

Abstract
Protein methylation is believed to occur extensively in creanarchaea. Recently, aKMT, a highly conserved crenarchaeal protein lysine methyltransferase, was identified and shown to exhibit broad substrate specificity in vitro. Here, we have constructed an aKMT deletion mutant of the hyperthermophilic crenarchaeon Sulfolobus islandicus. The mutant was viable but showed a moderately slower growth rate than the parental strain under non-optimal growth conditions. Consistent with the moderate effect of the lack of aKMT on the growth of the cell, expression of a small number of genes, which encode putative functions in substrate transportation, energy metabolism, transcriptional regulation, stress response proteins, etc, was differentially regulated by more than twofold in the mutant strain, as compared with that in the parental strain. Analysis of the methylation of total cellular protein by mass spectrometry revealed that methylated proteins accounted for ∼2/3 (1,158/1,751) and ∼1/3 (591/1,757) of the identified proteins in the parental and the mutant strains, respectively, indicating that there is extensive protein methylation in S. islandicus and that aKMT is a major protein methyltransferase in this organism. No significant sequence preference was detected at the sites of methylation by aKMT. Methylated lysine residues, when visible in the structure, are all located on the surface of the proteins. The crystal structure of aKMT in complex with S-adenosyl-l-methionine (SAM) or S-adenosyl homocysteine (SAH) reveals that the protein consists of four α helices and seven β sheets, lacking a substrate recognition domain found in PrmA, a bacterial homolog of aKMT, in agreement with the broad substrate specificity of aKMT. Our results suggest that aKMT may serve a role in maintaining the methylation status of cellular proteins required for the efficient growth of the organism under certain non-optimal conditions.
Protein methylation occurs in all three domains of life (1–6). In Eukarya, methylation of lysine and arginine residues in histones is involved in the control of gene expression (7–11). Nonhistone proteins, such as cytochrome c, Rubisco, ribosomal proteins, transcriptional factors p53, TAF10 and AML1, are also targets for lysine methylation, but the roles of these modifications are less clear (12–19). In Bacteria, flagellar and ribosomal proteins are found to be methylated. The methylated proteins appear to be more stable to proteolysis than unmethylated ones, but the functions of the modification remain to be understood (20–25).
In Archaea, especially in thermophilic crenarchaea, an increasing number of proteins have been shown to undergo post-translational methylation. Early examples include ferredoxin from Sulfolobus acidocaldarius, and glutamate dehydrogenase, aspartate aminotransferase, β-glycosidase and ribosomal proteins from S. solfataricus (26–30). Methylation of lysine residues in S. solfataricus glutamate dehydrogenase and β-glycosidase appears to enhance thermal stability of the enzymes or reduce their susceptibility to denaturation and aggregation (26, 28). The crenarchaeal chromatin proteins Sul7d and Cren7 are also shown to be methylated, and the level of lysine methylation of Sul7d is increased during heat shock (31–33). More recently, Botting et al. identified 21 methyllysine residues from nine subunits of S. solfataricus RNA polymerase and 52 methyllysine residues in 30 different proteins from Thermoproteus tenax, suggesting that lysine methylation may occur more extensively in crenarchaea than previously thought (6). The widespread protein methylation in crenarchaea has been speculated to represent an adaption of these organisms to growth in hyperthermal environments (6).
For a long time, little was known about the enzymes responsible for protein lysine methylation in Archaea. In the first report on protein methyltransferases in Archaea, a Su(var), Enhancer of zeste, Trithorax (SET) domain protein capable of methylating a lysine residue in the chromatin protein MC1-α was identified in the euryarchaeon Methanosarcina mazei (34). The homologues of this methyltransferase are also found in several other methanogens. However, no SET domain proteins have been found in the sequenced genomes of crenarchaea where lysine methylation is prevalent. Recently, we identified the first crenarchaeal protein lysine methyltransferase, designated as aKMT, from S. islandicus (35). This protein resembles methyltransferases of the eukaryotic Dot1 family (36). Notably, aKMT is capable of methylating several tested recombinant Sulfolobus proteins overproduced in Escherichia coli, exhibiting broad substrate specificity in vitro (35).
To gain more insight into the function of aKMT, we have now constructed an aKMT deletion mutant of S. islandicus. The mutant strain is viable but exhibits moderate growth defect under certain conditions. Expression of a small number of genes is significantly altered as a result of the deletion of the aKMT gene. We show that most of the cellular proteins from S. islandicus are methylated, and aKMT is responsible for the methylation at the majority of the target lysine residues in the organism. We have also determined the crystal structures of aKMT- S-adenosyl-L-methionine (SAM) 1 and aKMT-S-adenosyl homocysteine (SAH) complexes, providing the structural basis for the broad substrate specificity of the protein. Our results suggest that extensive protein methylation is not required for the adaptation of S. islandicus to growth at high temperature and may serve to enhance the ability of the organism to achieve efficient growth under certain non-optimal conditions.
EXPERIMENTAL PROCEDURES
Strains and Growth Conditions
S. islandicus Rey15A (37) and S. islandicus E233S (38) were generous gifts from Professor Qunxin She at Copenhagen University. Sulfolobus strains were grown at 65 or 75 °C with shaking at 150 rpm in SCVy or CVY-rich medium supplemented with d-arabinose (2 g/L) and uracil (20 μg/ml), as indicated (38, 39).
Construction of an aKMT Deletion Mutant and a Complementary Strain
The aKMT gene (denoted kmtA, Tg-arm) and its upstream and downstream sequences (In-arm and Out-arm, respectively) were amplified by PCR from the genomic DNA of S. islandicus Rey15A (for primer sequences, see supplemental Table S1 in the Supplemental Materials). The Tg-arm, In-arm and Out-arm fragments were digested with SalI/MluI, XhoI/SphI and NcoI/XhoI, respectively, and inserted into plasmid pMID (40). The resulting vector (pMID-aKMT) was transformed into E. coli strain DH5α. After growth, the plasmid was extracted from the cells and the sequences of the inserts were verified by DNA sequencing through primer walking. Plasmid pMID-aKMT was then linearized and introduced into S. islandicus E233S by electroporation, as described previously (38). Transformed cells were plated onto SCVy medium solidified with 0.8% (w/v) Gelrite (Sigma, St. Louis, MO) and incubated at 75 °C. After 7–10 days, colonies were stained with 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-Gal,2 mg/ml). Blue colonies were picked and grown on SCVy medium containing 5′-fluorooritic acid (50 μg/ml). Colony purification was repeated twice.
A strain that complemented the deletion of the genomic copy of the aKMT gene was prepared by amplifying the kmtA sequence from the S. islandicus DNA by PCR (supplemental Table S1). The PCR product was cleaved with NdeI/SalI, and the resulting fragment inserted into plasmid pSeSD (41), yielding the aKMT expression plasmid pSeSD-aKMT capable of replicating in both E. coli and S. islandicus. After propagation in E. coli DH5α, pSeSD-aKMT was transformed into the aKMT mutant strain. The transformed cells containing pSeSD-aKMT were purified by isolating single colonies on SCVy plates. Colony purification was repeated twice.
Thermal Stability of Cellular Proteins
Parental and mutant S. islandicus cells were grown to the exponential growth phase in SCVy medium at 75 °C, harvested by centrifugation, resuspended to the same cell density in 50 mm sodium phosphate buffer, pH 7.0, and sonicated. The thermal stability of cellular proteins in the cell-free extract was determined by monitoring the change of the sample in absorbance at 600 nm with an increase in temperature from 50 to 95 °C at a rate of 0.2 °C/min on a Shimadzu UV-2550 spectrophotometer (26). The thermal stability of the cellular proteins was also measured by incubating the extract for indicated lengths of time at various temperatures. Aggregation of the proteins following heat treatment was monitored by 90° light scattering at 488 nm on a Shimadzu RF5301PC spectrofluorimeter.
Identification of Methylated Lysine Residues
A sample (∼300 μg) of total cellular proteins from the parental or the mutant S. islandicus strain grown to an OD600 of ∼1.0 in SCVy medium at 75 °C was subjected to 15% SDS-PAGE. The gel was stained with Commassie brilliant blue R-250, and destained with 20% (v/v) ethanol/10% (v/v) acetic acid, instead of methanol, to avoid chemical methylation of the proteins. The protein-containing portion of the gel was horizontally cut into slices, and each slice was then cut into small pieces. Proteins in gel pieces were subjected to in-gel digestion as described previously (42, 43). Briefly, the gel pieces were destained, treated with dithiothreitol and iodoacetamide, and vacuum-dried. The proteins in the gel pieces were digested with 10 μl of trypsin (12.5 ng/μl; Roche, Switzerland) in 25 mm ammonium bicarbonate, pH 8.0, at 37 °C for overnight. The peptides were extracted from the gel pieces and vacuum-dried. The peptides were first separated on an EASY-nLCII integrated nano-HPLC system (Proxeon, Odense, Denmark) and then analyzed on a LTQ-Orbitrap Velos mass spectrometer (Thermo, Waltham, MA). Mobile phase A was 0.1% (v/v) formic acid and mobile phase B consisted of 100% (v/v) acetonitrile and 0.1% (v/v) formic acid. Peptide separation was performed for 105 min on a home-made fused silica capillary C18 column (3 μm, 75 μm x 150 mm; Upchurch, Middleboro, MA) at a flow rate of 300 nL/min using the following gradients successively: 2 to 6% B in 10 min, 6% to 25% B in 65 min, 25% B to 45% B in 20 min, and 45% to 100% B in 10 min. MS and MS/MS data acquisition was performed using Xcalibur in the data-dependent acquisition mode. We searched the data with SEQUEST search engine in Proteome Discoverer 1.4 software against a S. islandicus Rey15A database downloaded from NCBI (44). Percolator was used to calculate the FDR of each peptide. The decoy database was constructed by reversing all protein sequences in the original database. Search parameters were set to allow 20 ppm for ms tolerance and 0.8 Da for ms/ms tolerance. Mono-, di-, and tri-methylations of lysine were set as variable modification on Lys, oxidation as variable modification on Met, and Cys carbamidomethylation as a fixed modification, two missed cleavages were allowed for trypsin digestion. The false discovery rate (FDR) of the peptide was set to 0.01. The searched results were filtered by high confidence, and at least one matched peptide was required for each identified protein. For the identified methylated peptides, a manual check of randomly selected methylated peptides was carried out to ensure a high quality of spectrum. The raw data was also processed by using Proteome Discoverer 2.1 with a ptmRS node, which provided a confidence measure of the assignment of lysine methylation in peptide sequences. All peptides containing a methylated lysine residue at the C terminus were manually checked. The “Best Site Probabilities” values were obtained for the assignment of methylation in these peptides. To estimate the abundance of the methylated proteins, the raw data was further searched with Mascot 2.5 to obtain emPAI values for the proteins (45). Raw spectra have been submitted to the PRIDE database (http://www.ebi.ac.uk/pride/) via the ProteomeXchange with the data set identifier PXD003424 (46).
Global Analysis of Protein Lysine Methylation
The genomic information of S. islandicus REY15A was downloaded from the Integrated Microbial Genomes (IMG, http://img.jgi.doe.gov) (47). Two databases, termed parent and ΔaKMT, were constructed with methylated proteins identified in the parental and the mutant S. islandicus strains, respectively. Excel plotting was used in arCOG analysis. The position of a peptide in a protein from which the peptide was derived was determined by BLAST. A 15-residue sequence window containing seven amino acid residues on each side of a methylated site was determined. For cases where methylated sites were near the C- or N terminus, “-” was used to complete the sequence window. The characteristics of the secondary structure of each 15-residue amino acid sequence were predicted by using Garnier from the EMBOSS software package. The frequency of residues flanking a methylated lysine residue was analyzed by Weblogo (weblogo 3.0).
Experimental Design and Statistical Rationale
To compare the patterns of protein methylation in the parental and the aKMT deletion mutant strains, a sample (∼10 μg) from each strain, grown to an OD600 of ∼1.0 in SCVy medium at 75 °C, was loaded onto an SDS-PAGE gel for in-gel trypsin digestion and MS analysis. The experiment was then repeated once with a larger amount of proteins (∼300 μg) from a separately grown culture of each strain under the same conditions. The quality of each data set was ensured by setting the false discovery rate (FDR) to 0.01. Since the majority of the proteins (97.5% and 96.5% for the parental and the mutant strains, respectively) identified in the smaller sample were also found in the larger one and the two data sets showed similar patterns of protein methylation, the larger data set was presented in the present work.
Immunoblotting
Cells from the parental, the mutant or the complementary strain grown exponentially at 75 °C were harvested and resuspended in a calculated volume of the sample buffer for SDS-PAGE to the same cell density. Equal aliquots of each sample were loaded, in parallel, onto two 15% SDS-polyacrylamide gels. After electrophoresis, one of the gels was stained with Commassie brilliant blue R-250. The other gel was processed for immunoblotting. Proteins were transferred electrophoretically to a PVDF membrane (Merck Millipore, Darmstadt, Germany). The membrane was incubated with the rabbit anti-mono/dimethyllysine antibodies (Jingjie PTM Biolab, Hangzhou, China), which recognized mono- and dimethylated lysine residues but not a trimethylated, acetylated or unmodified lysine residue in a protein. An anti-rabbit IgG-HRP conjugate (Promega) was used as the secondary antibody and was detected by the chemiluminescent method.
Recombinant Wild-type and Mutant aKMT Proteins
The expression vector for aKMT, denoted pET30a-MT, was constructed as described previously (35). Expression vectors for aKMT mutant proteins containing a single point mutation (i.e. H6A, V7A, P8A, Y9A, V10A, P11A, T12A, D34K, G36R, Y9F, Y9W, Y9L, E59A, N87A, F88A, F102A, L103A, L104A, T105A, N106A, V107A, N108A, and E109A) were constructed by introducing a corresponding mutation into the wild-type aKMT gene on pET30a-MT by using a Fast Mutagenesis System (TransGen Biotech, Beijing, China) (see supplemental Table S1 for primer sequences). All expression vectors were transformed into E. coli Rosetta (DE3), and the inserted sequences were verified by DNA sequencing. Overproduction and purification of the wild-type and the mutant aKMT proteins were carried out as described previously (35).
To prepare selenomethionine (Se-Met) aKMT, the above aKMT expression strain was grown in M9 medium containing 0.2% glucose, 1 mm MgSO4, and 100 μg/ml ampicillin at 37 °C until the OD600 of the culture reached ∼0.8. Se-Met was added to the culture to a final concentration of 50 μg/ml. Se-Met aKMT was purified as described for the wile-type aKMT protein. Protein concentrations were determined by the Lowry method (48). In order to obtain the aKMT-SAH complex, aKMT copurified with SAH was incubated in 6 m guanidine-HCl, followed by incubation with SAH under renaturation conditions (49).
Protein Crystallization, Data Collection, and Structure Determination
Crystallization screening was performed using the sitting drop vapor diffusion method in 96-well plates with commercial screening kits from Hampton Research (Aliso Viejo, CA), Molecular Dimensions (Suffolk, UK) and Emerald BioSystems (Bainbridge Island, WA). A sample (0.3 μl, 20 mg/ml) of a Se-Met aKMT protein stock solution was mixed with 0.3 μl of reservoir solution using a Mosquito robot (TTP Labtech, Melbourn, UK) and equilibrated against 40 μl of reservoir solution at 16 °C. Initial hits were performed by mixing 1 μl of the protein mixture with 1 μl of reservoir solution in hanging drops (10 mm magnesium chloride hexahydrate, 0.1 m HEPES-NaOH, pH 7.0, 15% (w/v) polyethylene glycol 3,350, 5 mm nickel chloride hexahydrate) at 16 °C. Crystallization conditions for aKMT-SAH (20 mg/ml) in space group of P2 was 10 mm nickel chloride, 0.1 m Tris-HCl, pH 8.5, 20% (w/v) PEG 2,000 MME. The crystals were mounted on a nylon loop and cooled immediately in liquid nitrogen without any antifreeze. All data sets were indexed, integrated, and scaled using the HKL2000 software package (50). The initial phase was determined using the X2DF structure determination pipeline (51, 52). The model was manually improved in Coot (53). The aKMT-SAH structure was solved by molecular replacement (54) using our determined Se-Met aKMT structure as the search model. Refinement was carried out using REFMAC (55) and PHENIX (56) alternately. The quality of the final model was validated with MolProbity (57).
Methyltransferase Assays
The standard methyltransferase assay mixture (50 μl) contained 20 mm HEPES-NaOH, pH 8.0, 2 mm MgCl2, 250 nm methyltansferase, 8 μm Cren7, 3.8 μm SAM (Sigma), and 200 nm S-(methyl-3H)adenosyl-l-methionine (10 Ci/mmol; Perkin-Elmer, Waltham, MA). The assays were performed as described previously (35).
RNA Preparation and cDNA Synthesis for RNA-seq
The parental and the mutant S. islandicus strains were grown at 75 °C with shaking in SCVy medium, and harvested at an OD600 of ∼0.5. Total RNAs were extracted from the cells by using the TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. A sample (10 μg) of total RNA was treated with DNaseI (5 U; Takara, Dalian, China) at 37 °C for 30 min, and purified by using the RNeasy MinElute Cleanup Kit (Qiagen, Hilden, Germany). DNA-free RNA samples were fragmented by heating at 95 °C. A cDNA library was constructed from 100 ng of total RNA by using the RNA-Seq Library Preparation Kit for Whole Transcriptome Discovery (Gnomegen). The quality of the library was determined by agarose gel electrophoresis, and by using an NanoPhotometer® spectrophotometer (IMPLEN, München, Germany) and an Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA). Cluster generation of cDNA (10 ng) was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit (Illumina, CA) according to the manufacturer's protocol, and the library was subjected to sequencing in both directions on an Illumina HiseqTM2500 sequencer.
Analysis of RNA-seq data
Raw data in a FASTQ format was first processed by using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads containing sequencing adapters or of low quality (> 5 Ns, where N represents any of the unidentified bases) were removed. The number of reads mapped to each transcript was counted by HTSeq v0.5.3 (http://www-huber.embl.de/users/anders/HTSeq). To calculate the difference between the parental strain and ΔaKMT in the expression of each gene, MARS (a MA-plot-based method with a Random sampling model) from the DEGseq program package was employed (58). The difference was considered to be significant when FDR was no greater than 0.001. For each COG category, enrichment of differentially expressed transcripts compared with the entire genomic set of genes was determined by using the hypergeometric distribution statistics. Enrichment analysis of the KEGG pathways was performed in the same manner.
Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA (500 ng) was reverse transcribed into cDNA using M-MLV Reverse Transcriptase (Promega, Fitchburg, WI) according to the manufacturer's instruction. Gene-specific primers were designed using Primer Premier 5.0 (supplemental Table S1). Quantitative PCR (qPCR) reaction mixtures contained 2× KAPA SYBR® FAST qPCR Master Mix Universal (10 μl; KAPA Biosystems, Wilmington, DE), 50-fold diluted cDNA (1 μl), and 200 nm primers in a final volume of 20 μl. qPCR reactions were conducted on a LightCycler 480 II PCR machine (Roche, Basel, Switzerland) according to the manufacturer's protocol. Relative mRNA expression was calculated using the comparative threshold cycle (Ct) method (59). The level of 16S rRNA was used as a reference to normalize the expression data for target genes.
RESULTS
S. islandicus Defective in aKMT is Viable but Shows a Moderately Altered Growth Phenotype
To gain insights into the physiological role of aKMT, we deleted the gene encoding the protein in S. islandicus by introducing a deletion construct (pMID-aKMT) into S. islandicus strain E233S, referred to as the parental strain. A successful double crossover event would lead to the generation of an aKMT deletion mutant strain, denoted ΔaKMT. The deletion of the aKMT gene in the mutant strain was verified by PCR, Southern hybridization and immunoblotting with anti-aKMT antibodies (supplemental Fig. S1). Surprisingly, the antibodies, which were generated in rabbit with recombinant aKMT as the antigen and affinity purified with recombinant aKMT, recognized a number of S. islandicus proteins, in addition to aKMT, in the parental strain as well as the complementary strain, which was constructed by introducing an aKMT expression plasmid (pSeSD-aKMT) into ΔaKMT (supplemental Fig. S1B). Since the cross reaction was identical in both the parental and the complementary strains and was substantially reduced in ΔaKMT, we speculate that proteins recognized by the antibody shared antigenic determinants, possibly, including a methylated lysine, in which the addition of the methyl group was catalyzed by aKMT. A similar observation on a glycoprotein from a rumen bacterium was reported previously (57). Our results indicate that aKMT is dispensable for the growth of the cell.
In order to examine the growth phenotype of the aKMT mutant, we grew the parental strain, the deletion mutant and the complementary strain at 75 °C, the optimal growth temperature for S. islandicus, and 65 °C in either CVY-rich or SCVy medium. The two media were similar except that the former contained 40-fold more yeast extract than the latter. The cells of the three strains grew more slowly in SCVy medium than in CVY-rich medium, but reached similar maximum cell densities in the two media. As shown in Fig. 1, ΔaKMT showed a slower growth rate than the parental strain in both media, and the difference was most significant when they were grown at 65 °C in SCVy medium. The growth phenotype was partially restored in the complementary strain. This appears to be consistent with the finding that intracellular level of aKMT in the complementary strain was lower than that in the parental strain (supplemental Fig. S1B). It is also possible that the His6 tag attached to the C terminus of aKMT synthesized in the complementary strain reduced the methyltransferase activity of the enzyme. Our data suggest that the function of aKMT is required more for the growth of the organism under certain restrictive conditions than for that under optimal conditions.
Fig. 1.

Growth curves of the parental, the mutant and the complementary strains. The three strains were grown at 65 °C (A and B) or 75 °C (C and D) in CVY-rich medium (A and C) or SCVy medium (B and D). The OD600 values of the cultures were measured. All numbers are an average of three independent measurements.
To test further if the parental and the mutant cells would differ in sensitivity to treatment at temperatures higher than that optimal for growth, we first grew both strains at 75 °C in SCVy medium to the mid-exponential phase and subsequently incubated the two strains for two hours at 87, 90, or 95 °C. The fractions of the cells that survived the heat treatment were determined by plating. The parental and the mutant strains appeared to survive heat treatment similarly well at 87 and 90 °C with the survival rates of ∼100 and ∼90%, respectively (data not shown). However, both strains were unable to survive incubation for two hours at 95 °C. Therefore, the deletion of the aKMT gene does not appear to affect the thermal tolerance of the organism.
aKMT is Responsible for Methylation of the Bulk of Proteins in the Cell
In our previous study, we found that aKMT was capable of methylating in vitro several S. solfataricus proteins overproduced in E. coli (35). It was shown earlier that S. solfataricus RNA polymerase was highly methylated at lysine residues, suggesting that extensive protein methylation may occur in the organism (6). These observations prompted the suggestion that aKMT may play a major role in protein methylation in the cell. The availability of the aKMT deletion mutant permitted an analysis of the extent and the pattern of protein methylation catalyzed by the enzyme in vivo. In our preliminary experiments, anti-mono/dimethylated lysine antibodies (Jingjie PTM Biolab, China), which recognize proteins with mono- and dimethylated lysine residues, were employed to detect methylated proteins in the parental, the mutant and the complementary strains by immunoblotting. As shown in Fig. 2, the number of proteins recognized by the antibodies in the parental and the complementary strains far exceeded that in ΔaKMT, supporting the notion that aKMT is a major protein lysine methyltransferase in S. islandicus.
Fig. 2.

Detection of lysine methylation in total cellular proteins from the parental, the mutant and the complementary strains. Samples containing the same numbers of cells from each strain were loaded in parallel onto two 15% SDS-polyacrylamide gels. After electrophoresis, the two gels were subjected to staining with Commassie brilliant blue R-250 (A) and immunoblotting with anti-mono/dimethyllysine antibodies (B), respectively.
To learn more about protein methylation catalyzed by aKMT in vivo, we set out to compare the methylated proteins in the parental and the mutant strains. In a control experiment, the pattern of methylated proteins in the parental strain, as revealed by immunoblotting with anti-mono/dimethylated lysine antibodies, remained largely unchanged during the entire growth phase of the organism (supplemental Fig. S2). This finding agrees with the observation that the intracellular level of aKMT, as determined by immunoblotting using anti-aKMT antibodies, was nearly constant throughout the growth phase (data not shown; (35, 36)) as well as at various temperatures within a tested range. Therefore, we subjected samples containing the same number of either the parental or the mutant cells (OD600 = ∼1.0) to electrophoresis on a SDS-PAGE gel, followed by in-gel trypsin digestion and mass spectrometry. This experiment was repeated once. Similar observations were made in both experiments. The results of one of the experiments are shown in this report.
Among a total of 2535 proteins predicted to be encoded by the S. islandicus genome, 1751 and 1757 proteins were identified in the parental and the mutant strains, respectively, by mass spectrometry (Table I and supplemental Table S2). 1643 proteins were found in both strains. 1158 proteins (66.1% of the identified proteins) were found to be methylated in the parental strain, whereas 591 proteins (33.6%) were methylated in ΔaKMT. These results confirm the suggestion that there is extensive protein methylation in Sulfolobus (6). The observation that 591 methylated proteins remained in the mutant strain but they were not readily detected by anti-aKMT antibodies raised the possibility that these proteins were of low abundance. So we estimated the abundance of methylated proteins in the parental and the mutant strains by quantitative spectral counts (supplemental Table S2B). Our results suggest that the signal intensity on the immunoblot did not correlate with the amount of the proteins, and might primarily depend on the levels of methylation of the proteins. Given that the vast majority of the proteins identified in the parental and the mutant strains were the same, it may be inferred that the profile of the synthesis of cellular proteins was not drastically affected by protein methylation. Further analysis showed that 497 proteins were methylated in both strains, whereas 661 proteins were methylated only in the parental strain. In addition, 94 proteins were methylated only in ΔaKMT (supplemental Table S3), and 66 of these proteins were detected in an unmethylated form in the parental strain, suggesting that the deletion of the aKMT gene altered the pattern of protein methylation by the remaining methyltransferase(s) in the cell.
Table I. Number of methylated proteins and lysine residues as identified by mass spectrometry.
| Parent | ΔaKMT | Both strains | Parent onlya | ΔaKMT onlyb | |
|---|---|---|---|---|---|
| Total identified proteins | 1751 | 1757 | 1643 | 107 | 123 |
| Methylated proteins | 1158 | 591 | 497 | 661 | 94 |
| Total identified lysine residues | 17,784 | 16,941 | 14,347 | 3437 | 2594 |
| Monomethylated residues | 3013 | 274 | 113 | 2900 | 161 |
| Dimethylated residues | 746 | 566 | 210 | 536 | 356 |
| Trimethylated residues | 541 | 351 | 155 | 386 | 196 |
a Proteins and lysine residues were methylated in the parental strain and not in ΔaKMT.
b Proteins and lysine residues were methylated in ΔaKMT and not in the parental strain.
Since aKMT was initially identified in a search for enzyme(s) responsible for the methylation of Cren7 (Sire_1111), an abundant chromatin protein isolated from S. islandicus (35), we were interested in comparing the methylation state of Cren7 from the parental strain with that from the mutant strain. As reported previously, Cren7 from the parental strain was methylated at multiple lysine residues (32, 35). Monomethylated Lys11 and Lys16, dimethylatred Lys24 and Lys42, and trimethylated Lys16 were detected. By comparison, Cren7 from ΔaKMT appeared to be unmethylated, suggesting that the protein was methylated by aKMT in vivo.
A survey of methylated lysine residues revealed a far more drastic difference than that of methylated proteins between the parental and the mutant strains. 3718 and 1074 methylated residues were identified in the parental strain and ΔaKMT, respectively (Table I). Furthermore, there were 3003 monomethylated, 745 dimethylated, and 540 trimethylated lysine residues in the parental strain, and 274 monomethylated, 566 dimethylated, and 351 trimethylated lysine residues in ΔaKMT. Since there were over 10-fold more monomethylated residues but only slightly more dimethylated or trimethylated residues in the parental strain than those in ΔaKMT, aKMT is probably responsible primarily for the monomethylation of lysine residues. In addition, most of the proteins were methylated at multiple sites in the parental strain. The thermosome (SiRe_1214) was the most highly methylated polypeptide detected with 24 methylated lysine residues (Fig. 3). In comparison, most methylated proteins contained only one lysine residue in ΔaKMT. Based on these data, we conclude that aKMT is a major protein methyltransferase in S. islandicus.
Fig. 3.

Representative MS/MS spectra of methylated peptides derived from trypsin-digested thermosome (SiRe_1214). The amino acid sequences of the peptides are shown with methylated lysine residues labeled. Mono, monomethylation, di, dimethylation.
To determine if a lysine residue could be methylated to various extents, we investigated the methylation state of each lysine residue in the methylated proteins. The proteins identified in the parental strain and ΔaKMT contained 17,784 and 16,941 lysine residues, respectively. Approximately 20.9% (3, 718) of the identified lysine residues in the parental strain and 6.3% (1, 074) of those in ΔaKMT were methylated (Table I). Only 28.7% (1, 067) and 20.5% (220) of the methylated residues were detected in a single methylation state in the parental strain and ΔaKMT, respectively (Fig. 4). The majority of the methylated residues were found in more than one methylation forms. For example, 40 residues in the prental strain and 12 residues in ΔaKMT existed in all of the following four forms, i.e. monomethylated, dimethylated, trimethylated, and unmethylated forms. Differential methylation of the lysine residues may play a regulatory role in the cell, as in the case of eukaryotic histone methylation (60). However, it is worth noting that S. islandicus cells used in this study were not synchronized and, therefore, different methylation states for a given lysine residue may be related to cells in different phases of the cell cycle.
Fig. 4.

Schematic representation of lysine residues that were methylated to various extents. The extents of methylation on lysine residues, as identified by mass spectrometry in parent (A) or ΔaKMT (B), are depicted in a Venn diagram. Unmethylated, monomethylated, dimethylated or trimethylated lysine residues are indicated by none, methyl, dimethyl, or trimethyl, respectively.
To investigate if protein lysine methylation by aKMT shows sequence or structural specificity, amino acid sequences flanking the sites of methylation were analyzed (Fig. 5A). The secondary structures of the amino acid sequences were also predicted (Fig. 5B). No apparent bias in the flanking sequences was detected for the sites of methylation. The lack of sequence specificity is consistent with the ability of aKMT to catalyze methylation of a large number of proteins, often at multiple sites, in the cell. On the other hand, lysine methylation occurred preferentially in the helix regions or regions immediately adjacent to a helix on the C-terminal side in substrate proteins. To shed light on the location of methylated lysine residues in the three-dimensional structure of a protein, we conducted a survey on several S. solfataricus chromatin and replication proteins with a known or partially known structure, i.e. PCNA, Lig1, MCM, PriS/PriL, RFC, Cren7, and Sul7. These proteins are highly similar to their S. islandicus homologues whose structures are not yet available. Our data showed that these proteins were all methylated at multiple sites in the parental strains but were not methylated in ΔaKMT. We found that methylated residues, when visible in the structure, were all located on the surface of these proteins and were thus solvent exposed, as exemplified by Lig1, a monomeric protein, or PCNA, a trimeric protein complex (supplemental Fig. S9). However, not all surface-located lysine residues were detectably methylated. Therefore, our results indicate that lysine residues on the surface of a protein were targeted by aKMT for methylation using an unknown recognition mechanism in vivo.
Fig. 5.
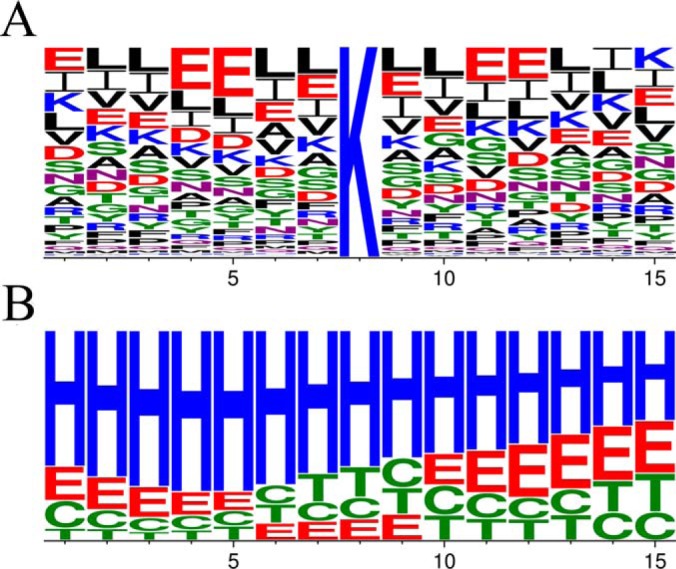
Sequence analysis of sites of methylation by aKMT. A, Primary structure. Different amino acid residues are shown in different colors. B, Predicted secondary structure. The secondary structures of the amino acid sequences containing the sites of methylation were predicted. Helix, sheet, turn and coil are represented by H, E, T and C, respectively. The frequencies of amino acid residues or secondary structure elements are generated by Weblogo.
We also performed an arCOG analysis on proteins, which were methylated in the parental strain but not in ΔaKMT and, therefore, probably methylated by aKMT (supplemental Table S4). These proteins were widespread without apparent bias in various functional categories (22 out of the 26 arCOG categories), suggesting that protein methylation catalyzed by aKMT is not restricted to specific cellular functions.
The Structural Basis of the Broad Substrate Specificity of aKMT
In order to understand the substrate recognition and the catalytic mechanism of aKMT, we sought to determine the crystal structure of the protein. Se-Met aKMT alone, aKMT alone and aKMT with added SAM were used in crystallization trials. Crystals were obtained for all three samples under the same crystallization conditions, and these structures were similar (supplemental Table S5). In addition, all three structures contained a SAM molecule in each protomer, indicating that an endogenous SAM molecule was bound by recombinant aKMT overproduced in E. coli.
As predicted from the sequence alignment, aKMT contains a conserved methyltransferase domain of the IPR025714-type but lacks the substrate recognition domain, as compared with the homologous L11 methyltransferase PrmA from the hyperthermophilic bacterium Thermus thermophilus (supplemental Fig. S3, (35)). The complex consists of four α helices (α1-α4) and a central seven-stranded β sheet (β1-β7), a structure typical of a number of SAM-dependent methyltransferases (61). In the β sheet, β1 through β6 strands are parallel to one another, whereas β7 is antiparallel to the others and inserted into the sheet between β5 and β6 (supplemental Fig. S4). A SAM molecule is bound in the SAM binding pocket within the aKMT molecule. The SAM binding pocket is negatively charged but contains a hydrophobic adenine binding region. The pocket has a wide opening and narrows down toward the center of the protein. The methionyl moiety of SAM inserts into the pocket, with the adenine group pointing outside of the pocket (Figs. 6A and 6B). Amino acid residues interacting with SAM are shown in Fig. 6C. Six of them, i.e. Thr12, Asp34, Gly36, Glu59, Asn87, and Phe88, interact with SAM via hydrogen bonding, and the remaining ten residues form hydrophobic interactions with SAM (Fig. 6C). Because of their structural roles, many of the residues (e.g. Asp34, Gly36, Glu59, Thr12, and Asn87) are conserved (35). The importance of these residues to the activity of aKMT is further verified by a site-directed mutagenesis experiment in which a single alanine substitution for Thr12, Asp34, Gly36, Glu59, or Asn87 resulted in over 90% loss of the methyltransferase activity of the protein (supplemental Fig. S5).
Fig. 6.
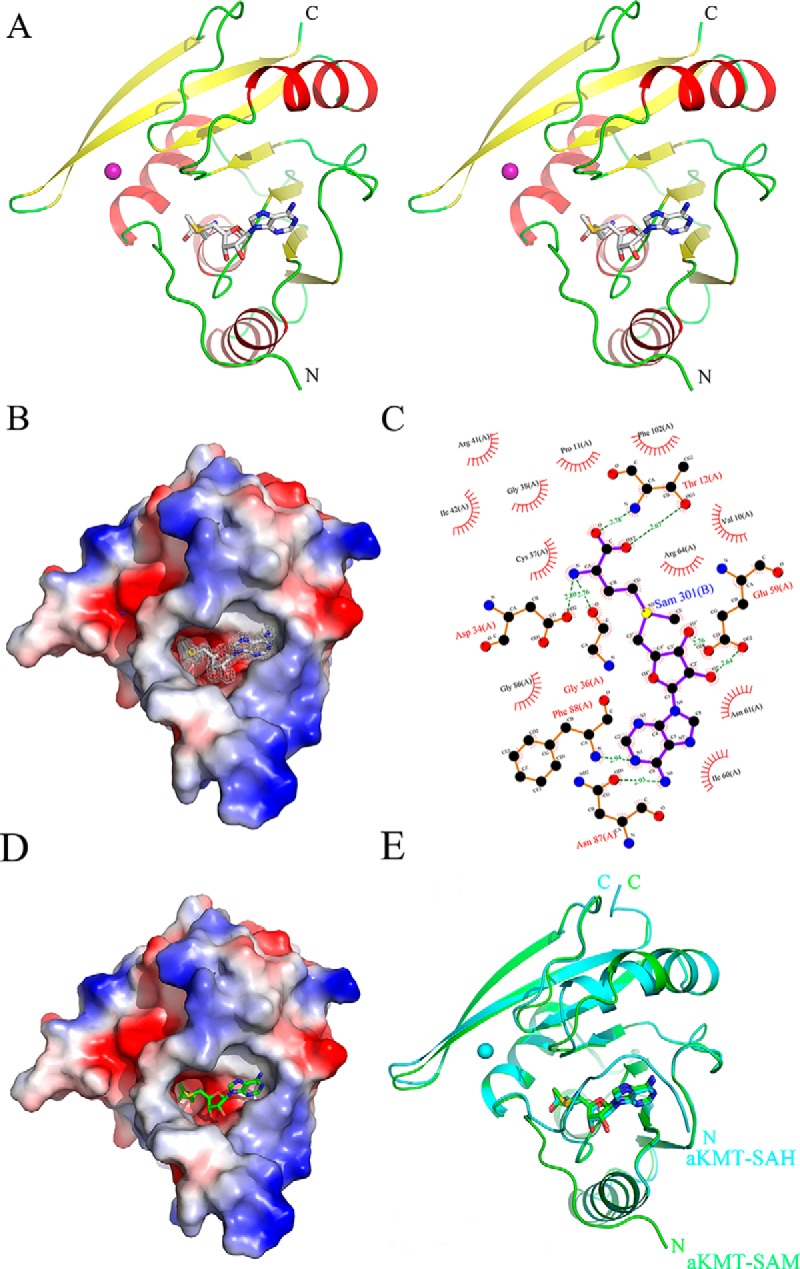
The crystal structures of aKMT-SAM and aKMT-SAH. A, Stereo view of the the aKMT-SAM structure. The structure is shown as a ribbon diagram with the α helices and the β sheets colored in red and yellow, respectively. The SAM molecule is shown as gray sticks. The purple sphere represents a magnesium ion. The N- and C termini are labeled with the respective letters. B, The solvent-accessible surface of aKMT in complex with SAM, colored according to electrostatic potential. Blue, positively charged; red, negatively charged; white, neutral. Electron density of a 2Fo-Fc simulated annealing (SA) omit map for SAM bound in the catalytic pocket contoured at 1.0σ is shown. The SAM molecule is shown as gray sticks. C, Schematic diagram summarizing the interactions between aKMT and SAM in the aKMT-SAM structure generated by LIGPLOT (67). Interacting atoms are connected by green dashed lines with bonding lengths indicated (in Å). Nonligand residues involved in direct hydrophobic contacts with SAM are shown as red semicircles with radiating spokes. D, The solvent-accessible surface of aKMT in complex with SAH, colored according to electrostatic potential. Blue, positively charged; red, negatively charged; white, neutral. The SAH molecule is shown as green sticks. E, Comparison of the structures of aKMT-SAH (cylan) and aKMT-SAM (green).
Because aKMT appears to be responsible primarily for the monomethylation, instead of di- or trimethylation of the proteins, we were interested in looking into the cycle of methyl transfer by the enzyme. We solved the crystal structure of aKMT containing the reaction product S-adenosyl homocysteine (SAH) in space group of P2 at 1.84 Å (Figs. 6D and 6E). The structures of aKMT-SAH and aKMT-SAM were highly superimposable with a root-mean-square deviation (RMSD) of 0.432Å, and identical residues were found to be interacting with both SAM and SAH. However, the N-terminal portion (1–8 residues) of the protein poses in very different positions in the two structures. In the aKMT-SAM complex, the N terminus of the protein extends outwards, creating a large opening for the pocket. Therefore, a substrate lysine residue may be readily inserted into the pocket and form a close contact with the methyl group of the SAM molecule. In comparison, the N-terminal seven residues (Ser2-Pro8) in the aKMT-SAH complex covers the opening of the pocket as the result of a 90°-turn of the main chain between Pro8 and Tyr9, drastically reducing the size of the entrance of the pocket. The SAH molecule is thus totally buried in the pocket of aKMT. It appears that aKMT may exist in two conformations, i.e. the open conformation (aKMT-SAM) and the closed conformation (aKMT-SAH). It is speculated that the N terminus serves as a switch between the two conformations in regulating the reaction process of the enzyme. Given its mode of action, along with its lack of the substrate recognition domain and, thus, its presumably low binding affinity for substrates, aKMT may catalyze methyl transfer in a distributive fashion.
Cellular Proteins from the Parental and the aKMT Deletion Mutant Strains Show Similar Thermal Stability
Methylation has been suggested to increase the stability of proteins from Eukarya and Archaea (12, 26, 62, 63). Because proteins are extensively methylated in S. islandicus, and aKMT is responsible for much of the protein lysine methylation in this organism, we were interested in learning if cellular proteins from the mutant differed from those from the parental strain in thermal stability. We first followed the change in optical density at 600 nm of cell-free extracts from the parental strain and ΔaKMT grown in the exponential growth phase with increasing temperature by using a spectrometric assay (26). As shown in Fig. 7A, cellular proteins from both the parental strain and ΔaKMT started to denature at ∼85 °C. We then compared the thermal stability of the cellular proteins from the parental strain with that from the mutant using a light scattering-based protein aggregation assay. Proteins from the two strains were similar in sensitivity to thermal denaturation (Fig. 7B). Taken together, these results reveal no significant differences in the thermal stability between cellular proteins from the parental strain and those from ΔaKMT.
Fig. 7.
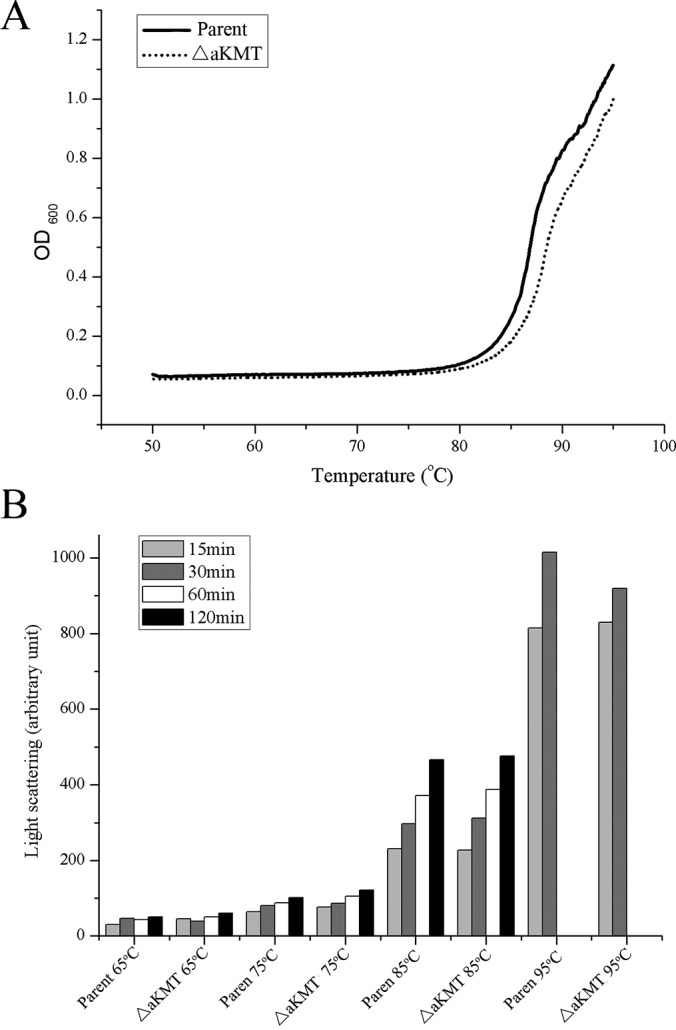
Thermal stability of total cellular proteins from the parental and the mutant strains. Cells from an exponentially grown culture of the parental strain or ΔaKMT were harvested, resuspended in 50 mm sodium phosphate buffer, pH 7.0, and sonicated. A, The clarified cell-free extracts were heated from 50 to 95 °C at a rate of 0.2 °C/min, and the increase in absorbance at 600 nm was recorded on a Shimadzu UV-2550 spectrophotometer. B, The cell-free extracts were incubated at 65, 75, 85, or 95 °C for 15, 30, 60, and 120 min. Protein aggregation was monitored by 90° light scattering at 488 nm on a Shimadzu RF5301PC spectrofluorimeter.
Transcriptomic Analysis of the Parental Strain and the aKMT Deletion Mutant
To elucidate the influence of the deletion of the aKMT gene on gene expression in S. islandicus, the transcriptomes of the parental and the mutant strains were compared. Both the parental strain and ΔaKMT were cultured in SCVy medium and harvested during the exponential growth phase, conditions where the difference between the two strains in growth was minimal. RNA-seq was employed to determine the levels of gene expression in the parental strain and ΔaKMT. 10,040,707 and 10,500,000 sequencing reads were obtained from the parental and the mutant samples, respectively (supplemental Table S6). Each transcript was covered by an average of 70 reads, and the transcripts of about 89% of the genes had a coverage of 90–100%. Reads mapped to open reading frames (ORFs), rRNAs and intergenic regions in the S. islandicus genome accounted for 14.86, 82.34, and 0.22% of the total reads, respectively (supplemental Fig. S7). Using a cutoff of a twofold difference in gene expression, the transcription levels of 43 or 42 genes increased or decreased, respectively, in ΔaKMT, as compared with those in the parental strain (Fig. 8, Tables II and supplemental Table S7). The number of these genes accounted for ∼3.3% of the total number of genes (∼2600) detected in this study. The expression of 4 and 8 genes was up- and down-regulated by more than fourfold. The changes in gene expression revealed by RNA-seq were confirmed by qRT-PCR performed on ten selected genes (supplemental Fig. S8).
Fig. 8.

A bar plot showing genes differentially expressed in the parental and the mutant strains. Transcriptional profiles for the parental strain and ΔaKMT were determined and compared. Genes whose expression differed by more than twofold (FDR < 0.001) in the two strains, are shown. Red and green bars indicate down- and up-regulated genes, respectively. Putative functions of proteins discussed in the text are indicated.
Table II. Genes differentially expressed in the parental strain and ΔaKMT.
| ORF | Description | log2(Fold_change) normalized | q-value |
|---|---|---|---|
| SiRe_0121 | flagellar protein F | 1.123885005 | 2.63E-20 |
| SiRe_0123 | hypothetical protein | 1.190823554 | 1.23E-80 |
| SiRe_0124 | flagellin | 1.167864412 | 0 |
| SiRe_0125 | hypothetical protein | 1.019799216 | 0.0001001 |
| SiRe_0128 | hypothetical protein | 1.041792647 | 4.57E-22 |
| SiRe_0130 | hypothetical protein | 2.535720072 | 0.0005146 |
| SiRe_0131 | CopG family transcriptional regulator | 1.331361573 | 1.43E-08 |
| SiRe_0159 | von Willebrand factor type A domain-containing protein | 1.0700565 | 4.68E-133 |
| SiRe_0227 | hypothetical protein | 1.151290559 | 1.16E-06 |
| SiRe_0301 | TetR family transcriptional regulator | 1.279706094 | 3.04E-06 |
| SiRe_0318 | carbon monoxide dehydrogenase subunit G | 1.079081668 | 0.0001399 |
| SiRe_0374 | VapB-type antitoxin | 1.353881749 | 1.98E-12 |
| SiRe_0379 | acetoacetate decarboxylase | 1.580807961 | 2.84E-10 |
| SiRe_0453 | ferritin Dps family protein | 1.229184027 | 5.32E-13 |
| SiRe_0552 | 5-oxoprolinase | 1.060539745 | 6.41E-135 |
| SiRe_0706 | 4-hydroxyphenylacetate 3-hydroxylase | 2.362183817 | 1.77E-67 |
| SiRe_0707 | 3,4-dihydroxyphenylacetate 2,3-dioxygenase | 2.960472726 | 2.21E-48 |
| SiRe_0975 | heterodisulfide reductase, subunit C (HdrC-1) | 1.363197959 | 3.93E-88 |
| SiRe_0978 | DsrE family protein | 1.274522325 | 1.73E-42 |
| SiRe_0981 | glutaredoxin-like protein | 1.254307137 | 8.16E-11 |
| SiRe_1012 | tRNA (guanine-N1-)-methyltransferase | 1.16400161 | 9.05E-13 |
| SiRe_1034 | hypothetical protein | 1.850761781 | 4.42E-39 |
| SiRe_1360 | radical SAM protein | 1.095492664 | 4.58E-19 |
| SiRe_1498 | hypothetical protein | 1.237638719 | 0.0007216 |
| SiRe_1528 | hypothetical protein | 1.098511192 | 1.75E-05 |
| SiRe_1536 | hypothetical protein | 1.046759391 | 9.19E-10 |
| SiRe_1549 | hypothetical protein | 1.193871283 | 1.28E-18 |
| SiRe_1752 | FAD dependent oxidoreductase | 1.487545478 | 6.03E-120 |
| SiRe_1753 | Adenylosuccinate synthase | 1.23141556 | 2.38E-76 |
| SiRe_2008 | peptidase M48 Ste24p | 1.021146899 | 0.00068 |
| SiRe_2014 | putative signal-transduction protein with CBS domains | 1.056110571 | 2.53E-06 |
| SiRe_2050 | Helix-turn-helix type 11 domain-containing protein | 1.045084954 | 0.000787 |
| SiRe_2095 | hypothetical protein | 1.618775812 | 1.39E-11 |
| SiRe_2149 | hypothetical protein | 1.071051805 | 8.91E-05 |
| SiRe_2250 | hypothetical protein | 1.332628207 | 1.66E-10 |
| SiRe_2380 | pyruvate flavodoxin/ferredoxin oxidoreductase domain-containing protein | 1.135873742 | 4.27E-161 |
| SiRe_2381 | pyruvate ferredoxin/flavodoxin oxidoreductase subunit delta | 1.018450272 | 2.31E-40 |
| SiRe_2444 | hypothetical protein | 1.304394526 | 2.50E-08 |
| SiRe_2462 | formate dehydrogenase family accessory protein FdhD | 1.313327651 | 1.04E-13 |
| SiRe_2463 | hypothetical protein | 1.542033846 | 6.32E-17 |
| SiRe_2464 | formate dehydrogenase subunit alpha | 1.170590661 | 3.55E-132 |
| SiRe_2603 | hypothetical protein | 3.013767369 | 1.06E-19 |
| SiRe_2663 | hypothetical protein | 1.78083257 | 2.62E-06 |
| SiRe_0072 | 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase | -1.193632338 | 5.58E-07 |
| SiRe_0427 | Rossmann fold nucleotide-binding protein | -1.132658437 | 8.34E-05 |
| SiRe_0429 | SAF domain-containing protein | -2.486647741 | 1.16E-09 |
| SiRe_0430 | d-galactarate dehydratase/altronate hydrolase domain-containing protein | -1.758463032 | 6.46E-19 |
| SiRe_0431 | mandelate racemase/muconate lactonizing family protein | -2.119631757 | 3.00E-10 |
| SiRe_0435 | citryl-CoA lyase | -1.17342184 | 4.37E-10 |
| SiRe_0471 | 6-phosphogluconate dehydrogenase | -1.006681967 | 5.17E-07 |
| SiRe_0546 | hypothetical protein | -1.021008748 | 1.83E-10 |
| SiRe_0587 | hypothetical protein | -2.703246574 | 6.28E-33 |
| SiRe_0588 | ABC transporter ATP-binding protein | -2.309478201 | 1.20E-26 |
| SiRe_0590 | beta-lactamase domain-containing protein | -1.089506299 | 2.04E-05 |
| SiRe_0591 | helicase domain-containing protein | -1.212281045 | 1.52E-32 |
| SiRe_0592 | hypothetical protein | -1.224684435 | 1.74E-07 |
| SiRe_0593 | ATPase (AAA+ superfamily)-like protein | -1.288354047 | 1.91E-70 |
| SiRe_0594 | hypothetical protein | -1.333846917 | 1.46E-87 |
| SiRe_0621 | cupin | -1.607237882 | 7.15E-05 |
| SiRe_0696 | hypothetical protein | -3.868002114 | 7.63E-27 |
| SiRe_0788 | hypothetical protein | -1.037169596 | 1.90E-05 |
| SiRe_0789 | amidohydrolase | -1.069394687 | 1.25E-33 |
| SiRe_0920 | hypothetical protein | -1.224949783 | 4.71E-20 |
| SiRe_0965 | Peptidase S53 propeptide | -1.360928471 | 0 |
| SiRe_1415 | histidinol-phosphate aminotransferase | -1.111683004 | 1.04E-24 |
| SiRe_1427 | magnesium-dependent phosphatase-1 | -1.408323522 | 0.0001571 |
| SiRe_1449 | methyltransferase | -7.879599176 | 3.86E-244 |
| SiRe_1450 | hypothetical protein | -2.200081589 | 3.77E-99 |
| SiRe_1598 | riboflavin synthase | -1.036303373 | 3.97E-05 |
| SiRe_1601 | amidohydrolase | -1.161717158 | 0.0001507 |
| SiRe_1708 | general substrate transporter | -1.110642973 | 5.20E-24 |
| SiRe_1905 | ribosomal protein L7Ae | -1.187504966 | 1.05E-75 |
| SiRe_1986 | hypothetical protein | -1.351565966 | 7.17E-25 |
| SiRe_2123 | raffinose synthase | -1.145744597 | 2.03E-73 |
| SiRe_2283 | ABC-type dipeptide/oligopeptide/nickel transport system, permease, DppC | -1.102353765 | 1.41E-11 |
| SiRe_2290 | hypothetical protein | -3.049242429 | 3.52E-06 |
| SiRe_2291 | radical SAM domain-containing protein | -1.712207442 | 0.0005369 |
| SiRe_2422 | 4Fe-4S ferredoxin | -1.617526188 | 3.26E-14 |
| SiRe_2423 | hypothetical protein | -1.488664087 | 3.70E-13 |
| SiRe_2424 | 4Fe-4S ferredoxin | -1.561354364 | 3.15E-38 |
| SiRe_2425 | molybdopterin oxidoreductase | -1.292619875 | 1.15E-172 |
| SiRe_2427 | polysulfide reductase NrfD | -1.053043999 | 1.75E-31 |
| SiRe_2482 | major facilitator superfamily protein | -1.339227631 | 1.52E-20 |
| SiRe_2537 | cellobiose binding protein | -1.713714636 | 0 |
| SiRe_2586 | family 5 extracellular solute-binding protein | -1.009435478 | 0 |
The down-regulated genes included those that were involved in membrane transportation, energy production and conversion (Tables II and supplemental Table S7). Expression of genes encoding the ATP-binding protein of an ABC transporter (SiRe_0588), a protein of the major facilitator superfamily (SiRe_2482), a general substrate transporter (SiRe_1708) and an ABC-type oligopeptide/nickel transport system (SiRe_2283) decreased by 5.0, 2.5, 2.2, and 2.1 fold, respectively, in the mutant strain, as compared with that in the parental strain. In addition, several genes encoding proteins with putative functions in redox reaction and electron transfer, e.g. 4Fe-4S ferredoxin (SiRe_2422, SiRe_2424), and molybdopterin oxidoreductase (SiRe_2425), and polysulfide reductase NrfD (SiRe_2427), were down-regulated in ΔaKMT. On the other hand, genes encoding archaellum proteins FlaB and FlaF were up-regulated in ΔaKMT (Table II). But, the mutant did not show an altered motility (data not shown). Also among the up-regulated genes in the mutant strain are those which encode transcriptional factors, e.g. CopG-like regulator (SiRe_0131), TetR protein (SiRe_0301), and stress response proteins, e.g. Dps family protein (SiRe_0453), VapB-type protein (SiRe_0374). Notably, genes encoding two enzymes involved in aromatic catabolism (SiRe_0706 and SiRe_0707) were up-regulated by 5–7 fold in ΔaKMT. In general, however, the expression of the vast majority of the genes was not significantly affected by aKMT-catalyzed protein methylation in S. islandicus.
DISCUSSION
Although it has been suggested that proteins are extensively methylated in crenarchaea (6), the present study shows for the first time the extent and pattern of protein lysine methylation in an archaeal proteome. Strikingly, methylated proteins accounted for 66% of the cellular proteins, and the methyllysines were about 21% of the lysine residues identified in S. islandicus. To the best of our knowledge, this organism harbors the most extensively methylated proteome that has ever been reported. The availability of a S. islandicus mutant defective in aKMT permits an analysis of potential roles of protein methylation in general, and aKMT in particular, in the cell. The proportions of the methylated proteins and lysine residues over the total proteins and lysine residues identified in the mutant were reduced to 33.4 and 6.3%, respectively. These results show that aKMT is a major protein methyltransferase responsible for the methylation of the majority of methylated cellular proteins in S. islandicus. The presence of the remaining methylated proteins in the mutant strain indicates that one or more additional protein methyltransferase(s) must exist. The unknown protein methyltransferase(s) appear to differ from aKMT in target site selection but the former also shares sites with the latter, as revealed by the comparison of patterns of protein methylation in the parental and the mutant strains. No genes encoding other protein lysine methyltransferases have been identified by genome sequence analysis so far. Notably, most of the methylated residues (2,651/3,718) were methylated to various extents from an unmethylated to a trimethylated form in the parental strain. This may indicate the presence of either a dynamic balance between methylation and demethylation or a continuing methylation process with trimethylation as the end point at the target residues. Protein demethylases required for the former process have yet to be identified but are likely present in Archaea. In any case, the presence of multiple methylation states in proteins may serve a regulatory role that remains to be understood.
No apparent sequence preference by aKMT was detected through the analysis of sequences flanking methylated lysine residues, in agreement with the previous finding that the enzyme exhibits broad substrate specificity (35, 36). Given the observation that ∼21% of the total lysine residues in cellular proteins were methylated and the report that lysine residues undergoing methylation were often found on the surface of a protein (6), most, if not all, lysine residues accessible to methyltransferases are presumably potential targets of posttranslational modification by protein methyltransferases in the cell. This is consistent with the observation that methylated lysine residues are loacated on the surface of the selected proteins. The structural basis for the broad substrate specificity of aKMT was explored by the crystallographic analysis of the aKMT-SAM and aKMT-SAH complexes. As revealed by sequence alignment (35), aKMT is structurally homologous to the C-terminal catalytic domain of bacterial ribosomal protein L11 methyltransferase (PrmA), but lacks the N-terminal substrate recognition domain of the bacterial protein. Therefore, unlike PrmA which specifically methylates L11, aKMT is able to catalyze methylation of a large number of proteins. By superimposing the structure of aKMT with that of bacterial PrmA in complex with its substrate L11, we identified a putative active site in the archaeal protein. Like its bacterial homolog, aKMT appears to possess an active site that provides a hydrophobic environment but contains no identifiable residue that could serve as a general base to facilitate the deprotonation of the substrate (24). The pKa of the substrate lysine residue would conceivably be lowered at the hydrophobic active site (64), permitting solvent-mediated deprotonation of the amino group during the methylation reaction.
Intriguingly, the N-terminal portion (the second to the eighth residues) of aKMT appears to adopt two distinct conformations. It extends outward from the aKMT-SAM complex (open conformation) and covers the bound SAH molecule in the aKMT-SAH complex (closed conformation). It may be speculated that the switch between the open and the closed conformations coincides with the cycle of methylation by aKMT. When aKMT binds SAM, the pocket is open and the methyl group of SAM molecule is available for transfer to the δ-amino group of a substrate lysine residue inserted. A successful methyl transfer reaction induces a change of the pocket into a closed conformation. The substrate is then released, and further interaction of the substrate with the enzyme is blocked until the SAH molecule is replaced by a new SAM molecule. This unique reaction cycle allows aKMT to modify a protein substrate in a nonprocessive mode.
Given the extensive protein methylation in S. islandicus and possibly in crenarchaea in general, it would be of great interest to understand the potential physiological role of the posttranslational modification. Protein methylation in bacteria and eukaryotes appears to be more restricted to subsets of proteins (such as ribosomal and flagellar proteins in bacteria and histone proteins in eukaryotes) and catalyzed by methyltransferases in a more specific manner than that in archaea. By comparison, proteins in various COG categories appear to be equally well methylated in S. islandicus. Therefore, it appears that, although protein methylation may play a regulatory role in specific processes in bacteria and eukaryotes, this post-translational modification affects primarily the overall biochemical properties of cellular proteins in archaea. As more methylated Sulfolobus proteins (e.g. glutamate dehydrogenase, aspartate aminotransferase) were reported over the years, it has been speculated that methylation enhances the thermal stability of a protein and represents an adaptation of the hyperthermophilic organism to growth in hot environments (26, 28). Examples in support of the contention include the observation that native (methylated) and recombinant (unmethylated) chromatin protein Sac7d, a member of the Sul7d family from S. acidocaldarius, differ by ∼6 °C in melting point temperature (Tm) (65). However, no significant differences were detected in thermal stability between the methylated and unmenthylated forms of Sso7d, a highly close homolog of Sac7d from S. solfataricus (66). Taking advantage of the availability of the aKMT deletion mutant, in which the level of protein methylation was substantially lower than that in the parental strain, we compared the thermal stability of the cellular proteins from the two strains. No significant differences were detected, suggesting the lack of contribution of methylation to the overall thermal stability of cellular proteins in S. islandicus. However, these results do not rule out the possibility that the thermal stability of some proteins may be affected by methylation. It remains to be investigated how methylation would affect cellular proteins in archaea, but a number of possibilities exist. For instance, methylation of the ε-amino group of a lysine residue in a protein would lower the pKa of the residue, increasing the hydrophobicity of the protein (26). This effect is probably substantial because a large proportion of the surface-accessible lysine residues in Sulfolobus proteins are potential targets for methylation. Therefore, protein lysine methylation may serve to modulate protein-protein and protein-nucleic acid interactions in Sulfolobus.
In agreement with the lack of the significant effect of methylation on the thermal stability of cellular proteins, the mutant strain was able to grow nearly as well as the parental strain at 75 °C in the nutrient-rich medium. Growth of the mutant was more significantly affected at 65 °C than at 75 °C, as compared with that of the parental strain, when they were both grown in the nutrient-poor medium. Therefore, the processes affected by the defect in protein methylation appeared to become growth-limiting under conditions where optimal growth was hindered. As expected from the growth phenotypes of the parental and the mutant strains, the transcriptional profiles of the two strains are in general similar with differentially expressed genes (>twofold difference in expression) accounting for only a small fraction of the total number of genes determined (85 out of ∼ 2600). Presumably, transcription of these differentially expressed genes is altered as a direct or indirect result of changes in the methylation state of upstream regulatory factors. It is observed that many of these differentially expressed genes are clustered in operons and the expression of genes in an operon is often changed in the same direction, supporting the above contention. The transcriptomic comparison of the parental and the mutant strains fails to provide a convincing interpretation of the differences between the two strains in growth, but it appears to yield some interesting clues. For example, several genes with putative functions in substrate transportation and energy metabolism were significantly down-regulated, whereas some genes encoding transcriptional regulators and stress response proteins were up-regulated in the mutant cells. However, it remains to be determined if these changes are related to the slower growth phenotype of the mutant strain.
Our study suggests that there is a distinct difference between bacteria/eukarya and archaea in the pattern and, possibly, the function of protein lysine methylation. As a well studied eukaryotic example, methylation of histones plays a key role in the epigenetic regulation of the chromatin structure and function. On the other hand, the finding in the present study supports a role for aKMT-mediated protein methylation in the growth of the organism under nutrient poor conditions. A better understanding of the physiological role of protein methylation in archaea clearly awaits further investigation.
Supplementary Material
Acknowledgments
We thank Qiu Xia, Li Liu, Xiaojun Ding, Yuanming Luo and Qian Wang for technical assistance.
Footnotes
Author contributions: Y. Chu, Z.L., S.O., and L.H. designed research; Y. Chu, Y.Z., Y. Chen, T.W., and S.O. performed research; Y. Chu, Y. Chen, W.L., Z.Z., D.L., J.M., H.D., Z.L., S.O., and L.H. analyzed data; Y. Chu and L.H. wrote the paper.
* This work was supported by National Natural Science Foundation of China grants 31130003, 30730003 and 31270123 to L. H, 31330019 to Z.J. L, and 31570875, 81590761 and 31200559 to S. O. S. O. was also supported by the Beijing Nova Program (Grant no. Z141102001814020) and Youth Innovation Promotion Association CAS (Grant no. 2013065). The PDB codes for the deposition of aKMT-SAM and aKMT-SAH are 5FA8 and 5FAD respectively.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SAM
- S-adenosyl-L-methionine
- SAH
- S-adenosyl homocysteine
- Se-Met
- Selenomethionine
- IMG
- Integrated Microbial Genomes
- FDR
- False discovery rate
- qRT-PCR
- Quantitative reverse transcription-PCR
- PTM
- Post-translational modifications
- ORFs
- Open reading frames
- SAD
- Single-wavelength anomalous dispersion
- RMSD
- Root-mean-square deviation
- SET
- (Su(var), Enhancer of zeste, Trithorax) domain
- X-gal
- 5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside
- PCNA
- proliferating cell nuclear antigen
- Lig1
- DNA ligase I
- MCM
- Minichromosome maintenance protein complex
- PriL
- DNA primase noncatalytic subunit
- PriS
- DNA primase catalytic subunit
- RFC
- Replication factor C.
REFERENCES
- 1. Eichler J., and Adams M. W. W. (2005) Posttranslational protein modification in Archaea. Microbiol. Mol. Biol. 69, 393–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lachner M., and Jenuwein T. (2002) The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 14, 286–298 [DOI] [PubMed] [Google Scholar]
- 3. Paik W. K., Paik D. C., and Kim S. (2007) Historical review: the field of protein methylation. Trends Biochem. Sci. 32, 146–152 [DOI] [PubMed] [Google Scholar]
- 4. Lanouette S., Mongeon V., Figeys D., and Couture J. F. (2014) The functional diversity of protein lysine methylation. Mol. Syst. Biol. 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moore K. E., and Gozani O. (2014) An unexpected journey: lysine methylation across the proteome. Biochim. Biophys. Acta 1839, 1395–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Botting C. H., Talbot P., Paytubi S., and White M. F. (2010) Extensive Lysine Methylation in Hyperthermophilic Crenarchaea: Potential Implications for Protein Stability and Recombinant Enzymes. Archaea, 2010, 106341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y., and Bedford M. T. (2013) Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 [DOI] [PubMed] [Google Scholar]
- 8. Lanouette S., Mongeon V., Figeys D., and Couture J. F. (2014) The functional diversity of protein lysine methylation. Mol. Systems Biol. 10, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clarke S. G. (2013) Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem. Sci. 38, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Black J. C., Van Rechem C., and Whetstine J. R. (2012) Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Lorenzo A., and Bedford M. T. (2011) Histone arginine methylation. FEBS Lett. 585, 2024–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chuikov S., Kurash J. K., Wilson J. R., Xiao B., Justin N., Ivanov G. S., McKinney K., Tempst P., Prives C., Gamblin S. J., Barlev N. A., and Reinberg D. (2004) Regulation of p53 activity through lysine methylation. Nature 432, 353–360 [DOI] [PubMed] [Google Scholar]
- 13. West L. E., and Gozani O. (2011) Regulation of p53 function by lysine methylation. Epigenomics 3, 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chakraborty S., Sinha K. K., Senyuk V., and Nucifora G. (2003) SUV39H1 interacts with AML1 and abrogates AML1 transactivity. AML1 is methylated in vivo. Oncogene 22, 5229–5237 [DOI] [PubMed] [Google Scholar]
- 15. Kluck R. M., Ellerby L. M., Ellerby H. M., Naiem S., Yaffe M. P., Margoliash E., Bredesen D., Mauk A. G., Sherman F., and Newmeyer D. D. (2000) Determinants of cytochrome c pro-apoptotic activity - The role of lysine 72 trimethylation. J. Biol. Chem. 275, 16127–16133 [DOI] [PubMed] [Google Scholar]
- 16. Kouskouti A., Scheer E., Staub A., Tora L., and Talianidis I. (2004) Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol. Cell 14, 175–182 [DOI] [PubMed] [Google Scholar]
- 17. Webb K. J., Al-Hadid Q., Zurita-Lopez C. I., Young B. D., Lipson R. S., and Clarke S. G. (2011) The ribosomal l1 protuberance in yeast is methylated on a lysine residue catalyzed by a seven-beta-strand methyltransferase. J. Biol. Chem. 286, 18405–18413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Polevoda B., Martzen M. R., Das B., Phizicky E. M., and Sherman F. (2000) Cytochrome c methyltransferase, Ctm1p, of yeast. J. Biol. Chem. 275, 20508–20513 [DOI] [PubMed] [Google Scholar]
- 19. Ying Z., Mulligan R. M., Janney N., and Houtz R. L. (1999) Rubisco small and large subunit N-methyltransferases. Bi- and mono-functional methyltransferases that methylate the small and large subunits of Rubisco. J. Biol. Chem. 274, 36750–36756 [DOI] [PubMed] [Google Scholar]
- 20. Ambler R. P., and Rees M. W. (1959) Epsilon-N-Methyl-Lysine in Bacterial Flagellar Protein. Nature 184, 56–57 [DOI] [PubMed] [Google Scholar]
- 21. Chang F. N., Chang C. N., and Paik W. K. (1974) Methylation of Ribosomal-Proteins in Escherichia-Coli. J. Bacteriol. 120, 651–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cameron D. M., Gregory S. T., Thompson J., Suh M. J., Limbach P. A., and Dahlberg A. E. (2004) Thermus thermophilus L11 methyltransferase, PrmA, is dispensable for growth and preferentially modifies free ribosomal protein L11 prior to ribosome assembly. J. Bacteriol. 186, 5819–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Demirci H., Gregory S. T., Dahlberg A. E., and Jogl G. (2007) Recognition of ribosomal protein L11 by the protein trimethyltransferase PrmA. EMBO J. 26, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Demirci H., Gregory S. T., Dahlberg A. E., and Jogl G. (2008) Multiple-site trimethylation of ribosomal protein L11 by the PrmA methyltransferase. Structure 16, 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bujnicki J. M. (2000) Sequence, structural, and evolutionary analysis of prokaryotic ribosomal protein L11 methyltransferases. Acta Microbiol. Polonica 49, 19–29 [PubMed] [Google Scholar]
- 26. Febbraio F., Andolfo A., Tanfani F., Briante R., Gentile F., Formisano S., Vaccaro C., Scire A., Bertoli E., Pucci P., and Nucci R. (2004) Thermal stability and aggregation of Sulfolobus solfataricus beta-glycosidase are dependent upon the n-epsilon-methylation of specific lysyl residues - Critical role of in vivo post-translational modifications. J. Biol. Chem. 279, 10185–10194 [DOI] [PubMed] [Google Scholar]
- 27. Minami Y., Wakabayashi S., Wada K., Matsubara H., Kerscher L., and Oesterhelt D. (1985) Amino-Acid Sequence of a Ferredoxin from Thermoacidophilic Archaebacterium, Sulfolobus-Acidocaldarius - Presence of an N-6-monomethyllysine and phyletic consideration of Archaebacteria. J. Biochem-Tokyo 97, 745–753 [DOI] [PubMed] [Google Scholar]
- 28. Maras B., Consalvi V., Chiaraluce R., Politi L., Derosa M., Bossa F., Scandurra R., and Barra D. (1992) The protein-sequence of glutamate-dehydrogenase from sulfolobus-solfataricus, a thermoacidophilic Archaebacterium - Is the presence of N-epsilon-methyllysine related to thermostability. Eur. J. Biochem. 203, 81–87 [DOI] [PubMed] [Google Scholar]
- 29. Ramirez C., Shimmin L. C., Newton C. H., Matheson A. T., and Dennis P. P. (1989) Structure and evolution of the L11, L1, L10, and l12 equivalent ribosomal-proteins in Eubacteria, Archaebacteria, and Eukaryotes. Can. J. Microbiol. 35, 234–244 [DOI] [PubMed] [Google Scholar]
- 30. Zappacosta F., Sannia G., Savoy L. A., Marino G., and Pucci P. (1994) Posttranslational modifications in aspartate-aminotransferase from sulfolobus-solfataricus - detection of N-epsilon-methyllysines by mass-spectrometry. Eur. J. Biochem. 222, 761–767 [DOI] [PubMed] [Google Scholar]
- 31. Baumann H., Knapp S., Lundback T., Ladenstein R., and Hard T. (1994) Solution structure and DNA-binding properties of a thermostable protein from the archaeon sulfolobus-solfataricus. Nat. Struct. Biol. 1, 808–819 [DOI] [PubMed] [Google Scholar]
- 32. Guo L., Feng Y. G., Zhang Z. F., Yao H. W., Luo Y. M., Wang J. F., and Huang L. (2008) Biochemical and structural characterization of Cren7, a novel chromatin protein conserved among Crenarchaea. Nucleic Acids Res. 36, 1129–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. White M. F., and Bell S. D. (2002) Holding it together: chromatin in the Archaea. Trends Genet. 18, 621–626 [DOI] [PubMed] [Google Scholar]
- 34. Manzur K. L., and Zhou M. M. (2005) An archaeal SET domain protein exhibits distinct lysine methyltransferase activity towards DNA-associated protein MCl-alpha. FEBS Lett. 579, 3859–3865 [DOI] [PubMed] [Google Scholar]
- 35. Chu Y. D., Zhang Z. F., Wang Q., Luo Y. M., and Huang L. (2012) Identification and characterization of a highly conserved crenarchaeal protein lysine methyltransferase with broad substrate specificity. J. Bacteriol. 194, 6917–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niu Y. L., Xia Y. S., Wang S. S., Li J. N., Niu C. Y., Li X., Zhao Y. H., Xiong H. Y., Li Z., Lou H. Q., and Cao Q. H. (2013) A prototypic lysine methyltransferase 4 from archaea with degenerate sequence specificity methylates chromatin proteins Sul7d and Cren7 in different patterns. J. Biol. Chem. 288, 13728–13740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Couture J. F., Collazo E., Hauk G., and Trievel R. C. (2006) Structural basis for the methylation site specificity of SET7/9. Nat. Struct. Mol. Biol. 13, 140–146 [DOI] [PubMed] [Google Scholar]
- 38. Deng L., Zhu H., Chen Z., Liang Y. X., and She Q. (2009) Unmarked gene deletion and host–vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 13, 735–746 [DOI] [PubMed] [Google Scholar]
- 39. Peng N., Xia Q., Chen Z., Liang Y. X., and She Q. (2009) An upstream activation element exerting differential transcriptional activation on an archaeal promoter. Mol. Microbiol. 74, 928–939 [DOI] [PubMed] [Google Scholar]
- 40. Zhang C. Y., Guo L., Deng L., Wu Y. X., Liang Y. X., Huang L., and She Q. X. (2010) Revealing the essentiality of multiple archaeal pcna genes using a mutant propagation assay based on an improved knockout method. Microbiol-Sgm 156, 3386–3397 [DOI] [PubMed] [Google Scholar]
- 41. Peng N., Deng L., Mei Y. X., Jiang D. Q., Hu Y. M., Awayez M., Liang Y. X., and She Q. X. (2012) A synthetic arabinose-inducible promoter confers high levels of recombinant protein expression in hyperthermophilic archaeon Sulfolobus islandicus. Appl. Environ. Microb. 78, 5630–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mao S., Luo Y., Zhang T., Li J., Bao G., Zhu Y., Chen Z., Zhang Y., Li Y., and Ma Y. (2010) Proteome reference map and comparative proteomic analysis between a wild type Clostridium acetobutylicum DSM 1731 and its mutant with enhanced butanol tolerance and butanol yield. J. Proteome Res. 9, 3046–3061 [DOI] [PubMed] [Google Scholar]
- 43. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 44. Guo L., Brugger K., Liu C., Shah S. A., Zheng H. J., Zhu Y. Q., Wang S. Y., Lillestol R. K., Chen L. M., Frank J., Prangishvili D., Paulin L., She Q. X., Huang L., and Garrett R. A. (2011) genome analyses of Icelandic strains of Sulfolobus islandicus, model organisms for genetic and virus-host interaction studies. J. Bacteriol. 193, 1672–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., and Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 46. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., and Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Markowitz V. M., Korzeniewski F., Palaniappan K., Szeto E., Werner G., Padki A., Zhao X. L., Dubchak I., Hugenholtz P., Anderson I., Lykidis A., Mavromatis K., Ivanova N., and Kyrpides N. C. (2006) The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34, D344–D348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 49. Dai P. G., Wang Y., Ye R. S., Chen L., and Huang L. (2003) DNA topoisomerase III from the hyperthermophilic Archaeon Sulfolobus solfataricus with specific DNA cleavage activity. J. Bacteriol. 185, 5500–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 51. Liu Z. J., Lin D., Tempel W., Praissman J. L., Rose J. P., and Wang B. C. (2005) Parameter-space screening: a powerful tool for high-throughput crystal structure determination. Acta Crystallogr. 61, 520–527 [DOI] [PubMed] [Google Scholar]
- 52. Ru H., Zhao L., Ding W., Jiao L., Shaw N., Liang W., Zhang L., Hung L. W., Matsugaki N., Wakatsuki S., and Liu Z. J. (2012) S-SAD phasing study of death receptor 6 and its solution conformation revealed by SAXS. Acta crystallogr. 68, 521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murshudov G. N., Skubak P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davis I. W., Murray L. W., Richardson J. S., and Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang L., Feng Z., Wang X., Wang X., and Zhang X. (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 [DOI] [PubMed] [Google Scholar]
- 59. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 60. Binder H., Steiner L., Przybilla J., Rohlf T., Prohaska S., and Galle J. (2013) Transcriptional regulation by histone modifications: towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 10, 026006 [DOI] [PubMed] [Google Scholar]
- 61. Cheng X., and Roberts R. J. (2001) AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 29, 3784–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Subramanian K., Jia D., Kapoor-Vazirani P., Powell D. R., Collins R. E., Sharma D., Peng J., Cheng X., and Vertino P. M. (2008) Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol. Cell 30, 336–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang X. D., Lamb A., and Chen L. F. (2009) Methylation, a new epigenetic mark for protein stability. Epigenetics-Us 4, 429–433 [DOI] [PubMed] [Google Scholar]
- 64. Toseland C. P., McSparron H., Davies M. N., and Flower D. R. (2006) PPD v1.0–an integrated, web-accessible database of experimentally determined protein pKa values. Nucleic Acids Res. 34, D199–D203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McAfee J. G., Edmondson S. P., Datta P. K., Shriver J. W., and Gupta R. (1995) Gene cloning, expression, and characterization of the Sac7 proteins from the hyperthermophile Sulfolobus acidocaldarius. Biochemistry-Us 34, 10063–10077 [DOI] [PubMed] [Google Scholar]
- 66. Knapp S., Karshikoff A., Berndt K. D., Christova P., Atanasov B., and Ladenstein R. (1996) Thermal unfolding of the DNA-binding protein Sso7d from the hyperthermophile Sulfolobus solfataricus. J. Mol. Biol. 264, 1132–1144 [DOI] [PubMed] [Google Scholar]
- 67. Wallace A. C., Laskowski R. A., and Thornton J. M. (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Engineering 8, 127–134 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.