

Abstract
Between-subject and within-subject variability is ubiquitous in biology and physiology, and understanding and dealing with this is one of the biggest challenges in medicine. At the same time, it is difficult to investigate this variability by experiments alone. A recent modelling and simulation approach, known as population of models (POM), allows this exploration to take place by building a mathematical model consisting of multiple parameter sets calibrated against experimental data. However, finding such sets within a high-dimensional parameter space of complex electrophysiological models is computationally challenging. By placing the POM approach within a statistical framework, we develop a novel and efficient algorithm based on sequential Monte Carlo (SMC). We compare the SMC approach with Latin hypercube sampling (LHS), a method commonly adopted in the literature for obtaining the POM, in terms of efficiency and output variability in the presence of a drug block through an in-depth investigation via the Beeler–Reuter cardiac electrophysiological model. We show improved efficiency for SMC that produces similar responses to LHS when making out-of-sample predictions in the presence of a simulated drug block. Finally, we show the performance of our approach on a complex atrial electrophysiological model, namely the Courtemanche–Ramirez–Nattel model.
Keywords: cardiac electrophysiology, population of models, Beeler–Reuter cell model, sequential Monte Carlo, Latin hypercube sampling, approximate Bayesian computation
1. Introduction
Many complex mathematical models, such as those for neural or cardiac excitability, are characterized by parameters living in a high-dimensional parameter space. In these applications, it is possible that only a small subset of this parameter space is required to explain the between-subject variability observed in experimental data.
A number of studies in systems electrophysiology have explicitly addressed the challenge of explaining between-subject variability in the response of specific neurons or particular cardiac muscle cells to an externally applied electrical stimulus by introducing the concept of ‘population of models’ (POM) [1–5]. The term ‘population of models’ arises from the notion that a mathematical model is typically considered to be the underlying set of model equations in conjunction with a specific set of model parameter values. In the statistical literature, however, this would be considered as being only a single model, with a set of parameter values sampled from some underlying population distribution. Such a model is often referred to as a random effects model.
In the POM approach, a particular system of equations is fixed (typically a given set of differential equations), and different combinations of the equation parameters are considered in order to generate a collection of models. These models are then calibrated, so that only the ones consistent with observed experimental data are kept in the final POM. The calibrated POM is then often used to make ‘out-of-sample’ predictions, such as predicting the cellular electrophysiological changes produced by a drug-induced conduction block [2] or by different expression levels of mRNA in failing human hearts [6].
In the POM literature, different methods have been used to sample from a multidimensional parameter space and to generate the initial collection of models. Gemmell et al. [7], for example, combine random sampling with a method known as clutter-based dimension reordering, enabling the visualization of a multidimensional parameter space in two dimensions, whereas Sánchez et al. [3] adopt a sampling method based on decomposition of model output variance, known as the extended version of the Fourier amplitude sensitivity test originally developed by Saltelli et al. [8]. Another option often found in the POM literature to explore the multidimensional parameter space and generate the initial collection of models is a method known as Latin hypercube sampling (LHS; see [2,9–11] for example). LHS is a technique first introduced by McKay et al. [12]. Suppose that the d dimensional parameter space is divided into n equally sized subdivisions in each dimension. A Latin hypercube trial is a set of n random samples with one from each subdivision; that is, each sample is the only one in each axis-aligned hyperplane containing it. McKay et al. [12] suggest that the advantage of LHS is that the values for each dimension are fully stratified, whereas Stein [13] shows that with LHS there is a form of variance reduction compared with uniform random sampling.
In this work, we focus on the POM method using a statistical framework, both in terms of methodological development and data analytic considerations. We show connections between a method originating in population genetics, known as approximate Bayesian computation (ABC), and POM, in particular, to develop a computationally efficient algorithm for POM. ABC has been developed in order to perform Bayesian statistical inference for models that do not possess a computationally tractable likelihood function [14]. ABC algorithms involve searching the parameter space until a certain number of parameter values are found that generate simulated data that are close to the observed data. ‘Close’ is typically defined as a user-specified discrepancy function involving some carefully chosen summary statistics of the data. The optimal choice of summary statistics represents a trade-off between information loss and dimensionality [15].
The output of ABC is a sample of parameter values from an approximate posterior distribution (the probability distribution of the parameters conditional on the data). The approximation depends on the choice of summary statistics and on how stringently the simulated data are matched with the observed data.
The original ABC rejection algorithm [14] involves repeated random sample selections from the parameter space until a certain number of sufficiently close samples is generated. If the posterior distribution is quite different from the prior distribution, then this method is known to be highly inefficient. To improve efficiency, Markov chain Monte Carlo (MCMC) methods can be used to sample from the approximate posterior (referred to as MCMC ABC [16]). MCMC involves proposing new parameter values locally via a Markov chain and accepting those proposals with a certain probability to ensure that the limiting distribution of the Markov chain is the posterior distribution of interest. However, because MCMC ABC relies on a single chain, it can sometimes get stuck in low posterior regions, so a collection of sequential Monte Carlo (SMC) methods has been developed (referred to as SMC ABC) [17–19].
The aims of this article are as follows. First, we develop a statistical framework for POM that provides insight into the outcomes of a POM approach and the impact of data summarization. We devise a novel computational algorithm for POM based on SMC using the developments in SMC ABC as guidance. We then compare this method with an existing approach based on LHS and random sampling. As a case study, we consider the Beeler–Reuter (BR) cardiac cell model [20]. We compare the performance of the POM methods (LHS and SMC) by analysing how certain biomarkers behave, based on the action potential, as a function of the ‘maximal current densities’ associated with four ion channels (see §3), and also for different summary statistic dimensions. We then make some predictions as to how the two approaches compare when we reduce the maximal current density of a selected potassium channel, thus mimicking the effect of a pharmaceutical drug block. As a by-product of our analysis, we also gain insights into how reliably parameter ranges of this model can be recovered based on what information of the solution profile is used.
2. Methods
2.1. A statistical framework for population of models
We generally assume that the phenomenon under study can be modelled by a mathematical model, denoted by m(θ), with θ being an unknown parameter of the model with parameter space  where d is the number of components in the parameter vector. We assume that the output of the model is deterministic with a solution function denoted by x(θ), where
where d is the number of components in the parameter vector. We assume that the output of the model is deterministic with a solution function denoted by x(θ), where 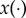 is a scalar function. The methods that we specify later still apply if
is a scalar function. The methods that we specify later still apply if 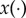 is a vector-valued function. We also assume that a highly accurate numerical solver for x(θ) is available, so that the numerical error is negligible.
is a vector-valued function. We also assume that a highly accurate numerical solver for x(θ) is available, so that the numerical error is negligible.
Data are collected on n subjects. We assume that data y from a subject have the same support as the model output x(θ). For simplicity, we assume that observations on the ith subject are replicated a sufficiently large number of times, so that the data y are assumed to be sample averages with negligible standard error. Under this assumption, it is reasonable to suggest that the averaged data from a particular subject may be explained by a differential equation with a certain parameter configuration. Further, we assume the data y are measured without error, which may not be realistic. We will consider the case of measurement error in future research. Interest then is in finding parameter values of this differential equation model in order to capture the between-subject variability in the averaged data. That is, it is assumed that the within-subject variability is negligible compared with the between-subject variability. Note that in actual experiments [2,3] it is unlikely that it is possible to replicate the experiment many times on an individual subject. For such cases, the objective is to capture both the within-subject and between-subject variability.
We assume that θ i denotes the (unobserved) parameter specific to the ith subject. The parameter for the ith subject is drawn from some probability distribution, i.e. 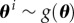 . Our objective is to estimate or learn the distribution g(θ) based on the observed data,
. Our objective is to estimate or learn the distribution g(θ) based on the observed data, 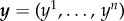 by finding a set of θ values that produces model outputs x(θ) close to y in some sense.
by finding a set of θ values that produces model outputs x(θ) close to y in some sense.
We assume there is an underlying population of subjects with a population density for y given by f(y) and that the data y are a random sample from the population with density f(y). We define the population distribution, h(x), of the model output x(θ) when θ is drawn from the distribution g(θ), that is, h(x) is the density of x(θ) when θ ∼ g(θ)
Estimation of g(θ) can be formulated in various ways, using ideas from the statistics literature. For example, an approach adopted from variational Bayes theory (e.g. Bishop [21, ch. 10]), where an intractable posterior distribution is approximated analytically, would involve finding g(θ) to minimize the Kullback–Leibler divergence (KLD) between 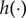 and
and  .
.
As 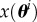 is a function, we cannot store or observe all the information from it for each subject. In the case of cardiac electrophysiology models, the action potential has a characteristic shape, and so a general functional form is known. Thus, it is common practice in the POM literature to summarize the data for the ith subject to give
is a function, we cannot store or observe all the information from it for each subject. In the case of cardiac electrophysiology models, the action potential has a characteristic shape, and so a general functional form is known. Thus, it is common practice in the POM literature to summarize the data for the ith subject to give 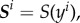 where
where 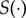 is a function that maps the action potential onto a vector of finite dimension K. Here,
is a function that maps the action potential onto a vector of finite dimension K. Here, 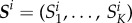 . The full set of observed summary statistics for all n subjects is denoted
. The full set of observed summary statistics for all n subjects is denoted 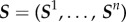 .
.
This dimension reduction of the full dataset leads to a loss of information and impacts on the estimation of g(θ). The summary statistic function 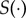 should be chosen carefully, so that the summary statistic carries most of the information regarding θ present in the data y. The ABC literature suggests choosing
should be chosen carefully, so that the summary statistic carries most of the information regarding θ present in the data y. The ABC literature suggests choosing  so that the summary statistics are sensitive to changes in θ. As is common practice in ABC, it might be necessary to run POM with different choices of
so that the summary statistics are sensitive to changes in θ. As is common practice in ABC, it might be necessary to run POM with different choices of 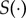 . To emphasize that the output of POM depends on
. To emphasize that the output of POM depends on  , we note that POM will produce an estimate of
, we note that POM will produce an estimate of 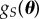 rather than g(θ). In the case study highlighted later in this section, we propose two different functions for
rather than g(θ). In the case study highlighted later in this section, we propose two different functions for 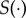 and present the outcomes in §3.
and present the outcomes in §3.
In the POM literature, a method is devised to generate samples from 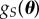 based on the information contained in S without having to propose some parametric form for
based on the information contained in S without having to propose some parametric form for 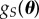 , which might be a common approach in the random effects statistics literature. Instead of using a closeness measure based on a functional metric such as the KLD, a common approach in the POM literature is to use a data analytic approach by keeping all θ values proposed that produce simulated summary statistics
, which might be a common approach in the random effects statistics literature. Instead of using a closeness measure based on a functional metric such as the KLD, a common approach in the POM literature is to use a data analytic approach by keeping all θ values proposed that produce simulated summary statistics  that satisfy a set of K constraints formulated from the observed data summary statistics S. Implicitly,
that satisfy a set of K constraints formulated from the observed data summary statistics S. Implicitly, 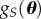 is defined uniformly over the set
is defined uniformly over the set 
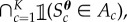 where
where 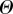 denotes the parameter space that is searched. Here,
denotes the parameter space that is searched. Here, 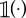 denotes the indicator function,
denotes the indicator function, 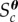 is the cth simulated summary statistic obtained from the model parameter θ and Ac is a set of values of the cth summary statistic formulated from the observed summary statistics
is the cth simulated summary statistic obtained from the model parameter θ and Ac is a set of values of the cth summary statistic formulated from the observed summary statistics 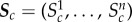 and defines a range for which the simulation is said to ‘match’ the cth summary statistic.
and defines a range for which the simulation is said to ‘match’ the cth summary statistic.
For example, Britton et al. [2] set 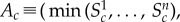
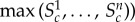 . We note that the min/max range generally increases with the sample size n, but it might be a reasonable choice when n is small. The repeated sampling probability coverage of this Ac, based on the range of the population distribution of Sc, is (n − 1)/(n + 1) [22]. Small sample sizes are common in cardiac electrophysiology experiments as they are expensive and subject to many ethical constraints.
. We note that the min/max range generally increases with the sample size n, but it might be a reasonable choice when n is small. The repeated sampling probability coverage of this Ac, based on the range of the population distribution of Sc, is (n − 1)/(n + 1) [22]. Small sample sizes are common in cardiac electrophysiology experiments as they are expensive and subject to many ethical constraints.
Here, we consider another way to form the constraints. Let 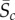 and
and 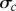 denote the sample mean and standard deviation of the data for the cth summary statistic. Then, we set
denote the sample mean and standard deviation of the data for the cth summary statistic. Then, we set 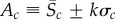 for some chosen value of k. Here, we set k = 2, indicating an approximate 95% confidence interval (CI) for the population mean of the cth summary statistic. The calibration of the value of k using the 95% CI does assume that each summary statistic is approximately normally distributed. Using an interval based on quantiles, such as the 95% central quantile interval of the data for Ac, would better accommodate asymmetry in the data. We note that the CI and quantile interval approaches are less dependent on the sample size, n, in the sense that the constraints will converge in probability to fixed quantities as the sample size increases and are therefore more stable. Our methodology can be applied to any form of the constraints, and for simplicity, we consider the range and CI constraints.
for some chosen value of k. Here, we set k = 2, indicating an approximate 95% confidence interval (CI) for the population mean of the cth summary statistic. The calibration of the value of k using the 95% CI does assume that each summary statistic is approximately normally distributed. Using an interval based on quantiles, such as the 95% central quantile interval of the data for Ac, would better accommodate asymmetry in the data. We note that the CI and quantile interval approaches are less dependent on the sample size, n, in the sense that the constraints will converge in probability to fixed quantities as the sample size increases and are therefore more stable. Our methodology can be applied to any form of the constraints, and for simplicity, we consider the range and CI constraints.
2.2. A sequential Monte Carlo algorithm for population of models
We consider three sampling approaches for POM. The first simply randomly samples parameter values from the specified parameter space (we refer to this as the random (RND) method). The second is LHS (see [2] for an example in electrophysiology). See appendix A of the electronic supplementary material for more details on our LHS approach. Below, we describe our novel method, based on SMC.
Here, we formulate the problem of finding parameter values that satisfy a set of K constraints into one of sampling from a probability distribution. The ‘target’ distribution, 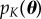 , is given by the following expression:
, is given by the following expression:
 |
2.1 |
The range of the probability distribution p(θ) defines the original parameter space to search. A natural choice for p(θ) is a uniform distribution over the specified range of the parameters. In Bayesian statistics, p(θ) is termed the prior distribution, and it is possible to incorporate information from previous experiments and expert opinion into this distribution. Note that the construction in (2.1) does not assume that the summary statistics are independent. For a particular θ to have non-zero probability mass, it must generate statistics Sθ that satisfy the constraints jointly.
The target distribution in (2.1) appears as a target distribution commonly encountered in ABC. ABC involves searching the parameter space of a stochastic model with a computationally intractable likelihood function until a certain set of parameter values are found that lead to simulated summary statistics that are close to the observed summary statistics. In ABC, the constraint might be defined as 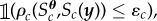 where
where  is the cth summary statistic of the observed data and
is the cth summary statistic of the observed data and  is some user-specified discrepancy function that measures the similarity between
is some user-specified discrepancy function that measures the similarity between 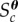 and
and 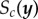 . In the context of ABC, interest is in bringing εc as close to 0 as possible. The motivation of our work is different, because we pre-specify some interval of allowable summary statistic values Ac in order to find sets of parameters that can explain the between-subject variability of the observed data. However, we may harness the methodological developments in SMC ABC to build an efficient computational algorithm to generate samples from (2.1).
. In the context of ABC, interest is in bringing εc as close to 0 as possible. The motivation of our work is different, because we pre-specify some interval of allowable summary statistic values Ac in order to find sets of parameters that can explain the between-subject variability of the observed data. However, we may harness the methodological developments in SMC ABC to build an efficient computational algorithm to generate samples from (2.1).
SMC methods have been developed for generating samples from a sequence of smoothly evolving probability distributions 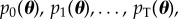 where p0(θ) is easy to sample and pT(θ) is the target distribution (assumed to be difficult to sample). It does this by traversing a set of N weighted samples (or ‘particles’) through the sequence of distributions by iteratively applying a set of importance sampling, re-sampling and perturbation steps. Importance sampling is used to move particles between distributions, the re-sampling step is used to maintain a reasonable effective sample size (ESS, the number of perfect samples that the weighted sample is worth) and the perturbation step ensures that a diverse set of particles represents each distribution. After the tth iteration of SMC, a set of properly weighted samples
where p0(θ) is easy to sample and pT(θ) is the target distribution (assumed to be difficult to sample). It does this by traversing a set of N weighted samples (or ‘particles’) through the sequence of distributions by iteratively applying a set of importance sampling, re-sampling and perturbation steps. Importance sampling is used to move particles between distributions, the re-sampling step is used to maintain a reasonable effective sample size (ESS, the number of perfect samples that the weighted sample is worth) and the perturbation step ensures that a diverse set of particles represents each distribution. After the tth iteration of SMC, a set of properly weighted samples 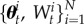 are obtained from the distribution
are obtained from the distribution 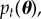 where
where 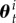 and
and 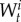 are the ith sample and weight from target t, respectively (see [23,24]). Note that SMC can provide a better exploration of the total parameter space than MCMC, and in SMC, it is possible to perform expensive computations associated with each particle in parallel.
are the ith sample and weight from target t, respectively (see [23,24]). Note that SMC can provide a better exploration of the total parameter space than MCMC, and in SMC, it is possible to perform expensive computations associated with each particle in parallel.
In the context of the target distribution in (2.1), a natural choice for the sequence of target distributions required for SMC is to introduce the constraints one at a time
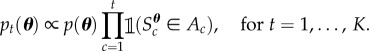 |
However, if a particular constraint results in a significant reduction in the plausible parameter space, then SMC may not be very robust. For example, there may be only a small number of particles that satisfy the next constraint (with all other particles having zero weight), implying that it could be difficult to construct an efficient proposal for the perturbation step (referred to as particle degeneracy). In the worst case, there may be no particles satisfying the next constraint, and the method breaks down completely (all particles have zero weight). Therefore, we allow the (novel) possibility of several intermediate distributions between distributions t and t + 1
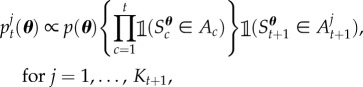 |
2.2 |
where  . Here,
. Here, 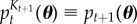 . In this sequence of targets, we do not enforce the algorithm to satisfy the (t + 1)th constraint straightaway, but allow it to move smoothly between distributions t and t + 1. We are able to determine the sets
. In this sequence of targets, we do not enforce the algorithm to satisfy the (t + 1)th constraint straightaway, but allow it to move smoothly between distributions t and t + 1. We are able to determine the sets 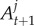 adaptively in the algorithm (see below). If a particular constraint does not reduce the plausible parameter space substantially, then it is likely that these intermediate distributions are not required. Our SMC algorithm is able to determine when the intermediate distributions are necessary. To illustrate, consider the specific example where Ac is the interval
adaptively in the algorithm (see below). If a particular constraint does not reduce the plausible parameter space substantially, then it is likely that these intermediate distributions are not required. Our SMC algorithm is able to determine when the intermediate distributions are necessary. To illustrate, consider the specific example where Ac is the interval  . The set of values in Ac is controlled by the value of k. In this case,
. The set of values in Ac is controlled by the value of k. In this case,  becomes
becomes  . Alternatively, if the range of the sample is used, then
. Alternatively, if the range of the sample is used, then  . Here, we set
. Here, we set  and we have that
and we have that 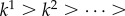
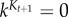 . The specific values of kj are selected so that there is a certain number of particles (here N/2) that satisfy the current set of constraints (that includes the constraint involving kj). When setting kj = k (CI constraints) or
. The specific values of kj are selected so that there is a certain number of particles (here N/2) that satisfy the current set of constraints (that includes the constraint involving kj). When setting kj = k (CI constraints) or 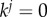 (range constraints) results in at least N/2 particles satisfying the current set of constraints, then we do not reduce kj further. Our method ensures that the SMC does not suffer from particle degeneracy.
(range constraints) results in at least N/2 particles satisfying the current set of constraints, then we do not reduce kj further. Our method ensures that the SMC does not suffer from particle degeneracy.
Note that Golchi & Campbell [25] consider SMC algorithms for sampling from constrained distributions. Our developments are specific to the POM application and include the novel aspect of introducing a sequence of distributions between adjacent distributions to improve the robustness.
The algorithm we propose here has similarities with the SMC ABC algorithm of Drovandi & Pettitt [18]. Denote the particle set at target t as  . First, the particles are re-weighted to reflect the next target t + 1. In our application, this involves removing the particles that do not satisfy the next constraint. If more than N/2 particles do not satisfy the next constraint, then the intermediate sequence in (2.2) is used where the
. First, the particles are re-weighted to reflect the next target t + 1. In our application, this involves removing the particles that do not satisfy the next constraint. If more than N/2 particles do not satisfy the next constraint, then the intermediate sequence in (2.2) is used where the 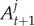 (here the values of k for our choice of Ac) are adaptively chosen, so that there are at least N/2 particles that satisfy the constraint. It is important to note that we never have
(here the values of k for our choice of Ac) are adaptively chosen, so that there are at least N/2 particles that satisfy the constraint. It is important to note that we never have  (i.e.
(i.e.  ) for any value of j. The ESS can be boosted to N by resampling from the remaining particles. Then, to help remove particle duplication, an MCMC kernel [23] with proposal density q is applied to the re-sampled particles. Owing to the way that the sequence of targets is formed via indicator functions, the ESS after each iteration of the SMC will always be N. Furthermore, the algorithm is of complexity O(N). However, the MCMC kernel does not guarantee that each particle will be moved, resulting in inevitable particle duplication. This issue can be mitigated by repeating the MCMC kernel several times on each particle. However, this decreases the efficiency as various particles will attempt to be moved even after they have satisfied the next set of constraints. On the other hand, it is possible to reject proposed parameter values by the MCMC kernel early if it does not satisfy the initial part (i.e. without the imposed constraints) of the Metropolis–Hastings ratio. Our SMC algorithm that uses an MCMC kernel for particle diversity is shown in algorithm 1 of appendix B of the electronic supplementary material.
) for any value of j. The ESS can be boosted to N by resampling from the remaining particles. Then, to help remove particle duplication, an MCMC kernel [23] with proposal density q is applied to the re-sampled particles. Owing to the way that the sequence of targets is formed via indicator functions, the ESS after each iteration of the SMC will always be N. Furthermore, the algorithm is of complexity O(N). However, the MCMC kernel does not guarantee that each particle will be moved, resulting in inevitable particle duplication. This issue can be mitigated by repeating the MCMC kernel several times on each particle. However, this decreases the efficiency as various particles will attempt to be moved even after they have satisfied the next set of constraints. On the other hand, it is possible to reject proposed parameter values by the MCMC kernel early if it does not satisfy the initial part (i.e. without the imposed constraints) of the Metropolis–Hastings ratio. Our SMC algorithm that uses an MCMC kernel for particle diversity is shown in algorithm 1 of appendix B of the electronic supplementary material.
For the case study described later, we assume that the model parameters are independent and uniformly distributed a priori between the specified lower (lb) and upper (ub) bounds, so our SMC algorithm samples over a re-parametrized space where each parameter is transformed based on
where θ is the model parameter and ϕ is the re-parametrized version. SMC can then search an unrestricted space while still enforcing that θ remains within its bounds. If θ is assumed uniform on (lb,ub), then the implied prior density on ϕ is
that is, the logistic density.
For q in the MCMC move step in SMC, we fit a three component normal mixture model to the re-sampled particle set and draw proposals independently from the mixture model. Note that other choices are possible for q. For example, a multivariate normal random walk centred on the current particle may be used with a covariance matrix estimated from the resampled particles. We find that using the independent mixture model for q works well for this application, but the multivariate normal random walk also works well (albeit slightly less efficient in general). We use N = 500 for the SMC algorithm and R = 3 when there is only one parameter of interest and R = 5 when there are two to four components in the parameter vector.
There are a number of tuning parameters in the SMC algorithm. First, we select the smallest fraction of particles to keep at each iteration as N/2, which is a common choice in the SMC literature. However, any fraction may be selected here. The smaller the fraction is, the less intermediate distributions will be required, leading to potentially fewer model simulations. However, there will be fewer particles to estimate the hyperparameter of the importance distribution q, which here consists of the normal mixture means, covariance matrices and weights. Further, there will be more particle duplication after the resampling step. Here, we fix the number of MCMC repeats, R, which works for our application. Drovandi & Pettitt [18] provide an approach to adapt the value of R based on the overall MCMC acceptance rate of the previous perturbation step. Elsewhere, we are investigating adapting the value of R at each perturbation step.
2.3. Case study: Beeler–Reuter model
The BR cell model [20] is a system of ordinary differential equations (ODEs) describing the excitability of ventricular myocardial fibres. This ODE system models the temporal evolution of the transmembrane potential of a single ventricular cell via the dynamics of certain ion channels present in the cell membrane in response to the application of a sufficiently strong electrical stimulus such that once the stimulus is removed, the action potential does not immediately return to rest but rather undergoes a large excursion, before eventually reaching the resting state. While this model has been superseded by much more complex cardiac electrophysiological models [26,27], it still produces a realistic action potential and is ideal for our study in this paper.
We assume there is between-subject variability in the amplitude of the ionic currents and generate the POMs of this study by varying four model parameters (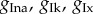 and
and 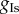 ) corresponding to the maximal current densities of the four ionic currents. A complete description of the model equations, the initial conditions, the stimulation protocol and implementation details are given in appendix C of the electronic supplementary material.
) corresponding to the maximal current densities of the four ionic currents. A complete description of the model equations, the initial conditions, the stimulation protocol and implementation details are given in appendix C of the electronic supplementary material.
Varying  has a direct influence on the shape of the action potential (AP). In order to characterize the data produced by a particular combination of these parameters, we compute a set of four biomarkers (figure 1) representing quantities of physiological interest from the AP profile generated by the model. Specifically, we consider the AP peak, the peak of the dome, the maximum upstroke velocity (defined as the maximum
has a direct influence on the shape of the action potential (AP). In order to characterize the data produced by a particular combination of these parameters, we compute a set of four biomarkers (figure 1) representing quantities of physiological interest from the AP profile generated by the model. Specifically, we consider the AP peak, the peak of the dome, the maximum upstroke velocity (defined as the maximum 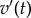 during the initial rapid depolarization of the cell membrane) and the action potential duration (APD90). The APD90 is defined as the difference between the repolarization and depolarization times. The depolarization time is measured as the time when the voltage reaches 10% of its full depolarization. The repolarization time is measured as the time when the voltage repolarizes to the same value, that is, the transmembrane potential reaches 90% of repolarization to its resting value. Linear interpolation is used to obtain better resolved time values.
during the initial rapid depolarization of the cell membrane) and the action potential duration (APD90). The APD90 is defined as the difference between the repolarization and depolarization times. The depolarization time is measured as the time when the voltage reaches 10% of its full depolarization. The repolarization time is measured as the time when the voltage repolarizes to the same value, that is, the transmembrane potential reaches 90% of repolarization to its resting value. Linear interpolation is used to obtain better resolved time values.
Figure 1.
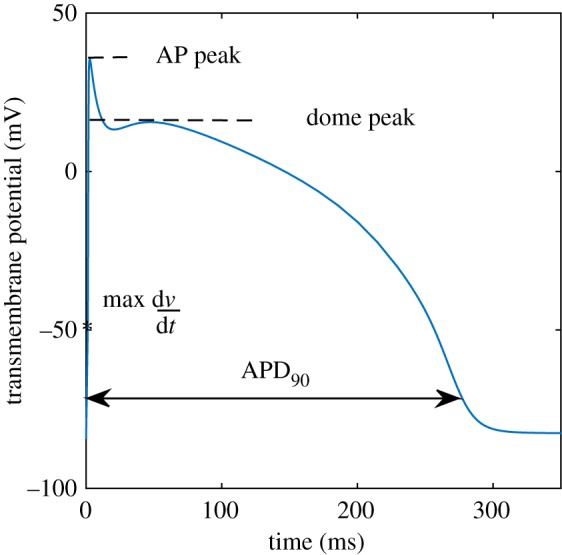
The action potential profile generated in the BR model by applying the stimulus 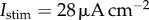 at t = 0 for two consecutive milliseconds. The four biomarkers are: the action potential peak, the dome peak, the APD90 and the maximum upstroke velocity (max
at t = 0 for two consecutive milliseconds. The four biomarkers are: the action potential peak, the dome peak, the APD90 and the maximum upstroke velocity (max 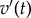 ) during the quick depolarization phase. (Online version in colour.)
) during the quick depolarization phase. (Online version in colour.)
As the exclusive use of these biomarkers may lead to a poor characterization of the AP profile, we additionally use a set of specific time points and their corresponding solution values to obtain a more informative characterization of the solution—see appendix C of the electronic supplementary material. One of our aims will be to see whether this additional information will help in the identification of the parameter distribution g(θ).
3. Results
For all results, we consider the approximate 95% CIs of the summary statistics to form the matching constraints unless otherwise specified.
3.1. Data
We simulate 10 trajectories from the model and assume they are 10 observations from different subjects in a population. We simulate the model by varying the four parameters 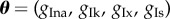 by ±10% around the vector of originally published values θT = (4, 0.35, 0.8, 0.09). We note the variability of the action potentials produced by this range of parameter values is typically less than what might be observed in experiments [2,3]. However, these simulated data are sufficient to allow us to perform a comparison of different methods for POM. To investigate how the POM methods perform with an increase in the dimension of the parameter vector, we consider four datasets where
by ±10% around the vector of originally published values θT = (4, 0.35, 0.8, 0.09). We note the variability of the action potentials produced by this range of parameter values is typically less than what might be observed in experiments [2,3]. However, these simulated data are sufficient to allow us to perform a comparison of different methods for POM. To investigate how the POM methods perform with an increase in the dimension of the parameter vector, we consider four datasets where 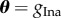 ,
, 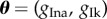 ,
, 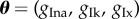 and
and  , where the datasets are generated by varying the relevant parameters and fixing the others at the values specified in θT. The resulting trajectories can be seen in grey in figure 2. We refer to these as parameter sets 1–4, respectively. For illustrative purposes, we set the original parameter space to search as ±50% of the values in θT, fixing parameters as necessary depending on the parameter set. In practice, this ‘prior’ range may be informed by experts.
, where the datasets are generated by varying the relevant parameters and fixing the others at the values specified in θT. The resulting trajectories can be seen in grey in figure 2. We refer to these as parameter sets 1–4, respectively. For illustrative purposes, we set the original parameter space to search as ±50% of the values in θT, fixing parameters as necessary depending on the parameter set. In practice, this ‘prior’ range may be informed by experts.
Figure 2.

Simulations from the BR model based on the SMC POM. The left-hand and right-hand columns are based on the biomarker statistics and biomarker/time statistics, respectively. From top to bottom shows results for parameter sets 1–4, respectively. The observed data are shown as grey trajectories, whereas the simulated trajectories from the POMs are shown in black.
To test the methods’ ability to handle an increasing number of constraints, we consider two sets of summary statistics. The first set consists of the four biomarkers presented in §2.3. The second option is to consider a set of summary statistics that includes the four biomarkers and the solution value corresponding to 28 selected time points for a total of 32 constraints. We refer to the first and second set of summaries as biomarker and biomarker/time statistics, respectively. The data are summarized in appendix D of the electronic supplementary material.
3.2. Performance comparison
The number of simulations required for SMC to generate N = 500 matches for the four different parameter sets is shown in table 1. Table 1 also shows the number of unique values out of 500 in the SMC particle sets. It can be seen that there is a steady increase in the number of simulations required as the number of parameters is increased. Further, more simulations are required for the biomarker/time statistics as the AP time statistics bring more information and a reduction in the parameter space consistent with the data.
Table 1.
Number of simulations required and number of unique values produced when running the SMC algorithm with N = 500. Shown are the mean values (and standard deviations in parentheses) from 10 independent runs and the estimated efficiency of the algorithm (mean number of simulations divided by N).
| dim(θ) | biomarker |
biomarker/time |
||||
|---|---|---|---|---|---|---|
| mean sims (s.d.) | mean unique (s.d.) | eff (%) | mean sims (s.d.) | mean unique (s.d.) | eff (%) | |
| 1 | 1792 (39) | 495 (3) | 28 | 2023 (98) | 495 (2) | 25 |
| 2 | 4531 (175) | 492 (3) | 11 | 5513 (116) | 494 (3) | 9 |
| 3 | 5131 (238) | 489 (2) | 10 | 6796 (118) | 488 (2) | 7 |
| 4 | 5587 (114) | 474 (5) | 9 | 8330 (157) | 473 (7) | 6 |
From tables 2–4, we can see that in terms of estimated efficiency, RND outperforms LHS over a one-dimensional parameter space. When the dimension of the parameter space is increased, the two methods become comparable in efficiency. It is evident that both the LHS and RND approaches do not scale to an increase in the parameter and summary statistic dimension compared with the SMC approach. For parameter set 4 and the biomarker/time statistics, the SMC approach has 6% efficiency, whereas it is only 1% for the other approaches.
Table 2.
Mean number of matches over 10 runs (with standard deviation shown in parentheses) using RND, along with the estimated efficiency.
| dim(θ) | biomarker |
biomarker/time |
||
|---|---|---|---|---|
| mean matches (s.d.) | eff (%) | mean matches (s.d.) | eff (%) | |
| 1 | 1322 (26) | 26 | 1196 (30) | 24 |
| 2 | 333 (20) | 7 | 256 (13) | 5 |
| 3 | 222 (13) | 4 | 115 (6) | 2 |
| 4 | 145 (10) | 3 | 41 (4) | 1 |
Table 3.
Mean number of matches over 10 runs of LHS (with standard deviation shown in parentheses) using a total of 5000 simulations, along with the estimated efficiency of the algorithm.
| dim(θ) | biomarker |
biomarker/time |
||
|---|---|---|---|---|
| mean matches (s.d.) | eff (%) | mean matches (s.d.) | eff (%) | |
| 1 | 1154 (4) | 23 | 756 (9) | 15 |
| 2 | 301 (14) | 6 | 222 (9) | 4 |
| 3 | 176 (13) | 4 | 80 (6) | 2 |
| 4 | 215 (15) | 4 | 66 (8) | 1 |
Table 4.
Mean number of matches over 10 runs of LHS (with standard deviation shown in parentheses) using a total of 10 000 simulations, along with the estimated efficiency of the algorithm.
| dim(θ) | biomarker |
biomarker/time |
||
|---|---|---|---|---|
| mean matches (s.d.) | eff (%) | mean matches (s.d.) | eff (%) | |
| 1 | 2306 (7) | 23 | 1510 (8) | 15 |
| 2 | 592 (23) | 6 | 434 (19) | 4 |
| 3 | 352 (11) | 4 | 151 (11) | 2 |
| 4 | 421 (19) | 4 | 133 (11) | 1 |
We also run the methods when the min/max range of the data are used to form the constraints instead of the approximate 95% CIs. We consider parameter set 4 and the biomarker/time statistics. We find that a smaller subset of the parameter space is consistent with these constraints, implying a more difficult sampling problem. This is likely due to the fact that we investigate only a small sample that produces a relatively narrow range. Here, n = 10, so that the range has expected coverage of size (n − 1)/(n + 1) or 9/11 of the population distribution of the corresponding statistic. Larger values of n would produce larger coverage probabilities. For a single run, the SMC approach needs 9700 simulations when N = 500. In contrast, with 50 000 simulations, LHS produces 129 matches. Thus, SMC produces an efficiency of roughly 5% versus 0.3% for LHS.
We also consider other datasets with larger variability ((−10%, +20%), (−10%, +40%), (−20%, +10%), (−20%, +20%), (−30%, +30%), (−40%, +10%), (−40%, +40%) around θT) and find that the efficiency gain of SMC is lost. In appendix E of the electronic supplementary material, we provide details of a more complex model with 12 parameters and a dataset with more realistic variability. The benefits of SMC become re-apparent. For the biomarker statistics, we obtain a 4.4% efficiency for SMC over 0.7% for LHS and for the biomarker/time statistics an efficiency of 3.7% for SMC over 0.01% for LHS. We expect the relative efficiency gain of SMC to increase as the relative volume of parameter space satisfying the constraints of the data decreases, which naturally occurs as the number of parameters increase and the variability in the data decreases.
3.3. Parameter range recovery
Owing to the increased efficiency of the SMC approach, we discuss the results obtained from the SMC algorithm in terms of the information content of the summary statistics in order to recover the parameter ranges used to generate the data. Note that if LHS and RND are run for long enough we find that the results they produce are similar to the output of SMC.
Figure 2 shows simulated trajectories (black) from parameter values kept from the SMC algorithm for both sets of summary statistics. It is evident from the left column that the four biomarkers are not capturing the majority of the information available in the observed trajectories (grey), because the between-subject variability of the simulated trajectories from the POM is substantially larger than that of the observed trajectories especially around the trough before the AP dome. Further, many parameter values are kept that lead to solution profiles that are well above the data between times 200–300 ms. The right column demonstrates that the set of parameter values kept when including additional constraints at various time points lead to solution profiles that follow the general shape of the observed trajectories well, but also capture the between-subject variability of the observed profiles.
Figure 3 shows the distributions of parameter values kept from SMC for the two sets of summary statistics for parameter set 1, and the distributions and bivariate scatter plots for parameter sets 2 and 3. The histograms and bivariate scatter plots for parameter set 4 are shown in figure 4. In general, there is a reduction in the parameter space consistent with the data when the time summary statistics are included, indicating that the four biomarkers do not carry all the information available in the data.
Figure 3.

Histograms and correlation plots from the SMC POM for parameter sets 1, 2 and 3 (top to bottom). In each histogram, the squares denote the interval from which the generated data for the considered parameter was sampled. The stars denote the actual minimum and maximum values of the parameter used to generate the data. The range of the x-axis gives the range of the parameter space explored. (a) Results obtained with the biomarker statistics. (b) Results obtained with the biomarker/time statistics. (Online version in colour.)
Figure 4.

Histograms and correlation plots from the SMC POM for parameter set 4. In each histogram, the squares denote the interval from which the generated data for the considered parameter was sampled. The stars denote the actual minimum and maximum values of the parameter used to generate the data. The range of the x-axis gives the range of the parameter space explored. (a) Results obtained with the biomarker statistics. (b) Results obtained with the biomarker/time statistics. (Online version in colour.)
The histograms in figures 3 and 4 show the ability of SMC to recover parameter ranges for the four parameter sets with appendix F of the electronic supplementary material showing the results for a single parameter. It is evident that when only one parameter is allowed to vary, the parameter values kept are constrained reasonably within the values used to generate the data.
From figures 3 and 4, we find that the first parameter, 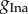 , is constrained to a similar range used to generate the data, regardless of how many parameters are varied and the choice of the summary statistics. It appears that the biomarkers may be sufficiently informative about
, is constrained to a similar range used to generate the data, regardless of how many parameters are varied and the choice of the summary statistics. It appears that the biomarkers may be sufficiently informative about 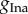 .
.
The results for the second parameter  can be seen in the second row of each correlation plot in figures 3a,b and 4a,b. When only two parameters are varying (second plot in columns of figure 3a,b), the biomarkers are able to constrain the parameters well. However, when three or four parameters are varying (bottom row of plots in figure 3a,b and both plots in figure 4), the biomarker/time statistics are necessary to constrain the parameters to a range similar to that used to simulate the data.
can be seen in the second row of each correlation plot in figures 3a,b and 4a,b. When only two parameters are varying (second plot in columns of figure 3a,b), the biomarkers are able to constrain the parameters well. However, when three or four parameters are varying (bottom row of plots in figure 3a,b and both plots in figure 4), the biomarker/time statistics are necessary to constrain the parameters to a range similar to that used to simulate the data.
The relevant rows of figures 3 and 4 demonstrate that in general there is a lack of information in the data to constrain the parameters 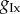 and
and 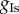 close to the ranges used to simulate the data. This phenomenon is often referred to as a lack of identifiability in the statistics literature. Here, we note that using the biomarker/time statistics provides improvements over just the biomarker statistics. One possible reason for the identifiability problem is that we observe only one out of the eight variables of the ODE model and observing more variables should improve identifiability. Another reason is that we expect complex relationships between parameters and the biomarker/time statistics which would, if known and exploited, provide improvements. Figures 3 and 4 show correlation plots and values for the parameters. For some combinations, plots suggest strong linear relations between parameters with correlation coefficients in excess of 0.8. In more complex situations, when the number of parameters exceeds four, it would be expected that a dimension reduction technique, such as principal components analysis or partial least-squares, could be applied to the parameter space to simplify and increase the robustness of the POM analysis.
close to the ranges used to simulate the data. This phenomenon is often referred to as a lack of identifiability in the statistics literature. Here, we note that using the biomarker/time statistics provides improvements over just the biomarker statistics. One possible reason for the identifiability problem is that we observe only one out of the eight variables of the ODE model and observing more variables should improve identifiability. Another reason is that we expect complex relationships between parameters and the biomarker/time statistics which would, if known and exploited, provide improvements. Figures 3 and 4 show correlation plots and values for the parameters. For some combinations, plots suggest strong linear relations between parameters with correlation coefficients in excess of 0.8. In more complex situations, when the number of parameters exceeds four, it would be expected that a dimension reduction technique, such as principal components analysis or partial least-squares, could be applied to the parameter space to simplify and increase the robustness of the POM analysis.
3.4. Out of sample predictions
POMs are often used to obtain out of sample predictions, that is, to test particular scenarios and observe how the variability of the calibrated population is affected under these particular circumstances. One example of interest in pharmacology is the case of conduction block of selected transmembrane ionic currents owing to drug action.
We assume that the injection of a suitable blocker significantly reduces the amplitude of one of the four ionic currents of the model (namely Ik) and observe the effects of this hypothesis on three populations of 200 models each, generated with the LHS, the SMC, and the RND methods, respectively. For all strategies, we start by selecting 200 values of θ generating matches with the considered experimental data when the min/max range is considered to constrain the biomarker/time statistics. In the first column of figure 5, we plot the solution trajectories corresponding to these three POMs (grey), and the 10 solution trajectories corresponding to our original dataset (black).
Figure 5.

Solution trajectories corresponding to the original dataset and the three calibrated populations of 200 models obtained with LHS, SMC and RND methods. The left-hand column represents the trajectories corresponding to the set of parameters θ (without drug block), whereas the right-hand column represents the trajectories corresponding to the set of parameters θmod (with drug block).
Under the drug block assumption, we then reduce by 75% the value of  in all the θ of our three POMs, obtaining three modified sets θmod of 200 parameters each. The solution trajectories corresponding to the new sets of parameters are shown in the right-hand column of figure 5 (grey). Once again, in each plot, we include 10 trajectories (black) corresponding to a reduction of 75% in the value of
in all the θ of our three POMs, obtaining three modified sets θmod of 200 parameters each. The solution trajectories corresponding to the new sets of parameters are shown in the right-hand column of figure 5 (grey). Once again, in each plot, we include 10 trajectories (black) corresponding to a reduction of 75% in the value of 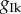 in the parameters corresponding to the original dataset.
in the parameters corresponding to the original dataset.
The 75% reduction in 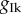 has some evident consequences on the solution profile: the resting membrane potential increases, the dome peak is much higher and the AP becomes significantly elongated, hence resulting in a larger APD90. However, a comparison of the three rows of figure 5 shows very minor differences in the results produced by the three POMs suggesting that the variability captured by the three strategies is essentially the same. The similarity of the results is further validated by looking at the effect of the drug block on the APD90 distribution produced by the three POMs as shown by figure 6a, and by considering the spread of the clouds of particles corresponding to the validated POM parameters (figure 6b).
has some evident consequences on the solution profile: the resting membrane potential increases, the dome peak is much higher and the AP becomes significantly elongated, hence resulting in a larger APD90. However, a comparison of the three rows of figure 5 shows very minor differences in the results produced by the three POMs suggesting that the variability captured by the three strategies is essentially the same. The similarity of the results is further validated by looking at the effect of the drug block on the APD90 distribution produced by the three POMs as shown by figure 6a, and by considering the spread of the clouds of particles corresponding to the validated POM parameters (figure 6b).
Figure 6.

APD90 distribution and scatter plot of parameter values for the three POMs considered in the study. (a) APD90 distribution for the three POMs considered, before (blue) and after (red) drug block. In each plot, we superimpose a kernel density estimate to the normalized histograms of the APD90 obtained from the solutions trajectories corresponding to the set of parameters θ and the set of modified parameters θmod. (b) Scatter plots and corresponding kernel density estimates for the location of the four components (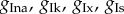 ) of the 200 parameters θmod obtained with the three POM strategies.
) of the 200 parameters θmod obtained with the three POM strategies.
When comparing our POMs before and after drug block, we observe a very interesting result. Although the constraints imposed on the summary statistics confine the solution trajectories of all three calibrated POMs, once drug block is applied a large proportion of the resulting solutions no longer falls within the limits given by the trajectories corresponding to the original dataset. The wider variety of AP shapes found in the drug block case is likely owing to all those parameters in the calibrated POMs that did not originally belong to the hypercube given by the min/max ranges of the data summary statistics. This does not have to be seen as a limitation of the POM approach. In fact, POMs are often used in electrophysiology to investigate whether abnormalities hide among experimental data and whether the application of a certain perturbation to the system (such as the injection of a drug) would lead to undesired electrophysiological effects such as delayed afterdepolarizations. This does not happen in the presented case, and all solutions remain reasonably smooth after the application of the drug. However, if more variability was observed in the experimental data and larger min/max ranges were used in the calibration of the POM, then different outcomes would be possible and abnormalities could easily arise. On the other hand, we believe that if data under the drug block assumption were available, then additional information obtained from these new trajectories could be used to further constrain the parameters of interest, thus improving our ability to recover the population distribution g(θ).
In appendix G of the electronic supplementary material, we show the results of the drug block test when calibrating the three POMs using the 95% CI constraints for the 32 biomarker/time statistics. This condition results in looser constraints than the ones given by the min/max range. Consequently, we find that the clouds of particles corresponding to the validated parameters are more spread out and cover a larger region of the parameter space (electronic supplementary material, figure S5b). Although more visible differences in the AP profiles (electronic supplementary material, figure S4) and in the APD90 distributions between the three POM strategies can be seen (electronic supplementary material, figure S5a), the overall behaviour before and after drug block remains unchanged.
4. Discussion
The three strategies considered for this study essentially differ in the way the search of the multidimensional parameter space is made when matches with the experimental data are sought. The results shown in the conduction block test revealed that all three strategies produced very similar POMs and captured a very similar degree of variability in AP responses both before and after drug block. However, this might not be the case when the experimental data are characterized by a larger degree of variability or when a parameter space of higher dimensionality is explored by each sampling strategy. Furthermore, the considerations made on the tightness of the validation criteria used to produce the calibrated POM highlight the need of a thorough investigation into the nature of the POM produced by different algorithms and into the capability of a given population to capture the variability of experimental data before a particular sampling strategy could be chosen over the many possible alternatives available in the literature.
We find that the SMC algorithm proposed in this paper is quite efficient in terms of the number of model simulations required when there is a moderate number of parameters and constraints, as for the BR model. The major disadvantage of the SMC approach adopted here is that it results in duplicated particles and also the MCMC step must be run for a fixed number of R iterations, even if a proposal has been accepted prior to the Rth iteration. The latter leads to some inefficiencies of the SMC approach. An alternative method to overcome both of these issues may be to use the Liu–West kernel [28] instead of an MCMC kernel.
It is possible that the ordering of the constraints may have an effect on the efficiency of SMC. In some applications, there may be a natural ordering of the constraints, as in time-series data. Using only the AP times as summary statistics is an example of this. In this case, it might be possible to save all information from the simulation run to the current time point and simulating forward to the next constraint without re-starting the simulation. However, in general, there is currently no method for determining the optimal ordering of constraints.
We selected the AP times based on a visual inspection of the data profiles in an attempt to include most of the information. We have not yet considered a more principled method for selecting these. We note, however, that our SMC method is robust to the inclusion of unnecessary constraints; if a constraint brings little additional information about the parameters, then very few (or even 0) of the particles will have zero weight and need to be replenished, resulting in little extra model simulations.
We will also consider emulating cardiac electrophysiological models using Gaussian processes [29] as well as using ideas from Sarkar & Sobie [30], who use a partial least-squares linear model as an emulator. We also plan to further compare the performance and the variability accounted for by POMs generated with the LHS and the SMC algorithms for more complex cardiac atrial electrophysiology models (see appendix E of the electronic supplementary material for the initial investigation on the Courtemanche–Ramirez–Nattel model).
Supplementary Material
Acknowledgements
K.B. thanks Blanca Rodriguez, Alfonso Bueno-Orovio, Ollie Britton and the Computational Cardiovascular group in the Department of Computer Science at the University of Oxford for on-going and deep discussions about populations of models.
Data accessibility
All data required to reproduce the results in this paper can be found in the appendices.
Authors' contributions
C.C.D. developed the SMC algorithm, performed analyses, interpreted results and drafted the article. N.C. performed analyses, interpreted results and drafted the article. S.P. and B.A.J.L. performed analyses and helped draft the article. A.N.P. designed the study, interpreted results and helped draft the article. K.B. designed the study, interpreted results and critically reviewed the article. P.B. interpreted results and critically reviewed the article. All authors gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
C.C.D. was supported by an Australian Research Council's Discovery Early Career Researcher Award funding scheme (DE160100741). K.B. was supported by an Australian Research Council Discovery Project (DP120103770). A.N.P. was supported by an Australian Research Council Discovery Project (DP110100159).
References
- 1.Marder E, Taylor AL. 2011. Multiple models to capture the variability in biological neurons and networks. Nat. Neurosci. 14, 133–138. ( 10.1038/nn.2735) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ, Rodriguez B. 2013. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc. Natl Acad. Sci. USA 110, E2098–E2105. ( 10.1073/pnas.1304382110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sánchez C, Bueno-Orovio A, Wettwer E, Loose S, Simon J, Ravens U, Pueyo E, Rodriguez B. 2014. Inter-subject variability in human atrial action potential in sinus rhythm versus chronic atrial fibrillation. PLoS ONE 9, e105897 ( 10.1371/journal.pone.0105897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Leary T, Marder E. 2014. Mapping neural activation onto behavior in an entire animal. Science 344, 372–373. ( 10.1126/science.1253853) [DOI] [PubMed] [Google Scholar]
- 5.Sarkar AX, Christini DJ, Sobie EA. 2012. Exploiting mathematical models to illuminate electrophysiological variability between individuals. J. Physiol. 590, 2555–2567. ( 10.1113/jphysiol.2011.223313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walmsley J, Rodriguez JF, Mirams GR, Burrage K, Efimov IR, Rodriguez B. 2013. mRNA expression levels in failing human hearts predict cellular electrophysiological remodelling: a population based simulation study. PLoS ONE 8, e56359 ( 10.1371/journal.pone.0056359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gemmell P, Burrage K, Rodriguez B, Quinn TA. 2014. Population of computational rabbit-specific ventricular action potential models for investigating sources of variability in cellular repolarisation. PLoS ONE 9, e90112 ( 10.1371/journal.pone.0090112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saltelli A, Tarantola S, Chan KP-S. 1999. A quantitative model-independent method for global sensitivity analysis of model output. Technometrics 41, 39–56. ( 10.1080/00401706.1999.10485594) [DOI] [Google Scholar]
- 9.Zhou X, Bueno-Orovio A, Orini M, Hanson B, Hayward M, Taggart P, Lambiase PD, Burrage K, Rodriguez B. 2013. Population of human ventricular cell models calibrated with in vivo measurements unravels ionic mechanisms of cardiac alternans. Proc. Comput. Cardiol. Conf. (CINC) 40, 855–858. [Google Scholar]
- 10.Muszkiewicz A, Bueno-Orovio A, Liu X, Casadei B, Rodriguez B. 2014. Constructing human atrial electrophysiological models mimicking a patient-specific cell group. Proc. Comput. Cardiol. Conf. (CINC) 41, 761–764. [Google Scholar]
- 11.Burrage K, Burrage P, Donovan D, Thompson B. 2015. Populations of models, experimental designs and coverage of parameter space by Latin hypercube and orthogonal sampling. Proc. Comput. Sci. 51, 1762–1771. ( 10.1016/j.procs.2015.05.383) [DOI] [Google Scholar]
- 12.McKay MD, Beckman RJ, Conover WJ. 1979. A comparison of three methods for selecting values of input variables in the analysis of output from a computer code. Technometrics 21, 239–245. [Google Scholar]
- 13.Stein M. 1987. Large sample properties of simulations using Latin hypercube sampling. Technometrics 29, 143–151. ( 10.1080/00401706.1987.10488205) [DOI] [Google Scholar]
- 14.Beaumont MA, Zhang W, Balding DJ. 2002. Approximate Bayesian computation in population genetics. Genetics 162, 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blum MGB, Nunes MA, Prangle D, Sisson SA. 2013. A comparative review of dimension reduction methods in approximate Bayesian computation. Stat. Sci. 28, 189–208. ( 10.1214/12-STS406) [DOI] [Google Scholar]
- 16.Marjoram P, Molitor J, Plagnol V, Tavaré S. 2003. Markov chain Monte Carlo without likelihoods. Proc. Natl Acad. Sci. USA 100, 15 324–15 328. ( 10.1073/pnas.0306899100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sisson SA, Fan Y, Tanaka MM. 2007. Sequential Monte Carlo without likelihoods. Proc. Natl Acad. Sci. USA 104, 1760–1765. ( 10.1073/pnas.0607208104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drovandi CC, Pettitt AN. 2011. Estimation of parameters for macroparasite population evolution using approximate Bayesian computation. Biometrics 67, 225–233. ( 10.1111/j.1541-0420.2010.01410.x) [DOI] [PubMed] [Google Scholar]
- 19.Vo BN, Drovandi CC, Pettitt AN, Pettet GJ. 2015. Melanoma cell colony expansion parameters revealed by approximate Bayesian computation. PLoS Comput. Biol. 11, e1004635 ( 10.1371/journal.pcbi.1004635) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beeler G, Reuter H. 1977. Reconstruction of the action potential of ventricular myocardial fibres. J. Physiol. 268, 177–210. ( 10.1113/jphysiol.1977.sp011853) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishop CM. 2006. Pattern recognition and machine learning. New York, NY: Springer. [Google Scholar]
- 22.Gibbons JD. 1971. Nonparametric statistical inference. New York, NY: McGraw-Hill. [Google Scholar]
- 23.Del Moral P, Doucet A, Jasra A. 2006. Sequential Monte Carlo samplers. J. R. Stat. Soc. B (Stat. Methodol.) 68, 411–436. ( 10.1111/j.1467-9868.2006.00553.x) [DOI] [Google Scholar]
- 24.Chopin N. 2002. A sequential particle filter method for static models. Biometrika 89, 539–551. ( 10.1093/biomet/89.3.539) [DOI] [Google Scholar]
- 25.Golchi S, Campbell D. 2016. Sequentially constrained Monte Carlo. Stat. Comput. 97, 98–113. ( 10.1016/j.csda.2015.11.013) [DOI] [Google Scholar]
- 26.O'Hara T, Virág L, Varró A, Rudy Y. 2011. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7, e1002061 ( 10.1371/journal.pcbi.1002061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grandi E, Pasqualini FS, Bers DM. 2010. A novel computational model of the human ventricular action potential and Ca transient. J. Mol. Cell Cardiol. 48, 112–121. ( 10.1016/j.yjmcc.2009.09.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, West M. 2001. Combined parameter and state estimation in simulation-based filtering. In Sequential Monte Carlo methods in practice (eds Doucet A, de Freitas N, Gordon N), pp. 197–223. New York, NY: Springer; ( 10.1007/978-1-4757-3437-9_10) [DOI] [Google Scholar]
- 29.Chang ETY, Strong M, Clayton RH. 2015. Bayesian sensitivity analysis of a cardiac cell model using a Gaussian process emulator. PLoS ONE 10, e0130252 ( 10.1371/journal.pone.0130252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar AX, Sobie EA. 2010. Regression analysis for constraining free parameters in electrophysiological models of cardiac cells. PLoS Comput. Biol. 6, e1000914 ( 10.1371/journal.pcbi.1000914) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data required to reproduce the results in this paper can be found in the appendices.