data about species outside Theria. For that reason we focus on genes that are expressed in opossum ESF but not in the reconstructed transcriptome of the most recent common ancestor of the boreotherian clade, which is the most recent common ancestor of the eutherian species included in this study. Below we will call these genes “Monodelphis-specific genes” or “paleo-ESF-specific genes” meaning genes expressed in opossum ESF but not in the reconstructed ESF transcriptome of the boreotherian ancestor. These names of course are somewhat inaccurate because it certainly also includes genes that are scored as expressed or nonexpressed due to experimental and biological variation as well as due to genetic change in the Monodelphis lineage.
The set of “Monodelphis-specific genes” consists of 679 members and is highly enriched for many GO and pathway categories (fig. 6A and supplementary table S3, Supplementary Material online). The enriched molecular function and cellular component categories suggest that the difference between paleo-ESF and neo-ESF is mostly one of cell–cell communication. The five most enriched molecular function categories, with q-values between 5.5 × 10−9 and 2.5 × 10−3, are all related to cell–cell signaling: “Receptor activity” GO:0004872, “transmembrane signal receptor activity” GO:0004888, “signal receptor activity” GO:0038023, “signal transducer activity” GO:0004871, and “receptor binding” GO:0005102. This signal is also reflected in the cellular localization GO enrichment where the seven most enriched categories are molecules located in the plasma membrane with q-values between 1.9 × 10−11 and 4.0 × 10−8.
Fig. 6.—

Visualization of biological process GO terms in sematic space by REVIGO. GO terms overrepresented in the list of genes specific to the opossum ESF transcriptome are shown in (A), whereas those overrepresented in the list of genes specific to the ancestral neo-ESF transcriptome are shown in (B). Likewise, GO terms overrepresented in the list of genes present or absent in the cow ESF transcriptome are shown in (C) and (D), respectively. The color of each circle corresponds to log 10 P values according to the color scale shown at the bottom left of each figure. The size of each circle is proportional to the size of GO terms.
Paleo-ESF-specific genes are enriched for two clusters of pathways: Inflammation/immune response as well as cell movement and adhesion. Genes expressed in paleo-ESF suggest that opossum uterine fibroblasts are part of the mucosal immune defense, as is the case with fibroblasts in other organs, and that this function was attenuated in the evolution of eutherian neo-ESF. For instance, the GO category “immune response” (GO:0006955) has 69 members and a q-value of 6.74 × 10−4 . The GO category “inflammatory response” (GO:0006954) has 44 members in the paleo-ESF-specific genes (q = 1.63 × 10−3).
We considered the possibility that genes related to inflammation and immune response could be detected due to contamination with leukocytes, which also stain positive for vimentin. We concluded that at least a considerable number of these genes are in fact expressed by the opossum ESF (fig. 7A). First, the level of CD45 RNA (aka PTPRC, Protein Tyrosine Phosphatase Receptor Type C), a pan leukocyte marker, is low (on average ∼3 TPM) and very low in one of the analyzed samples (=0.86 TPM). It is notable that the expression level of many of the genes in the inflammatory/immune response categories is high in the sample with very low CD45 mRNA level. These include CCRL2 (115 TPM in the low CD45 sample) a receptor for CCL19, modulating the recruitment of lymphocytes, LBP (53 TPM) the lipopolysaccharide receptor (LPS receptor), HRH4 a receptor for histamine active in peripheral tissues (39.7 TPM), SYK (Spleen Tyrosine Kinase at 48.2 TPM) a nonreceptor type kinase that transduces signals from immune receptors and many more. The high level of expression of these genes is not easily explained by leukocyte contamination, in particular in samples with essentially no CD45 RNA. There is also substantial expression of F2 (F2/Trombin), even though RNA for the platelet marker SELP (Selectin P) is essentially absent in all samples (<1 TPM). These data suggest that the paleo-ESF has the capability to participate in the immune response of the uterine mucosa of opossum, similar to fibroblasts in other organs (Smith et al. 1997). This finding further suggests that the evolution of neo-ESF and implantation required de-emphasizing the role mucosal fibroblasts play in the inflammatory response. In particular, the relatively high level of expression of the LPS receptor is strongly indicative that opossum ESF is able to detect bacterial infections.
Fig. 7.—

Expression patterns of select genes. (A) Expression patterns of select inflammation/immune response-related genes in the opossum ESF. The TPM values for a classical lymphocyte marker (PTPRC, a.k.a. CD45) as well as a platelet marker (SELP) are low, except for a moderate level of CD45 expression in MdESF2. Note that even in a sample with minimal CD45 expression (MdESF1), high levels of pro-inflammatory gene expression are observed. (B) Expression patterns of cell cycle genes. Neo-ESF express various cell proliferation-related genes that are not expressed in the opossum ESF, including FOXM1. (C) Expression patterns of WNT signaling-related genes. A number of WNT signaling-related genes are expressed in both opossum and cow ESFs, although they are absent or inconsistently expressed in other mammalian ESF transcriptomes. (D) Expression patterns of neo-ESF-specific transcription factors. Red dashed lines represent threshold hold expression values. Note that in (B)–(D) the vertical axis represents square-root TPM values instead of raw TPM values.
The Ancestral Neo-ESF Transcriptome
There are 442 genes that are classified as nonexpressed in opossum and reconstructed as expressed in the most recent common ancestor of boreoeutherians. These genes show a very strong overrepresentation of genes related to cell division and proliferation (fig. 6B and supplementary table S3, Supplementary Material online). The top ten GO biological role categories with q-values between 1.23 × 10−6 and 8.02 × 10−4 belong to “organelle fission” (GO:0048285), “cell cycle process” (GO:0022402), and others like it. Pathway enrichment analysis also confirms this result with Ractome pathway “mitotic cell cycle” containing 23 members and enriched at a q-value of 0.018 and others more.
Among the cell cycle-related genes (fig. 7B) the expression pattern across species of FOXM1, a transcription factor implicated in the regulation of proliferation, is particularly interesting. It is not expressed in opossum (0.62 TPM on average) but consistently expressed in all eutherian species between at moderate 18 TPM (mink) and high levels, for example, at 453 TPM in human ESF. This gene has recently been identified as critically involved in cell cycle regulation during decidual differentiation as well as decidual differentiation itself (Gao et al. 2015; Jiang et al. 2015). It is regulated by HoxA10 and affects the expression of STAT3 in decidual cells. Hence, the recruitment of FOXM1 could have been a key step in the evolution of the neo-ESF cell type identity which is distinguished from paleo-ESF by their ability to decidualize.
Gene Expression Evolution in the Bovine Lineage
The lineage leading to the cow includes an interesting reversal in the fetal–maternal interface, namely the re-evolution of epitheliochoreal, that is, a noninvasive, placentation (Mess and Carter 2006; Wildman et al. 2006). The cow shares this feature with other Laurasiatheria, such as the sheep, pig, horse, and others (Ramsey 1982). It is thus interesting to ask whether there are biologically meaningful changes in the ESF transcriptome related to this transition in fetal–maternal biology.
There are 333 genes that are reconstructed as having gained ESF expression in the bovine lineage. Both molecular function and biological process GO term enrichment suggest that significant changes in the cell–cell communication network (fig. 6C and supplementary table S3, Supplementary Material online), showing enrichment for GO categories such as “secretion” (GO:0046903; q-value = 1.03 × 10−3) and “cell–cell signaling” (GO:0007267; q-value = 1.51 × 10−3). Next we asked whether the genes that gained ESF expression in the bovine lineage have any relationship with genes that are present in paleo-ESF, that is, expressed in opossum ESF, and in decidual human cells.
There is considerable overlap (N = 100) between the 333 genes gained in the bovine lineage and the 679 genes specific to opossum ESF. The overlap represents a 4.54-fold enrichment over random expectation and has a P value of 2.85 × 10−6. The overlap set is moderately enriched for signal transduction (GO:0007165, P = 6.97 × 10−5, but only q = 0.88). Among the signaling genes a number of WNT-signaling-related genes are shared between opossum and cow (fig. 7C). These are DKK2, a WNT antagonist, WNT6, WNT2B, HS3ST2, heparan sulfate 3-O-Sufotransferase 2 (an enzyme involved in the synthesis of a WNT-binding component of the extracellular matrix), SFRP2, Secreted Frizzle-related Protein 2, a soluble WNT-binding domain, as well as WISP2, WNT1 Inducible Signaling Pathway Protein 2, which is a matricellular protein inducing canonical WNT signaling in mesenchymal cells. WISP2 is known for inhibiting invasive migration of cancer cells (Banerjee et al. 2008; Fritah et al. 2008). The latter gene is highly expressed in both opossum (650 TPM) as well as cow (129 TPM) but completely absent in human ESF (av = 0.37 TPM) and even in human decidual cells (Fig. 7C).
There is also a significant overlap with genes that are induced in human ESF upon decidualization. Genes that are below 3 TPM in human ESF and above 5 TPM upon decidualization have 16 genes in common with the genes acquired in the cow lineage. This represents a 3.3-fold enrichment over random expectation (P = 4.4 × 10−4). Among them highly expressed genes are somatostatin (SST) and arylsulfatase G (ARSG).
Equally interesting are genes that lost ESF expression in the bovine lineage. There are 231 genes included in this set and the enriched biological process GO categories include taxis (GO:0042330, q = 8.37 × 10−4), and regulation of localization (GO:0040012, q = 1.2 × 10−2), suggesting a decreased role of cell migration for bovine ESF (fig. 6D and supplementary table S3, Supplementary Material online). In pathway analysis the three most enriched pathways are axon guidance (KEGG, ten members and P = 5.23 × 10−5; q = 2.04 × 10−2), TNF signaling pathway (KEGG, seven members, P = 1.12 × 10−3; q = 1.69 × 10−1) and prostaglandin I2 and E2 receptors PTGIR and PTGER4, where only the latter is highly expressed in almost all other mammals save mink. The most interesting loss of gene expression is the loss of genes related to axon guidance. Axon guidance genes are also engaged in the regulation of blood vessel growth (Carmeliet 2003; Klagsbrun and Eichmann 2005) and this enrichment may indicate an evolutionary change in the regulation of endometrial vascularization in bovine endometrium. The ten genes in this category include four semaphorins (SEMA3D, SEMA3E, SEMA6C, and SEMA6D), a semaphoring coreceptor (PLXNA2 and plexin A2), ephrin A5 (EFNA5), and two ephrin receptors (EPHB1 and EPHB1) as well as netrin G1 (NTNG1).
Evolution of the Expressed Transcription Factor Inventory
In the across species comparison, 634 genes were identified as transcription factor genes and mapped in all six species. We found that there were nine transcription factor genes that were consistently expressed in eutherian neo-ESF but not in Monodelphis paleo-ESF. Those genes were FOXM1, TSC22D3, MYBL2, TCFL5, MYBL1, MXD3, and PRDM8 (fig. 7D). To our knowledge, only FOXM1 is currently known to be involved in endometrial biology (Gao et al. 2015; Jiang et al. 2015). Below we consider two specific issues, the expression of FOXO1 in opossum and an inference about the core gene regulatory network of ESF.
One surprising result of this study was the expression of FOXO1 mRNA in opossum ESF. In a previous study, we reported that FOXO1 protein was not detected in opossum ESF (Kin et al. 2014). In the transcriptomic data, there was substantial expression of FOXO1 mRNA in Monodelphis ESF ∼ 20 TPM. We considered the possibility that the FOXO1 mRNA was not translated. To test whether FOXO1 protein is expressed in isolated Monodelphis ESF we performed immnocytochemical detection as well as Western blotting in cultured opossum ESF and did not find staining (data not shown), consistent with the in vivo immohistochemical results. We further considered the possibility that the relatively high expression of FOXO1 mRNA in cultured opossum ESF could be a consequence of removing the cells from their native environment. We tested this hypothesis by extracting RNA from laser microdissection of Monodelphis ESC. RT-PCR clearly showed the presence of FOXO1 mRNA in the endometrial stroma isolated from its native environment (fig. 8). We thus conclude that in opossum ESF FOXO1 mRNA expression is not strongly influenced by removal from its in vivo context and that the FOXO1 protein expression is very low, likely due posttranscriptional mechanisms as it is the case in other fibroblast populations.
Fig. 8.—
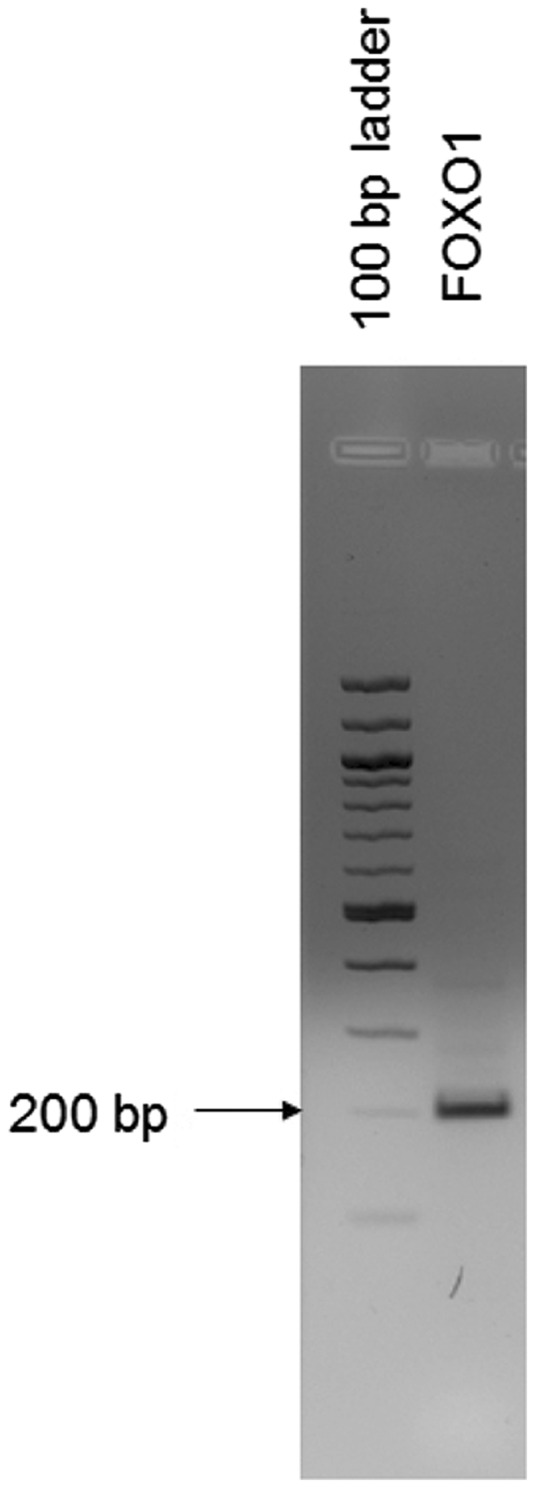
RT-PCR of FOXO1 mRNA isolated from laser microdissected Monodelphis ESF. The amplified fragment of FOXO1 is observed at the expected size. This result suggests that the expression of FOXO1 mRNA observed in cultured endometrial stromal fibroblasts is not an artifact of isolating these cells from their native context.
Another unexpected result was the absence of FOXO1 mRNA in the transcriptome of rabbit ESF, even though FOXO1 transcript was detected in all the other species. The most parsimonious interpretation is that the absence of FOXO1 mRNA in rabbit ESF is a derived condition in the rabbit lineage, after the most recent common ancestor of rabbit and mouse, or an artifact caused by cell isolation.
The Gene Inventory of the ESF Core Gene Regulatory Network
To probe our data for gene regulatory features shared among the ESF from all therian species involved, we first identified 298 transcription factor genes that were expressed in all six species examined. We expect that this list of genes included genes that are broadly shared with other cell types, like other mesenchymal cells, as well as genes that were uniquely shared among therian ESF. To obtain a candidate list of ESF cell type identity genes, we then intersected the set of consistently expressed genes with the list of 224 genes that were inferred to be recruited after the cell type lineage of ESF split from that of FDCs (Kin et al. 2015). The intersection yielded five transcription factor genes: PGR, GATA2, HOXD9, HOXD10, and HOXB2; all of which are known to be involved in human ESF biology (Kin et al. 2015). We suggest that these genes are part of the gene regulatory network that conveys ESF identity.
Discussion
Rate of Genome-Wide Gene Expression Change for a Defined Cell Tyoe
In this study, we investigated the rate and mode of transcriptome evolution at the cell type level using ESFs from six mammalian species. The degree of gene expression divergence observed in this study was within the range of gene expression divergence previously reported for various organs in mammals (Brawand et al. 2011). The plot of transcriptome divergence reached a plateau at 70 Myr (Fig. 3). This pattern of transcriptome divergence implies relatively strong stabilizing selection. In fact, a simple mathematical model of transcriptome evolution suggests that there are about 5,500 expressed genes that are under stabilizing selection. This number closely matches the number of genes that are found to be expressed in all five species. Hence the saturation of transcriptome divergence can be explained by the limited number of genes that can change without affecting ESF cell identity and function, estimated to be approximately 2,600. The expression profile of these genes appears to be randomized across the species compared. This set of genes certainly includes genes with expression levels close to the operational threshold for expression calls and thus represent differences that are largely due to experimental and biological noise. But there is also a considerable number of genes that have moderate to high average expression levels across species, and thus the set of “evolvable” genes likely includes functionally relevant genes.
Reconstruction of Transcriptome Evolution
The Brownian motion-threshold model was applied to reconstruct the ancestral transcriptomes. This model assumes that the discrete traits (in this case presence or absence of gene expression) are determined by an underlying, unobserved continuous trait called “liability” (Revell 2014). In this model, the trait value changes when liability crosses fixed thresholds. As the change in gene expression does not usually occur by a single mutation but by contributions of multiple mutations, the threshold model seems to be more realistic and appropriate for transcriptome evolution than other evolutionary models for discrete characters such as stochastic mapping (Huelsenbeck et al. 2003) which assumes instantaneous change of character states (Revell 2014). On the other hand, the model has some limitations. As always, the results of ancestral gene expression state reconstruction should be evaluated in the light of available biological background knowledge.
For instance, the model assumes that the evolution of liability is a Brownian random walk, but this assumption is likely to be violated on some branches because of natural selection acting on the mode of placentation and thus on gene expression in ESF. In addition, one difficulty in interpreting the results of the threshold algorithm is the reconstruction of the character state at the root node. The model does not discriminate between ingroup and outgroup lineages and assumes random directional evolution. In contrast, in our data set the five eutherian species represent derived character states with respect to female reproductive biology: Invasive placentation and longer gestation that plausibly have been maintained by natural selection, rather than by chance. There is also unbalanced taxon sampling of the ingroup (eutherian mammals, n = 5) and the outgroup (marsupial, n = 1). For example, when the gene expression state of a certain gene is 0 (not expressed) for the opossum and 1 (expressed) for all eutherian mammals, the algorithm reconstructs the ancestral state of the therian root node to be 1 (present), although this cannot be determined in principle without additional information from species outside therians. Thus, the gene expression changes reconstructed to be “gained” on the opossum branch (branch #10, fig. 5) might actually be “lost” on the stem lineage of eutherian mammals (branch #1, fig. 5) with the gene expression unchanged on the opossum branch and vice versa. For these reasons, we use ancestral state reconstruction only to infer states within the eutherian clade and for the comparison with opossum we limit our discussion to gene expression differences between the opossum ESF and the reconstructed ancestor of the eutherian clade.
Evolution of the ESFs
The comparative analysis of ESF transcriptomes suggests an evolutionary scenario of the function and identity of ESF during the origin of eutherian pregnancy. Here, we want to outline the evolution of the ESF cell suggested by our findings without repeating the empirical results summarized above.
In this scenario, we assume that there was an ancestral ESF present in the therian ancestor and may be even in egg laying amniotes (fig. 1). These could be the tissue fibroblast from the shell gland which is homologous to the uterus of mammals. We call this cell type paleo-ESF and define it as the cell type that is homologous to the ESF of eutherians but ancestrally unable to differentiate into decidual cells. This cell was a tissue fibroblast with a role in maintaining tissue integrity and immune defense. This scenario is suggested by the fact that tissue fibroblasts are part of the innate immune system in other organs as well (Smith et al. 1997). These cells can assume roles similar to those of macrophages in eliciting inflammatory reactions to tissue injury. Furthermore, this scenario is supported by the expression, in opossum, of immune- and inflammatory function genes, which are not expressed in eutherian ESF. Most intriguing is the strong expression of LBP, a receptor for LPS which, in monocytes, is involved in the rapid acute phase response to G-negative bacteria. This activity is performed in cooperation with CD14 delivering the LPS to TLR4, which is also expressed in opossum ESF, to initiate the innate immune response (Alexander and Rietschel 2001). Incidentally, we also find CD14 expressed in opossum ESF but not in eutherian ESF.
The finding that the genes related to immune response are not inferred to have been present in the ancestral boreotherian ESF suggests that the evolution of the neo-ESF from paleo-ESF involved a loss or reduction of the role of ESF in immune response. This scenario makes sense in that the ancestral role of the derived DSC cell type is to control the implantation-related inflammatory response (Chavan et al. 2016). In addition our data suggest that neo-ESF are more proliferative, as they became a precursor for the production of DSC during pregnancy or the secretory phase during the menstrual cycle. The origin of neo-ESF during the evolution of pregnancy is also associated with massive changes in the cell–cell signaling network, as judged by the differences between opossum and ancestral boreoeutherian transcriptomes. Among those genes that have been recruited into neo-ESF and that may have been critical for the evolution of decidualization is FOXM1 which is strongly expressed in all eutherian ESF examined but not in opossum ESF. This gene has recently been shown to be critical for the decidualization of mouse and human ESF (Gao et al. 2015; Jiang et al. 2015), and is thus a strong candidate for a gene causally involved in the evolution of the decidualization. It does not seem to be a decidual cell identity gene, as it is only transiently expressed during decidualization (Jiang et al. 2015). Rather this gene seems to have been recruited into neo-ESF to enable them to proliferate and to undergo decidualization. It is thus a potential marker gene to distinguish neo-ESF from paleo-ESF.
Another major event in the evolution of the fetal–maternal interface was the secondary noninvasive placenta of artiodactyls and perissodactyls. Our data include the ESF of the cow, an epitheliochorial artiodactyl species, suggest several trends in the gene expression of ESF associated with the re-evolution of a secondarily noninvasive placenta. Among them are 1) the re-evolution of expression of genes that are otherwise only found in opossum; 2) expression in ESF of some genes induced in human decidual cells, such as somatostatin; and 3) the loss of expression of genes related to axon guidance and angiogenesis. Among the first group of genes, the ones shared with opossum, the most interesting is WISP2, which has been shown to be involved in the suppression of invasiveness of several human cancers (Banerjee et al. 2008; Fritah et al. 2008). It may thus be plausible that it has a similar role in preventing invasion of trophoblast cells in bovines and marsupials, in particular given the high expression levels in cow ESF (129 TPM) and opossum ESF (650 TPM). If this inference is correct this would incidentally imply that the marsupial conceptus is invasive, rather than primarily noninvasive as has been shown in Pseudemoia entrecasteauxii, an Australian skink species (Griffith et al. 2013). A similar inference is supported by the fact that in the fat-tailed Dunnart the luminal epithelial cells reinforce their anchoring to the underlying stroma during apposition of the conceptus (Laird et al. 2015). It will be important to test these inferences with blastocyst transplantation experiments to extrauterine sites in basal marsupial species. Another important implication of this finding is that these anti-invasiveness genes might help explain the curious fact that cows and horses are much less vulnerable to malignancy of certain cancers than humans (D'Souza and Wagner 2014).
ESF Cell Type Identity Genes
Cell type identity is caused by the activation of a core gene regulatory network by some external signals and which regulate the “realizer” genes that determine the cell phenotype. Because cell type identity genes are expected to be transcription factor genes, transcriptional cofactors or regulatory ncRNA that are jointly necessary for the realization of a cell fate, one would predict that they are expressed in the homologous cell types in all species that have that cell type. We identified 473 transcription factor genes that are expressed in all six species examined in this study. Clearly this set does include genes that are shared among a larger group of cell types, for example, all mesenchymal cell types, and cannot all be considered cell type identity genes for ESF. For that reason we intersected the set of shared expressed transcription factor genes with the transcription factor genes that have been inferred to be recruited in ESF after the split from FDCs, a cell types closely related to ESF (Munoz-Fernandez et al. 2006; Kin et al. 2015). The resulting list contains five genes: PGR, GATA2, HOXD9, HOXD10, and HOXB2. All of them were previously shown to be essential for endometrial stromal cell biology: PGR (Lydon et al. 1995; Large and DeMayo 2012), GATA2 (Dyson et al. 2014; Kin et al. 2015), HOXD9 and HOXD10 (Raines et al. 2013; Kin et al. 2015). HOXB2 is a homeodomain transcription factor gene involved in hindbrain patterning (Maconochie et al. 1997) and in vitro decidualization (Kin et al. 2015).
This list does not include other transcription factors that are known to be essential for ESF biology. Notably, HOXA11 is expressed in mouse, human, and opossum ESF and known to be necessary for mouse ESF development (Hsieh-Li et al. 1995). This gene has also been recruited to ESF after the split from FDC (Kin et al. 2015). Another important gene is HOXA10 (Taylor et al. 1998; Das 2010). The absence of these genes from our list is due to the limitations of genome annotation as HOXA genes are not annotated in some of the genomes compared here. Nevertheless, HOXA11 is clearly necessary in mouse and human endometrial stromal cells, and inspection of primary mapping results shows that it is expressed in all animals except mink SV40-immortalized cell line, which is prone to artifacts.
In addition, there are genes necessary for endometrial decidualization and also widely expressed in other mesenchymal cell types, such as CEBPB (Lynch et al. 2011; Kin et al. 2015). This gene is involved in regulation of decidual PRL expression in humans (Pohnke et al. 1999; Lynch et al. 2011), and is expressed in all mesenchymal cells examined. Thus, CEBPB is not specifically recruited at the evolutionary origin of ESF (Kin et al. 2015). This suggests that core gene regulatory networks of novel cell types evolve through modification (addition and subtraction of genes) from the core networks of ancestral cell types. Hence, transcription factor genes important in the identity and activity of a certain cell type can be parts of the ancient core regulatory network inherited from their ancestral cell types and thus shared with related cell types.
The inferences discussed above suggest that we need to distinguish between genes that are part of the core gene regulatory network of a cell types but are not specific cell type identity genes. “Cell type identity genes” could be understood as a subset of regulatory genes within the core network. We suggest that one may call those genes “cell type identity genes” if they fulfill two criteria: 1) Genes that are part of the core gene regulatory network of the cell type, that is, are necessary for the realization of the cell type identity, and 2) genes that have been recruited into the core network at the time when the respective cell type originated in evolution. Under these criteria a preliminary list of ESF cell type identity genes would include: PGR, GATA2, HOXD9, HOXD10, HOXA11, and HOXB2. For all of them, experimental evidence shows that they have a role in the biology of endometrial stromal cell differentiation, are consistently expressed in the ESF of therian species (this study), and likely have been recruited to ESF expression after the split from the FDC lineage (Kin et al. 2015).
The Role of FOXO1 in the Evolution of Decidual Cell Identity
FOXO1 is a transcription factor necessary for decidual differentiation of ESF in humans. It interacts with other ESF transcription factors, including HOXA11 (Brayer et al. 2011) and CEBPB (Pohnke et al. 1999; Lynch et al. 2011), and activates decidual PRL expression as well as other genes. Previously we reported and confirmed in this study that FOXO1 protein is not expressed in Monodelphis ESF, and thus suggested that the recruitment of FOXO1 may have been a key step in the evolution of decidual cells (Kin et al. 2014). In our samples, FOXO1 mRNA was consistently expressed in eutherian species, with the exception of rabbit. In addition, we found that FOXO1 mRNA was expressed in Monodelphis ESF, both in vitro as well as in vivo at comparable levels, but FOXO1 protein could not be detected by immunohistochemistry or Western blotting, even though the antibody reacted with forced expressed Monodelphis FOXO1 protein (Kin et al. 2014). It is thus questionable that FOXO1 plays a role in opossum ESF cell identity. Also, in human ESF, FOXO1 mRNA is expressed at lower amounts than in decidual cells and FOXO1 protein is not found in the nucleus of human ESF. FOXO1 protein in human endometrial stromal cells is stabilized and retained in the nucleus only in response to decidualizing stimuli (Christian et al. 2011). We thus suggest that FOXO1 translation is posttranscriptionally suppressed in opossum ESF, and, even though FOXO1 mRNA is produced, it is likely not active in these cells. A possible role of FOXO1 expression in paleo-ESF is their role in inflammatory reactions. In mouse skin fibroblasts FOXO1 protein gets stabilized upon stimulation with TNF, a key inflammatory mediator (Alikhani et al. 2005). Hence the FOXO1 mRNA expression may thus be part of the mechanisms of paleo-ESF for mediating inflammatory reactions, like many other tissue fibroblasts.
Conclusions
The comparative study of ESF transcriptomes reveals key events in the evolutionary origin of eutherian pregnancy, in particular the evolution of a fetal–maternal interface that is compatible with an invasive conceptus. We identify several key differences between paleo-ESF, those that do not decidualize, and neo-ESF, those that can decidualize, and are characteristic of eutherian uterus. Paleo-ESF expresses genes that suggest that they play a role in innate immune response, like other fibroblasts. The expression of these genes was likely lost in the origin of eutherian mammals as is the expression of genes known to oppose tissue invasion such as WISP2 and others. Some of these genes have been re-expressed in the ESF of secondarily noninvasive species, such as the cow.
Supplementary Material
Supplementary methods, figure S1, and tables S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgment
Financial support provided by a grant from the John Templeton Foundation (Grant #54860) and the Yale University Science Development Fund is gratefully acknowledged. The opinions expressed in this article are not necessarily those of the John Templeton Foundation. The NG sequencing has been done at the Yale Center for Genome Analysis.
Literature Cited
- Alexander C, Rietschel ET. 2001. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 7:167–202. [PubMed] [Google Scholar]
- Alikhani M, Alikhani Z, Graves DT. 2005. FOXO1 functions as a master switch that regulates gene expression necessary for tumor necrosis factor-induced fibroblast apoptosis. J Biol Chem. 280:12096–12102. [DOI] [PubMed] [Google Scholar]
- Arendt D. 2008. The evolution of cell types in animals: emerging principles from molecular studies. Nat Rev Genet. 9:868–882. [DOI] [PubMed] [Google Scholar]
- Banerjee S, et al. 2008. CCN5/WISP-2 expression in breast adenocarcinoma is associated with less frequent progression of the disease and suppresses the invasive phenotypes of tumor cells. Cancer Res. 68:7606–7612. [DOI] [PubMed] [Google Scholar]
- Brawand D, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478:343–348. [DOI] [PubMed] [Google Scholar]
- Brayer KJ, Lynch VJ, Wagner GP. 2011. Evolution of a derived protein-protein interaction between HoxA11 and Foxo1a in mammals caused by changes in intramolecular regulation. Proc Natl Acad Sci U S A. 108:E414–E420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. 2003. Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet. 4:710–720. [DOI] [PubMed] [Google Scholar]
- Chavan ARC, Bhullar BAS, Wagner G. 2016. What was the ancestral function of decidual stromal cells? A model for the evolution of eutherian pregnancy. Placenta 40:40–51. [DOI] [PubMed] [Google Scholar]
- Christian M, Lam EW, Wilson MS, Brosens JJ. 2011. FOXO transcription factors and their role in disorders of the female reproductive tract. Curr Drug Targets. 12:1291–1302. [DOI] [PubMed] [Google Scholar]
- Das SK. 2010. Regional development of uterine decidualization: molecular signaling by Hoxa-10. Mol Reprod Dev. 77:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, et al. 2012. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc Biol Sci. 279:3491–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza AW, Wagner GP. 2014. Malignant cancer and invasive placentation: a case for positive pleiotropy between endometrial and malignancy phenotypes. Evol Med Public Health. 2014:136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson MT, et al. 2014. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet. 10:e1004158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot MG, Crespi BJ. 2009. Phylogenetic evidence for early hemochorial placentation in eutheria. Placenta 30:949–967. [DOI] [PubMed] [Google Scholar]
- Emera D, Romero R, Wagner G. 2012. The evolution of menstruation: a new model for genetic assimilation: explaining molecular origins of maternal responses to fetal invasiveness. Bioessays 34:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 2012. A comparative method for both discrete and continuous characters using the threshold model. Am Nat. 179:145–156. [DOI] [PubMed] [Google Scholar]
- Freyer C, Zeller U, Renfree MB. 2003. The marsupial placenta: a phylogenetic analysis. J Exp Zool A Comp Exp Biol. 299:59–77. [DOI] [PubMed] [Google Scholar]
- Fritah A, et al. 2008. Role of WISP-2/CCN5 in the maintenance of a differentiated and noninvasive phenotype in human breast cancer cells. Mol Cell Biol. 28:1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Bian F, Ma X, Kalinichenko VV, Das SK. 2015. Control of regional decidualization in implantation: role of FoxM1 downstream of Hoxa10 and cyclin D3. Sci Rep. 5:13863.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellersen B, Brosens JJ. 2014. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr Rev. 35:851–905. [DOI] [PubMed] [Google Scholar]
- Graf T, Enver T. 2009. Forcing cells to change lineages. Nature 462:587–594. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Van Dyke JU, Thompson MB. 2013. No implantation in an extra-uterine pregnancy of a placentotrophic reptile. Placenta 34:510–511. [DOI] [PubMed] [Google Scholar]
- Hobert O. 2011. Regulation of terminal differentiation programs in the nervous system. Annu Rev Cell Dev Biol. 27:681–696. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, et al. 1995. Hoxa 11 structure, extensive antisense transcription, and function in male and female fertility. Development 121:1373–1385. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Nielsen R, Bollback JP. 2003. Stochastic mapping of morphological characters. Syst Biol. 52:131–158. [DOI] [PubMed] [Google Scholar]
- Jiang Y, et al. 2015. FoxM1 directs STAT3 expression essential for human endometrial stromal decidualization. Sci Rep. 5:13735.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GA, et al. 2003. Osteopontin expression in uterine stroma indicates a decidualization-like differentiation during ovine pregnancy. Biol Reprod. 68:1951–1958. [DOI] [PubMed] [Google Scholar]
- Kamburov A, et al. 2011. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 39:D712–D717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin K. 2015. Inferring cell type innovations by phylogenetic methods-concepts, methods, and limitations. J Exp Zool B Mol Dev Evol. 324:653–661. [DOI] [PubMed] [Google Scholar]
- Kin K, Maziarz J, Wagner GP. 2014. Immunohistological study of the endometrial stromal fibroblasts in the opossum, Monodelphis domestica: evidence for homology with eutherian stromal fibroblasts. Biol Reprod. 90:111.. [DOI] [PubMed] [Google Scholar]
- Kin K, Nnamani MC, Lynch VJ, Michaelides E, Wagner GP. 2015. Cell-type phylogenetics and the origin of endometrial stromal cells. Cell Rep. 10:1398–1409. [DOI] [PubMed] [Google Scholar]
- Klagsbrun M, Eichmann A. 2005. A role for axon guidance receptors and ligands in blood vessel development and tumor angiogenesis. Cytokine Growth Factor Rev. 16:535–548. [DOI] [PubMed] [Google Scholar]
- Laird MK, Turancova M, McAllan BM, Murphy CR, Thompson MB. 2015. Unlocking amniote live birth: the “other” mammalian model. J Proc R Soc New South Wales. 148:52–59. [Google Scholar]
- Large MJ, DeMayo FJ. 2012. The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol Cell Endocrinol. 358:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Consortium F, Forrest AR, Wagner GP. 2015. The statistical geometry of transcriptome divergence in cell-type evolution and cancer. Nat Commun. 6:6066.. [DOI] [PubMed] [Google Scholar]
- Lydon JP, et al. 1995. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9:2266–2278. [DOI] [PubMed] [Google Scholar]
- Lynch VJ, May G, Wagner GP. 2011. Regulatory evolution through divergence of a phosphoswitch in the transcription factor CEBPB. Nature 480:383–386. [DOI] [PubMed] [Google Scholar]
- Maconochie MK, et al. 1997. Cross-regulation in the mouse HoxB complex: the expression of Hoxb2 in rhombomere 4 is regulated by Hoxb1. Genes Dev. 11:1885–1895. [DOI] [PubMed] [Google Scholar]
- Martin RD. 2008. Evolution of placentation in primates: implications of mammalian phylogeny. Evol Biol. 35:125–145. [Google Scholar]
- Mess A, Carter AM. 2006. Evolutionary transformations of fetal membrane characters in Eutheria with special reference to Afrotheria. J Exp Zool B Mol Dev Evol. 306:140–163. [DOI] [PubMed] [Google Scholar]
- Munoz-Fernandez R, et al. 2006. Follicular dendritic cells are related to bone marrow stromal cell progenitors and to myofibroblasts. J Immunol. 177:280–289. [DOI] [PubMed] [Google Scholar]
- Musser JM, Wagner GP. 2015. Character trees from transcriptome data: origin and individuation of morphological characters and the so-called “species signal.” J Exp Zool B Mol Dev Evol. 324:588–604. [DOI] [PubMed] [Google Scholar]
- Nilsson MA, et al. 2010. Tracking marsupial evolution using archaic genomic retroposon insertions. PLos Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohnke Y, Kempf R, Gellersen B. 1999. CCAAT/enhancer-binding proteins are mediators in the protein kinase A-dependent activation of the decidual prolactin promoter. J Biol Chem. 274:24808–24818. [DOI] [PubMed] [Google Scholar]
- Raines AM, et al. 2013. Recombineering-based dissection of flanking and paralogous Hox gene functions in mouse reproductive tracts. Development 140:2942–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey EM. 1982. The placenta: human and animal. New York: Praeger Publisher. [Google Scholar]
- Renfree MB. 2010. Marsupials: placental mammals with a difference. Placenta 31:S21–S26. [DOI] [PubMed] [Google Scholar]
- Revell LJ. 2014. Ancestral character estimation under the threshold model from quantitative genetics. Evolution 68:743–759. [DOI] [PubMed] [Google Scholar]
- Smith RS, Smith TJ, Blieden TM, Phipps RP. 1997. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol. 151:317–322. [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HS, Arici A, Olive D, Igarashi P. 1998. HOXA10 is expressed in response to sex steroids at the time of implantation in the human endometrium. J Clin Invest. 101:1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine JW, Collins AG, Meyer CP. 1994. Morphological complexity increase in metazoans. Paleobiology 20:131–142. [Google Scholar]
- Vilella AJ, et al. 2009. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP. 2014. Homology, genes, and evolutionary innovation. Princeton, NJ: Princeton University Press. [Google Scholar]
- Wagner GP, Kin K, Lynch VJ. 2012. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131:281–285. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Lynch VJ. 2013. A model based criterion for gene expression calls using RNA-seq data. Theory Biosci. 132:159–164. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Muglia L, Pavlicev M. 2014. Evolution of mammalian pregnancy and the origin of the decidual stromal cell. Int J Dev Biol. 58:117–126. [DOI] [PubMed] [Google Scholar]
- Wildman DE, et al. 2006. Evolution of the mammalian placenta revealed by phylogenetic analysis. Proc Natl Acad Sci U S A. 103:3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.