

Abstract
Background
Rhizobia are soil bacteria that establish symbiotic relationships with legumes and fix nitrogen in root nodules. We recently reported that several nitrogen-fixing rhizobial strains, belonging to Rhizobium phaseoli, R. trifolii, R. grahamii and Sinorhizobium americanum, were able to colonize Phaseolus vulgaris (common bean) seeds. To gain further insight into the traits that support this ability, we analyzed the genomic sequences and proteomes of R. phaseoli (CCGM1) and S. americanum (CCGM7) strains from seeds and compared them with those of the closely related strains CIAT652 and CFNEI73, respectively, isolated only from nodules.
Results
In a fine structural study of the S. americanum genomes, the chromosomes, megaplasmids and symbiotic plasmids were highly conserved and syntenic, with the exception of the smaller plasmid, which appeared unrelated. The symbiotic tract of CCGM7 appeared more disperse, possibly due to the action of transposases. The chromosomes of seed strains had less transposases and strain-specific genes. The seed strains CCGM1 and CCGM7 shared about half of their genomes with their closest strains (3353 and 3472 orthologs respectively), but a large fraction of the rest also had homology with other rhizobia. They contained 315 and 204 strain-specific genes, respectively, particularly abundant in the functions of transcription, motility, energy generation and cofactor biosynthesis. The proteomes of seed and nodule strains were obtained and showed a particular profile for each of the strains. About 82 % of the proteins in the comparisons appeared similar. Forty of the most abundant proteins in each strain were identified; these proteins in seed strains were involved in stress responses and coenzyme and cofactor biosynthesis and in the nodule strains mainly in central processes. Only 3 % of the abundant proteins had hypothetical functions.
Conclusions
Functions that were enriched in the genomes and proteomes of seed strains possibly participate in the successful occupancy of the new niche. The genome of the strains had features possibly related to their presence in the seeds. This study helps to understand traits of rhizobia involved in seed adaptation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3053-z) contains supplementary material, which is available to authorized users.
Keywords: Nitrogen fixation, Comparative genomics, Proteome
Background
Rhizobia are saprophytic soil bacteria commonly studied for their ability to enter into nitrogen-fixing symbioses with legumes. The establishment of these symbioses by rhizobia, a collective term for strains from genera such as Rhizobium, Sinorhizobium, Mesorhizobium and Bradyrhizobium, involves the formation of organ-like structures on the legume roots (for recent reviews see references [1] and [2]). The rhizobia in the nodules are present in a metabolically differentiated form called bacteroids, which perform the reduction of atmospheric dinitrogen into ammonium. In exchange for dicarboxylic acids supplied from the plant, the bacteroids export the ammonium to the plant. Rhizobia have also been found inside legume non-nodular tissues such as roots, stems and pods [3–5]. There are also reports of endophytic rhizobia associated with Arabidopsis, wheat, maize, sugar cane, and rice [6–9]. Strains of endophytic Rhizobium were recently isolated from the tree species Populus euphratica and P. deltoides [10, 11].
Previously, we described several nitrogen-fixing rhizobial strains isolated from the interior of common bean seeds (Phaseolus vulgaris) [12]. We postulated that the vertical transmission of effective rhizobacteria in seeds expands the spectrum of their beneficial interactions with the host plants and has potential biotechnological application.
Given the increasing number of endophytic rhizobial isolates, it is worth determining which genetic traits are responsible for their ability to persist in plant tissues and discover if genomic differences exist among isolates able to persist in seeds. Despite the difficulties in assigning functions to novel genes, these analyses can measure changes in cellular physiology in response to genetic or environmental adaptations [13]. The model of our previous study was to compare closely related strains with different lifestyles by analyzing their genomes in addition to other approaches, with the aim of understanding how the strains have adapted to new niches. In a previous work, we reported an initial analysis of two genomes of seed-borne rhizobia corresponding to Rhizobium phaseoli (strain CCGM1) and Sinorhizobium americanum (strain CCGM7) species [12], but a thorough analysis was pending. The first species belongs to the symbiont most preferred by P. vulgaris, and the second to a recently described Phaseolus symbiont.
In this work we report the new genome sequence of an S. americanum strain, CFNEI73, isolated from nodules, and the improved sequence of strain CCGM7. We also compared the strains obtained from the interior of bean seeds, CCGM1 and CCGM7, with the strains from nodules, R. phaseoli CIAT652 [14] and S. americanum CFNEI73 [15], respectively. We analyzed their genomic sequences to infer the prevalence, identity and function of their orthologs, and also performed proteomic analyses to compare the abundance and function of proteins in seed-borne strains, compared with those from nodules.
Methods
Genome sequencing, assembly, and annotation of strain CFNEI73 and re-sequencing of CCGM7
DNA of strain CFNEI73 was extracted according to standard protocols and sequenced by Macrogen (Seoul, South Korea). A 3 kilobase pair (kb)-library was prepared and run on an Illumina HiSeq sequencer to obtain 100 base pair (bp)-mated pair reads. A total of 10,599,614 paired reads were obtained, and 4,629,584 remained after trimming. A second sequencing protocol was done with PacBio at the Duke Center for Genomic and Computational Biology (Durham, NC, USA) with a 10 kb-library, obtaining 731,017,143 reads, filtered to 612,800,193. Sequences obtained were mixed with the Illumina reads to enhance the accuracy of the final assembly with genome coverage of 71×. Assembly was performed with SMRT Analysis v2.3.0 (Pacific Biosciences) and SPAdes v3.5.0 [16]. Annotation was conducted with RAST v4.0 [17], with manual curation. Strain CCGM7 was re-sequenced with PacBio at the Duke Center for Genomic and Computational Biology, with a 10 kb-library, obtaining 1,147,065,864 reads, filtered to 998,800,19. Reads were mixed with those obtained previously with Illumina [12], with genome coverage of 121×. Assembly and annotation were done as for strain CFNEI73.
Comparative genomic analysis
The comparison was performed in two ways: (i) by pairwise analysis in the case of strain-specific genes and (ii) by group analysis to calculate relatedness and sequence identity. The genome sequences were downloaded from GenBank with the following assembly accession numbers: R. phaseoli strains CIAT652 (GCA_000020265.1), CNPAF512 (GCA_000194195.2), and CCGM1 (GCA_000705615.1), R. etli CFN42 (GCA_000092045.1), S. fredii strains NGR234 (GCA_000018545.1), USDA257 (GCA_000265205.2) and HH103 (GCA_000283895.1). The most closely related species to R. phaseoli was R. etli and to S. americanum was S. fredii. Additional searches for rhizobial homologs were done with the nonredundant (nr) database. For the group comparison we used OrthoMCL version 2.0 [18] with default parameters, with BLAST (E value 1e−5; 30 % identity and 70 % overlap). Predicted ORFs with lengths <300 nt were discarded from the analysis of strain-specific genes. For the study of paralog families, the inparalog files were used. Synteny was determined with an in-house Perl program using the ortholog files from OrthoMCL as described previously [19]. Function was assigned using the extended annotation of clusters of orthologous groups (COG) tool [20]. Genomic average nucleotide identity (ANIm) was calculated with JSpecies [21]. Phylogeny of nodD was obtained with PhyML server (http://www.atgc-montpellier.fr/phyml/) using default parameters.
Proteomic analysis
The strains were grown in liquid minimal medium (MM) containing succinate (10 mM) and ammonium chloride (10 mM) as carbon and nitrogen sources, respectively, for 8 h at 30 °C with 200 rpm shaking. The methods used for sample preparation, analytical and preparative two-dimensional (2D) polyacrylamide gel electrophoresis (PAGE), and image analysis were as described previously [22]. Briefly, pH gradients were determined by using a 2D SDS-PAGE standard (Sigma, United States). For the first dimension approximately 500 μg of total protein was loaded. The gels were stained with Coomassie blue R-250, and protein spots on the gels were detected at a resolution of 127 × 127 μm using a PDI image analysis system and PD-Quest software (Protein Databases, Inc., Huntington Station, NY). We were interested in spots that showed at least a 2-fold change with the corresponding protein in the other strain, and met the conditions of a statistical Student test (level of significance, 95 %). Fifty spots were selected per strain from Coomassie blue-stained preparative 2D gels, excised manually and prepared for mass spectrometry analysis [22]. Experiments were performed three times. Mass spectra were obtained using a Bruker Daltonics Autoflex (Bruker Daltonics, Billerica, MA) operated in the delayed extraction and reflectron mode. Spectra were externally calibrated using a peptide calibration standard (Bruker Daltonics 206095). Peak lists of the tryptic peptide masses were generated and searched against the NCBI nr databases or with Rhizobase (http://bacteria.kazusa.or.jp/rhizo/) using the Mascot search program (Matrix Science, Ltd., London United Kingdom). The isoelectric point and molecular weight of the proteins were calculated. Each of the proteins with spot concentration under the detection level was revised manually. A global proteome correlation between strains was calculated and expressed as percentage of similar proteins. Enrichment of gene ontology (GO) terms was done through the EVPedia server (http://student4.postech.ac.kr/evpedia2_xe/xe/) using the TopGO program v2.14.0 [23], with default parameters. Only the first five or 6 classes with the most significant P values, and exclusive terms for each strain, were included in the Table 4. The participation of the abundant proteins in metabolic pathways was graphed using the Biocyc site (http://biocyc.org/overviewsWeb/celOv.shtml#).
Table 4.
Gene ontology (GO) term enrichment for the abundant proteins of the proteomes of rhizobial strains
| Strain GO ID | GO term | Annotated orthologous groups (OGs) | Annotated OGs in this list | Expected annotated OGs by random | P-value |
|---|---|---|---|---|---|
| CCGM1 | Biological processes | ||||
| GO:0046395 | Carboxylic acid catabolic process | 425 | 12 | 0.4 | 3.60E-15 |
| GO:0009063 | Cellular amino acid catabolic process | 221 | 10 | 0.21 | 8.50E-15 |
| GO:0046365 | Monosaccharide catabolic process | 189 | 9 | 0.18 | 1.30E-13 |
| GO:0051188 | Cofactor biosynthetic process | 624 | 12 | 0.59 | 3.30E-13 |
| GO:0006732 | Coenzyme metabolic process | 710 | 12 | 0.67 | 1.50E-12 |
| Molecular function | |||||
| GO:0048037 | Cofactor binding | 993 | 12 | 0.88 | 3.50E-11 |
| GO:0051287 | NAD binding | 69 | 5 | 0.06 | 4.40E-09 |
| GO:0050662 | Coenzyme binding | 665 | 9 | 0.59 | 5.30E-09 |
| GO:0016740 | Transferase activity | 7189 | 21 | 6.35 | 8.20E-08 |
| GO:0016491 | Oxidoreductase activity | 3415 | 15 | 3.01 | 8.60E-08 |
| CIAT652 | Biological processes | ||||
| GO:0005996 | Monosaccharide metabolic process | 404 | 9 | 0.31 | 1.70E-11 |
| GO:0046394 | Carboxylic acid biosynthetic process | 1074 | 11 | 0.83 | 2.70E-10 |
| GO:0006790 | Sulfur compound metabolic process | 583 | 9 | 0.45 | 4.30E-10 |
| GO:0044275 | Cellular carbohydrate catabolic process | 156 | 6 | 0.12 | 2.20E-09 |
| GO:0009117 | Nucleotide metabolic process | 1446 | 11 | 1.12 | 5.90E-09 |
| Molecular function | |||||
| GO:0016787 | Hydrolase activity | 7866 | 21 | 5.7 | 3.00E-09 |
| GO:0016462 | Pyrophosphatase activity | 1346 | 9 | 0.97 | 3.30E-07 |
| GO:0042626 | ATPase activity, coupled to transmembrane movement of substances | 392 | 6 | 0.28 | 3.50E-07 |
| GO:0016835 | Carbon-oxygen lyase activity | 411 | 6 | 0.3 | 4.60E-07 |
| GO:0004022 | Alcohol dehydrogenase (NAD) activity | 23 | 3 | 0.02 | 6.10E-07 |
| GO:0004812 | Aminoacyl-tRNA ligase activity | 106 | 4 | 0.08 | 1.10E-06 |
| CCGM7 | Biological processes | ||||
| GO:0046483 | Heterocycle metabolic process | 2459 | 17 | 1.72 | 3.50E-14 |
| GO:0009117 | Nucleotide metabolic process | 1446 | 12 | 1.01 | 9.40E-11 |
| GO:0015980 | Energy derivation by oxidation of organic compounds | 652 | 9 | 0.46 | 4.30E-10 |
| GO:0010035 | Response to inorganic substance | 944 | 10 | 0.66 | 4.90E-10 |
| GO:0006950 | Response to stress | 5245 | 18 | 3.67 | 5.80E-10 |
| GO:0046686 | Response to cadmium ion | 271 | 7 | 0.19 | 6.60E-10 |
| Molecular function | |||||
| GO:0008266 | Poly(U) RNA binding | 22 | 4 | 0.01 | 1.10E-09 |
| GO:0008187 | Poly-pyrimidine tract binding | 29 | 4 | 0.02 | 3.50E-09 |
| GO:0046872 | Metal ion binding | 5534 | 16 | 3.63 | 4.80E-08 |
| GO:0043169 | Cation binding | 5559 | 16 | 3.65 | 5.20E-08 |
| GO:0003727 | Single-stranded RNA binding | 67 | 4 | 0.04 | 1.10E-07 |
| GO:0017076 | Purine nucleotide binding | 1824 | 10 | 1.2 | 1.40E-07 |
| CFNEI73 | Biological processes | ||||
| GO:0044283 | Small molecule biosynthetic process | 1489 | 13 | 1.04 | 6.30E-12 |
| GO:0016054 | Organic acid catabolic process | 425 | 9 | 0.3 | 9.70E-12 |
| GO:0008652 | Cellular amino acid biosynthetic process | 491 | 9 | 0.34 | 3.50E-11 |
| GO:0009081 | Branched-chain amino acid metabolic process | 55 | 5 | 0.04 | 4.00E-10 |
| GO:0043648 | Dicarboxylic acid metabolic process | 148 | 6 | 0.1 | 8.30E-10 |
| Molecular function | |||||
| GO:0050662 | Coenzyme binding | 665 | 8 | 0.45 | 1.10E-08 |
| GO:0050660 | Flavin adenine dinucleotide binding | 153 | 5 | 0.1 | 6.20E-08 |
| GO:0016462 | Pyrophosphatase activity | 1346 | 9 | 0.91 | 1.80E-07 |
| GO:0048037 | Cofactor binding | 993 | 8 | 0.67 | 2.40E-07 |
| GO:0016817 | Hydrolase activity, acting on acid anhydrides | 1456 | 9 | 0.99 | 3.40E-07 |
| GO:0051287 | NAD binding | 69 | 3 | 0.05 | 1.40E-05 |
Only exclusive terms for each strain are shown
Plasmid visualization by pulsed field gel electrophoresis (PFGE) and in Eckhardt gels
High-molecular-weight plasmids were visualized by PFGE, basically as described previously [24]. Gel electrophoresis was done in a Bio-Rad CHEF-DRIII system with the following conditions: one-sixth of the plug; initial switch time 800 s, final switch time 800 s, temperature 13.5 °C, field angle 106°, run time 64 h at 2.2 V cm−1. Plasmids were also visualized by the Eckhardt technique, as modified by Hynes and McGregor [25].
Nucleotide accession numbers
The CFNEI73 genome has the following accession numbers at GenBank: CP013107 to CP013110 for chromosome, and plasmids a, b and c, respectively. For CCGM7, the sequences were registered under the accession numbers CP013051 to CP013054 for chromosome, and plasmids a, b and c, respectively.
Results
The main objective of the work in the seed-borne rhizobial strains was to find specific differences that allow the bacteria to persist in legume seeds. The seed prevalence is very interesting due to the vertical transmission of the bacteria and its biotechnological potential, and represents a new paradigm in the Rhizobium-legume interaction. We consider that seed isolates form a distinctive new group of rhizobial strains that are adapted to endophytic life. Some strains still have the complete set of genes for nodulation and nitrogen fixation, but others lack some symbiotic genes (unpublished results). Apparently these strains are in an initial process of diversification and thus, must have phenotypic and genomic features that enable them to occupy the new niche.
The selection of strains for this study was based upon several factors. For example, the R. phaseoli CCGM1 seed strain belongs to the most common symbiont species of P. vulgaris, is a biotin auxotroph, has low pyruvate dehydrogenase (PDH) activity, shows decreased growth in subcultures of minimal medium, and high sensitivity in normal laboratory and storage conditions, but has normal symbiotic ability [12]. The S. americanum CCGM7 seed strain is a biotin prototroph, presents good stress resistance, high PDH activity, no growth dcrease in subcultures, and has high symbiotic performance [12]. Extraordinarily, this strain has genes new to symbiotic rhizobia such as nifV gene (encoding homocitrate synthase) that make it the first candidate for fixing nitrogen in free-living state. The nodule strains selected for comparison were the most closely related available. For R. phaseoli, the nodule strain CIAT652 was sequenced previously by us [26], and has been well characterized in our laboratory [14]. It is used as a biofertilizer for beans in Mexico and Central America. The S. americanum strain CFNEI73 was isolated in our Center from Acacia trees, and is also able to nodulate P. vulgaris and Leucaena leucocephala [15]. We sequenced this strain given that no S. americanum genome was available.
In the genomic studies, we firstly performed genomic comparisons of seed and nodule strains, looking for gene differences. A structural study was done to detect genome rearrangements. The presence of strain-specific genes and transposases was evaluated to explain genome rearrangements. Then, the genomic comparison helped to detect orthologs and strain-specific genes. A functional analysis on these genes was done to detect functions that possibly participate in the capability for seed persistance. The study of paralogs arose from the analysis of shared genes to detect signals of differential evolutionary trends. Finally, we obtained the proteomes of these strains, grown in minimal medium, to identify which proteins were most abundant in the seed strains, to deduce their function and participation in metabolism.
Genome sequence of S. americanum CFNEI73
We obtained the genome sequence of S. americanum strain CFNEI73, characterized previously [15]. This strain is efficient for nodulation and nitrogen fixation with bean plants (data not shown). Strain CFNEI73 was reported to have three plasmids [15], but we observed only the two smallest by pulsed-field gel electrophoresis (PFGE) (Additional file 1: Figure S1). The CFNEI73 assembled genome consisted of a 3.7 Mb chromosome and three plasmids: pSamCFNEI73c (2177 Kb), pSamCFNEI73b or pSym (586 Kb), and pSamCFNEI73a (222 Kb), with 6466 predicted genes in total. Previously, we did not detect plasmids in the S. americanum strain CCGM7 [12]. Considering the CFNEI73 plasmid arrangement, we re-analyzed and re-sequenced strain CCGM7 and three plasmids were assembled: pSamCCGM7c (2249 Kb), pSamCCGM7b or pSym (547 Kb) and pSamCCGM7a (405 Kb) The two smallest were observed by PFGE (Additional file 1: Figure S1). The genomic features of strains CFNEI73 and CCGM7 are shown on the Table 1.
Table 1.
Genomic features of S. americanum CCGM7 and CFNEI73 strains
| Feature | CCGM7 | CFNEI73 |
|---|---|---|
| Genome size in bp (CDSa) | 6,853,050 (6601) | 6,751,508 (6466) |
| Size per replicon in bp (CDS): | ||
| Plasmid a | 405,481 (379) | 222,756 (226) |
| Plasmid b (pSym) | 547,106 (491) | 586,526 (529) |
| Plasmid c | 2,249,899 (2168) | 2,177,502 (2045) |
| Chromosome | 3,650,564 (3563) | 3,764,724 (3666) |
| %G + C content (pa, pb, pc, chr) | 60.0, 58.8, 62.1, 63.0 | 60.0, 59.0, 62.2, 63.0 |
| % CDS with assigned function | 77.3 | 76.3 |
| Average CDS length, bp | 887 | 898 |
a CDS coding sequence
Genomic comparison of seed-borne and nodule strains
Structural comparison of the genomes of S. americanum strains
We obtained the ordered sequence of replicons of both S. americanum strains CCGM7 and CFNEI73 and performed a fine synteny analysis to detect rearrangements of genes with relative changed position. Figure 1 shows the ORF prediction for each replicon of the genomes and a synteny comparison. This analysis showed that the chromosomes were almost completely conserved between the strains and that the megaplasmids (plasmids c) and the symbiotic plasmids (plasmids b) showed extensive synteny, with 70–85 % total coverage. However, the smallest plasmid, pSamCFNEI73a, had only 18 % synteny with the CCGM7 genome, with the rest of the genes being strain-specific. The pSamCCGM7a was related to a segment of megaplasmid c of CFNEI73 (with 69 % synteny coverage). We also found that the cluster of genes for symbiosis on plasmid b were more dispersed in CCGM7, in comparison with CFNEI73, due to the insertion of strain-specific segments (see the positions of the numbers 1 to 4 in Fig. 1).
Fig. 1.
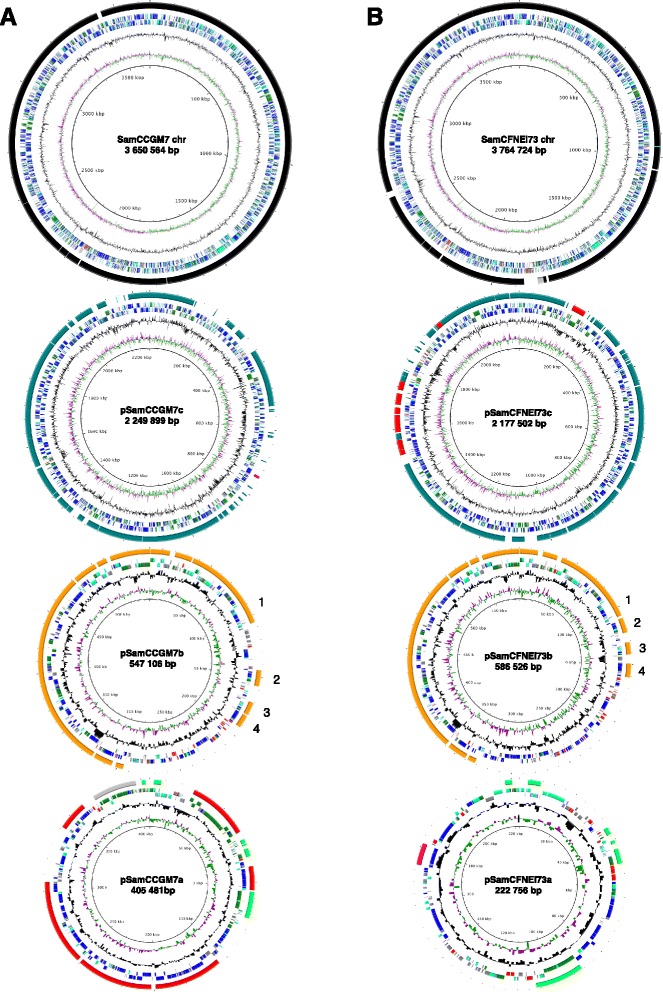
Schematic representation of the genomes of S. americanum strains CCGM7 and CFNEI73. a CCGM7. b CFNEI73. The circles represent, from top to bottom, the chromosome, plasmid c (megaplasmid), plasmid b (symbiotic plasmid) and plasmid a of each. From the innermost circle: GC skew, %GC content, ORF prediction with direction of transcription (color code according to the function: dark blue metabolism, light blue cellular processes, green information, gray unkown, red transposases and other mobile elements), structural comparison by synteny. Color code for synteny: black, synteny between chromosomes; turquoise, synteny between plasmids c; orange, synteny between plasmids b; green, syntenic segments between plasmids a. Other syntenic segments between replicons, as follow: CCGM7 pa with CFNEI73 chromosome gray; CCGM7 pc with CFNEI73 pa pink; and CCGM7 pa with CFNEI73 pc red
Abundance of transposases and strain-specific genes
The transposases are a key factor in the loss of synteny [19, 27, 28]. We found that the chromosomes of both seed-borne strains (CCGM1 and CCGM7) contained fewer transposases and integrases than the typical strains from nodules (Table 2). The plasmids contained from five to ten times more transposases, per megabase, than the chromosomes. As observed, the proportion of transposases per megabase remained almost constant in the plasmids, but was more reduced in the chromosomes of seed-strains. Since the incorporation of strain-specific genes could arise from recent events of transposition, we evaluated the number of these genes by pair comparisons (Table 2). The chromosomes of seed-borne strains had one-half to one-third less of strain-specific genes than the nodule strains, while the plasmids contained similar numbers. The pair of Rhizobium strains had twice the number of strain-specific genes in comparison to Sinorhizobium. Together, the results revealed a tendency to reduce both the number of transposases and the incorporation of strain-specific genes into the chromosomes of seed-borne strains.
Table 2.
Transposases and strain-specific genes deduced by pairwise genome comparison in rhizobial strains
| Transposases (Tn/Mb) a | CCGM1 | CIAT652 | CCGM7 | CFNEI73 |
|---|---|---|---|---|
| Genome | 67 (9.7) | 86 (13.3) | 85 (12.4) | 96 (14.2) |
| By type of replicon: | ||||
| Chromosome | 11 (2.5) | 37 (8.2) | 15 (4.1) | 29 (7.7) |
| Plasmids | 56 (23.4) | 49 (25.3) | 70 (21.9) | 67 (22.4) |
| Homologsb | 5402 (84.0) | 5202 (84.6) | 5783 (87.6) | 5721 (88.5) |
| Strain-specific genesc | 604 (9.4) | 668 (10.9) | 373 (5.7) | 364 (5.6) |
| By type of replicond: | ||||
| Chromosome | 282 (6.6) | 420 (9.7) | 47 (1.3) | 96 (2.6) |
| Plasmids | 322 (14.7) | 248 (14.5) | 326 (10.7) | 268 (9.6) |
aInclude integrases
b% of genome in parenthesis
cThe rest of genes corresponded to short genes (<300 nt) which were discarded, by strain: CCGM1, 422; CIAT652, 279; CCGM7, 445; CFNEI73, 381
d% of replicon(s)
Ortholog detection and identity analysis
A comparative analysis of the genomes of the isolates from bean seeds with related strains was performed. Strain CCGM1 was compared with R. phaseoli closest strains CIAT652 and CNPAF512 and then followed by strain CFN42 from the relative species R. etli. Strain CCGM7 was compared with S. americanum strain CFNEI73 and then with strains NGR234, HH103 and USDA257, from the closely related species S. fredii. Orthologs shared in each group were deduced. Figure 2a shows the number of orthologs shared among the strains and strain-specific genes. Only half of the genome of each organism had orthologs with the others; but many of the remaining genes also had homologs in several strains of Rhizobiales (not shown). In the tested groups, only 315 and 204 genes were found exclusively and without homologs in strains CCGM1 and CCGM7, respectively (Additional file 2: Table S1). Many of them had hypothetical function and others apparently are isozymes. The seed-strains showed lesser strain-specific genes than nodule strains.
Fig. 2.
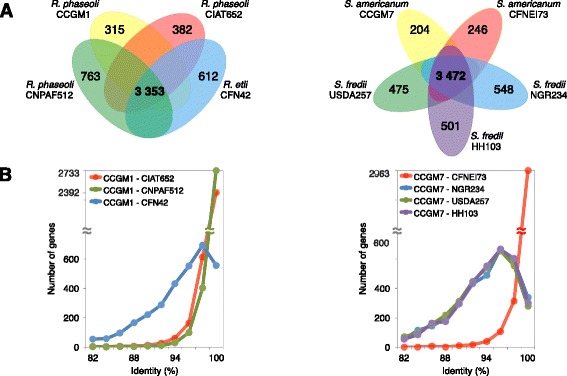
Gene content comparison between seed-borne rhizobial strains and nodule strains. a Venn diagram showing the number of gene clusters of shared orthologs and strain-specific genes among the genomes of Rhizobium phaseoli-R. etli and Sinorhizobium americanum-S. fredii strains. b Frequency distribution of identity percentages of shared orthologs, by group
To discern the relatedness of strains and the global identity of the shared orthologs, their sequency identities were calculated and a frequency distribution was obtained (Fig. 2b). The strain most closely related to CCGM1 was CNPAF512, with 98.4 % identity on average, followed by CIAT652 (97.9 %), and CFN42 (92.8 %). For CCGM7, the closest strain was CFNEI73 with average identity of 99.0 %, followed by the S. fredii strains HH103 (92.2 %), NGR234 (92.0 %) and USDA257 (91.8 %). We calculated the global average nucleotide identity (ANI) of the strains and the values obtained for these comparisons were in good concordance with the identity of shared orthologs (Additional file 3: Table S2) [29].
Function of orthologs and strain-specific genes
A functional distribution was determined for orthologs and strain-specific genes of the seed-borne and nodule strains (Fig. 3). The proportions of shared orthologs of both comparisons had similar functional profile. The strain-specific genes of R. phaseoli CCGM1 were particularly abundant in transcription (COG class K) and cell motility (N) and for CIAT652 in defense (V) and energy generation (C). The strain-specific genes of CCGM7 were enriched in cofactor biosynthesis (H). These enriched functions possibly are important in the seed niche. Strain-specific genes of CFNEI73 appeared to have increased proportion only for replication and recombination (L).
Fig. 3.
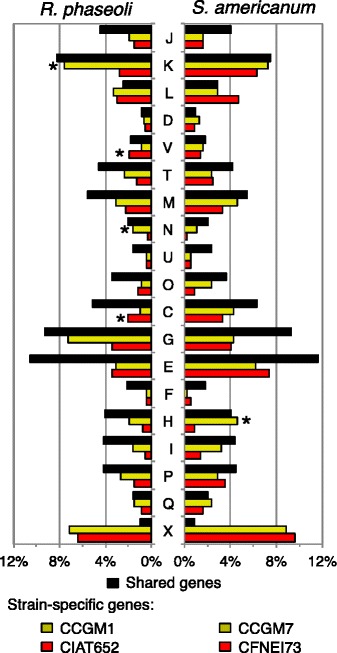
Functional classification of orthologs and strain-specific genes. Left, Rhizobium phaseoli. Right, Sinorhizobium americanum. Yellow, seed-borne strain. Red, nodule strain. Black, orthologs between the pair of strains. Distribution by COG functional categories. Letters: J Translation, ribosomal structure and biogenesis, K Transcription, L Replication, recombination and repair, D Cell cycle control, cell division, chromosome partitioning, V Defense mechanisms, T Signal transduction mechanisms, M Cell wall/membrane/envelope biogenesis, N Cell motility, U Intracellular trafficking, secretion, and vesicular transport, O Post-translational modification, protein turnover, and chaperones, C Energy production and conversion, G Carbohydrate transport and metabolism, E Amino acid transport and metabolism, F Nucleotide transport and metabolism, H Coenzyme transport and metabolism, I Lipid transport and metabolism, P Inorganic ion transport and metabolism, Q Secondary metabolites biosynthesis, transport, and catabolism, and X mobile elements. Genes with general function (R), poorly characterized (S) and not in COGs (−), were not included. Asterisks denote significant difference between seed-borne and nodule strains, with p > 0.05
Analysis of families of paralogs
We found that CFNEI73 contained some genes that we initially described as unique in the CCGM7 genome [12]. For examples, the cluster of genes for hydrogenase (hydrogen uptake), the two RuBisCO genes (one of them associated to a complete cluster of genes of the Calvin cycle) and the nifV gene for the synthesis of homocitrate (the cofactor for nitrogenase, only found in free-living nitrogen fixers). Other unusual gene reiterations reported in strain CCGM7 were five nodD and three nodA reiterations, also present in the CFNEI73 genome. A phylogenetic tree showing the relatedness of the nodD reiterations is shown in Fig. 4a. Two paralogs appeared identical and the other three very similar. Given that, we extended the analysis to the genome content of both pairs of strains, looking for the families of paralogs and their identity level. In strain CCGM7 we found 156 groups of paralogs and 145 groups in CFNEI73. For CCGM1 and CIAT652, we found 101 groups and 86 groups, respectively. The identity among members of each group was calculated and a frequency distribution is shown in Fig. 4b and c. Despite a slight tendency to higher identity in the families of paralogs in the seed-borne strains, no significant statistical differences were found (ANOVA and Kruskal-Wallis H’s).
Fig. 4.
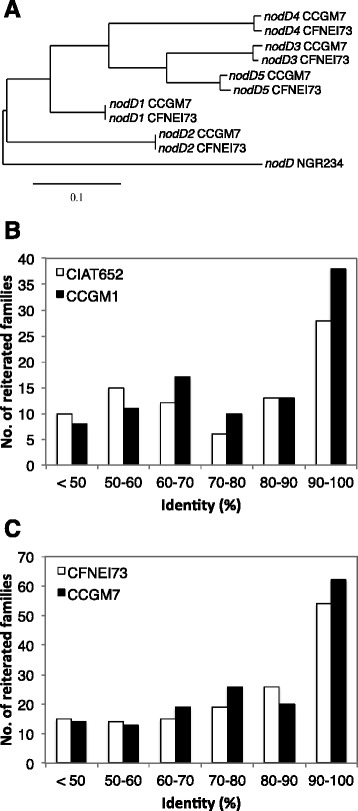
Phylogeny of nodD and identity of families of paralogs of the rhizobial strains. a Phylogenetic tree of nodD gene reiterations of Sinorhizobium americanum strains. nodD of S. fredii NGR234 was used as outgroup. b Frequency distribution of identity percentages, in classes of 10 %, among the members of families of paralogs in R. phaseoli strains CCGM1 and CIAT652. c Frequency distribution of identity percentages, in classes of 10 %, among the members of families of paralogs in S. americanum strains CCGM7 and CFNEI73
Proteomes of the rhizobial seed isolates
We made proteome comparisons from cells grown in exponential phase (MM succinate-ammonium) to look for abundant specific proteins in the seed-borne strains in comparison with their close nodule relatives. CCGM1 and CIAT652 expressed about 725 and 710 proteins, or spots, respectively. By comparison, around 420 spots were the same in both strains; in contrast, 305 spots were found only in CCGM1 and 290 found only in CIAT652 (Additional file 4: Figure S2). The global correlation between the proteomes was 84 %. Table 3 lists 40 of the proteins with higher differential intensity identified in each strain. As can be observed, more proteins belonging to energy generation and translation appeared in CIAT652 strain; in contrast, more proteins for coenzyme and cofactor metabolism were abundant in CCGM1. Two hypothetical proteins were found in each strain and only five abundant proteins of strain CIAT652 had signals in CCGM1. We used the gene ontology (GO) enrichment terms tool to analyze the functional profiles of the abundant proteins in each of the strains. We found interesting differential profiles, as shown in the Table 4. For example, in CCGM1, enriched terms were cofactor and coenzyme binding and metabolism, transferases and oxidoreductases. For CIAT652, carbohydrate metabolism and sulfur compound metabolism with ATPase and pyrophosphatase, hydrolases and amino acyl-tRNA activities. The participation of abundant proteins in metabolic pathways was analyzed (Additional file 5: Figure S3A). As can be observed, pathways such as biosynthesis of carbohydrates, fatty acids and cofactors were better covered by the seed strain CCGM1, and the nodule strain CIAT652 had abundant proteins in the pathways of aminoacyl tRNA charging and carbohydrate degradation.
Table 3.
Abundant proteins in the proteomes of seed-borne strains compared with nodule strains
| Proteins of R. phaseoli CCGM1 | ||||||||||
| Spot no. | Gene ID | Protein | Function | COG | Mascot score | Sequence coverage | Matched peptides | Spot concentrationa | Ratio | |
| CIAT652 | CCGM1 | |||||||||
| 24 | RLPCCGM1_c3018 | SucB | Dihydrolipoamide succinyltransferase | C | 115 | 27 | 13/28 | - | 679.8 | - |
| 41 | RLPCCGM1_c0918 | PdhA | Pyruvate dehydrogenase (acetyl-transferring) protein subunit alpha | C | 71 | 25 | 9/36 | 503.7 | 1013.7 | 2.0 |
| 8 | RLPCCGM1_p0657 | RepA | Plasmid partitioning protein RepAb | D | 73 | 28 | 10/32 | 21.8 | 77.3 | 3.5 |
| 5 | RLPCCGM1_c0705 | AldA | Alanine dehydrogenase | E | 150 | 34 | 13/29 | 143.1 | 442.6 | 3.1 |
| 7 | RLPCCGM1_c1335 | SufS | Cysteine desulfurase | E | 72 | 19 | 7/19 | 14.5 | 46.9 | 3.2 |
| 14 | RLPCCGM1_p1587 | FdhA | Glutathione-independent formaldehyde dehydrogenase | E | 118 | 42 | 16/32 | 3.3 | 62.8 | 19.0 |
| 25 | RLPCCGM1_c1870 | IlvI | Acetolactate synthase 3 catalytic subunit | E | 73 | 17 | 8/30 | - | 59.4 | - |
| 29 | RLPCCGM1_c3812 | ArgD | Acetylornithine transaminase | E | 143 | 49 | 14/35 | - | 97.4 | - |
| 6 | RLPCCGM1_c0701 | CpdB | Bifunctional 2′,3′-cyclic nucleotide 2′-phosphodiesterase/3′-nucleotidase periplasmic protein | F | 96 | 18 | 11/27 | - | 46.5 | - |
| 11 | RLPCCGM1_c3249 | PurH | Bifunctional phosphoribosylaminoimidazolecarboxamide formyltransferase/IMP cyclohydrolase | F | 105 | 31 | 11/31 | 6.7 | 41.5 | 6.2 |
| 2 | RLPCCGM1_c2616 | Pgk | Phosphoglycerate kinase | G | 118 | 36 | 11/25 | 17.5 | 41.3 | 2.4 |
| 9 | RLPCCGM1_c2662 | PykA | Pyruvate kinase | G | 86 | 29 | 9/43 | 9.6 | 37.6 | 3.9 |
| 34 | RLPCCGM1_c1984 | - | Family 1 extracellular solute-binding protein | G | 130 | 45 | 13/35 | - | 127.5 | - |
| 38 | RLPCCGM1_c3763 | FrcB | Fructose ABC transporter substrate-binding protein | G | 75 | 25 | 5/8 | - | 156 | - |
| 40 | RLPCCGM1_c2613 | Gap | Glyceraldehyde 3-phosphate dehydrogenase | G | 67 | 35 | 8/29 | - | 191.9 | - |
| 3 | RLPCCGM1_c4187 | Dxs | 1-deoxy-D-xylulose-5-phosphate synthase | H | 108 | 26 | 12/24 | 9.7 | 25.8 | 2.7 |
| 23 | RLPCCGM1_c3273 | CoaE | Dephospho-CoA kinase | H | 57 | 23 | 5/23 | - | 53 | - |
| 26 | RLPCCGM1_c4205 | RibA | Riboflavin biosynthesis protein | H | 69 | 24 | 7/21 | - | 75 | - |
| 36 | RLPCCGM1_c3299 | AhcY | S-adenosyl-L-homocysteine hydrolase | H | 142 | 34 | 13/28 | - | 146.6 | - |
| 39 | RLPCCGM1_c2569 | SerC | Phosphoserine aminotransferase | H | 113 | 43 | 12/39 | - | 190.4 | - |
| 17 | RLPCCGM1_c3650 | FadB | Enoyl-CoA hydratase | I | 70 | 35 | 8/30 | - | 20.8 | - |
| 18 | RLPCCGM1_c3059 | - | Oxidoreductase | I | 87 | 44 | 12/55 | - | 21 | - |
| 35 | RLPCCGM1_c3416 | FabB | 3-oxoacyl-ACP synthase | I | 89 | 33 | 12/49 | - | 127.6 | - |
| 1 | RLPCCGM1_c1345 | TyrS | Tyrosyl-tRNA synthetase | J | 101 | 27 | 12/37 | 20.7 | 44 | 2.1 |
| 15 | RLPCCGM1_c2760 | - | Acetyltransferase | J | 40 | 37 | 10/44 | - | 19.1 | - |
| 13 | RLPCCGM1_c1935 | MurD | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase | M | 139 | 31 | 13/26 | - | 15.2 | - |
| 33 | RLPCCGM1_c2681 | ExoN | UTP--glucose-1-phosphate uridylyltransferase | M | 162 | 51 | 14/40 | - | 122.2 | - |
| 10 | RLPCCGM1_c3962 | FlaC | Flagellin C protein | N | 80 | 32 | 8/29 | 29.6 | 178.8 | 6.0 |
| 21 | RLPCCGM1_p1412 | HtpG | Heat shock protein 90 | O | 103 | 45 | 13/35 | - | 31.3 | - |
| 22 | RLPCCGM1_c1051 | PpiA | Peptidyl-prolyl cis-trans isomerase A | O | 55 | 30 | 4/29 | - | 38.1 | - |
| 31 | RLPCCGM1_p1186 | - | Glutathione S-transferase YghU | O | 60 | 30 | 6/23 | - | 113.8 | - |
| 4 | RLPCCGM1_p1923 | KatG | Catalase | P | 89 | 23 | 12/35 | 26.8 | 81.6 | 3.0 |
| 16 | RLPCCGM1_c0709 | SseA | Thiosulfate sulfurtransferase | P | 138 | 55 | 12/34 | - | 20.7 | - |
| 27 | RLPCCGM1_c2174 | - | Methanol dehydrogenase regulator MoxR-like protein | R | 108 | 52 | 14/48 | - | 76.8 | - |
| 28 | RLPCCGM1_c3573 | - | Oxidoreductase | R | 80 | 37 | 12/48 | - | 76.8 | - |
| 30 | RLPCCGM1_c2292 | IdhA | Myo-inositol 2-dehydrogenase | R | 87 | 44 | 12/55 | - | 102.8 | - |
| 19 | RLPCCGM1_c4033 | - | Isoprenylcysteine carboxyl methyltransferase | S | 62 | 35 | 6/54 | - | 21.8 | - |
| 37 | RLPCCGM1_c2285 | - | Hypothetical protein | S | 76 | 42 | 9/39 | - | 153.1 | - |
| 12 | RLPCCGM1_p0713 | - | Sensory box/GGDEF family protein | T | 54 | 10 | 9/29 | 32.6 | 314.2 | 9.6 |
| 20 | RLPCCGM1_p2036 | CpaC | Pilus assembly protein | U | 135 | 32 | 12/47 | - | 25.2 | - |
| 32 | RLPCCGM1_p1459 | - | Hypothetical protein | - | 67 | 22 | 6/16 | 23 | 119.3 | 5.2 |
| Proteins of R. phaseoli CIAT652 | ||||||||||
| Spot no. | Gene ID | Protein | Function | COG | Mascot score | Sequence coverage | Matched peptides | Spot concentrationa | Ratio | |
| CIAT652 | CCGM1 | |||||||||
| 47 | RHECIAT_PC0000173 | - | Putative oxidoreductase | C | 132 | 21 | 15/27 | 6.5 | - | - |
| 62 | RHECIAT_CH0002031 | PdhA2 | Pyruvate dehydrogenase subunit beta | C | 114 | 37 | 18/39 | 51.5 | - | - |
| 67 | RHECIAT_CH0004343 | - | Aldehyde dehydrogenase | C | 52 | 17 | 6/31 | 86.1 | - | - |
| 69 | RHECIAT_CH0001680 | NuoE1 | NADH dehydrogenase subunit E | C | 147 | 51 | 17/45 | 119.3 | - | - |
| 75 | RHECIAT_CH0000039 | PckA | Phosphoenolpyruvate carboxykinase | C | 99 | 33 | 13/38 | 400.7 | - | - |
| 76 | RHECIAT_CH0002032 | PdhC | Dihydrolipoamide S-acetyltransferase | C | 101 | 33 | 12/25 | 909.1 | - | - |
| 49 | RHECIAT_PC0000423 | - | Putative oligopeptide ABC transporter substrate-binding protein | E | 56 | 8 | 7/23 | 7.6 | 2.9 | 2.6 |
| 50 | RHECIAT_CH0003659 | PotD | Spermidine/putrescine ABC transporter substrate-binding protein | E | 60 | 25 | 7/24 | 243.5 | 23.9 | 10.2 |
| 60 | RHECIAT_CH0002150 | - | Peptide ABC transporter substrate-binding protein | E | 218 | 44 | 22/38 | 39.9 | - | - |
| 72 | RHECIAT_CH0001992 | MetC | Cystathionine beta-lyase | E | 81 | 38 | 9/22 | 190.4 | - | - |
| 73 | RHECIAT_CH0000879 | GuaB | Inosine 5′-monophosphate dehydrogenase | F | 192 | 44 | 21/47 | 210.1 | - | - |
| 51 | RHECIAT_CH0003979 | LacZ2 | Beta-D-galactosidase | G | 129 | 18 | 11/14 | 16.8 | - | - |
| 55 | RHECIAT_CH0000213 | GpmA | Phosphoglyceromutase | G | 214 | 84 | 18/52 | 23.7 | - | - |
| 61 | RHECIAT_CH0003393 | XylF | Xylose ABC transporter substrate-binding protein | G | 113 | 32 | 10/41 | 46.7 | - | - |
| 64 | RHECIAT_CH0003248 | - | Omega amino acid--pyruvate transaminase | H | 147 | 47 | 14/26 | 67.8 | - | - |
| 42 | RHECIAT_CH0001955 | AccC | Acetyl-CoA carboxylase biotin carboxylase subunit | I | 77 | 33 | 9/26 | 795.2 | 364.4 | 2.2 |
| 53 | RHECIAT_CH0003282 | BdhA | D-beta-hydroxybutyrate dehydrogenase | I | 81 | 28 | 6/8 | 19.3 | - | - |
| 48 | RHECIAT_PA0000110 | FusAa | Elongation factor G | J | 55 | 16 | 8/27 | 6.8 | - | - |
| 54 | RHECIAT_CH0004024 | PrfA | Peptide chain release factor 1 | J | 119 | 41 | 14/40 | 19.9 | - | - |
| 58 | RHECIAT_CH0001897 | ValS | Valyl-tRNA synthetase | J | 85 | 12 | 8/12 | 26.5 | - | - |
| 59 | RHECIAT_CH0002285 | ThrS | Threonyl-tRNA synthetase | J | 124 | 23 | 16/34 | 31.3 | - | - |
| 65 | RHECIAT_CH0000980 | GlyS | Glycyl-tRNA synthetase subunit beta | J | 192 | 38 | 23/37 | 71.6 | - | - |
| 66 | RHECIAT_CH0002242 | MetS | Methionyl-tRNA synthetase | J | 105 | 26 | 10/22 | 72.1 | - | - |
| 57 | RHECIAT_CH0001733 | - | Nucleoside-diphosphate-sugar epimerase | M | 57 | 19 | 5/22 | 26.4 | - | - |
| 74 | RHECIAT_CH0003488 | NoeJ | Mannose-1-phosphate guanylyltransferase (GDP) protein | M | 145 | 48 | 18/52 | 243.4 | - | - |
| 46 | RHECIAT_CH0001879 | - | Peroxidase | O | 67 | 35 | 8/29 | 119.2 | 23.7 | 5.0 |
| 52 | RHECIAT_CH0001895 | Pcm1 | Protein-L-isoaspartate(D-aspartate) O-methyltransferase | O | 93 | 38 | 6/16 | 19 | - | - |
| 63 | RHECIAT_CH0002260 | PpiD1 | Peptidyl-prolyl cis-trans isomerase D signal peptide protein | O | 57 | 16 | 6/14 | 67.7 | - | - |
| 70 | RHECIAT_CH0004147 | - | Nitrate/sulfonate/bicarbonate ABC transporter substrate-binding protein | P | 106 | 42 | 11/29 | 127.6 | - | - |
| 71 | RHECIAT_CH0002820 | - | Ferrichrome ABC transporter substrate-binding protein | P | 135 | 52 | 12/23 | 146.6 | - | - |
| 56 | RHECIAT_PC0000681 | VbsO | L-lysine 6-monooxygenase (NADPH) protein | Q | 105 | 32 | 11/23 | 25.5 | - | - |
| 43 | RHECIAT_CH0004212 | - | ABC transporter substrate-binding protein | R | 99 | 36 | 9/29 | 360.8 | - | - |
| 44 | RHECIAT_CH0004405 | - | Hypothetical protein | S | 89 | 33 | 8/38 | 253.5 | 111.4 | 2.3 |
| 68 | RHECIAT_CH0002473 | - | Hypothetical protein | S | 109 | 31 | 14/38 | 94.5 | - | - |
| 45 | RHECIAT_CH0004199 | TypA | GTP-binding protein TypA/BipA | T | 151 | 34 | 21/42 | 215.3 | - | - |
| Proteins of S. americanum CCGM7 | ||||||||||
| Spot no. | Gene ID | Protein | Function | COG | Mascot score | Sequence coverage | Matched peptides | Spot concentrationa | Ratio | |
| CFNEI73 | CCGM7 | |||||||||
| 2 | SAMCCGM7_Ch3507 | AcnA | Aconitate hydratase | C | 69 | 17 | 11/31 | - | 185.6 | - |
| 3 | SAMCCGM7_Ch3374 | SucB | Dihydrolipoamide succinyltransferase | C | 99 | 25 | 11/21 | - | 127 | - |
| 4 | SAMCCGM7_Ch1973 | FumC | Fumarate hydratase class II | C | 100 | 21 | 10/20 | - | 105.8 | - |
| 8 | SAMCCGM7_pC0039 | TctC | Tricarboxylate transport protein | C | 64 | 30 | 6/11 | - | 45.4 | - |
| 11 | SAMCCGM7_Ch0047 | PckA | Phosphoenolpyruvate carboxykinase | C | 88 | 20 | 11/30 | 6.5 | 143.9 | 22.1 |
| 12 | SAMCCGM7_Ch2624 | Ald | Aldehyde dehydrogenase | C | 65 | 17 | 7/18 | - | 17.3 | - |
| 20 | SAMCCGM7_Ch3356 | AtpD | ATP synthase subunit beta | C | 111 | 38 | 13/34 | 41.4 | 407.9 | 9.9 |
| 28 | SAMCCGM7_Ch1381 | NuoG | NADH-quinone oxidoreductase subunit G | C | 111 | 23 | 13/27 | 9.3 | 52.4 | 5.6 |
| 29 | SAMCCGM7_Ch2523 | AcoD | Acetaldehyde dehydrogenase 2 | C | 133 | 27 | 14/33 | 65.9 | 259.8 | 3.9 |
| 31 | SAMCCGM7_Ch3358 | AtpA | ATP synthase subunit alpha | C | 97 | 29 | 14/41 | 78.5 | 290.4 | 3.7 |
| 16 | SAMCCGM7_Ch1849 | CarB | Carbamoyl-phosphate synthase large subunit | E | 74 | 10 | 11/20 | 7.5 | 103.4 | 13.8 |
| 17 | SAMCCGM7_Ch2552 | LeuA | 2-isopropylmalate synthase | E | 118 | 27 | 13/23 | - | 11.4 | - |
| 18 | SAMCCGM7_pA0276 | - | Homoserine dehydrogenase | E | 72 | 22 | 9/28 | - | 11.2 | - |
| 26 | SAMCCGM7_Ch1267 | GlyA | Pyridoxal-phosphate-dependent serine hydroxymethyltransferase | E | 85 | 21 | 9/22 | 11.9 | 75.8 | 6.4 |
| 19 | SAMCCGM7_Ch3100 | - | 5′-nucleotidase | F | 122 | 22 | 12/19 | 11.1 | 120.3 | 10.8 |
| 35 | SAMCCGM7_Ch3605 | PurH | Bifunctional phosphoribosylaminoimidazolecarboxamide formyltransferase/IMP cyclohydrolase | F | 80 | 23 | 10/31 | 140.6 | 278.9 | 2.0 |
| 24 | SAMCCGM7_Ch3149 | Pgm | Phosphoglucomutase Pgm | G | 61 | 12 | 5/7 | 6.4 | - | - |
| 30 | SAMCCGM7_Ch2630 | ChvE | Multiple sugar-binding periplasmic receptor | G | 55 | 24 | 6/25 | 34.2 | 127.5 | 3.7 |
| 32 | SAMCCGM7_Ch3057 | FbaB | Fructose-bisphosphate aldolase | G | 70 | 32 | 9/34 | 27.2 | 95.8 | 3.5 |
| 33 | SAMCCGM7_Ch3054 | Gap | Glyceraldehyde-3-phosphate dehydrogenase | G | 80 | 29 | 7/15 | 33.7 | 109.3 | 3.2 |
| 21 | SAMCCGM7_Ch0041 | AhcY | S-adenosyl-L-homocysteine hydrolase | H | 81 | 26 | 10/29 | 21.2 | 169 | 8.0 |
| 14 | SAMCCGM7_Ch1196 | FabF | 3-oxoacyl-ACP synthase | I | 66 | 16 | 7/17 | - | 16.8 | - |
| 1 | SAMCCGM7_Ch2527 | RplY | 50S ribosomal protein L25 | J | 199 | 72 | 17/32 | - | 498.9 | - |
| 7 | SAMCCGM7_Ch1428 | GatB | Aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase subunit B | J | 72 | 18 | 7/19 | - | 59.6 | - |
| 23 | SAMCCGM7_Ch0231 | Pnp | Polyribonucleotide nucleotidyltransferase | J | 122 | 29 | 17/38 | 35.5 | 270.8 | 7.6 |
| 9 | SAMCCGM7_Ch0831 | GroEL | Chaperonin | O | 95 | 28 | 14/36 | 12.8 | 380.2 | 29.7 |
| 25 | SAMCCGM7_pC0433 | ClpV | Protease | O | 63 | 15 | 10/32 | - | 7 | - |
| 5 | SAMCCGM7_Ch0999 | CysN | Sulfate adenylyltransferase subunit 1 | P | 121 | 45 | 15/44 | - | 65.2 | - |
| 13 | SAMCCGM7_pC1570 | - | Iron ABC transport system, solute-binding protein | P | 89 | 19 | 17/11 | - | 17.1 | - |
| 22 | SAMCCGM7_pC1145 | - | Ferrichrome-iron receptor | P | 149 | 19 | 13/16 | - | 7.8 | - |
| 27 | SAMCCGM7_pC1571 | - | Iron ABC transport system, solute-binding protein | P | 105 | 26 | 11/23 | 20.6 | 123.5 | 6.0 |
| 34 | SAMCCGM7_pC1766 | Fct | Ferrichrysobactin receptor | P | 174 | 35 | 18/31 | 2.6 | 7.4 | 2.8 |
| 6 | SAMCCGM7_Ch2381 | - | ABC transporter ATP-binding protein | R | 126 | 31 | 17/30 | - | 60.4 | - |
| 10 | SAMCCGM7_pA0100 | VirB10 | Type IV secretion system protein | U | 150 | 32 | 12/19 | - | 23.8 | - |
| 15 | SAMCCGM7_pC0430 | ImpC | Type VI secretion system protein | U | 96 | 23 | 10/21 | 14 | 206 | 14.7 |
| Proteins of S. americanum CFNEI73 | ||||||||||
| Spot no. | Gene ID | Protein | Function | COG | Mascot score | Sequence coverage | Mached peptides | Spot concentrationa | Ratio | |
| CFNEI73 | CCGM7 | |||||||||
| 37 | SAMCFNEI73_Ch3492 | LpdA | Dihydrolipoamide dehydrogenase | C | 134 | 37 | 13/23 | 48.2 | - | - |
| 44 | SAMCFNEI73_Ch3194 | - | Hypothetical protein | C | 93 | 44 | 9/22 | 21.6 | - | - |
| 47 | SAMCFNEI73_Ch0872 | AtpF | ATP synthase subunit B’ | C | 94 | 35 | 7/18 | 51 | 3.4 | 15.0 |
| 53 | SAMCFNEI73_Ch1106 | - | Electron transfer flavoprotein-ubiquinone oxidoreductase | C | 105 | 24 | 11/22 | 10.4 | - | - |
| 54 | SAMCFNEI73_Ch0062 | GlcB | Malate synthase G | C | 156 | 27 | 14/19 | 15.1 | 1.7 | 8.9 |
| 57 | SAMCFNEI73_Ch0755 | MmsA | Methylmalonate-semialdehyde dehydrogenase | C | 132 | 33 | 17/29 | 66.6 | 12.8 | 5.2 |
| 70 | SAMCFNEI73_Ch1382 | NuoD | NADH-quinone oxidoreductase subunit D | C | 135 | 42 | 20/54 | 45.7 | 21 | 2.2 |
| 72 | SAMCFNEI73_Ch3505 | SdhA | Succinate dehydrogenase flavoprotein subunit | C | 96 | 20 | 10/15 | 42.5 | 19.9 | 2.1 |
| 36 | SAMCFNEI73_pC0436 | FliY | Cystine-binding periplasmic protein | E | 63 | 26 | 6/17 | 63.4 | - | - |
| 43 | SAMCFNEI73_Ch1350 | ArgC | N-acetyl-gamma-glutamyl-phosphate reductase | E | 66 | 29 | 7/27 | 23.5 | - | - |
| 49 | SAMCFNEI73_pC0934 | HutU | Urocanate hydratase | E | 93 | 21 | 13/34 | 13.2 | - | - |
| 51 | SAMCFNEI73_Ch2492 | IlvB | Acetolactate synthase large subunit | E | 82 | 14 | 7/10 | 12.1 | - | - |
| 52 | SAMCFNEI73_Ch0061 | - | Hydantoinase/oxoprolinase family protein | E | 71 | 17 | 7/16 | 10.5 | - | - |
| 59 | SAMCFNEI73_Ch3399 | MetH | Methionine synthase | E | 208 | 25 | 24/29 | 4.6 | - | - |
| 63 | SAMCFNEI73_Ch0442 | DapD | 2,3,4,5-tetrahydropyridine-2,6-carboxylate N-succinyltransferase | E | 55 | 20 | 7/42 | 38.3 | 10.6 | 3.6 |
| 65 | SAMCFNEI73_Ch2207 | MetH2 | Methionine synthase | E | 79 | 28 | 8/37 | 25.4 | 7.9 | 3.2 |
| 69 | SAMCFNEI73_Ch0208 | - | Extracellular solute-binding protein, family 5 | E | 74 | 21 | 12/47 | 30 | 11 | 2.7 |
| 71 | SAMCFNEI73_Ch1748 | - | Peptide ABC transporter substrate-binding protein | E | 93 | 24 | 13/34 | 32.5 | 15.1 | 2.2 |
| 62 | SAMCFNEI73_Ch3182 | Pgk | Phosphoglycerate kinase | G | 113 | 41 | 12/26 | 146.7 | 39.3 | 3.7 |
| 66 | SAMCFNEI73_Ch3273 | GlgX | Glycogen debranching protein | G | 131 | 23 | 12/18 | 6.7 | 2.1 | 3.2 |
| 58 | SAMCFNEI73_pC1978 | ThiC | Phosphomethylpyrimidine synthase | H | 113 | 33 | 17/37 | 23.9 | 5 | 4.8 |
| 64 | SAMCFNEI73_Ch1162 | NadB | L-aspartate oxidase | H | 156 | 31 | 15/23 | 33.7 | 9.7 | 3.5 |
| 48 | SAMCFNEI73_pC0946 | MmgC | Acyl-CoA dehydrogenase | I | 83 | 32 | 13/40 | 13.9 | - | - |
| 46 | SAMCFNEI73_Ch1475 | FusA | Elongation factor G | J | 93 | 22 | 12/24 | 15.8 | - | - |
| 42 | SAMCFNEI73_Ch1028 | - | ATPase component BioM of energizing module of biotin ECF transporter | L | 136 | 40 | 15/36 | 23.8 | - | - |
| 39 | SAMCFNEI73_pC1683 | RkpQ | N-acylneuraminate-9-phosphate synthase | M | 202 | 55 | 16/39 | 25.6 | - | - |
| 50 | SAMCFNEI73_Ch1571 | KdsA | 2-dehydro-3-deoxyphosphooctonate aldolase | M | 106 | 43 | 16/54 | 12.4 | - | - |
| 67 | SAMCFNEI73_Ch2467 | MurE | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate--2,6-diaminopimelate ligase | M | 166 | 34 | 13/19 | 26.3 | 8.7 | 3.0 |
| 40 | SAMCFNEI73_Ch1644 | Pcm | Protein-L-isoaspartate O-methyltransferase | O | 140 | 57 | 11/20 | 24.9 | - | - |
| 61 | SAMCFNEI73_Ch1368 | LonA | Endopeptidase La | O | 62 | 12 | 9/20 | 55 | 14.3 | 3.8 |
| 68 | SAMCFNEI73_pC1783 | - | Phosphate ABC transporter substrate-binding protein | P | 78 | 27 | 7/24 | 104.4 | 36.3 | 2.9 |
| 45 | SAMCFNEI73_Ch3296 | - | Cobalamin synthesis protein/P47K family protein | R | 69 | 25 | 9/37 | 17.9 | - | - |
| 55 | SAMCFNEI73_Ch3670 | - | Hypothetical protein | R | 72 | 16 | 8/19 | 8.2 | - | - |
| 38 | SAMCFNEI73_pB0529 | TraI | Autoinducer synthesis protein | T | 157 | 75 | 15/39 | 30.5 | - | - |
| 60 | SAMCFNEI73_Ch1589 | NtrC | Nitrogen assimilation regulatory protein | T | 63 | 19 | 7/20 | 26.2 | 6 | 4.4 |
| 56 | SAMCFNEI73_pB0525 | TrbE | Conjugal transfer protein | U | 177 | 27 | 20/32 | 7.9 | - | - |
| 41 | SAMCFNEI73_pC1268 | - | Peroxiredoxin | V | 95 | 37 | 8/22 | 24.8 | - | - |
aIn OD units. No value means the spot concentration was under the level of detection
For COG class definition, see legend of Fig. 3
The proteomes of strains CCGM7 and CFNEI73 showed 715 and 713 spots, respectively, sharing 491 spots. In this case, 224 were unique to CCGM7 and 222 unique to CFNEI73. The correlation between the proteomes was 82.6 %. Spots with differential abundance profiles were identified (Table 3). Only one protein with hypothetical function was found in CFNEI73. The GO analysis of the abundant proteins showed enriched terms in CCGM7 for stress response, energy generation and metal detoxification and polyU, pyrimidine and purine metabolisms and metal binding, and single strand RNA-binding (Table 4). For CFNEI73, enriched terms included branched amino acid and dicarboxylic acid metabolisms, with binding of coenzyme, flavin, cofactor and NAD as biological activities. The abundant proteins of the seed strain CCGM7 participated more in pathways of the biosynthesis of carbohydrates and fatty acids and glycolysis (Additional file 5: Figure S3B). The nodule strain CIAT652 had abundant proteins for amino acid biosynthesis, cell structure and carbohydrate degradation.
The analyzed strains showed in the proteomes about 700 proteins each, shared up to 490 with the related strain and from 222 to 305 were considered unique proteins. Based on the pair comparisons, about 50 of the most abundant proteins were chosen for each strain and analyzed. Only 5 proteins had hypothetical function. Even when the strains were growing at the same exponential rate, the proteins had different metabolic functions.
Discussion
The seed-borne strains R. phaseoli CCGM1 and S. americanum CCGM7 described in this work were obtained through assays with noninoculated bean plants that nodulated and fixed nitrogen [12]. As described previously, the first non efficient strain tested was an Agrobacterium tumefaciens devoid of pTi and carrying instead a pSym derived from R. etli strain CFN42 [12]. Using these procedures, ten strains were isolated that showed plasmid profiles not observed previously. We reported that strain CCGM1 encoded several prophages (the firsts reported in Rhizobium), toxin/antitoxin pairs, queuosine, cellulosome anchoring system and other genes possibly related to the interaction with the plants [12]. The strain was a biotin auxotroph that showed a growth decline in serial subcultures, accumulated poly-beta-hydroxybutyrate (PHB) and had low pyruvate dehydrogenase (PDH) activity (as typical of some strains of its species), yet had optimal nodulation and nitrogen fixation ability [12].
Here, we compared the genomes and proteomes of rhizobial strains isolated only from nodules with isolates from bean seeds. The R. phaseoli strains were CIAT652 and CCGM1, respectively, and the S. americanum strains were CCGM7 and CFNEI73 [15]. This last strain was sequenced twice and, together with CCGM7 resequencing, allowed us to make a fine structural genome comparison (Fig. 1). The S. americanum strains each have three plasmids: a megaplasmid of about 2 Mb, the symbiotic plasmid ranging from 450 to 550 Kb, and a smaller plasmid between 200 and 400 kb. The main observation of the structural study was the high synteny of the chromosomes and the megaplasmids. However, their symbiotic plasmids showed important differences in the region surrounding the symbiotic gene clusters. Furthermore, the smallest plasmid of CCGM7 apparently derived from a segment of the megaplasmid, but the smallest plasmid of CFNEI73 was almost completely unrelated. The plasmids in these S. americanum strains were difficult to observe. However, it was easy to observe the plasmids of other strains of S. americanum, CCBAU051121 and CCBAU051127 [30]. In our previous report, we did not observe plasmids in strain CCGM7 [12]. However, given the genome assembly and the report that CFNEI73 contained three plasmids [15], additional efforts were made to detect its plasmids. To avoid the action of nucleases that possibly degrade the nucleic acids when the cells are lysed, the protocols were modified as described in Methods.
CFNEI73 also had some features that we previously found only in strain CCGM7: a nifV gene for homocitrate synthesis, hydrogenase uptake genes (hup) and two RubisCO clusters. The strains shared the three nodA and the five nodD reiterations (see the phylogeny in Fig. 4a), with some of them being identical and others having slight differences. The expansion of genetic families appears as an adaptative trait, as observed in Leishmania [31]. Also, we observed that chromosomes of seed-borne strains had less transposases and strain-specific genes in comparison to the typical strains, indicating reduced potential for rearrangement and possibly gene loss as a requisite for seed prevalence. This could be analogous to the genome reduction observed in obligate intracellular bacteria [32, 33].
The S. americanum strains had interesting metabolic abilities. CCGM7 had high PDH activity, grew without decline in serial cultures of minimal medium and, like CFNEI73, had the complete gene set for biotin synthesis, thus making them biotin prototrophs [12]. In the proteome analysis of abundant proteins, CCGM7 showed a protein set enriched for energy generation, response to stress, metal detoxification, translation and carbohydrate and ion metabolisms (Table 3). CFNEI73 proteins appeared enriched for amino acid transport and metabolism. The abundant proteins of seed strains participated in the metabolic pathways of biosynthesis of carbohydrates, fatty acids and cofactors. On the other hand, the nodule strains had better coverage of amino acyl tRNA charging, and biosyntheses of amino acids and cell structures (Additional file 5: Figure S3).
It is important to mention that only five hypothetical proteins were abundant in the proteins analyzed (from total 173). Apparently the main difference between the strains is related to the form in which the metabolism is performed, using the same main pathways. Also, 136 proteins had names, with specific function, and the rest 36 only generic functions. Only two proteins were abundant in both seed strains, namely PurH (Bifunctional phosphoribosyl amino imidazole carboxamide formyl transferase/IMP cyclo hydrolase) and SucB (Dihydrolipoamide succinyl transferase). This pair of proteins may be considered specific markers of the seed strains in minimal medium.
The differential metabolic functions of the identified abundant proteins were found even when the strains were growing at the same rate. Although the physiological meaning of these particular proteins in each strain can be matter of speculation, the data contribute to the characterization of the peculiarities of the strains. For example, the majority of abundant proteins (93 out 173) had no signal in the other strain, thus appearing as specific traits for each one. In E. coli it has been found that the core proteome is significantly enriched in nondiferentially expressed genes and depleted in differentially expressed genes [34].
The nodule strain CIAT652 had abundant proteins for energy generation, translation, and more dehydrogenases (related to redox and energy processes). We previously performed symbiotic and physiological characterization of this strain, qualifying it as a highly efficient strain [14, 26]. The abundant proteins of seed strain CCGM1 were enriched for synthesis of coenzymes and cofactors (Tables 3 and 4). The seed bacteria must develop great adaptative traits because the spermosphere is a new niche with high competition between seed borne and soil microorganisms occurring at the time of seed emergence [35].
It will be of interest to determine the host range of the S. americanum strains because they are relatively newly described species and their closest relatives belong to the very broad host range S. fredii strains NGR234 and USDA257, which can nodulate up to 112 and 79 legume species, respectively [36]. We have so far determined that CCGM7 can nodulate and fix nitrogen with P. vulgaris and Medicago truncatula; CFNEI73 can nodulate Acacia farnesiana [15] and P. vulgaris. As mentioned, the strains present five nodD reiterations and possibly these have a role in the host range. In a relevant recent paper, Del Cerro et al. determined that the five nodD genes of R. tropici CIAT899 were necessary to engage the microsymbiont in nodulation with different legume plants [37].
A factor that might be crucial for our ability to isolate rhizobia from seed was that the seeds were cropped from plants irrigated previously with nitrogen. Apparently, the seed strain is more adapted to the presence of nitrogen. On the other hand, nodulation is the main process by which Rhizobium colonizes the plants, but the seed niche is a less constrained environment that relaxes the selective pressure on the symbiotic genes. Although the strain persistence in seeds can represent an advantage for the plants given their potential metabolic capabilities, the seed rhizobia can also lose the symbiotic capability in the seed environment, without apparent consequences for the plant. Thus, originally the nodulation ability was a necessary feature for entry into the plants, but in the seed isolates it is not an essential feature.
Conclusion
The seed-borne, nitrogen-fixing rhizobia strains represent an extended symbiotic model of the interaction with legume plants. Genomic differences such as rearrangement and reduction of transposases in the chromosomes possibly resulted from the adaptation to the seeds. Some functions such as stress response and biosynthesis of coenzymes, cofactors, carbohydrates and fatty acids appeared enriched in the seed strains. Comprehensive genomic studies, such as those presented here help to reveal global differences between the rhizobial seed strains and those isolated only from nodules.
Acknowledgments
We thank Dr. En Tao Wang (IPN, Mexico City) by the gift of S. americanum CCBAU051121 and CCBAU051127 strains; and from CCG-UNAM Dr. Luis Bolaños and Gabriela Guerrero by help in bioinformatics, Sandra Contreras by preparing samples for proteome, Paz Salas by technical support, Magdalena Hernández by preparation of proteomics data, Dr. Julio Martínez by help in statistics and Dr. Michael F. Dunn by careful reviewing of the manuscript.
Funding
This project was partially supported by grants by the Consejo Nacional de Ciencia y Tecnología-Mexico (213606 and 152776) and by DGAPA-PAPIIT-UNAM (IN206914 and IN208216). The agencies had no role in the design of study, analysis or interpretation of results.
Availability of data and materials
The strains mentioned in this work are available. Send inquiries to Prof. Jaime Mora, Centro de Ciencias Genomicas, UNAM, Apdo. postal 565-A, Cuernavaca, Morelos, Mexico CP 62220 or contact at jmora@ccg.unam.mx. The datasets supporting the conclusions of this work are available at our web server ftp://kanan.ccg.unam.mx/PGFP/genomicstudies and at public repositories. The CFNEI73 genome was registered in the GenBank (http://www.ncbi.nlm.nih/GenBank) with the following accession numbers: CP013107 to CP013110 for chromosome, and plasmids a, b and c, respectively, and the CCGM7 genome with the accession numbers CP013051 to CP013054 for chromosome, and plasmids a, b and c, respectively. The mass spectrometry proteomics data are available at ProteomeXchange Consortium (http://www.proteomecentral.proteomexchange.org), with identifier PXD004702.
Authors’ contributions
JM and HP designed the study. AA and HP performed the computational analyses. YM, RD, ES and CV-L participated in the characterization of the strains. GM-B and SE performed the proteomic analyses. LG and EM contributed with interpretation and discussion of the results. HP and JM wrote the manuscript. All authors contributed with the revision of the work. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not apply.
Ethics approval and consent to participate
Not apply.
Abbreviations
- ANIm
Genomic average nucleotide identity
- Bp
Base pairs
- COG
Cluster of orthologous groups
- GO
Gene ontology
- Kb
Kilo base pairs
- NR
Nonredundant
- PAGE
Polyacrylamide gel electrophoresis
- PFGE
Pulsed-field gel electrophoresis
Additional files
Plasmids of S. americanum strains visualized by pulsed field gel electrophoresis (PFGE). 1, CCGM7, 2 and 3, molecular weight marker (chromosomes of Saccharomyces cerevisiae, only some are denoted). 4, CFNEI73. Only the smallest plasmids were visible. (PDF 717 kb)
Strain-specific genes of strains isolated from bean seeds, deduced by pairwise comparison with close relatives. Short genes (<300 nt) were discarded. (PDF 156 kb)
Average nucleotide identity (ANIm) among the genomes of selected Sinorhizobium and Rhizobium strains. (PDF 34 kb)
Proteomes of R. phaseoli strains. A, CCGM1. B, CIAT652. Spots taken for analysis are encircled, in red, abundant proteins in the strain which were not visible in the other; in green, abundant proteins in the strain with counterpart in the other. Lines with arrows denote the direction of the 2D runs. (PDF 460 kb)
Metabolic reconstruction with BioCyc using the most abundant proteins of the strains detected in the proteome. A, Rhizobium phaseoli strains. Enzymes participating in metabolic pathways. Green boxes, the most abundant proteins from CCGM1 strain. Red boxes, the most abundant proteins from CIAT652 strain. B, Sinorhizobium americanum strains. Enzymes participating in metabolic pathways. Green boxes, the most abundant proteins from CCGM7 strain. Red boxes, the most abundant proteins from CFNEI73 strain. Brown were both coincided. The lists of proteins enable their use directly on the BioCyc page with the denoted organism. (−) denotes that no homolog was found in the strain. (PDF 355 kb)
Contributor Information
Humberto Peralta, Email: peralta@ccg.unam.mx.
Alejandro Aguilar, Email: aaguilar@ccg.unam.mx.
Rafael Díaz, Email: rdiaz@ccg.unam.mx.
Yolanda Mora, Email: ymora@ccg.unam.mx.
Gabriel Martínez-Batallar, Email: angelmb@ccg.unam.mx.
Emmanuel Salazar, Email: esalazar@ccg.unam.mx.
Carmen Vargas-Lagunas, Email: cvargas@ccg.unam.mx.
Esperanza Martínez, Email: emartine@ccg.unam.mx.
Sergio Encarnación, Email: encarnac@ccg.unam.mx.
Lourdes Girard, Email: girard@ccg.unam.mx.
Jaime Mora, Phone: (+52) 777 3114661, Email: jmora@ccg.unam.mx.
References
- 1.Haag FA, Arnold MF, Myka KK, Kerscher B, Dall’Angelo S, Zanda M, Mergaert P, Ferguson GP. Molecular insights into bacteroid development during Rhizobium–legume symbiosis. FEMS Microbiol Rev. 2013;37(3):364–383. doi: 10.1111/1574-6976.12003. [DOI] [PubMed] [Google Scholar]
- 2.Oldroyd GE, Murray JD, Poole PS, Downie JA. The rules of engagement in the legume-rhizobial symbiosis. Annu Rev Genet. 2011;45:119–144. doi: 10.1146/annurev-genet-110410-132549. [DOI] [PubMed] [Google Scholar]
- 3.Gutiérrez-Zamora ML, Martínez-Romero E. Natural endophytic association between Rhizobium etli and maize (Zea mays L.) J Biotechnol. 2001;91:117–126. doi: 10.1016/S0168-1656(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 4.López-López A, Rogel MA, Ormeño-Orrillo E, Martínez-Romero J, Martínez-Romero E. Phaseolus vulgaris seed-borne endophytic community with novel bacterial species such as Rhizobium endophyticum sp. nov. Syst App Microbiol. 2010;33:322–327. doi: 10.1016/j.syapm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Rosenblueth M, Martínez-Romero E. Rhizobium etli maize populations and their competitiveness for root colonization. Arch Microbiol. 2004;181:337–344. doi: 10.1007/s00203-004-0661-9. [DOI] [PubMed] [Google Scholar]
- 6.Truyens S, Weyens N, Cuypers A, Vangronsveld J. Changes in the population of seed bacteria of transgenerationally Cd-exposed Arabidopsis thaliana. Plant Biol (Stuttg) 2012;15(6):971–981. doi: 10.1111/j.1438-8677.2012.00711.x. [DOI] [PubMed] [Google Scholar]
- 7.Sharma PK, Sarita S, Prell J. Isolation and characterization of an endophytic bacterium related to Rhizobium/Agrobacterium from wheat (Triticum aestivum L.) roots. Curr Sci. 2005;89:608–610. [Google Scholar]
- 8.Beneduzi A, Moreira F, Costa PB, Vargas LK, Lisboa BB, Favreto R, Baldani JI, Passaglia LMP. Diversity and plant growth promoting evaluation abilities of bacteria isolated from sugarcane cultivated in the South of Brazil. App Soil Ecol. 2013;4:94–104. doi: 10.1016/j.apsoil.2012.08.010. [DOI] [Google Scholar]
- 9.Crook MB, Mitra S, Ané J-M, Sadowsky MJ, Gyaneshwar P. Complete genome sequence of the Sesbania symbiont and rice growth-promoting endophyte Rhizobium sp. strain IRBG74. Genome Announc. 2013;1(6):e00934–00913. doi: 10.1128/genomeA.00934-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rozahon M, Ismayil N, Hamood B, Erkin R, Abdurahman M, Mamtimin H, Abdukerim M, Lal R, Rahman E. Rhizobium populi sp. nov., an endophytic bacterium isolated from Populus euphratica. Int J Syst Evol Microbiol. 2014;64(9):3215–3221. doi: 10.1099/ijs.0.061416-0. [DOI] [PubMed] [Google Scholar]
- 11.Brown SD, Utturkar SM, Klingeman DM, Johnson CM, Martin SL, Land ML, Lu TY, Schadt CW, Doktycz MJ, Pelletier DA. Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoides. J Bacteriol. 2012;194:5991–5993. doi: 10.1128/JB.01243-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mora Y, Díaz R, Vargas-Lagunas C, Peralta H, Guerrero G, Aguilar A, Encarnación S, Girard L, Mora J. Nitrogen-fixing rhizobial strains isolated from common bean seeds: phylogeny, physiology, and genome analysis. App Environ Microbiol. 2014;80(18):5644–5654. doi: 10.1128/AEM.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martins dos Santos VAP, Heim S, Moore ERB, Strätz M, Timmis KN. Insight into the genomic basis of niche specificity of Pseudomonas putida KT2440. Environ Microbiol. 2004;6:1264–1286. doi: 10.1111/j.1462-2920.2004.00734.x. [DOI] [PubMed] [Google Scholar]
- 14.Peralta H. Regulatory elements of the transcription of reiterated nitrogenase operons. Cuernavaca: National University of Mexico; 2004. [Google Scholar]
- 15.Toledo I, Lloret L, Martínez-Romero E. Sinorhizobium americanus sp. nov., a new Sinorhizobium species nodulating native Acacia spp. in Mexico. Syst App Microbiol. 2003;26:54–64. doi: 10.1078/072320203322337317. [DOI] [PubMed] [Google Scholar]
- 16.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comp Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Stoeckert CJJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrero G, Peralta H, Aguilar A, Díaz R, Villalobos MA, Medrano-Soto A, Mora J. Evolutionary, structural and functional relationships revealed by comparative analysis of syntenic genes in Rhizobiales. BMC Evol Biol. 2005;5:55. doi: 10.1186/1471-2148-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucl Acids Res. 2015;43(D1):D261–D269. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106(45):19126–31. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Encarnación S, Guzmán Y, Dunn MF, Hernández M, del Carmen Vargas M, Mora J. Proteome analysis of aerobic and fermentative metabolism in Rhizobium etli CE3. Proteomics. 2003;3(6):1077–1085. doi: 10.1002/pmic.200300427. [DOI] [PubMed] [Google Scholar]
- 23.Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 24.Mavingui P, Flores M, Guo X, Dávila G, Perret X, Broughton WJ, Palacios R. Dynamics of genome architecture in Rhizobium sp. strain NGR234. J Bacteriol. 2002;184(1):171–176. doi: 10.1128/JB.184.1.171-176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hynes MF, McGregor NF. Two plasmids other than the nodulation plasmid are necessary for formation of nitrogen-fixing nodules by Rhizobium leguminosarum. Mol Microbiol. 1990;4:567–574. doi: 10.1111/j.1365-2958.1990.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 26.González V, Acosta JL, Santamaría RI, Bustos P, Fernández JL, Hernández González ILH, Díaz R, Flores M, Palacios R, Mora J, et al. Conserved symbiotic plasmid DNA sequences in the multireplicon pangenomic structure of Rhizobium etli. Appl Environ Microbiol. 2010;76(5):1604–1614. doi: 10.1128/AEM.02039-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, Hao B, Li J, Gu H, Peng J, Xie F, Zhao X, Frech C, Chen N, Ma B, et al. Whole-genome sequencing of Mesorhizobium huakuii 7653R provides molecular insights into host specificity and symbiosis island dynamics. BMC Genomics. 2014;15(1):440. doi: 10.1186/1471-2164-15-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iida T, Itakura M, Anda M, Sugawara M, Isawa T, Okubo T, Sato S, Chiba-Kakizaki K, Minamisawa K. Symbiosis island shuffling with abundant insertion sequences in the genomes of extra-slow-growing strains of soybean bradyrhizobia. Appl Environ Microbiol. 2015;81(12):4143–4154. doi: 10.1128/AEM.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YM, Li YJ, Chen WF, Wang ET, Tian CF, Li QQ, Zhang YZ, Sui XH, Chen WX. Soybean plants grown in the north China plain. Appl Environ Microbiol. 2011;77(18):6331–6342. doi: 10.1128/AEM.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valdivia HO, Reis-Cunha JL, Rodrigues-Luiz GF, Baptista RP, Baldeviano GC, Gerbasi RV, Dobson DE, Pratlong F, Bastien P, Lescano AG, et al. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genomics. 2015;16:715. doi: 10.1186/s12864-015-1928-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottlieb Y, Lalzar I, Klasson L. Distinctive genome reduction rates revealed by genomic analyses of two Coxiella-like endosymbionts in ticks. Genome Biol Evol. 2015;7(6):177. doi: 10.1093/gbe/evv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casadevall A. Evolution of intracellular pathogens. Annu Rev Microbiol. 2008;62:19–33. doi: 10.1146/annurev.micro.61.080706.093305. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Tana J, O’Brien EJ, Monk JM, Kim D, Li HJ, Charusanti P, Ebrahim A, Lloyd CJ, Yurkovich JT, et al. Systems biology definition of the core proteome of metabolism and expression is consistent with high-throughput data. Proc Natl Acad Sci U S A. 2015;112(34):10810–10815. doi: 10.1073/pnas.1501384112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barret M, Briand M, Bonneau S, Préveaux A, Valière S, Bouchez O, Hunault G, Simoneau P, Jacquesa M-A. Emergence shapes the structure of the seed microbiota. Appl Environ Microbiol. 2015;81(4):1257–1266. doi: 10.1128/AEM.03722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pueppke SG, Broughton WJ. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol Plant Micr Interact. 1999;12(4):293–318. doi: 10.1094/MPMI.1999.12.4.293. [DOI] [PubMed] [Google Scholar]
- 37.Del Cerro P, Rolla-Santos AAP, Gomes DF, Marks BB, Espuny MR, Rodríguez-Carvajal MA, Soria-Díaz MA, Nakatani AS, Hungria M, Ollero FJ, et al. Opening the “black box” of nodD3, nodD4 and nodD5 genes of Rhizobium tropici strain CIAT 899. BMC Genomics. 2015;16:864. doi: 10.1186/s12864-015-2033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The strains mentioned in this work are available. Send inquiries to Prof. Jaime Mora, Centro de Ciencias Genomicas, UNAM, Apdo. postal 565-A, Cuernavaca, Morelos, Mexico CP 62220 or contact at jmora@ccg.unam.mx. The datasets supporting the conclusions of this work are available at our web server ftp://kanan.ccg.unam.mx/PGFP/genomicstudies and at public repositories. The CFNEI73 genome was registered in the GenBank (http://www.ncbi.nlm.nih/GenBank) with the following accession numbers: CP013107 to CP013110 for chromosome, and plasmids a, b and c, respectively, and the CCGM7 genome with the accession numbers CP013051 to CP013054 for chromosome, and plasmids a, b and c, respectively. The mass spectrometry proteomics data are available at ProteomeXchange Consortium (http://www.proteomecentral.proteomexchange.org), with identifier PXD004702.