

Summary
Viral replication in the liver is generally detected by cellular endosomal Toll‐like receptors (TLRs) and cytosolic helicase sensors that trigger antiviral inflammatory responses. Recent evidence suggests that surface TLR2 may also contribute to viral detection through recognition of viral coat proteins but its role in the outcome of acute viral infection remains elusive. In this study, we examined in vivo the role of TLR2 in acute infections induced by the highly hepatotrophic mouse hepatitis virus (MHV) type 3 and weakly hepatotrophic MHV‐A59 serotype. To address this, C57BL/6 (wild‐type; WT) and TLR2 knockout (KO) groups of mice were intraperitoneally infected with MHV3 or MHV‐A59. MHV3 infection provoked a fulminant hepatitis in WT mice, characterized by early mortality and high alanine and aspartate transaminase levels, histopathological lesions and viral replication whereas infection of TLR2 KO mice was markedly less severe. MHV‐A59 provoked a comparable mild and subclinical hepatitis in WT and TLR2 KO mice. MHV3‐induced fulminant hepatitis in WT mice correlated with higher hepatic expression of interferon‐β, interleukin‐6, tumour necrosis factor‐α, CXCL1, CCL2, CXCL10 and alarmin (interleukin‐33) than in MHV‐A59‐infected WT mice and in MHV3‐infected TLR2 KO mice. Intrahepatic recruited neutrophils, natural killer cells, natural killer T cells or macrophages rapidly decreased in MHV3‐infected WT mice whereas they were sustained in MHV‐A59‐infected WT mice and MHV3‐infected TLR2 KO. MHV3 in vitro infection of macrophagic cells induced rapid and higher viral replication and/or interleukin‐6 induction in comparison to MHV‐A59, and depended on viral activation of TLR2 and p38 mitogen‐activated protein kinase. Taken together, these results support a new aggravating inflammatory role for TLR2 in MHV3‐induced acute fulminant hepatitis.
Keywords: coronavirus, inflammation, Toll‐like receptor‐2, viral hepatitis
Abbreviations
- ALT
alanine transaminase
- AST
aspartate transaminase
- CCL
chemokines CXCL
- Fgl‐2
fibrinogen‐like protein
- FHF
fulminant hepatic failure
- HBC
hepatitis B virus
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HPRT
hypoxanthine ribosyltransferase
- IFN‐α/β
interferon‐α/β
- IL
interleukin
- i.p.
intraperitoneally
- KO
knockout
- LSECs
liver sinusoidal endothelial cells
- MAPK
mitogen‐activated protein kinase
- MDA‐5
melanoma differentiation‐associated protein
- MHV
mouse hepatitis virus
- MNCs
mononuclear cells
- m.o.i.
multiplicity of infection
- NK cells
natural killer cells
- NK‐T cells
natural killer‐T cells
- p.i.
post‐infection
- PRR
pattern recognition receptor
- RIG‐1
retinoic acid inducible gene
- siRNA
small interfering RNA
- TGF‐β
transforming growth factor β
- TLR
Toll‐like receptor
- TNF‐α
tumour necrosis factor α
- WT
wild‐type
Introduction
Surface or endosomal toll‐like receptors (TLR) are key pattern recognition receptors (PRRs) of infectious microorganisms in innate immunity. Host immune recognition of viruses during infections relies mainly on a combination of endosomal Toll‐like receptors (TLR3, ‐7/8, ‐9) and cytosolic helicases retinoic acid inducible gene (RIG‐1) and melanoma differentiation‐associated protein (MDA‐5) that sense viral RNA or DNA and trigger signalling pathways leading to inflammatory cytokines, chemokines and antiviral type 1 interferon (IFN‐α/β) production (reviewed in ref. 1).
Increasing evidence has shown that surface TLRs, such as TLR2 or TLR4, may also trigger acute inflammatory responses against viral infections through recognition of viral coat or core proteins.2, 3, 4, 5 Activation of surface TLR‐dependent signalling pathways leads to the production of various chemokines involved in the recruitment of natural killer (NK) cells, macrophages, neutrophils and B‐ and T‐cell subsets (reviewed in ref. 5). Higher expression of TLR2 and/or TLR4 and up‐regulation of inflammatory factors have been observed in macrophages in vitro infected by some viruses such as human immunodeficiency virus (HIV),6, 7, 8 influenza virus,9 hepatitis C virus (HCV),4 hepatitis B virus (HBV),10 severe acute respiratory syndrome virus11 and several herpes viruses.12, 13, 14 TLR2 and/or TLR4 have also been involved in the induction of high inflammatory responses during acute viral infections. Indeed, TLR2 and TLR4 increase the susceptibility to rotavirus infections in infants15 and TLR2 was suggested as an aggravating inflammatory factor in herpes virus infections.12, 14
Recent studies have also reported an over‐expression of TLR2 in the liver and on monocytes from HCV‐ or HCV/HIV‐infected patients correlating with hepatic inflammation and damages, suggesting a role for TLR2 in hepatitis‐associated inflammation.16, 17 HCV and HBV infections present subclinical to severe acute‐phase patterns that may lead to viral clearance or evolve towards chronic infections (reviewed in ref. 18). For reasons that are not well understood, few cases of acute infections progress into fulminant hepatic failure, characterized by extensive necrosis and hepatocellular dysfunction, exacerbated inflammation and high mortality rate.19 Hepatic lesions occurring during viral infections are primarily related to virally triggered host inflammatory responses (reviewed in ref. 20) and depend on a poorly elucidated balance between innate immune cell–virus interactions and the control of viral replication that is critical in the outcome of hepatitis. The contribution of TLR2 in antiviral defences and inflammatory responses during acute hepatic viral infections is unknown and needs investigation.
The mouse hepatitis viruses (MHV), belonging to the coronavirus group, induce acute or subclinical hepatitis, neurological, respiratory or/and enteric diseases in mice according to serotypes.21 The highly hepatotrophic MHV3 serotype is a relevant model for studying virus‐induced inflammatory disorders during acute hepatic infections as it induces fulminant lethal hepatitis in C57BL/6 mice within 4 days post‐infection (p.i.)22 whereas the weakly hepatotrophic MHV‐A59 serotype induces a subclinical hepatitis and is rapidly cleared in the liver.23 Within 72 hr of infection, liver dysfunction in MHV3‐infected mice results from several foci of extensive necrosis24 in contrast to that observed in the liver of mice infected with the MHV‐A59 serotype.25 All MHV serotypes use the CEACAM1a molecule as viral receptor for infection of host cells through interaction with their viral surface (S) protein.26 It was previously demonstrated that the differential levels of viral replication and hepatitis induced by the MHV serotypes were largely related to the viral S protein,27 suggesting that interactions of the S protein with molecules other than CEACAM1a may reflect their virulence for liver.
Peritoneal macrophages are the first viral cell target during MHV infection, followed by liver cells such as hepatocytes, macrophages, liver sinusoidal endothelial cells (LSECs) and Ito cells.26, 28 Intrahepatic macrophages, LSECs as well as NK and NK T cells are considered as major contributors to antiviral responses and release of cytokines/chemokines in the liver upon viral infection.29 During MHV3 acute infection, inflammatory mediators such as tumour necrosis factor‐α (TNF‐α), interleukin‐1 (IL‐1), transforming growth factor‐β (TGF‐β), leukotriene B4 and mouse fibrinogen‐like 2 (Fgl‐2) protein are strongly produced by infected macrophages, correlating with the severity of hepatitis.30 Accordingly, depletion of macrophages was reported to protect against the fulminance of hepatitis,31 suggesting that exacerbated inflammatory responses are an aggravating factor during MHV3 infection. In addition, the intrahepatic tolerance sustained by IL‐4, IL‐10, TGF‐β and prostaglandin E2 is also disturbed.32 In contrast, it has been suggested that macrophages would rather contribute to the lower gravity of hepatitis and viral clearance during MHV‐A59 infection, presumably through a rapid type 1 IFN‐dependent suppression of viral replication.33 It was demonstrated that the induction of IL‐6 and TNF‐α in peritoneal macrophages infected by MHV3 depended on the fixation of the viral surface (S) protein to TLR2.34 Previous in vivo studies also revealed higher levels of IL‐6 and TNF‐α in livers from MHV3‐infected C57BL/6 mice than TLR2 knockout (KO) mice, suggesting an inflammatory role for TLR2 in MHV3 infection.34 Hence, the ability of MHVs to ligate and activate TLR2‐dependent inflammatory pathways in the liver may represent one determining and differential factor involved in their virulence.
In this study, we demonstrated that the severe acute hepatitis provoked by the highly hepatotrophic MHV3 but not by the weakly hepatotrophic MHV‐A59 serotype, is aggravated by TLR2 in the liver, as demonstrated by significantly higher mortality, liver injury, viral replication, levels of inflammatory cytokines and chemokines as well as disturbances in intrahepatic neutrophils, macrophages, NK and NK T‐cell recruitment in MHV3‐infected wild‐type (WT) but not infected TLR2 KO mice or MHV‐A59‐infected WT mice. Through in vitro infection of macrophagic cells, we showed that rapid and higher viral replication and/or IL‐6 induction by MHV3, in comparison to MHV‐A59, depended on activation of TLR2 and p38 mitogen‐activated protein kinase (MAPK), pointing out macrophages as one source of TLR2‐dependent inflammatory responses in MHV3 infection.
Material and methods
Mice
Female C57BL/6 (Charles River, St Constant, Qc, Canada) and TLR2 KO (C57BL/6 background, Jackson Laboratory, Bar Harbor, MA) mice were housed in a HEPA‐filtered air environment. All experiments were conducted with mice between 8 and 10 weeks of age in compliance with the regulations of the Animal Committee of the University of Quebec at Montreal (CIPA‐no.641).
Viruses and cells
Highly hepatotrophic MHV3 is a cloned substrain isolated from the liver of MHV3‐infected DBA2 mice.35 Weakly hepatotrophic MHV‐A59 serotype was obtained from the American Type Culture Collection (Rockville, MD). Both viruses were produced in L2 cells as previously described34 and used within three passages. Pathogenic properties of MHV3 were assessed routinely. The mouse fibroblastic L2 cell line used for virus production and titration was grown in RPMI‐1640 supplemented with 10% fetal calf serum (GIBCO Laboratories, Grand Island, NY) and antibiotics. Murine macrophagic cells J774A.1 (TIB‐67™) were grown in RPMI‐1640 supplemented with l‐glutamine, antibiotics (GIBCO Laboratories) and 5% fetal bovine serum (Gemini Bio‐Products, Woodland, CA). All cells were passaged before reaching 85% confluence.
In vivo viral infections
Groups of six or seven WT C57BL/6 and TLR2 KO mice were infected intraperitoneally (i.p.) with 103 TCID50 of MHV3 or MHV‐A59. Mock‐infected mice received a similar volume of PBS. Clinical signs and survival percentages were recorded and mice were killed when clinical signs reached limit points as determined by CIPA regulations. In other experiments, mice were killed by CO2 anoxia at 24, 48 and 72 hr p.i. without regard for clinical signs. Liver and blood samples were collected and processed for further analyses.
Histopathological, transaminase activity and immunohistochemical analyses
The histopathological analysis of liver was performed using haematoxylin & eosin–safran staining. Levels of serum alanine and aspartate transaminases (ALT and AST) were assessed according to the IFCC primary reference procedures using Olympus AU2700 Autoanalyser® (Olympus Optical, Tokyo, Japan). Immunolocalization of IL‐33 or CXCL10 was performed by histochemical staining using primary goat anti‐mouse‐IL‐33 and anti‐CXCL10 (R&D Systems, Minneapolis, MN) and secondary horseradish peroxidase‐conjugated rabbit anti‐goat antibody for IL‐33 (Dako, Markham, Ont., Canada) and OmniMap anti‐Rabbit‐horseradish peroxidase (RUO) for CXCL‐10 followed by haematoxylin counterstaining in a Ventana machine (Ventana Medical Systems, Inc. Tucson, AZ), as previously described.36 Counting of necrotic areas and inflammatory foci was carried out on liver areas of 15–30 mm2 using the NDP.2 view image analysis software (Hamamatsu Photonics K.K., Japan).
In vitro viral infections
J774A.1 cells were infected with MHV‐A59 or MHV3 at a multiplicity of infection (m.o.i.) of 0·1–1 or treated with the synthetic bacterial ligand for TLR1/2 (Pam3CSK4) (InvivoGen, San Diego, CA). Infections were conducted in a minimal volume of fresh complete medium for the first 2 hr and then incubated at 37° in 5% CO2 atmosphere for various times p.i. according to experiments. Supernatants were collected and kept at −80° for subsequent viral titration and/or ELISA tests. Total cell RNA was extracted from cell culture and prepared for subsequent RT‐PCR analysis.
RNA interference treatments
J774A.1 cells were plated in 24‐well plates at 6 × 104 cells per well and transfected with 25 nm of mouse CEACAM1 small interfering RNA (siRNA) FlexiTube premix (Qiagen, Cambridge, MA) (Mm_Ceacam1_3: CACACTCATGCATTCACTCTA) and/or mouse TLR2 (Mm_Tlr2_4: CTCGTTCTCCAGCATTTAAA) for at least 24 hr before infection. Negative and positive siRNA controls (AllStars Negative Control siRNA and Ctrl_AllStars_3,respectively, Qiagen), were added to all transfection experiments. Synthetic bacterial ligand for TLR1/2 (Pam3CSK4) (InvivoGen San Diego, CA) was used as TLR2‐positive control for cytokine production.
RNA isolation and quantitative RT‐PCR
Total RNA from frozen liver samples of C57BL/6 and TLR2 KO mice was extracted using TRIzol reagent (Invitrogen, Burlington, Ont., Canada) and residual genomic DNA was removed with the Turbo DNA‐free kit (Ambion, Austin, TX). Cell culture RNA was extracted with the NucleoSpin® RNA II kit (Macherey‐Nagel GmbH & Co. KG, Düren, Germany). One microgram of RNA was retro‐transcribed into cDNA using the high‐capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Real‐time PCR amplification was carried out on 25 ng cDNA using the HotStart‐IT™ SYBR® Green qPCR Master Mix (USB Corporation, Cleveland, OH) on an ABI 7300 system (Applied Biosystems). Threshold cycle values (Ct) were collected and used for ‘ΔΔCt’ analysis. Specific primers for hypoxanthine phosphoribosyltransferase (HPRT), MHV‐nucleocapsid, TLR2, TLR3, TLR4, TLR7, MDA‐5, RIG‐1, IFN‐β, IL‐6, TNF‐α, IL‐33, Fgl‐2, CXCL1, CXCL10 and CCL2 were used (Table 1). The relative gene expression was normalized to HPRT as endogenous control and expressed as a ratio to gene expression in mock‐infected mice livers (arbitrarily taken as 1). The specificity of the PCR products was confirmed by melting curve analyses.
Table 1.
Primer sets used for quantitative RT‐PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| HPRT | 5′‐GAAAGACTTGCTCGAGATGTCATG‐3′ | 5′‐CACACAGAGGGCCACAATGT‐3′ |
| IFN‐β | 5′‐CGGACTTCAAGATCCCTATGGA‐3′ | 5′‐TGGCAAAGGCAGTGTAACTCTTC‐3′ |
| IL‐6 | 5′‐TCGGAGGCTTAATTACACATGTTC‐3′ | 5′‐TGCCATTGCACAACTCTTTTCT‐3′ |
| TNF‐α | 5′‐TCCCAGGTTCTCTTCAAGGGA‐3′ | 5′‐GGTGAGGAGCACGTAGTCGG‐3′ |
| CCL2 | 5′‐GCAGCAGGTGTCCCAAAGAA‐3′ | 5′GGTCAGCACAGACCTCTCTCTTG‐3′ |
| CXCL10 | 5′‐GGCCATAGGGAAGCTTGAAAT‐3′ | 5′‐TCGTGGCAATGATCTCAACAC‐3′ |
| TLR2 | 5′‐CCCTGTGCCACCATTTCC‐3′ | 5′‐CCACGCCCACATCATTCTC‐3′ |
| TLR3 | 5′‐TGGGCTGAAGTGGACAAATCT‐3′ | 5′‐TGCCGACATCATGGAGGTT‐3′ |
| TLR4 | AGCTTCAATGGTGCCATCATT | CCAGGTGCTGCAGCTCTTCT |
| TLR7 | 5′‐CAGTGAACTCTGGCCGTTGA‐3′ | 5′‐CAAGCCGGTTGTTGGAGAA‐3′ |
| MHV‐N | 5′‐TGGAAGGTCTGCACCTGCTA‐3′ | 5′‐TTTGGCCCACGGGATTG‐3′ |
| RIG‐I | 5′‐GCCAGAGTGTCAGAATCTCAGTCAG‐3′ | 5′‐GAGAACACAGTTGCCTGCTGCTCA‐3′ |
| MDA‐5 | 5′‐GCCCTCTCCTTCCTCTGAGACT‐3′ | 5′‐GCTGGAGGAGGGTCAGCAA‐3′ |
| IL‐33 | 5′‐GCTGCGTCTGTTGACACATTG‐3′ | 5′‐GGGAGGCAGGAGACTGTGTTAA‐3′ |
| Fgl‐2 | 5′‐ CGTTGTGGTCAACAGTTTGGA‐3′ | 5′‐ GATGTTGAACCGGCTGTGACT‐3′ |
| CXCL1 | 5′‐CCGAAGTCATAGCCACACTCAA‐3′ | 5′‐CAAGGGAGCTTCAGGGTCAA‐3′ |
ELISAs
Frozen liver samples from C57BL/6 and TLR2 KO mice were weighted and homogenized in Nonidet‐P40 lysis buffer (Invitrogen) completed with a protease inhibitor cocktail and 1 mm PMSF (Sigma Aldrich, St Louis, MA) for protein extraction. Liver suspension was kept on ice for 30 min and centrifuged for 10 min at 1000g. Determination of IFN‐β (PBL, Piscataway, NJ), IL‐6, TNF‐α (BD Biosciences, Mississauga, Ont., Canada), and CXCL1, CXCL10, CCL2 (eBiosciences, San Diego, CA) levels in liver lysates or cell culture supernatants was carried out according to the manufacturers’ procedures.
p38 MAPK assay
The p38 MAPK activity has been evaluated in J774A.1 cells plated in 24‐well plates at 3 × 104 cells per well 24 hr before infections with MHV‐A59 or L2‐MHV3 at an m.o.i. of 5. At 5, 15, 30, 45 or 60 min p.i., supernatants were collected and frozen at −80°, and the cells were washed three times with cold PBS once before RNA and protein extraction. Activity of p38 MAPK in in vitro infected cells was evaluated by phosphorylation (T180/Y182) levels with the ELISA ONE™ kit (TGR BioSciences, Thebarton, Australia) according to the manufacturer's indications. Values are expressed as percentage of phosphorylated p38/ total p38 relative to cellular control.
Virus titration
Frozen liver samples from 24‐hr and 72‐hr MHV3‐ or MHV‐A59‐infected C57BL/6 or TLR2 KO mice were weighted and homogenized in cold PBS. Suspension was then centrifuged at 1000g for 30 min, 10‐fold serial‐diluted and tested for viral detection on L2 cells cultured in 96‐well plates. Cytopathic effects were recorded at 72 hr p.i. and virus titres were determined according to the Reed–Muench method and expressed as log10 TCID50.
Cytofluorometric studies
Livers were perfused with PBS through the portal vein to remove blood cell contamination before dissection. After homogenization of liver tissue and elimination of hepatocytes by sedimentation, immune cells were purified using 35% Percoll gradient (Sigma Aldrich) and red blood cells were lysed with a Tris‐buffered ammonium chloride solution. Mononuclear cells (MNCs; 106 cells) were incubated with anti‐CD16/32 antibodies (BD Biosciences) to block non‐specific binding. Cells were then incubated with optimal dilutions of anti‐CD3‐V500, anti‐Gr1‐V450, anti‐CD11b‐phycoerythrin‐Cy7, anti‐CD19‐allophycocyanin, anti‐CD4‐FITC, anti‐NK1.1‐Peridinin chlorophyll protein‐Cy‐5.5 and anti‐CD8‐allophycocyanin‐Cy7 antibodies (BD Biosciences) and fixed in PBS containing 2% fetal calf serum, 0·01 m sodium azide and 2% formaldehyde. Stained cells were analysed on a FACS Aria II® flow cytometer using BD FACS diva software (BD Bioscience) and the data were processed using CXP software (Beckman Coulter, Mississauga, Ont, Canada). Dead cells and doublet cells were excluded on the basis of forward and side scatter and analyses were performed on 10 000 events recorded. Myeloid cells, gated by high side scatter, were assessed for CD11b and Gr1 to enumerate macrophages (CD11b+ Gr1inter) and neutrophils (CD11b+ Gr1high). Lymphoid cells were gated according to forward and side scatter and first assessed for NK1.1 and CD3 expression to discriminate NK from NKT cells. CD3+ NK1.1− T cells were further gated to allow determination of CD4+ and CD8+ subpopulations. B lymphocytes were determined by CD19+ CD3− expression (see Supplementary material, Fig. S1).
Statistical analyses
Data obtained from in vivo experiments were expressed as means ± SEM. Multiple group analyses for PCR, ELISA and viral titres data were evaluated by one‐way analyses of variance test with post‐hoc Tukey test using PASW statistics software (PASW version 18, IBM SPSS Inc. Chicago, IL). Survival curve comparisons were performed using the Log Rank test. Analysis of in vitro results was performed using Student's t‐test to evaluate the statistical significance of differences between infected or treated cells and uninfected or untreated cells. All results are shown as means ± standard error. Values of P ≤ 0·05 were considered as significant.
Results
Higher hepatic damage and viral replication in the liver of MHV3‐infected than MHV‐A59‐infected mice
To confirm that the highly hepatotrophic MHV3 induces more dramatic hepatic lesions than the weakly virulent MHV‐A59 serotype following i.p. inoculation, groups of C57BL/6 mice were infected with both viruses, livers were collected at 24–72 hr p.i. and histopathological analysis, viral replication and IFN‐β production levels were recorded. Histopathological analysis of livers from MHV3‐infected mice revealed few inflammatory cells surrounding necrotic foci as soon as 24 hr p.i. [Fig. 1a (i)]. Presence of inflammatory cells, however, was reduced from 48 hr p.i. whereas necrotic foci extended until 72 hr p.i. [Fig. 1a (ii, iii)]. No inflammatory foci were observed in livers from mock‐infected mice (results not shown). Larger inflammatory foci, however, were observed in liver from MHV‐A59‐infected mice up to 48 hr p.i. without extensive necrosis areas as seen in MHV3‐infected mice (P ≤ 0·01) [Fig. 1a (iv–vii)]. Extensive hepatic lesions in mice infected by MHV3 correlated with higher levels of AST and ALT at 72 hr p.i. than in MHV‐A59‐infected mice [Fig. 1b (i, ii)] (P ≤ 0·001).
Figure 1.
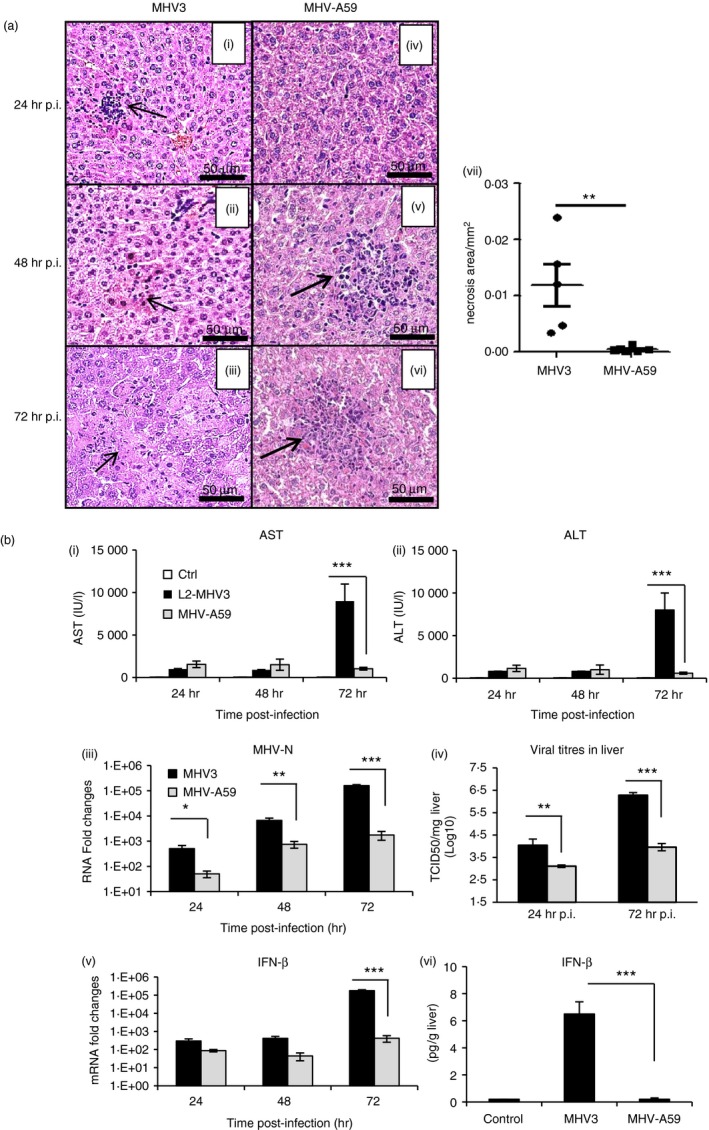
Mortality, hepatic damages and viral replication during murine hepatitis virus 3 (MHV3) and MHV‐A59‐induced hepatitis in C57BL/6 mice. Groups of six or seven C57BL/6 mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3 or MHV‐A59. Percentages of surviving mice were recorded at various times post‐infection (p.i.). (a) (i–vi) Histopathological analysis was conducted on livers at 24, 48 and 72 hr p.i. (vii) Histological analysis of inflammatory foci in liver from MHV3‐ or MHV‐A59 infected C57BL/6 mice. (b) Serum samples from infected mice were assayed for aspartate transaminase (AST) and alanine transaminase (ALT) activity from 24 to 72 hr p.i. (i, ii). MHV3 or MHV‐A59 replication in livers of infected mice was determined by analysis of the nucleoprotein (MHV‐N) RNA expression from 24 to 72 hr p.i. by quantitative RT‐PCR, and values represent fold change in gene expression relative to mock‐infected mice after normalization with HPRT expression (iii). Viral titration (TCID50) in liver was assayed at 24 and 72 hr p.i. (iv). mRNA fold increases for interferon‐β (IFN‐β) were evaluated by quantitative RT‐PCR in livers of MHV3‐ or MHV‐A59 infected mice. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression (v). Production levels of IFN‐β were quantified by ELISA test at 72 hr p.i. in livers (vi). Results are representative of two different experiments. (*P < 0·05; **P < 0·01; ***P < 0·001).
Viral replication, as evidenced by nucleocapsid RNA levels and viral titres, increased more in the liver of MHV3‐infected mice when compared with mice infected with MHV‐A59 (P ≤ 0·05 to P ≤ 0·001) [Fig. 1b (iii, iv)]. MHV3 replication in the liver increased throughout infection despite higher IFN‐β production (P ≤ 0·001) [Fig. 1b (v, vi)]. Such lower viral replication of MHV‐A59, however, did not result from an increase in IFN‐β, since lower mRNA levels and production of IFN‐β were detected in the liver of MHV‐A59‐infected. No increase in IFN‐α transcription has been observed in both MHV3‐ and MHV‐A59‐infected mice (results not shown).
Higher expression of TLR2 over other TLRs and helicases in the liver of MHV3‐ than MHV‐A59‐infected mice
Several endosomal TLRs and helicases are simultaneously activated and up‐regulated upon viral infection, triggering inflammatory responses (reviewed in ref. 1). We explored the hypothesis that MHV3 infection may induce higher expression of TLRs or helicase genes in the liver than MHV‐A59. Hence, kinetics of transcription levels of surface TLR2 and TLR4, endosomal TLR3 and TLR7, and helicase RIG‐1 and MDA‐5 genes have been assessed by quantitative RT‐PCR in the liver of infected mice from 24 to 72 hr p.i. and expressed as mRNA fold changes relative to levels in mock‐infected mice. As shown in Fig. 2(a), TLR2 gene expression steadily increased over the course of infection with MHV3 reaching over 120‐fold the expression in mock‐infected mice at 72 hr p.i. whereas it remained barely increased and drastically lower in MHV‐A59‐infected mice (P ≤ 0·05 to P ≤ 0·001). Levels of TLR4 and TLR7 mRNA expression were not affected by either MHV3 or MHV‐A59 infections (Fig. 2b,c). Endosomal TLR3 or RIG‐1 and MDA‐5 gene expression levels also increased in MHV3‐infected mice, albeit markedly less than TLR2, and were similarly lower or not induced in mice infected with MHV‐A59 (P ≤ 0·05 to P ≤ 0·001) (Fig. 2d–f).
Figure 2.
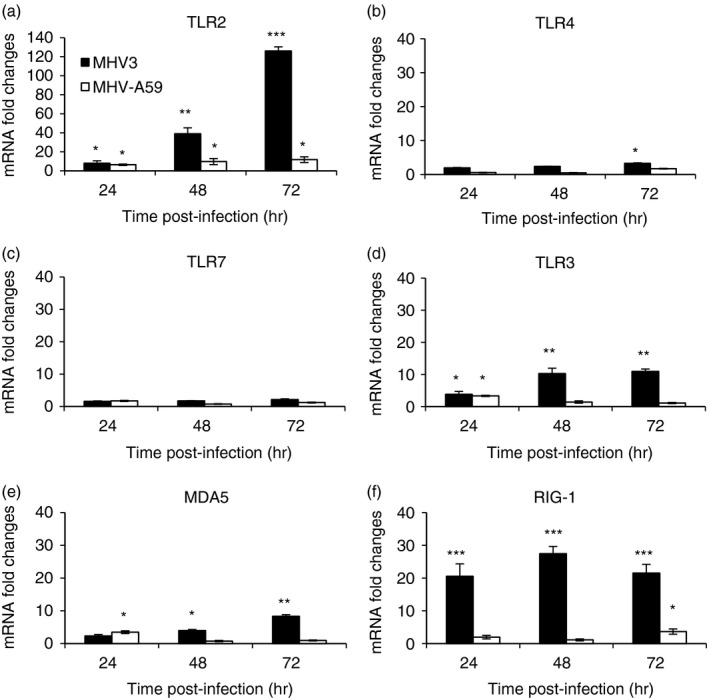
Messenger RNA levels of Toll‐like receptor 2 (TLR2), TLR4, TLR3, TLR7 and helicases retinoic acid inducible gene (RIG‐1) and melanoma differentiation‐associated protein (MDA‐5) genes in the liver of murine hepatitis virus (MHV) 3‐ and MHV‐A59‐infected mice. Groups of six or seven C57BL/6 (wild‐type; WT) mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3 or MHV‐A59. At 24, 48 or 72 hr post‐infection (p.i.), livers from each group were collected and Toll‐like receptors (TLR2, ‐3, ‐4, ‐7) (a–d) and helicases (MDA‐5 and RIG‐1) (e and f) mRNA fold changes were analysed by quantitative RT‐PCR. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Results are representative of two different experiments. (*P < 0·05; **P < 0·01; ***P < 0·001).
These data suggest that MHV3 but not MHV‐A59 infection strongly induces expression of TLR2, over other TLRs and helicases, in the liver of infected mice.
Higher expression of inflammatory cytokines, IL‐33 (alarmin) and Fgl‐2 in the liver of MHV3‐infected than MHV‐A59‐infected mice
Activation of TLRs, mainly surface TLR2, is involved in the release of pro‐inflammatory cytokines such as TNF‐α and IL‐6 (reviewed in ref. 1). Higher induction of hepatic TLR2 expression by MHV3 than MHV‐A59 infection suggests that higher levels of TNF‐α or IL‐6 may occur in the liver of MHV3‐infected mice. To investigate this hypothesis, levels of TNF‐α and IL‐6 expression were evaluated by quantitative RT‐PCR and ELISA tests in livers from infected mice. As shown in Fig. 3(a,b), IL‐6 mRNA and secretion levels increased only in MHV3‐infected mice (P ≤ 0·05 and P ≤ 0·001). Higher expression of TNF‐α was also found in the liver of MHV3‐ than MHV‐A59‐infected mice (P ≤ 0·01 to P ≤ 0·001) (Fig. 3c) and correlated with higher secretion at 72 hr p.i. (P ≤ 0·001) (Fig. 3d).
Figure 3.
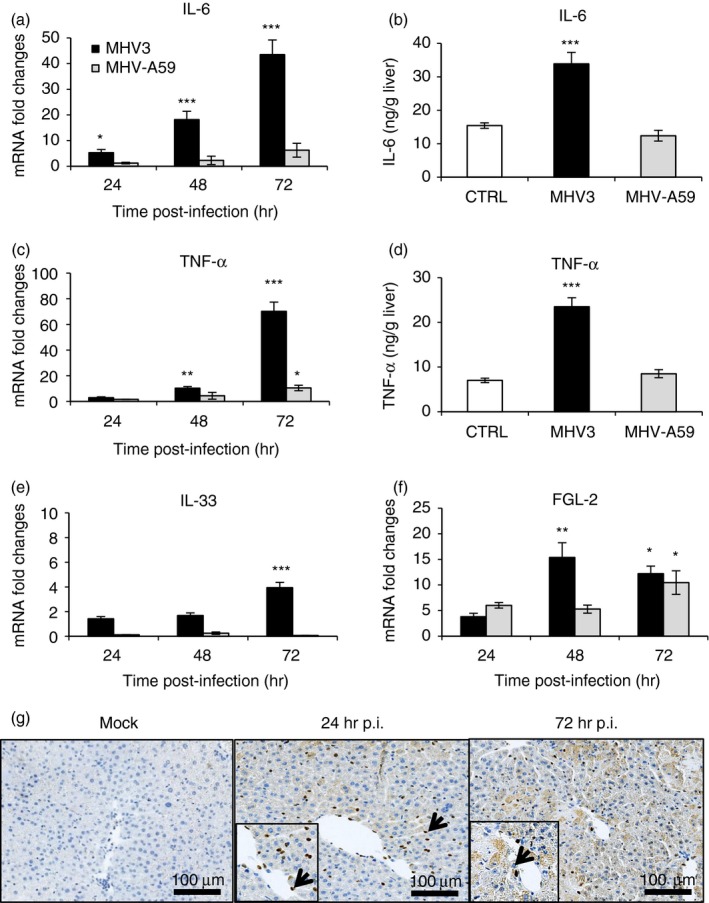
Messenger RNA levels and production of inflammatory cytokines, interleukin‐33 (IL‐33) and fibrinogen‐like 2 (Fgl‐2) in the liver of murine hepatitis virus (MHV) 3‐ and MHV‐A59‐infected mice. Groups of six or seven C57BL/6 mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3 or MHV‐A59. At 24, 48 or 72 hr post‐infection (p.i.), livers from each group were collected and mRNA fold increases for interleukin‐6 (IL‐6) (a), tumour necrosis factor‐α (TNF‐α) (c), IL‐33 (e) and Fgl‐2 (f) were evaluated by quantitative RT‐PCR in livers of infected mice. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Production levels of IL‐6 (b) and TNF‐α (d) were quantified by ELISA test at 72 hr p.i. in livers. Expression of IL‐33 determined by immunohistochemistry in livers from mock‐infected and MHV3‐infected WT mice at 24 and 72 hr p.i. (g). Some positive IL‐33 cells are indicated by arrows. Results are representative of two different experiments. (*P < 0·05; **P < 0·01; ***P < 0·001).
We have previously demonstrated that expression of IL‐33, a new alarmin, was up‐regulated in the liver of MHV3‐infected C57BL/6 mice correlating with an increase of inflammatory cytokines.36 Messenger RNA levels and immunolocalization for IL‐33 were assayed in the livers of MHV3‐infected and/or MHV‐A59‐infected mice. As shown in Fig. 3(e), gene expression of IL‐33 increased only in the liver of MHV3‐infected mice (P ≤ 0·001) and IL‐33 was localized mainly in cells lining sinusoids and to a lesser extent in hepatocytes at 24 and 72 hr p.i. in MHV3‐infected mice (Fig. 3g).
It was previously demonstrated that fulminance of MHV3‐induced hepatitis correlated with levels of Fgl‐2 produced by liver sinusoidal endothelial cells.37, 38 As expected, Fgl‐2 expression increased sooner, at 48 hr p.i., in the liver of MHV3‐infected mice than MHV‐A59‐infected mice (P ≤ 0·05 and P ≤ 0·01).
Activation of TLR2, as other TLRs, is also involved in chemokine production in acute and chronic liver diseases39 (reviewed in ref. 1). We hypothesized that MHV3 infection may favour greater release of chemokines in the liver than MHV‐A59 infection. Hence, mRNA and protein levels of CCL2, CXCL1 and CXCL10 were evaluated by quantitative RT‐PCR and ELISA tests, respectively, in the livers of infected mice. As shown in Fig. 4, transcription levels of CCL2 and CXCL10 genes increased over infection time in MHV3‐infected mice and reached higher levels at 72 hr p.i. than in MHV‐A59‐infected mice (P ≤ 0·05 to P ≤ 0·001) (Fig. 4a,c). CCL2 was highly produced in liver from MHV3‐infected mice (P ≤ 0·01 and P ≤ 0·001) (Fig. 4b). Production of CXCL10, however, was higher in the liver of MHV‐A59‐infected than MHV3‐infected mice although gene expression was lower (P ≤ 0·01 and P ≤ 0·001) (Fig. 4d). Levels of mRNA and protein for CXCL1 were more increased in the liver of MHV3‐infected mice than in MHV‐A59‐infected mice (P ≤ 0·05 to P ≤ 0·001) (Fig. 4e,f).
Figure 4.
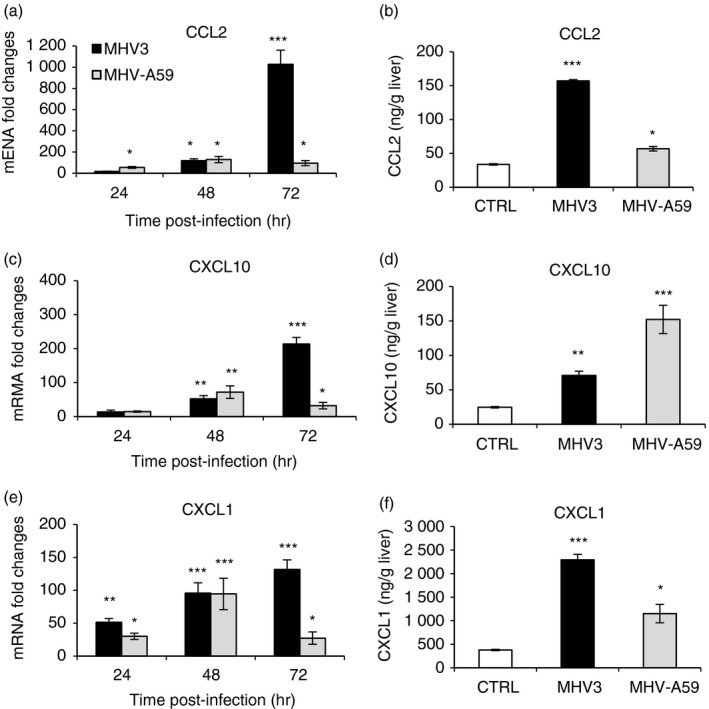
Messenger RNA expression and production of chemokines CCL2, CXCL1 and CXCL10 in the livers of murine hepatitis virus (MHV) 3‐ and MHV‐A59‐infected mice. Groups of six or seven C57BL/6 mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3 or MHV‐A59. At 24, 48 or 72 hr post‐infection (p.i.), livers from each group were collected. mRNA expression levels for CCL2 (a), CXCL10 (c) and CXCL1 (e) were evaluated by quantitative RT‐PCR at 24, 48 and 72 hr p.i in livers from MHV3‐ or MHV‐A59‐infected mice. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Protein levels of CCL2 (b), CXCL10 (d) and CXCL1 (f) were quantified in livers by ELISA tests at 72 hr p.i. (*P < 0·05; **P < 0·01; ***P < 0·001).
Transient and lower recruitment of neutrophils, NK cells and macrophages in the livers of MHV3‐ and MHV‐A59‐infected mice
Higher CXCL1, CCL2 and lower CXCL10 levels in the livers of MHV3‐infected mice suggest higher recruitment of neutrophils, macrophages, NK and NK T cells rather than lymphoid cells (reviewed in ref. 40). Smaller or lower inflammatory foci, however, were observed in the livers of MHV3‐infected mice compared with MHV‐A59‐infected mice [Fig. 1a (ii–vi)] suggesting that inflammatory cell subsets are differentially recruited during MHV3 and MHV‐A59 infections. To verify this hypothesis, intrahepatic MNCs were isolated from the livers of MHV3‐ and MHV‐A59‐infected mice at 24 and 48 hr p.i., immunolabelled and then phenotyped by cytofluorometric analysis. Percentages and numbers of NK‐T (NK1.1+ CD3+) and NK (NK1.1+ CD3−) cells, neutrophils (CD11bhi Gr1hi), macrophages (CD11b+ Gr1int) as well as B (CD19+), CD4 (CD3+ CD4+) and CD8 (CD3+ CD8+) lymphocyte subsets were compared between groups of mice. As shown in Fig. 5(a), percentages of neutrophils (CD11bhi Gr1hi) and macrophages (CD11b+ Gr1int) rapidly increased at 24 hr p.i. in MHV3‐infected mice in spite of high individual variation for neutrophils (P ≤ 0·05 to P ≤ 0·001). Percentages of NK cells (NK1.1+ CD3−) increased only at 48 hr p.i (P ≤ 0·001) and NK‐T (NK1.1+ CD3+) cell percentage decreased at 24 hr p.i. only (P ≤ 0·001). Percentages of CD4, CD8 and B lymphocytes decreased in the livers of MHV3‐infected mice at 24 and/or 48 hr p.i. (P ≤ 0·05 to P ≤ 0·001) (Fig. 5b), as previously reported.41
Figure 5.
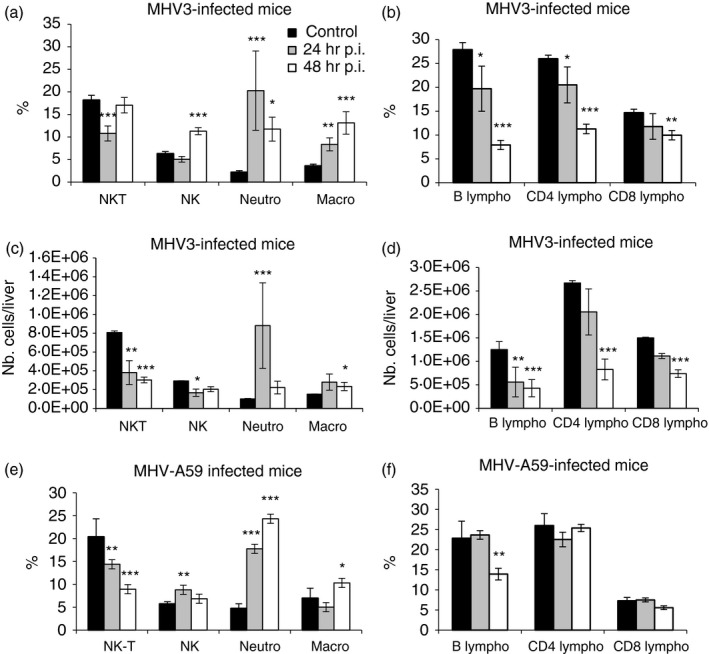
Analysis of intrahepatic mononculear cells (MNCs) in the liver of murine hepatitis virus (MHV) 3‐ and MHV‐A59‐infected C57BL/6 mice. Intrahepatic MNCs were isolated from groups of six mock‐infected, MHV3‐ and MHV‐A59‐infected C57BL/6 mice at 24 and 48 hr p.i., immunolabelled with NK1.1, CD3, Gr1, CD11b, CD19, CD4 and CD8 monoclonal antibodies and analysed by cytofluorometry. Percentages of natural killer T (NK‐T) (NK1.1+ CD3+), natural killer (NK) (NK1.1+ CD3−), neutrophils (Gr1hi CD11bhi) and macrophages (Gr1+ CD11bint) cells were evaluated in the livers of MHV3 (a) and MHV‐A59 (e) ‐infected mice. Percentages of b (CD19+) and CD4 (CD3+ CD4+) and CD8 (CD3+ CD8+) subpopulations of CD3+ NK1.1− cells were similarly analysed (b and f). Absolute numbers for each cell subset were calculated in using respective percentages reported to total number of isolated MNC in the liver of MHV3‐ (c and d). (*P < 0·05; **P < 0·01; ***P < 0·001).
Analyses of cell‐subset percentages, however, are not fully representative of recruited inflammatory cells because total number of isolated MNCs strongly decreased with time in the liver of MHV3‐infected mice.41 To properly reflect the evolution of cell recruitment in infected mice, absolute numbers of each inflammatory cell subset have been calculated using the percentage of each cell subset reported to total number of isolated MNCs. As shown in Fig. 5(c), the transient increase of neutrophil percentage observed in the liver of MHV3‐infected mice was in accordance with the increase in neutrophil numbers at 24 hr p.i. only (P ≤ 0·001). However, low increase of absolute number of macrophages occurred at 48 hr p.i. only (P ≤ 0·05). In addition, both NK‐T and NK cell numbers decreased (P ≤ 0·05 to P ≤ 0·001) in contrast to that seen in percentages. The decreases in numbers of CD4, CD8 and B cells were magnified when compared with percentages (P ≤ 0·01 and P ≤ 0·001) (Fig. 5b,d). The extensive cell necrosis in the liver of MHV3‐infected mice at 72 hr p.i., however, did not allow us to isolate sufficient numbers of MNCs for accurate immunolabellings and cytofluorometric analysis.
In livers from MHV‐A59‐infected mice, however, percentages of neutrophils strongly increased as soon as 24 hr p.i. (P ≤ 0·001) whereas NK cells transiently increased (P ≤ 0·05) and NK‐T cells decreased (P ≤ 0·01 and P ≤ 0·001) (Fig. 5e). Macrophage percentage slightly increased at 48 hr p.i. only (P ≤ 0·05). In contrast to that seen in the liver of MHV3‐infected mice, only the B‐cell percentage decreased slightly at 48 hr p.i. (P ≤ 0·01) whereas CD4 and CD8 lymphocyte percentages remained unchanged (Fig. 5f). The number of MNCs isolated in the liver of MHV‐A59‐infected mice did not significantly differ that in from mock‐infected mice, in contrast with that seen in MHV3‐infected mice. In consequence, variations in absolute numbers of cell subsets did not differ from percentages (results not shown).
Lower liver damage and viral replication in MHV3‐infected TLR2 KO mice
We have previously observed that MHV3‐induced acute hepatitis was less severe in TLR2 KO mice.34 The comparative study between MHV3 and MHV‐A59 infections regarding severity of hepatitis, TLR2 expression level, and inflammatory cytokines and chemokines in liver from infected mice support this hypothesis and suggest that higher virulence of MHV3 may be related to TLR2‐dependent exacerbated inflammatory responses. To verify whether TLR2 is involved in aggravated hepatic damages and viral replication during acute MHV3 infection, groups of WT C57BL/6 and TLR2 KO mice were i.p. infected with MHV3 or MHV‐A59 and survival rates, liver injury and viral load in the liver were monitored from 24 to 72 hr p.i. As shown in Fig. 6(a), mortality rate of MHV3‐infected TLR2 KO mice was delayed when compared with infected WT mice as it occurred from only 96 to 120 hr p.i. in contrast to WT mice, for which mortality reached more than 70% of mice at 72 hr p.i. (P ≤ 0·001). Considering the fulminance of hepatitis, such a statistically significant delay can support a role for TLR2 in the pathogenic processes of hepatitis. As expected, no mortality was observed in WT and TLR2 KO mice infected with MHV‐A59 (see Supplementary material, Fig. S2A). Histopathological analysis of livers from MHV3‐infected TLR2 KO revealed inflammatory foci only by 48 hr p.i. and fewer necrotic foci at 72 hr p.i. when compared with liver from MHV3‐infected WT mice (Fig. 6b compared with Fig. 1a). In livers from MHV‐A59‐infected TLR2 KO mice, however, few large inflammatory foci, similar to those observed in MHV‐A59‐infected WT mice, were noted (see Supplementary material, Fig. S2B). Accordingly, serum AST and ALT levels were lower in MHV3‐infected TLR2 KO than in infected WT mice (Fig. 6c,d) (P ≤ 0·05 and P ≤ 0·001). Hepatic lesions, comparable to those seen in livers from MHV3‐infected WT mice, occurred later in infected TLR2 KO mice (results not shown), leading to a delayed mortality of mice.
Figure 6.
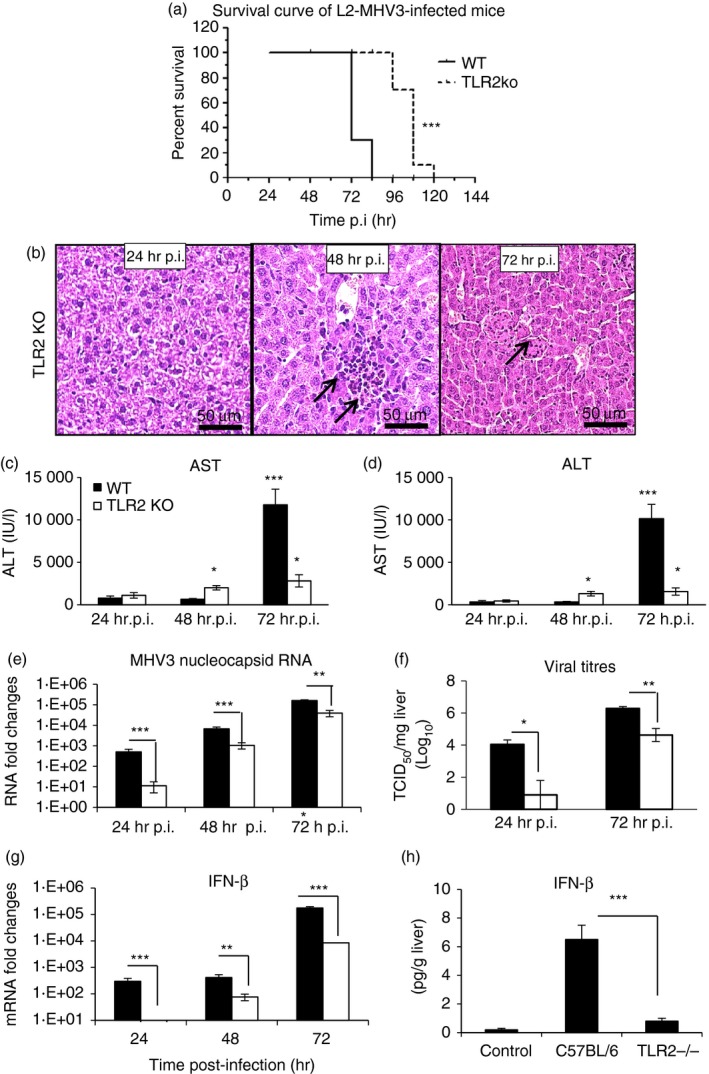
Hepatic damages, viral replication and interferon‐β (IFN‐β) production in the liver of murine hepatitis virus (MHV) 3‐infected wild‐type (WT) and Toll‐like receptor 2 knockout (TLR2 KO) mice. Groups of six to seven C57BL/6 (WT) and TLR2 KO mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3. Survival curve of MHV3‐infected WT and TLR2 KO mice (a). Histopathological analysis was conducted on livers from MHV3‐infected TLR2 KO at 24, 48 and 72 hr post‐infection (p.i.) (b). Serum samples from MHV3‐infected WT and TLR2 KO mice were assayed for aspartate transaminase (AST) (c) and alanine transaminase (ALT) (d) activity at 24–72 hr p.i. Replication of MHV3 in livers of infected mice was determined by analysis of the nucleoprotein (MHV‐N) RNA expression at 24, 48 and 72 hr p.i. by quantitative RT‐PCR, and by viral titration (TCID50) at 24 and 72 hr p.i. (e, f). Messenger RNA fold increases for IFN‐β were analysed in livers from MHV3‐infected mice by quantitative RT‐PCR at 24, 48 and 72 hr p.i. (g). Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Protein levels of IFN‐β in the liver were quantified by ELISA test at 72 hr p.i. (h). (*P < 0·05; **P < 0·01; ***P < 0·001).
Viral replication of MHV3 in the liver of infected TLR2 KO, as determined by the expression of the viral nucleoprotein RNA (quantitative RT‐PCR) and viral titres, was also lower and delayed when compared with replication in MHV3‐infected WT mice (Fig. 6e,f) (P ≤ 0·05 to P ≤ 0·001). MHV‐A59 replication in livers from TLR2 KO mice, however, was not altered (see Supplementary material, Fig. S2C) when compared with infected WT mice.
Dietrich et al.42 have previously reported that TLR2 signalling may lead to IFN‐α/β production when TLR2 is internalized in endosomes. As shown in Fig. 6(g,h), transcription and production of IFN‐β occurred later and reached lower levels in MHV3‐infected TLR2 KO mice than in WT mice (P ≤ 0·001). No significant increase of IFN‐α transcription has been observed in both MHV3‐infected mouse strains (results not shown).
TLR2 is involved in aggravation of MHV3‐induced hepatitis instead of other TLRs or helicase genes
To support the hypothesis that severity of MHV3‐induced hepatitis mainly involves TLR2 rather than other PRRs, kinetics of transcription levels of surface TLR2 and TLR4, endosomal TLR3 and TLR7, as well as helicase RIG‐1 and MDA‐5, genes were compared in livers from MHV3‐infected WT and TLR2 KO mice. As observed above, an increase of surface TLR2 but not TLR4 gene expression levels over the course of infection by MHV3 was confirmed in the liver of WT mice (P ≤ 0·05 to P ≤ 0·001) (Fig. 7a,b) whereas absence of TLR2 induction was validated in infected TLR2 KO mice. Endosomal TLR3 or RIG‐1 and MDA‐5, but not TLR7, gene expression levels similarly increased in livers from both WT and TLR2 KO mice infected with MHV3 (P ≤ 0·05 to P ≤ 0·001) (Fig. 7c–f). These data support that MHV3 infection specifically increases the expression of TLR2 in the liver of WT mice because other TLRs and helicases are similarly expressed in infected WT and TLR2 KO mice.
Figure 7.
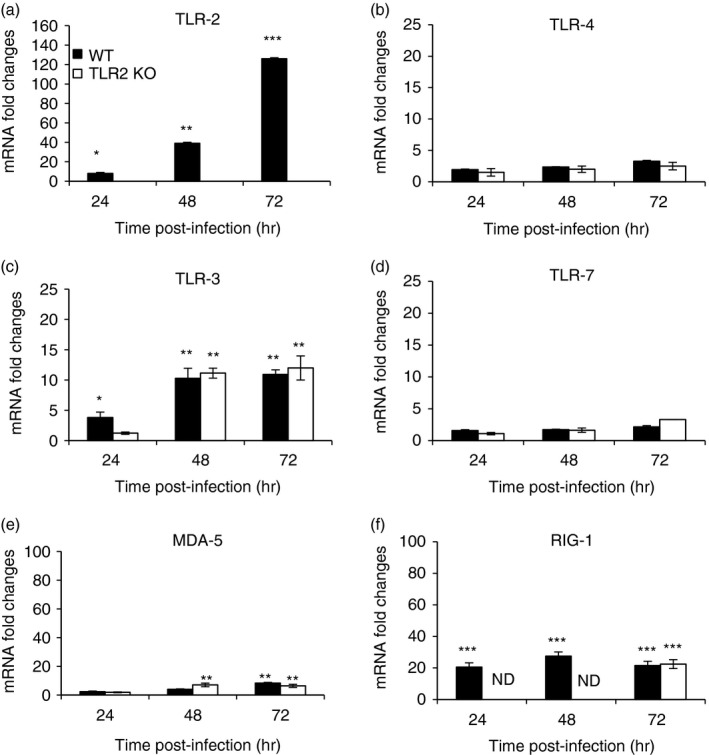
Messenger RNA levels of Toll‐like receptors (TLRs) and helicases in the livers of murine hepatitis virus (MHV) 3‐infected wild‐type (WT) and TLR2 knockout (KO) mice. Groups of six or seven C57BL/6 (WT) and TLR2 KO mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3. At 24, 48 or 72 hr post‐infection (p.i.), livers from each group were collected and Toll‐like receptors (TLR2, ‐3, ‐4, ‐7) (a–d) and helicases [retinoic acid inducible gene (RIG‐1) and melanoma differentiation‐associated protein (MDA‐5)] (e and f) mRNA fold changes were analysed by quantitative RT‐PCR. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. (*P < 0·05; **P < 0·01; ***P < 0·001). N.D. not done.
Decreases of inflammatory cytokines, alarmin IL‐33 but not Fgl‐2 gene expression in the liver of MHV3‐infected TLR2 KO mice
We have previously reported that IL‐6 and TNF‐α production by peritoneal macrophages in vitro infected with MHV3 depends on TLR2 activation by the viral glycoprotein S.34 In order to verify whether TLR2 is specifically involved in the exacerbation of the inflammatory cytokine response during MHV3 infection, as previously observed in Fig. 3, levels of TNF‐α and IL‐6 expression were compared by quantitative RT‐PCR and ELISA tests in MHV3‐infected WT and TLR2 KO mice. As shown in Fig. 8(a), mRNA levels of IL‐6 increased throughout MHV3 infection in WT mice (P ≤ 0·01 and P ≤ 0·001) but were impaired in infected TLR2 KO mice as demonstrated by fourfold lower levels at 72 hr p.i. (P ≤ 0·05). Interleukin‐6 secretion increased only in the liver of MHV3‐infected WT mice (P ≤ 0·001) (Fig. 8b). Similarly, lower expression of TNF‐α was found in the livers of MHV3‐infected TLR2 KO mice than WT mice (P ≤ 0·05 and P ≤ 0·001) (Fig. 8c). Such defects in TNF‐α mRNA expression correlated with lower production at 72 hr p.i. (P ≤ 0·001) (Fig. 8d).
Figure 8.
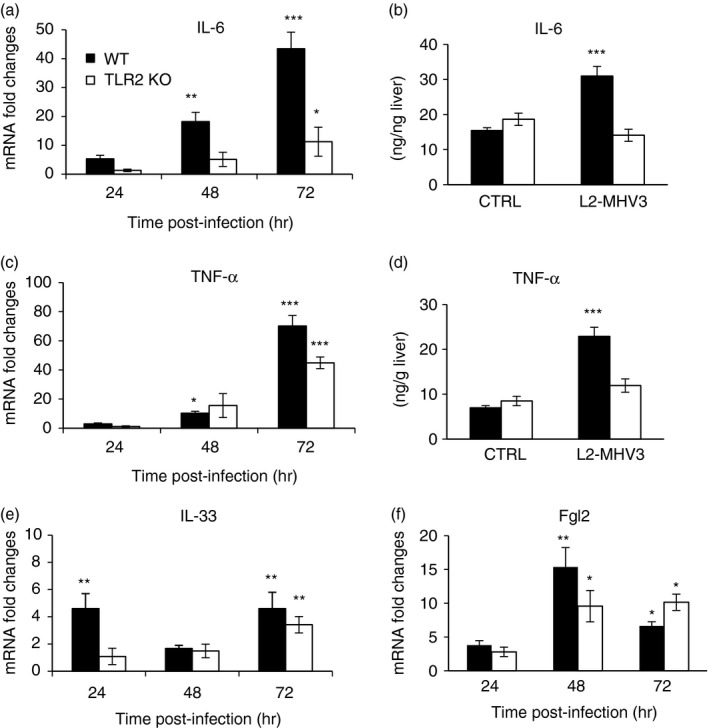
Expression levels of cytokines, interleukin‐33 (IL‐33) and fibrinogen‐like 2 (Fgl‐2) gene in the liver of murine hepatitis virus (MHV) 3‐infected wild‐type (WT) and Toll‐like receptor 2 (TLR2) knockout (KO) mice. Groups of six or seven C57BL/6 (WT) and TLR2 KO mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3. At 24, 48 or 72 hr p.i., livers from each group were collected. mRNA fold increases for IL‐6 (a), tumour necrosis factor‐α (TNF‐α) (c), IL‐33 (e), and the Fgl‐2 (f) were evaluated by quantitative RT‐PCR in livers of infected mice. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Production levels of IL‐6 (b) and TNF‐α (d) were quantified in livers by ELISA tests at 72 hr p.i. (*P < 0·05; **P < 0·01; ***P < 0·001).
The IL‐33 gene expression level was delayed to 72 hr p.i. in MHV3‐infected TLR2 KO mice whereas it was induced as early as 24 hr p.i. in WT mice (P ≤ 0·01) (Fig. 8e). mRNA levels for Fgl‐2, however, increased similarly up to 48 hr p.i. in the livers of both WT and TLR2 KO infected mice (P ≤ 0·05 and P ≤ 0·01) (Fig. 8f).
Decreases in CXCL1, CCL2 and CXCL10 levels in the livers of MHV3‐infected TLR2 KO mice
It was recently shown that the TLR2 signalling network is essential for inflammatory cell recruitment in acute liver injury.39 Accordingly, we hypothesized that TLR2 activation may be involved in the induction of the high chemokine levels observed during MHV3 infection (Fig. 4) but expected that it would be decreased in liver from infected TLR2 KO mice. As shown in Fig. 9(a,b), mRNA and protein levels of CXCL1 increased sooner and higher in the liver of MHV3‐infected WT mice than TLR2 KO mice (P ≤ 0·05 to P ≤ 0·001). Transcription and production levels of CCL2 and CXCL10 also increased over infection time in infected WT mice but were dramatically impaired and delayed in infected TLR2 KO mice (P ≤ 0·05 to P ≤ 0·001) (Fig. 9c–f). Immunolocalization of CXCL10 showed lower expression in hepatocytes of MHV3‐infected TLR2 KO than WT mice at 72 hr p.i. (Fig. 9g).
Figure 9.
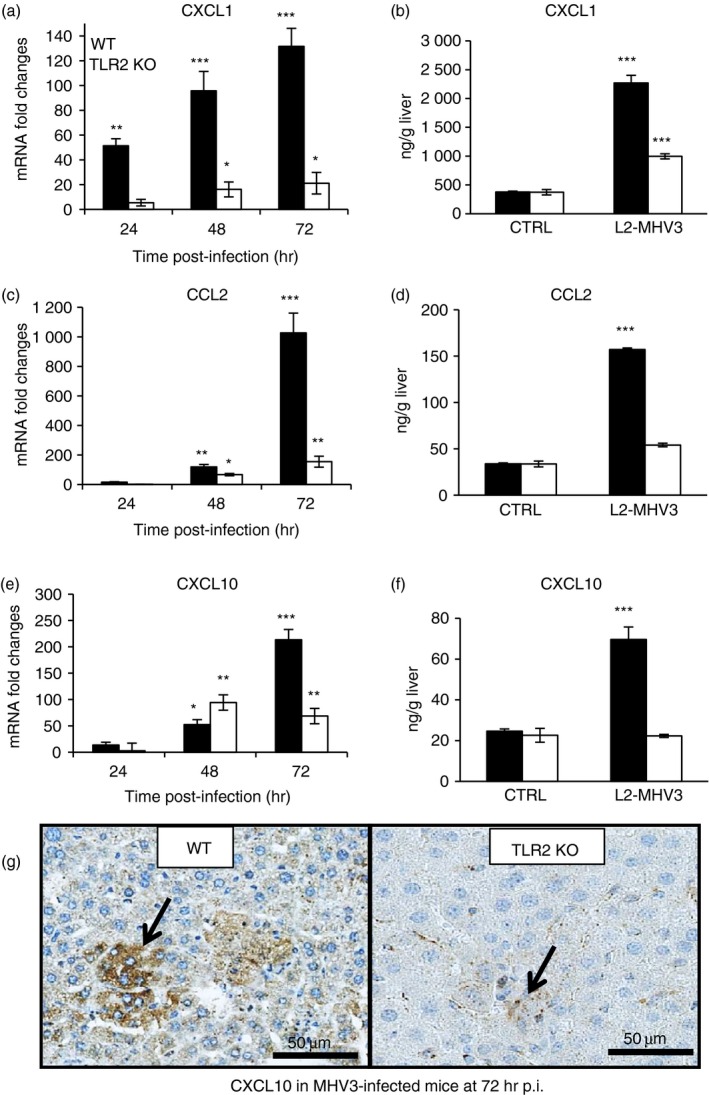
Messenger RNA expression levels and production of CCL2, CXCL1 and CXCL10 in the livers of murine hepatitis virus (MHV) 3‐infected wild‐type (WT) and Toll‐like receptor knockout (TLR2 KO) mice. Groups of six or seven C57BL/6 (WT) and TLR2 KO mice were intraperitoneally (i.p.) infected with 1000 TCID50 of MHV3. At 24, 48 or 72 hr post‐infection (p.i.), livers from each group were collected. mRNA expression for CXCL1 (a), CCL2 (c) and CXCL10 (e) genes was evaluated by quantitative RT‐PCR in livers from MHV3‐infected WT and TLR2 KO mice. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Protein levels of CXCL1 (b), CCL2 (d) and CXCL10 (f) in the liver were quantified by ELISA test at 72 hr p.i. Immunolocalization of CXCL10 in the liver determined by histochemistry staining in MHV3‐infected WT and TLR2 KO mice (g). (*P < 0·05; **P < 0·01; ***P < 0·001).
Delayed recruitment of neutrophils, NK cells and macrophages in the liver of MHV3‐infected TLR2 KO mice
To verify whether the reduced production of CXCL1, CCL2 and/or CXCL10 in the liver of MHV3‐infected TLR2 KO mice involved lower recruitment of inflammatory cells, intrahepatic MNCs were isolated from the livers of mock‐infected and MHV3‐infected WT and TLR2 KO mice at 24 and 48 hr p.i., immunolabelled and then phenotyped by cytofluorometric analysis, as indicated above. As shown in the Supplementary material (Fig. S3A), percentages of neutrophils and macrophages increased at 48 hr p.i. only in the liver of MHV3‐infected TLR2 KO mice (P ≤ 0·001) whereas NK and NKT cell percentages remained unchanged. Both B and CD8 cell percentages, however, decreased in the liver of infected TLR2 KO mice (P ≤ 0·01 and P ≤ 0·001) (see Supplementary material, Fig. S3B). As no major reduction in total isolated MNCs was observed over time in livers from MHV3‐infected TLR2 KO mice in contrast to that seen in MHV3‐infected WT mice, changes in percentages for each cell subset reflect similar changes in cell numbers (results not shown).
Taken together, these results suggest that TLR2 favours greater hepatic damage, more viral replication and a stronger inflammatory response as well as the transient increase in inflammatory cells and losses of NK and NKT cells in the liver only in MHV3‐infected WT mice, not in MHV‐A59‐infected WT mice or MHV3‐infected TLR2 KO mice, supporting an aggravating role for TLR2 in acute hepatitis.
TLR2‐dependent viral replication and inflammatory responses in in vitro MHV3‐infected macrophages
We aimed to identify the cells involved in the exacerbated TLR2‐dependent inflammatory responses in the liver of MHV3‐infected mice. We first investigated whether TLR2 expression was increased in in vitro infected macrophages, hepatocytes and LSECs. Preliminary data revealed that both LSECs (Bleau et al. manuscript in revision) and macrophages expressed higher TLR2 expression levels upon MHV3 infection. It has been previously demonstrated that macrophages are the first target cells following MHV infection and that lower virulence of MHV‐A59 is due to suppression of viral replication by these cells.22, 33 In addition, induction of TNF‐α and IL‐6 in peritoneal macrophages infected with MHV3 was shown to depend on surface (S) viral protein fixation to TLR2 and heparan sulphate34 in contrast to MHV‐A59.43 These observations suggest that macrophages may be one cellular source participating in TLR2‐dependent inflammatory responses during MHV3 infection.
We first verified whether MHV‐A59 and MHV3 differentially replicate and induce cytokine production in macrophages. J774A.1 macrophages were infected with both viruses at 0·1–1 m.o.i. for 4–24 hr p.i. and MHV nucleoprotein (N‐MHV) and IL‐6 expression were then evaluated. As shown in Fig. 10(a), the nucleoprotein RNA expression occurred sooner and reached higher levels in cells infected by MHV3 than MHV‐A59 (P ≤ 0·01 to P ≤ 0·001). In accordance, viral titres were also higher in MHV3‐infected cells (results not shown). Rapid and higher IL‐6 transcription levels occurred in MHV3‐infected cells (Fig. 10b) except at 24 hr p.i. (P ≤ 0·001) and correlated with higher production levels (MHV3: 430 ± 45 pg/ml; MHV‐A59: 82 ± 15 pg/ml; P ≤ 0·001). To verify whether higher replication of MHV3 in macrophages was associated with faster viral entry into cells, immediate‐early levels (within 60 min p.i.) of N‐MHV RNA levels were evaluated in MHV3 and MHV‐A59‐infected J774.1 cells. Higher levels of N‐MHV mRNA were detected in MHV3‐infected cells than in MHV‐A59‐infected cells, from 5 min p.i. until 60 min p.i. (P ≤ 0·05 to P ≤ 0·001) (Fig. 10c).
Figure 10.
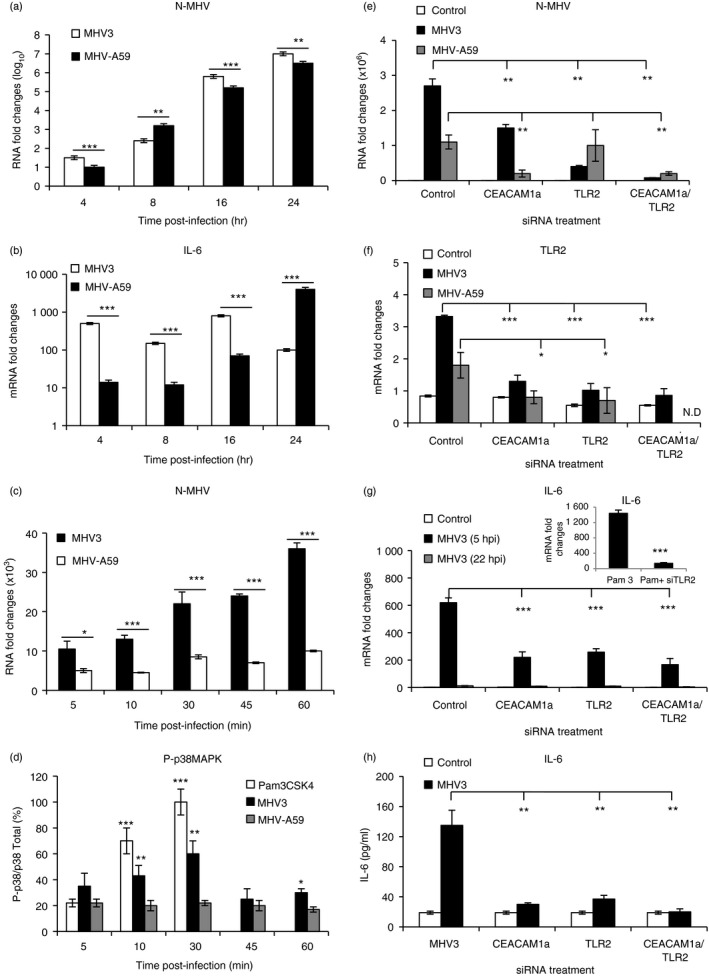
In vitro murine hepatitis virus (MHV) 3 and MHV‐A59 infections of macrophages untreated or treated with small interfering (si) RNA for Toll‐like receptor 2 (TLR2) and CEACAM1a. J774A.1 macrophages were infected with 0·1–1 multiplicity of infection (m.o.i.) of MHV3 or MHV‐A59 and incubated for various times post‐infection (p.i.) according to experiments (a–d). Cells were also transfected with mouse CEACAM1 and/or TLR2 siRNA for at least 24 hr before infections in other experiments (e and f). Nucleocapsid RNA (a, c and e), TLR2 (f), and interleukin‐6 (IL‐6) (b and g) mRNA fold changes were analysed by quantitative RT‐PCR at various times p.i. Values represent fold change in gene expression relative to mock‐infected mice (arbitrary value of 1) after normalization with HPRT expression. Phosphorylated p38 mitogen‐activated protein kinase (MAPK) levels were evaluated by ELISA test and expressed as percentages of total p38 MAPK (d). The synthetic bacterial ligand for TLR2–TLR1 (Pam3CSK4) was used as TLR2 positive control for the detection of phosphorylated p38 MAPK and IL‐6 expression (d and g). IL‐6 secretion was quantified by ELISA test in MHV3‐infected cells at 5 hr p.i. (h). Results are representative of two different experiments. (*P < 0·05; **P < 0·01; ***P < 0·001). N.D. Not done.
Since early p38 MAPK activation is involved in early secretion of IL‐6 in MHV3‐infected peritoneal macrophages,34 the kinetics of p38 MAPK phosphorylation was evaluated within the first 60 min p.i. by ELISA test. A TLR2 agonist (Pam3C5K4) was used as positive control for TLR2‐dependent induction of the p38 MAPK signalling pathway. Results shown in Fig. 10(d) indicate that phosphorylated p38 MAPK rapidly increased in cells treated with TLR2 agonist and peaked within 30 min (P ≤ 0·001). Similar increase, peaking at 30 min, also occurred in MHV3 infected cells (P ≤ 0·05 to P ≤ 0·01) whereas no increase of phosphorylated p38 MAPK was detected in MHV‐A59‐infected cells. These results suggest that TLR2 may be involved in rapid and higher viral replication and IL‐6 secretion in MHV3‐infected cells.
To verify this hypothesis, J774.1 cells were next treated with siRNAs for TLR2 and/or CEACAM1a genes before infection with MHV3 at an m.o.i of 0·1–1·0 for 22 hr. MHV RNA, TLR2 and CEACAM1a fold changes were then evaluated by quantitative RT‐PCR and infectious viruses were titrated. Results indicate that MHV‐RNA expression decreased in MHV3‐infected cells treated with siRNA for CEACAM1a and/or TLR2 (P ≤ 0·01) (Fig. 10e). Similarly to N‐MHV RNA levels, infectious MHV3 virus titres in supernatants of infected J774.1 cells decreased in siCEACAM1a‐ or siTLR2‐ or both siCEACAM1a/siTLR2‐treated J774.1 cells when compared with untreated infected cells (1·2 × 106 ± 0·3 × 106; 4·5 × 105 ± 0·3 × 105 and 1·5 × 104 ± 0·3 × 104 compared with 2·5 × 106 ± 0·8 × 106 TCID50/ml, respectively; P ≤ 0·05 to P ≤ 0·01). MHV‐A59 RNA, however, decreased only in cells treated with siRNA for CEACAM1a (alone or in combination with siRNA for TLR2) (P ≤ 0·01)(Fig. 10e). As expected, TLR2 expression levels increased higher in MHV3‐infected macrophages, and such an increase was also inhibited following knockdown of one or both CEACAM1a and TLR2 genes (P ≤ 0·001) (Fig. 10f).
To confirm the role of TLR2 in the early induction of the IL‐6 response by MHV3, IL‐6 mRNA fold changes and secretion were assessed in macrophages treated with siRNA for TLR2 and/or CEACAM1a and infected for 5 or 22 hr p.i. As shown in Fig. 10(g,h), knockdown of CEACAM1a and/or TLR2 genes strongly decreased IL‐6 transcription and secretion levels by MHV3‐infected macrophages, as also shown at 5 hr p.i. (P ≤ 0·001), indicating that IL‐6 production depends on both CEACAM1a and TLR2 molecules. The efficiency of siTLR2 treatment and the involvement of TLR2 in IL‐6 induction was confirmed by significant decreases of IL‐6 expression in cells activated by the TLR2 agonist Pam3CSK4 (P ≤ 0·01) (Fig. 10g).
Discussion
In this work, we demonstrated for the first time an aggravating role for TLR2 in the acute phase of hepatitis in comparing the fulminant MHV3‐induced hepatitis to milder hepatitis induced by the closely related MHV‐A59 infection in mice. The involvement for TLR2 in the fulminance of MHV3‐induced acute hepatitis was shown by earlier mortality and higher hepatic lesions and viral replication in infected WT mice than TLR2 KO mice whereas subclinical hepatitis induced by MHV‐A59 infection was not influenced by TLR2 as mortality rate, hepatic damages and viral titres were comparable in both infected WT and TLR2 KO mice. The severity of hepatitis in MHV3‐infected WT mice correlated with higher expression of TLR2, IFN‐b inflammatory cytokines and chemokines and alarmin IL‐33 in the liver. Despite higher chemokine levels, neutrophils, NK, NK‐T cells or macrophages were only transiently or weakly recruited in the liver of MHV3‐infected WT mice in contrast to delayed but sustained inflammatory cells recruitment in MHV3‐infected TLR2 KO or MHV‐A59‐infected WT mice.
It is the first report showing higher TLR2 expression over other PRRs in acute viral hepatitis during the first days of infection. Many PRRs are activated after exposure to hepatotrophic viruses, such as endosomal TLR3 or TLR7 and intracytoplasmic helicases RIG‐1 or MDA‐5, to activate IFN type 1 and inflammatory factors. In the livers from MHV3‐ but not MHV‐A59‐infected WT mice, we have observed greater expression of TLR2 over other PRRs such as TLR3, RIG‐1, MDA‐5 or TLR4 and TR7, suggesting that MHV3 preferentially activates TLR2 transcription. Higher expression of TLR2 has been recently associated with disease progression of hepatitis C.44 Levels of hepatic inflammation in HCV‐ and HCV/HIV‐infected patients, also correlated with higher transcription of TLR2 and TLR4 genes in the liver17 suggesting a pro‐inflammatory role for TLR2 in hepatitis. Accordingly, blocking of TLR2 was recently shown to attenuate Concanavalin A‐induced experimental hepatitis in mice.45 We have observed fewer hepatic lesions, less viral replication and lower inflammatory responses in MHV3‐infected TLR2 KO mice, indicating that TLR2 may act as an aggravating factor in fulminant hepatitis. However, TLR2 KO mice were not protected from MHV3‐induced lethal hepatitis, but survival was significantly improved due to delayed occurrence of hepatic necrosis (observations not shown), supporting that other TLR2‐independent mechanisms are also involved in the outcome of MHV3 infection. Unlike MHV3, the weakly hepatotrophic MHV‐A59 showed no ability to increase TLR2 expression in the liver and induced comparable mild hepatitis in WT or TLR2 KO mice, strengthening the importance of TLR2 in MHV3‐induced hepatitis.
Activation of TLR2 through the MyD88 /nuclear factor‐κB‐dependent pathway leads to up‐regulation of numerous genes involved in innate host defence such as TNF‐α, IL‐1β, IL‐6, IFN‐γ, chemokines and TLR expression (reviewed in ref. 46). We have observed that TNF‐α and IL‐6 increased sooner and more in the liver of MHV3‐infected WT mice than in TLR2 KO or MHV‐A59‐infected mice. It was previously reported that TNF‐α activity significantly increases as soon as 24 hr p.i. during MHV3 infection, even before the virus is detectable in the liver,47 supporting an early activation through an unidentified signalling pathway. Following i.p. infection, MHV3 primarily replicates in peritoneal macrophages and then within liver Kupffer cells and LSECs.48, 49 A role for macrophages in MHV3‐induced TLR2‐dependent inflammatory responses is supported by in vitro infections. Our results indicate that both CEACAM1a and TLR2 are involved in viral replication and early IL‐6 expression (as soon as 5 hr p.i.) in macrophages. Jacques et al.34 have previously demonstrated that TLR2 and heparan sulphate were involved in the induction of inflammatory cytokines in MHV3‐infected macrophages. Our results suggest that higher and earlier TLR2‐dependent IL‐6 induction by MHV3 may be related to higher and earlier activation of the p38 MAPK pathway by the virus. In addition, since a very early role for the p38 MAPK pathway (within 30 min) was reported in viral replication of MHV3 in J774.1 macrophages,50 higher and earlier replication of MHV3 than MHV‐A59 may also result from differential activation of this pathway by the viruses. No role for TLR2, however, was reported in viral replication and induction of IL‐6 by MHV‐A59.43, 51 Such a difference may explain the lower viral load and inflammatory responses in the liver of MHV‐A59‐infected mice. In addition, preliminary data have shown that UV‐inactivated MHV3 viral particles bound more rapidly to macrophage surface (<10 min) than UV‐inactivated MHV‐A59, suggesting that S protein from MHV3 may express higher affinity for TLR2 than MHV‐A59.
Hepatocytes and non‐parenchymal hepatic cells were also reported to express both the viral receptor CEACAM1a26 and TLR2.52, 53 In vitro MHV3 infection in hepatocytes leads to rapid cell death (less than 24 hr p.i.) and low viral infectious titres and inflammatory cytokine levels, which are barely influenced by siTLR2 treatments (results not shown), suggesting that the exacerbated inflammatory response in the liver of MHV3‐infected mice does not mostly depend on infected hepatocytes, in spite of extensive necrosis foci. Vascular and tolerant properties of LSECs, however, were disturbed when in vitro infected with MHV3 (Bleau et al. manuscript in revision). Hence, we can hypothesize that TLR2 fixation of viral infectious particles or free viral S proteins on MHV3 permissive cells, such as Kupffer cells and LSECs, may favour both viral replication and exacerbated inflammatory responses that may contribute to higher liver injury in MHV3‐infected mice. Indeed, high levels of TNF‐α in the liver are generally associated with extensive necrosis,54 and absence of TNF‐α, but not IL‐6, was recently shown to significantly reduce hepatic lesions (AST/ALT) and increase the survival of MHV3‐infected mice.55
Following viral replication in macrophages and LSECs, viruses reach hepatocytes leading to rapid extensive syncytia and cell lysis.28 The fulminance of liver lesions indicates that innate antiviral mechanisms cannot control viral infection and inflammatory responses. Type I IFN production by infected cells is the most important antiviral mechanism acting in the first days of infection. Higher IFN‐β production, however, was found in the liver of MHV3‐infected WT mice than TLR2 KO mice or MHV‐A59‐infected WT mice. The positive correlation between IFN‐β production and viral replication levels may reflect the spreading of viral replication in the liver rather than the efficiency of antiviral effects. Indeed, it was already reported that higher levels of type I IFN were produced simultaneously with higher MHV3 titres by peritoneal macrophages from C57BL/6 mice,56 supporting a negligible role of type I IFN in the control of MHV3 replication. Such explanation is also supported by a positive correlation between low levels of IFN‐β and low viral replication in MHV3‐infected TLR2 KO mice or MHV‐A59‐infected WT mice, indicating that lower viral replication does not result from higher production of antiviral IFN‐β. In addition, no significant IFN‐α expression was induced in the liver of all infected groups of mice (results not shown), indicating no major role for type I IFN in the control of MHV replication and hepatitis. In addition, type I IFN response in mouse coronavirus infections depends mainly on TLR3 and helicases RIG‐1 or MDA‐5 engagement by viral RNAs.57, 58, 59 Transcription levels of TLR3, RIG‐1 and MDA‐5 were comparable in livers from both MHV3‐infected WT and TLR2 KO mice and less transcribed in MHV‐A59‐infected WT mice, suggesting that higher induction of IFN‐β by MHV3 infection did not depend on these PRRs. We can therefore postulate that higher levels of IFN‐β in the livers of MHV3‐infected WT mice may result from extensive hepatic infection and/or TLR2 engagement by MHV3. Indeed, Dietrich et al.42 have demonstrated that translocation of TLR2 in endolysosomal compartments following ligand engagement can trigger IFN‐β production via the MyD88/interferon regulatory factor (IRF)‐1/IRF‐7‐dependent pathway in macrophages. Preliminary data revealed increased expression of IRF‐7 in livers from MHV3‐infected WT mice, suggesting activation of this signalling pathway in some hepatic cells. Future work should address this hypothesis. Nevertheless, such an interesting potential new role for TLR2 in IFN‐β regulation may not be of major importance in MHV3 infection as no protection against hepatitis seems to be provided by high IFN‐β levels.
TLR2 activation also leads to production of chemokines involved in inflammatory cell recruitment. Earlier and higher production of CXCL1, CCL2 and CXCL10, as seen in the liver of MHV3‐infected WT mice, may involve rapid recruitment of neutrophils, macrophages and NK or T‐cell subsets, respectively (reviewed in ref. 40). As expected, CXCL1 production and concomitant neutrophil recruitment occurred sooner in the liver of MHV3‐infected WT mice, suggesting that increase in neutrophils may result from early CXCL1 release, as demonstrated by Moles et al.39 The subsequent loss of neutrophils at 48 hr p.i., however, did not depend on a decrease of CXCL1 production because levels increased up to 72 hr p.i. We can hypothesize that recruited neutrophils may serve as a new cell target for viral infection leading to cell apoptosis, such as previously demonstrated for NK cells.60 Work is in progress to clarify the loss of these cells during MHV3 infection. Our data regarding neutrophils slightly differ from those of Xu et al.61 who reported percentage increases of neutrophils up to 48 hr p.i in the liver of MHV3‐infected mice. Such apparent discrepancy results from the dramatic decrease in total intrahepatic MNCs, including NK, B and CD4 cells, leading to an apparent increase in neutrophil percentages in spite of a reduction in their absolute number, such as is demonstrated in the present work. These authors also linked neutrophil infiltration to a TNF‐α‐dependent Fgl‐2 production. Our results do not support this hypothesis as chemokine production and neutrophil infiltration in the liver occurred earlier than Fgl‐2 induction in MHV3‐infected WT mice. Total counts of MNCs in the livers of both MHV3‐infected TLR2 KO mice and MHV‐A59‐infected WT mice were not highly altered over infection time and neutrophil numbers steadily increased up to 48 hr p.i. supporting a recruitment of these cells despite lower levels of CXCL1. The delayed and preserved neutrophil pool may therefore favour viral clearance and contribute to controlling acute hepatitis, as suggested by lower viral titres, inflammation and damages in these mice. Recent work regarding the respiratory rat coronavirus infection showed that neutrophils were required for an effective antiviral response but could also contribute to lung pathology.62 On the other hand, we cannot exclude that higher levels of chemokines in the liver of MHV3‐infected WT mice may also contribute to hepatotoxicity without respect to neutrophil infiltration.63
We have similarly observed lower macrophage recruitment in the livers of MHV3‐infected WT mice than MHV3‐infected TLR2 KO and MHV‐A59‐infected WT mice despite higher production of CCL2. It was previously demonstrated that macrophage expansion in the liver is related to influx of peripheral monocytes facilitated by high levels of CCL2 rather than to increase of Kupffer cells64 (reviewed in ref. 65). However, resident and recruited macrophages are permissive to MHV3 infection48, 49 and we have shown that viral replication in macrophages is increased by TLR2, suggesting that low number of macrophages in the livers of MHV3‐infected WT mice result from higher viral replication and subsequent cell lysis. Accordingly, the low presence of inflammatory cells in necrosis foci observed in livers from MHV3‐infected WT mice may result from virus‐induced cell lysis of recruited MNCs. Lower levels of CXCL‐1 and CCL2 in the livers of MHV3‐infected TLR2 KO and MHV‐A59‐infected WT mice involved lower but sustained recruitment of neutrophils and macrophages, suggesting that these cells might be protective rather than deleterious in acute hepatitis process. In agreement, depletion of macrophages in MHV‐A59‐infected mice has been shown to promote lethal fulminant hepatitis within 4 days p.i.23
In addition, NK, NK‐T, B and T lymphocytes decreased only in the liver of MHV3‐infected WT mice despite higher levels of CXCL10. Such a reduction may result directly from permissivity of NK and B cells to viral infection, and indirectly from virally induced lysis of thymic dendritic cells, as was previously reported.60, 66, 67 The larger inflammatory foci observed in the livers of MHV‐A59‐infected WT mice and MHV3‐infected TLR2 KO mice may therefore reflect the sustained recruitment of MNCs. On the other hand, high amounts of CXCL10 in the liver have already been associated with apoptosis of human and murine hepatocytes,68 suggesting that high levels of CXCL10 induced by MHV3 may also contribute to hepatic lesions.
Some other inflammatory factors induced by MHV3 infection were less dependent, or not dependent, on TLR2. Indeed, we have observed that IL‐33 release was less influenced by TLR2 and rather reflected the severity of liver damage, as was previously reported.69, 70 We have recently shown that IL‐33 expression was up‐regulated in the liver of MHV3‐infected C57BL/6 mice, mainly in LSECs, vascular endothelial cells and hepatocytes in the first 24–32 hr p.i.36 This study also demonstrated that TLR3 was involved in the up‐regulation of IL‐33 in poly(I:C)‐treated mice. However, IL‐33 expression was more strongly induced in the livers of MHV3‐infected WT mice than in poly(I:C)‐treated mice, suggesting that other PRRs might be involved in IL‐33 release during MHV3 infection. Our observations suggest that TLR2 might be another candidate involved in early IL‐33 induction. The mechanism(s) by which MHV3 favour(s) IL‐33 secretion is pending and need(s) further investigation. The induction of the vascular factor Fgl‐2, a pro‐thrombinase promoting microvascular thrombosis and hepatocyte necrosis during MHV3 infection,37, 38 was comparable in WT and TLR2 KO mice and remained low or delayed in MHV‐A59‐infected WT mice, indicating that this factor is not regulated by TLR2 and not involved in TLR2‐exacerbated hepatic damage, in spite of its worsening role in hepatitis outcome.
The present work with animal models of viral hepatitis induced by two closely related serotypes of MHVs, highlights the complex interactions between surface TLR2 and intracellular PRRs in the recognition of viral infections and the induction of protective or worsening inflammatory responses during acute infections. The use of the MHV3 model provided new insights into the aggravating role of surface TLR2 in acute viral diseases. Activation of surface TLR signalling pathways has been recently associated with pathogenic processes in several viral infections. Indeed, TLR2 and/or TLR4 are involved in the induction of inflammatory response in severe acute respiratory syndrome virus, several herpesviruses (herpes simplex virus 1, varicella virus, cytomegalovirus), influenza, HIV, HBV and HCV infections.6, 7, 8, 9, 10, 11, 12, 13, 14 In most cases, TLR2‐promoted inflammatory TNF‐α, IL‐6 or chemokine IL‐8 responses are mediated by macrophages.6, 7, 12, 13, 17 The viral mechanisms involved in the activation of TLR2 inflammatory pathways are not clearly elucidated. The ability of MHV3 viral proteins to bind and activate TLR2 signalling is not unique to coronaviruses. Indeed, HBV and HCV core proteins were reported to induce TLR2‐dependent activation of nuclear factor‐κB and p38 MAPK and subsequent production of TNF‐α, IL‐6 and IL‐12 in macrophages.3, 71 Hence, one could presume that activation of TLR2 by HCV/HBV core proteins could aggravate hepatic inflammation and damage. In agreement with this hypothesis, an up‐regulation of TLR2 in the liver and on monocytes, correlating with higher TNF‐α levels and necroinflammatory activity in the liver, was reported in patients with hepatitis C.16, 17 Our data demonstrate for the first time that up‐regulating the inflammatory activity of TLR2 in the acute phase of viral hepatitis favours fulminant hepatitis. In humans, fulminant hepatic failure (FHF) is an uncommon clinical condition characterized by extensive hepatic necrosis, severe impairment of liver function and a high mortality rate.19 The most recognized causal agents of FHF are hepatitis viruses (especially HBV) but several non‐hepatic herpesviruses (herpes simplex virus, cytomegalovirus and varicella virus) and drugs were also associated with FHF.19 The pathophysiology of FHF is unclear but increasing evidence suggests that regardless of the aetiological cause of FHF, the host's inflammatory response contributes to liver microcirculatory disorders and injury. Accordingly, macrophage activation and inflammatory cytokines were shown to play a key role in FHF.72, 73 Moreover, the core protein from HBV was suggested as a potential initiating factor in patients with fulminant hepatitis B but mechanisms are still elusive.74, 75 Hence, we can propose that strong TLR2‐dependent activation of macrophages in the liver by viral proteins from hepatitis or non‐hepatitis viruses during acute infection may predispose to FHF induction.
Work is in progress to further determine the mechanisms involved in TLR2‐dependent increase of inflammatory responses and viral replication in infected intrahepatic resident and recruited cell populations.
Author contributions
C. Bleau, M. Burnette, and A. Filliol performed the experiments. C. Piquet‐Pellorce has designed cytofluorometric experiments. L. Lamontagne and C. Bleau have designed the study. C. Bleau and L. Lamontagne have written the paper and all authors contributed to revision and corrections of the manuscript.
Disclosures
The authors indicated no financial or commercial conflict of interest.
Supporting information
Figure S1. Gating strategies used for cytofluorometric studies.
Figure S2. Mortality, hepatic damages and viral replication in murine hepatitis virus (MHV) ‐A59‐infected Toll‐like receptor 2 knockout mice.
Figure S3. Analysis of intrahepatic mononuclear cells in the liver of murine hepatitis virus 3‐infected Toll‐like receptor 2 knockout mice.
Acknowledgements
The authors want to acknowledge Pascale Bellaud and Eric Massicotte for their technical assistance within histochemistry and cytofluorometry analyses, and Dr Anthony Karelis for revising the manuscript. This work was funded by a grant from NSERC from Government of Canada (no. 2895‐2009). Christian Bleau and Melanie Burnette were supported by NSERC fellowships.
References
- 1. Broering R, Lu M, Schlaak JF. Role of Toll‐like receptors in liver health and disease. Clin Sci (Lond) 2011; 121:415–26. [DOI] [PubMed] [Google Scholar]
- 2. Villalba M, Hott M, Martin C, Aguila B, Valdivia S, Quezada C et al Herpes simplex virus type 1 induces simultaneous activation of Toll‐like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes. Med Microbiol Immunol 2012; 201:371–9. [DOI] [PubMed] [Google Scholar]
- 3. Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt‐Jones E et al Hepatitis C core and nonstructural 3 proteins trigger toll‐like receptor 2‐mediated pathways and inflammatory activation. Gastroenterology 2004; 127:1513–24. [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann M, Zeisel MB, Jilg N, Paranhos‐Baccalà G, Stoll‐Keller F, Wakita T et al Toll‐like receptor 2 senses hepatitis C virus core protein but not infectious viral particles. J Innate Immun 2009; 1:446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mercin A, Kluwe J, Schwabe RF. Toll‐like receptors as target on chronic liver disease. Gut 2009; 58:704–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heggelund L, Müller F, Lien E, Yndestad A, Ueland T, Kristiansen KI et al Frøland SS Increased expression of toll‐like receptor 2 on monocytes in HIV infection: possible roles in inflammation and viral replication. Clin Infect Dis 2004; 39:264–9. [DOI] [PubMed] [Google Scholar]
- 7. Heggelund L, Damås JK, Yndestad A, Holm AM, Mūller F, Lien E et al Stimulation of toll‐like receptor 2 in mononuclear cells from HIV‐infected patients induces chemokine responses: possible pathogenic consequences. Clin Exp Immunol 2004; 138:116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gekonge B, Giri MS, Kossenkov AV, Nebozyhn M, Yousef M, Mounzer K et al Constitutive gene expression in monocytes from chronic HIV‐1 infection overlaps with acute Toll‐like receptor induced monocyte activation profiles. PLoS ONE 2012; 7:e41153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karlström A, Heston SM, Boyd KL, Tuomanen EI, McCullers JA. Toll‐like receptor 2 mediates fatal immunopathology in mice during treatment of secondary pneumococcal pneumonia following influenza. J Infect Dis 2011; 204:1358–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao RR, Yang XF, Dong J, Zhao YY, Wei X, Huang CX et al Toll‐like receptor 2 promotes T helper 17 cells response in hepatitis B virus infection. Int J Clin Exp Med 2015; 8:7315–23. [PMC free article] [PubMed] [Google Scholar]
- 11. Dosch SF, Mahajan SD. Collins ARSARS coronavirus spike protein‐induced innate immune response occurs via activation of the NF‐κB pathway in human monocyte macrophages in vitro . Virus Res 2009; 142:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurt‐Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R et al Herpes simplex virus 1 interaction with Toll‐like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 2004; 101:1315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang JP, Kurt‐Jones EA, Shin OS, Manchak MD, Levin MJ, Finberg RW. Varicella‐zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll‐like receptor 2. J Virol 2005; 79:12658–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol 2006; 177:7094–102. [DOI] [PubMed] [Google Scholar]
- 15. Xu J, Yang Y, Sun J, Ding Y, Su L, Shao C et al Expression of Toll‐like receptors and their association with cytokine responses in peripheral blood mononuclear cells of children with acute rotavirus diarrhoea. Clin Exp Immunol 2006; 144:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riordan SM, Skinner NA, Kurtovic J, Locarnini S, McIver CJ, Williams R et al Toll‐like receptor expression in chronic hepatitis C: correlation with pro‐inflammatory cytokine levels and liver injury. Inflamm Res 2006; 55:279–85. [DOI] [PubMed] [Google Scholar]
- 17. Berzsenyi MD, Roberts SK, Preiss S, Woollard DJ, Beard MR, Skinner NA et al Hepatic TLR2 & TLR4 expression correlates with hepatic inflammation and TNF‐α in HCV & HCV/HIV infection. J Viral Hepat 2011; 18:852–60. [DOI] [PubMed] [Google Scholar]
- 18. Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 2005; 5:215–29. [DOI] [PubMed] [Google Scholar]
- 19. Liu M, Chan WY, McGilvray I, Ning Q, Levy GA. Fulminant viral hepatitis: molecular and cellular basis, and clinical implications. Expert Rev Mol Med 2001; 3:1–19. [DOI] [PubMed] [Google Scholar]
- 20. Ramadori G, Moriconi F, Malik I, Dudas J. Physiology and pathophysiology of liver inflammation, damage and repair. J Physiol Pharmacol 2008; 59:107–17. 21. [PubMed] [Google Scholar]
- 21. Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res 2011; 81:85–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le Prevost C, Levy‐Leblond E, Virelizier JL, Dupuy JM. Immunopathology of mouse hepatitis virus type 3 infection. Role of humoral and cell‐mediated immunity in resistance mechanisms. J Immunol 1975; 114:221–5. [PubMed] [Google Scholar]
- 23. Wijburg OL, Heemskerk MH, Boog CJ, Van Rooijen N. Role of spleen macrophages in innate and acquired immune reponses against mouse hepatitis virus strain A59. Immunology 1997; 92:252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lamontagne L, Descoteaux JP, Jolicoeur P. Mouse hepatitis virus 3 replication in T and B lymphocytes correlate with viral pathogenicity. J Immunol 1989; 142:4458–65. [PubMed] [Google Scholar]
- 25. Lavi E, Gilden DH, Highkin MK, Weiss SR. The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 1986; 36:130–5. [PubMed] [Google Scholar]
- 26. Godfraind C, Coutelier JP. Morphological analysis of mouse hepatitis virus A‐59‐induced pathology with regard to viral receptor expression. Histol Histopathol 1998; 13:181–9. [DOI] [PubMed] [Google Scholar]
- 27. Navas S, Seo SH, Chua MM, Das Sarma J, Lavi E, Hingley ST et al Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J Virol 2001; 5:2452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin JP, Chen W, Koehren F, Pereira CA. The virulence of mouse hepatitis virus 3, as evidenced by permissivity of cultured hepatic cells toward escape mutants. Res Virol 1994; 145:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol 2009; 36:4–12. [DOI] [PubMed] [Google Scholar]
- 30. Pope M, Rotstein O, Cole E, Sinclair S, Parr R, Cruz B et al Pattern of disease after murine hepatitis virus strain 3 infection correlates with macrophage activation and not viral replication. J Virol 1995; 69:5252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schindler L, Brücher J, Kirchner H. Protection of mice against infection with mouse hepatitis virus type 3 by injection of silica. Immunobiology 1984; 166:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jacques A, Bleau C, Martin JP, Lamontagne L. Intrahepatic endothelial and Kupffer cells involved in immunosuppressive cytokines and natural killer (NK)/NK T cell disorders in viral acute hepatitis. Clin Exp Immunol 2008; 152:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cervantes‐Barragán L, Kalinke U, Züst R, König M, Reizis B, López‐Macías C et al Type I IFN‐mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J Immunol 2009; 182:1099–106. [DOI] [PubMed] [Google Scholar]
- 34. Jacques A, Bleau C, Turbide C, Beauchemin N, Lamontagne L. Macrophage interleukin‐6 and tumour necrosis factor‐α are induced by coronavirus fixation to Toll‐like receptor 2/heparan sulphate receptors but not carcinoembryonic cell adhesion antigen 1a. Immunology 2009; 128:e181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dupuy JM, Rodrigue D. Heterogeneity in evolutive patterns of inbred mice infected with a cloned substrain of mouse hepatitis virus type 3. Intervirology 1981; 16:114–7. [DOI] [PubMed] [Google Scholar]
- 36. Arshad MI, Patrat‐Delon S, Piquet‐Pellorce C, L'helgoualc'h A, Rauch M, Genet V et al Pathogenic mouse hepatitis virus or poly(I:C) induce IL‐33 in hepatocytes in murine models of hepatitis. PLoS ONE 2013; 8:e74278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ding JW, Ning Q, Liu MF, Lai A, Leibowitz J, Peltekian KM et al Fulminant hepatic failure in murine hepatitis virus strain 3 infection: tissue‐specific expression of a novel fgl2 prothrombinase. J Virol 1997; 71:9223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marsden PA, Ning Q, Fung LS, Luo X, Chen Y, Mendicino M et al The Fgl2/ fibroleukin prothrombinase contributes to immunologically mediated thrombosis in experimental and human viral hepatitis. J Clin Invest 2003; 112:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ et al A tlr2/S100a9/Cxcl‐2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol 2014; 60:782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oo YH, Shetty S, Adams DH. The role of chemokines in the recruitment of lymphocytes to the liver. Dig Dis 2010; 28:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lamontagne L, Lusignan S, Page C. Recovery from mouse hepatitis virus infection depends on recruitment of CD8+ cells rather than activation of intrahepatic CD4+ αβ‐TCRinter or NK‐T cells. Clin Immunol 2001; 101:345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dietrich N, Lienenklaus S, Weiss S, Gekara NO. Murine Toll‐like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS ONE 2010; 5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou H, Zhao J, Perlman S. Autocrine interferon priming in macrophages but not in dendritic cells results in enhanced cytokine and chemokine production after coronavirus infection. MBio 2010; 1:e00219–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tarantino G, Di Cristina A, Pipitone R, Almasio PL, Di Vita G, Craxi A et al In vivo liver expression of TLR2, TLR3 and TLR7 in chronic hepatitis C. J Biol Regul Homeost Agents 2013; 27:233–9. [PubMed] [Google Scholar]
- 45. Zhou M, Zhu X, Ye S, Zhou B. Blocking TLR2 in vivo attenuates experimental hepatitis induced by concanavalin A in mice. Int Immunopharmacol 2014; 21:241–6. [DOI] [PubMed] [Google Scholar]
- 46. Melchjorsen J. Learning from the messengers: Innate sensing of viruses and cytokine regulation of immunity — clues for treatments and vaccines. Viruses 2013; 5:470–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Devictor D, Décimo D, Sebire G, Tardieu M, Haddchouel M. Enhanced tumor necrosis factor α in coronavirus but not in paracetamol‐induced acute hepatic necrosis in mice. Liver 1992; 12:205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Décimo D, Boespflug O, Meunier‐Rotival M, Hadchouel M, Tardieu M. Genetic restriction of murine hepatitis virus type 3 expression in liver and brain: comparative study in BALB/c and C3H mice by immunochemistry and hybridization in situ . Arch Virol 1993; 130:269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pereira CA, Steffan AM, Kirn A. Interaction between mouse hepatitis viruses and primary cultures of Kupffer and endothelial liver cells from resistant and susceptible inbred mouse strains. J Gen Virol 1984; 65:1617–20. [DOI] [PubMed] [Google Scholar]
- 50. McGilvray ID, Lu Z, Wei AC, Dackiw AP, Marshall JC, Kapus A et al Murine hepatitis virus strain 3 induces the macrophage prothrombinase fgl‐2 through p38 mitogen‐activated protein kinase activation. J Biol Chem 1998; 273:32222–9. [DOI] [PubMed] [Google Scholar]
- 51. Mazaleuskaya L, Veltrop R, Ikpeze N, Martin‐Garcia J, Navas‐Martin S. Protective role of Toll‐like Receptor 3‐induced type I interferon in murine coronavirus infection of macrophages. Viruses 2012; 4:901–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schwabe RF, Seki E, Brenner DA. Toll‐like receptor signaling in the liver. Gastroenterology 2006; 130:1886–1900. [DOI] [PubMed] [Google Scholar]
- 53. Matsumura T, Ito A, Takii T, Hayashi H, Onozaki K. Endotoxin and cytokine regulation of toll like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J Interferon Cytokine Res 2000; 20:915–21. [DOI] [PubMed] [Google Scholar]
- 54. Schümann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N et al Importance of Kupffer cells for T‐cell‐dependent liver injury in mice. Am J Pathol 2000; 157:1671–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu J, Tan Y, Zhang J, Zou L, Deng G, Xu X et al C5aR, TNF‐α, and FGL2 contribute to coagulation and complement activation in virus‐induced fulminant hepatitis. J Hepatol 2015; 62:354–62. [DOI] [PubMed] [Google Scholar]
- 56. Schindler L, Engler H, Kirchner H. Activation of natural killer cells and induction of interferon after injection of mouse hepatitis virus type 3 in mice. Infect Immun 1982; 35:869–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao L, Rose KM, Elliott R, Van Rooijen N, Weiss SR. Cell‐type‐specific type I interferon antagonism influences organ tropism of murine coronavirus. J Virol 2011; 85:10058–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roth‐Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 2008; 82:9829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ireland DD, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J Virol 2008; 82:300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lehoux M, Jacques A, Lusignan S, Lamontagne L. Murine viral hepatitis involves NK cell depletion associated with virus‐induced apoptosis. Clin Exp Immunol 2004; 137:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu H, Li H, Cao D, Wu Y, Chen Y. Tumor necrosis factor α (TNF‐α) receptor‐I is required for TNF‐α‐mediated fulminant virus hepatitis caused by murine hepatitis virus strain‐3 infection. Immunol Lett 2014; 158:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Haick AK, Rzepka JP, Brandon E, Balemba OB, Miura TA. Neutrophils are needed for an effective immune response against pulmonary rat coronavirus infection, but also contribute to pathology. J Gen Virol 2014; 95:578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stefanovic L, Brenner DA, Stefanovic B. Direct hepatotoxic effect of KC chemokine in the liver without infiltration of neutrophils. Exp Biol Med 2005; 230:573–86. [DOI] [PubMed] [Google Scholar]
- 64. Zimmermann HW, Seidler S, Nattermann J, Gassler N, Hellerbrand C, Zernecke A et al Functional contribution of elevated circulating and hepatic non‐classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS ONE 2010; 5:e11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Karlmark KR, Wasmuth HE, Trautwein C, Tacke F. Chemokine‐directed immune cell infiltration in acute and chronic liver disease. Expert Rev Gastroenterol Hepatol 2008; 2:233–42. [DOI] [PubMed] [Google Scholar]
- 66. Jolicoeur P, Lamontagne L. Mouse hepatitis virus 3 pathogenicity expressed by a lytic viral infection in bone marrow 14.8+ mu+ B lymphocyte subpopulations. J Immunol 1989; 143:3722–30. [PubMed] [Google Scholar]
- 67. Lamontagne L, Jolicoeur P. Mouse hepatitis virus 3‐thymic cell interactions correlating with viral pathogenicity. J Immunol 1991; 146:3152–9. [PubMed] [Google Scholar]
- 68. Sahin H, Borkham‐Kamphorst E, Doo NT, Berres ML, Kaldenbach M, Schmitz P et al Proapoptotic effects of the chemokine, CXCL 10 are mediated by the noncognate receptor TLR4 in hepatocytes. Hepatology 2013; 57:797–805. [DOI] [PubMed] [Google Scholar]
- 69. Wang J, Zhao P, Guo H, Sun X, Jiang Z, Xu L et al Serum IL‐33 levels are associated with liver damage in patients with chronic hepatitis C. Mediators Inflamm 2012;2012:819636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang J, Cai Y, Ji H, Feng J, Ayana DA, Niu J et al Serum IL‐33 levels are associated with liver damage in patients with chronic hepatitis B. J Interferon Cytokine Res 2012; 32:248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cooper A, Tal G, Lider O, Shaul Y. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol 2005; 175:3165–76. [DOI] [PubMed] [Google Scholar]
- 72. Ando K, Moriyama T, Guidotti LG, Wirth S, Schreiber RD, Schlicht HJ et al Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J Exp Med 1993; 178:1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. de la Mata M, Meager A, Rolando N, Daniels HM, Nouri‐Aria KT, Goka AK et al Tumour necrosis factor production in fulminant hepatic failure: relation to aetiology and superimposed microbial infection. Clin Exp Immunol 1990; 82:479–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mondelli M, Eddleston AL. Mechanisms of liver cell injury in acute and chronic hepatitis B. Semin Liver Dis 1984; 4:47–58. [DOI] [PubMed] [Google Scholar]
- 75. Aritomi T, Yatsuhashi H, Fujino T, Yamasaki K, Inoue O, Koga M et al Association of mutations inthe core promoter and precore region of hepatitis virus with fulminant and severe acute hepatitis in Japan. J Gastroenterol Hepatol 1998; 13:1125–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gating strategies used for cytofluorometric studies.
Figure S2. Mortality, hepatic damages and viral replication in murine hepatitis virus (MHV) ‐A59‐infected Toll‐like receptor 2 knockout mice.
Figure S3. Analysis of intrahepatic mononuclear cells in the liver of murine hepatitis virus 3‐infected Toll‐like receptor 2 knockout mice.