

Abstract
Protein misfolding is implicated in neurodegenerative diseases such as ALS, where mutations of superoxide dismutase 1 (SOD1) account for about 20% of the inherited mutations. Human SOD1 (hSOD1) contains four cysteines, including Cys57 and Cys146, which have been linked to protein stability and folding via forming a disulfide bond, and Cys6 and Cys111 as free thiols. But the roles of the cellular oxidation-reduction (redox) environment in SOD1 folding and aggregation are not well understood. Here we explore the effects of cellular redox systems on the aggregation of hSOD1 proteins. We found that the known hSOD1 mutations G93A and A4V increased the capability of the thioredoxin and glutaredoxin systems to reduce hSOD1 compared with wild-type hSOD1. Treatment with inhibitors of these redox systems resulted in an increase of hSOD1 aggregates in the cytoplasm of cells transfected with mutants but not in cells transfected with wild-type hSOD1 or those containing a secondary C111G mutation. This aggregation may be coupled to changes in the redox state of the G93A and A4V mutants upon mild oxidative stress. These results strongly suggest that the thioredoxin and glutaredoxin systems are the key regulators for hSOD1 aggregation and may play critical roles in the pathogenesis of ALS.
Keywords: ALS (Lou Gehrig disease), reduction-oxidation (redox), oxidative stress, protein aggregation, thiol, thioredoxin, cysteine 111, SOD1, glutaredoxin, glutathione
Introduction
ALS is a neurodegenerative disease in which the main feature is a loss of motoneurons and muscle atrophy. About 10% of cases are inherited and described as familial form of ALS (fALS).4 fALS is associated with mutations in the genes C9orf72, SOD1, TDP-43, and FUS, with C9orf72 being the most prevalent and followed by SOD1 (1–3). In fact, the first reported gene related to fALS is Cu,Zn superoxide dismutase 1 (SOD1) (4), and since then, more than a hundred mutations associated with fALS have been identified in human SOD1 (5, 6). Among them, the point mutation A4V is one of the most invasive and prevalent mutations associated with human ALS (5, 7), and G93A is used to produce a widely employed transgenic mouse model (8, 9). SOD1 is a cytosolic ubiquitous enzyme that scavenges superoxide radicals in every cell type of the human body, including erythrocytes (1, 5). However, mutations induce exclusively cell death of motoneurons and, to a lesser extent, other neurons (10). In a metal-coordinated state, SOD1 forms a homodimer to accomplish its full enzymatic activity. Copper is necessary for the enzymatic activity, whereas zinc mainly works in maintaining the protein structure (5) (Fig. 1). A conformational change in mutant SOD1 may increase the accessibility of other substrates to copper in the protein to generate reactive oxygen or nitrogen species. Because most mutations associated so far with fALS do not show a reduction in SOD1 enzymatic activity, it has been postulated that the deleterious action of mutated SOD1 is probably due to a gain of toxic function rather than the expected increase in the concentration of free radicals (4). Several cellular mechanisms, such as mitochondrial dysfunction, NMDA-mediated excitoxicity, altered axonal transport, proteasome inhibition, endoplasmic reticulum stress, and so on have been suggested to initiate and promote neuronal death (2, 10, 11).
FIGURE 1.

Representation of the Cu,Zn human SOD1 molecule. A, linear representation of SOD1, indicating the position of cysteines. Each subunit coordinates one atom of each of copper and zinc. The disulfide bond between Cys57 and Cys146 determines a flexible loop that contains Cys111. B, left panel, the dimeric form of human SOD1, PDB code 2C9V, is represented using VMD software. Each monomer shows the eight β sheets. One monomer is shown in magenta and the other in cyan. Cu2+ is depicted in red and Zn2+ in blue. Cysteines are shown in yellow. Notice that cysteines are located face-to-face in the interface between dimers. The right panel corresponds to the same representation, but the molecules have been turned 180° over its x axis. C, surface representation of the molecule. Cys111 is the unique cysteine that has access to the surface of the molecule. The right panel, as in B, shows the molecule after a rotation of 180°.
A hallmark in neurodegenerative disease is the presence of protein aggregates in the nervous tissue of patients. Indeed, SOD1 makes aggregates in the spinal cord of fALS patients and transgenic mice mimicking ALS-carrying mutations of SOD1 (8). Although it is not fully clear whether the aggregates are toxic or represent a kind of self-inactivation system, it is well known that they are the results of protein misfolding (12). Metals are essential for the correct folding of SOD1. The apoenzyme is prone to aggregation (13). Additionally, cysteines have been implicated to be involved in aggregation. SOD1 mutants are susceptible to have more aggregation propensity (14, 15), and they are also prone to disulfide reduction, unfolding, and misfolding (16, 17). SOD1 has four cysteines in positions 6, 57, 111, and 146. According to the crystal structure of SOD1, there is an intramolecular disulfide bond between Cys57 and Cys146 (Fig. 1). When cysteines were substituted by site-directed mutagenesis to other amino acids, the intracellular SOD1 aggregates decreased (18–20). Cys111 is located on the surface of the molecule, where it can be oxidized, changing the molecular conformation of the protein (Fig. 1) (21), and it contributes to keeping the Cu2+ in the correct position (22).
The quaternary structure and stability of SOD1 are controlled by the intramolecular disulfide bond. The equilibrium between oxidized and reduced forms of the disulfide bond of SOD1 should be highly dependent on the redox potential of the cytosolic environment, which is mainly controlled by Trx or Grx systems (23). The Trx system is composed of NADPH, thioredoxin reductase (TrxR), and Trx, whereas the Grx system is composed of NADPH, glutathione reductase (GR) and GSH, and Grx (23, 24). Changes in the cytoplasmic redox potential would influence the reduction or the oxidation of the disulfide bond of WT or mutant SOD1 proteins, rendering reactive free cysteines that could interact with thiol groups of other molecules forming new aggregates. Human Grx1 was reported to be able to reduce apo-hSOD1 (25, 26), and overexpression of Grx2 prevents the aggregation of mutant SOD1 in mitochondria, preserves mitochondrial function, and protects neuronal cells against apoptosis induced by mutant SOD1 (27).
Using force-clamp atomic force spectroscopy, we have shown previously that a nucleophilic attack of Cys111 toward either Cys57 or Cys146 in a SOD1 single molecule can occur. The reactivity of Cys111 toward the conserved disulfide bond in WT and G93A or A4V mutant SOD1 is different (28). Our aim here is to investigate the effects of the Trx and Grx systems on the redox state of wild-type and G93A and A4V mutant hSOD1 to evaluate the contribution of cellular redox environment change to SOD1 aggregation and ALS progression.
Results
Reduction of hSOD1 by the Thioredoxin and GSH-Grx Systems in Vitro
To detect whether the mutations in ALS-linked SOD1 affect the susceptibility of SOD1 to reductants, we investigated the reduction of the WT and mutants G93A and A4V of hSOD1 by the GSH-Grx and Trx systems (Fig. 2A). When native oxidized hSOD1 (Fig. 2A, lane 6) was incubated with strong reductants, including a high concentration of GSH (10 mm) (Fig. 2A, lane 1) or DTT (10 mm) (Fig. 2A, lane 11) the proteins were fully reduced, which is consistent with a previous report (25). A lower concentration of GSH (1 mm) alone could partially reduce disulfides in mutant G93A hSOD1 but not in WT hSOD1 (Fig. 2A, lane 2). GR plus NADPH did not affect the redox state of SOD1 proteins and could not increase the reduction efficiency of 1 mm GSH for the oxidized WT and mutant SOD1 proteins (Fig. 2A, lanes 3 and 4). The presence of Grx1 in the GSH-Grx system enhanced the reduction of disulfide in G93A SOD1 protein. In contrast, the whole GSH-Grx system had a little reduction of disulfide in WT and A4V SOD1 proteins (Fig. 2A, lane 5).
FIGURE 2.

The effects of the mutation in SOD1 on its reduction by the GSH-Grx or Trx systems and catalytic activity. A, after WT, G93A, and A4V hSOD1 proteins were incubated with components of the GSH-Grx and Trx systems for 1 h, the proteins were separated on a non-reducing SDS-PAGE, and SOD1 was detected by Western blotting. Lane 1, 10 mm GSH; lane 2, 1 mm GSH; lane 3, 1 mm GSH plus 100 nm GR and 200 μm NADPH; lane 4, 100 nm GR and 200 μm NADPH; lane 5, 1 mm GSH, 100 nm GR, and 200 μm NADPH plus 2 μm Grx1; lane 6, control; lane 7, 100 nm TrxR1 and 200 μm NADPH; lane 8, 100 nm TrxR1 and 200 μm NADPH plus 10 μm Trx1; lane 9, 100 nm TrxR1 and 200 μm NADPH plus 20 μm Trx1; lane 10, 100 nm TrxR1 and 200 μm NADPH plus 40 μm Trx1; lane 11, 10 mm DTT. Red, Reduced; ox, oxidized. B, effects of mutation in SOD1 on its activity. SOD1 activity was assayed by detecting the scavenging effects on superoxide generated by xanthine and xanthine oxidase. Open circles correspond to xanthine oxidase activity to produce superoxide. Open squares, open triangles, and inverted open triangles correspond to the activity of 0.1, 1, and 10 units of human erythrocytes SOD1, respectively. The activity of recombinant hSOD1 is represented as red circles (WT), blue circles (G93A), and green circles (A4V). The figure is a representative of three experiments.
None of the oxidized SOD1 proteins were substrates of TrxR1 (Fig. 2A, lane 7); only the whole Trx system could reduce them. The Trx system reduced the oxidized A4V and G93A mutant SOD1 proteins efficiently but only gave a slight reduction for oxidized WT hSOD1 (Fig. 2A, lanes 8–10). Increasing the concentration of Trx1 did not enhance the reduction capacity (Fig. 2A, lanes 8–10). Mutation of SOD1 did not result in significant SOD1 activity change (Fig. 2B). The activities of recombinant proteins hSOD1 were 5.6 ± 0.6 units/μg for WT, 4.3 ± 1.8 units/μg for G93A, and 8.3 ± 1.4 units/μg for A4V, respectively. The presence of thiol-dependent redox systems did not change the enzymatic activity of hSOD1. Neither GSH nor the individual reductants of the Trx system affected the SOD1 activity of the G93A or A4V mutants (data not shown).
Regulation of SOD Aggregation in the Cells by the Thioredoxin and Grx Systems
To explore whether the Trx and GSH-Grx systems are key regulators of the aggregation of hSOD1 in cells, we generated RT4-D6P2T schwannoma cells stably expressing hSOD1-GFP fusion proteins to visualize their cellular location. We used aurothioglucose (ATG) to inhibit the cytosolic TrxR1 and buthionine sulfoximine (BSO), which blocks the synthesis of GSH in cells, to see the effect of the Trx and Grx systems on SOD1 aggregation. Three different fluorescent cells, WT hSOD1-GFP, G93A hSOD1-GFP, and A4V hSOD1-GFP, were used to observe insoluble protein accumulations (Fig. 3A). Under resting conditions, fluorescence is spread over the cytoplasm (Fig. 3B, top panels). In all untreated cells, very few fluorescent bright spots in the cytoplasm were observed (Fig. 3B, top panels).
FIGURE 3.

The effect of inhibition of Trx1 by ATG or depletion of GSH by BSO on hSOD1 aggregation in cultured cells. A, SOD1 amount in transfected cell lines. B, SOD1-GFP stably transfected cells expressing WT, G93A, and A4V SOD1 were treated with 100 μm ATG or 100 μm BSO for 24 h. GFP aggregation in the cells was detected with a confocal microscope. C, the amounts of aggregates in each cell were analyzed by florescence microscopy at higher magnification (×400). Shown are mean values found in the WT (n = 69), WT with BSO (n = 91), WT with ATG (n = 103), A4V (n = 70), A4V with BSO (n = 168), A4V with ATG (n = 95), G93A (n = 85), G93A with BSO (n = 91), and G93A with ATG (n = 85). *, p < 0.05; **, p < 0.01; treated cells versus untreated cells.
ATG inhibits TrxR1 by targeting the essential selenocysteine residue, and BSO blocks GSH synthesis by inhibiting glutathione synthase, and thus treatment with 100 μm ATG or BSO for 24 h caused an alteration of the reducing capacity of the Trx and GSH systems, respectively. These treatments did not affect cell viability (data not shown). Using a confocal microscope, no remarkable morphologic changes were observed in the WT hSOD1-GFP cell line. In contrast, in cells overexpressing G93A hSOD1-GFP and A4V hSOD1-GFP, we detected condensed GFP fluorescent spots, mostly spherical, with sizes ranging from 0.2–20 μm2 and apparently located in the cytoplasm with no special distribution (Fig. 3B, center and bottom panels). The cells were analyzed with confocal florescence microscopy at a higher magnification (×400) to see the contents of the aggregates in cells (Fig. 3C). In WT cells, treatment with ATG did not show any increase in the aggregate amount, but treatment with BSO resulted in a slight increase. The aggregates in cells expressing G93A hSOD1-GFP and A4V hSOD1-GFP showed a significant increase upon treatment with BSO and ATG. These results indicated that the cellular redox environment changes that are regulated by the Trx and GSH systems are important for mutant hSOD1 aggregation in the cytoplasm.
To further understand how the two cellular thiol-dependent redox systems change upon treatment with ATG and BSO, we detected protein levels of TrxR1, Trx1, and GR in the cells using Western blotting (Fig. 4). In all cells, the expression level of TrxR1 was elevated upon treatment with BSO but not in cells treated with ATG (Fig. 4, first lane). No significant differences were detected in terms of GR level in all cells (Fig. 4, fifth lane). Interestingly, treatment with BSO did not cause dramatic changes for Trx1 in cells expressing WT SOD1 but resulted in the enhancement of Trx1 in cells expressing mutant SOD1. Treatment with ATG induced an increase of Trx1 in cells overexpressing A4V SOD1 but not in cells overexpressing WT SOD1 (Fig. 4, third lane). These results indicate that the inhibition of electron transfer in one pathway may activate the expression of the other system. Cells overexpressing WT SOD1 also exhibited some difference in terms of the response to oxidative stress compared with cells expressing mutant SOD1.
FIGURE 4.
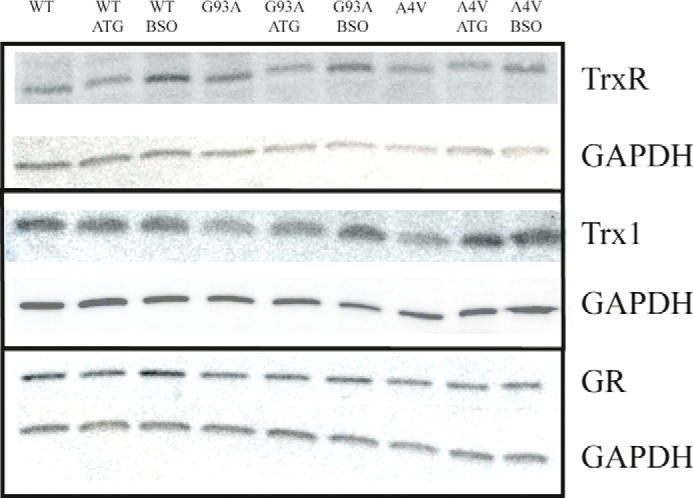
Alteration of protein level of the thiol-dependent redox system caused by treatment with ATG or BSO. Stably transfected cells expressing WT, G93A, or A4V SOD1 were treated with 100 μm ATG or 100 μm BSO for 24 h. Then the cells were harvested, and the proteins were detected by Western blotting.
Cys111 Is Critical for hSOD1 Aggregation upon Alteration of the Trx and GSH Systems
Because Cys111 has been shown to be a critical amino acid residue in the redox regulation of SOD1 and may affect the aggregation and formation of inclusion body-like structures of mutant SOD1 (29, 30), we prepared stably transfected cell lines with expression of SOD1 harboring a Cys111 mutation to detect the effects of alteration of the Trx and GSH systems on SOD1 aggregation (Fig. 5). Compared with the results shown in Fig. 3, similar to cells expressing WT hSOD1, cells expressing C111G hSOD1 had little aggregates upon treatment with BSO and ATG (Fig. 5A). For cells expressing G93A and A4V mutant hSOD1, treatment with ATG or BSO showed an increase in aggregation (Fig. 3), whereas mutations in Cys111 prevented aggregate formation upon treatment with ATG and BSO in all cells (Fig. 5A, center and right panels). The amount of aggregates per cell in G93A/C111G- and A4V/C111G-expressing cells stayed at the same level upon treatment with BSO and ATG (Fig. 5B). This cumulative plot of probability of the size of hSOD1 aggregate in cells showed that treatment with BSO and ATG enlarged the probability of aggregates with a bigger area in the mutations G93A and A4V (Fig. 5C, top panels), but when Cys111 was mutated, the size of aggregates decreased in both cases and the aggregates were distributed as under untreated conditions (Fig. 5C, bottom panels). These results indicate that Cys111 in hSOD1 is a critical amino acid residue for the formation of SOD1 aggregate during thiol-dependent redox environment change.
FIGURE 5.

The effect of Cys111 in hSOD on hSOD1 aggregation upon treatment with ATG or BSO. A, SOD1-GFP stably transfected cells expressing C111G, G93A/C111G, and A4V/C111G SOD1 were treated with 100 μm ATG or 100 μm BSO for 24 h. GFP aggregation in the cells was detected with a confocal microscope. B, the amounts of aggregates in each cell were analyzed by fluorescence microscopy at higher magnification (×400). Mean values were counted at C111G (n = 49), C111G with BSO (n = 78), C111G with ATG (n = 82), A4V/C111G (n = 70), A4V/C111G with BSO (n = 68), A4V/C111G with ATG (n = 105), G93A/C111G (n = 115), G93A with BSO (n = 104), and G93A with ATG (n = 89). C, cumulative plot of probability of the size of hSOD1aggregates. The probability of finding a specific interval size of aggregate was plotted accumulatively. Black lines correspond to untreated conditions, red lines to ATG treatment, and blue lines to BSO treatments.
ROS Level Detection under GSH Depletion
Treatment with BSO and ATG can cause a redox environment change, which may result in an alteration of ROS production. We used luminol chemiluminescence to detect the ROS level upon treatment with BSO. Superoxide reacts with luminol, which is oxidized, and light is emitted and detected (31). After the treatment with BSO, the ROS level significantly increased in cells expressing WT hSOD1, C111G, A4V, and A4V/C111G (Fig. 6).
FIGURE 6.
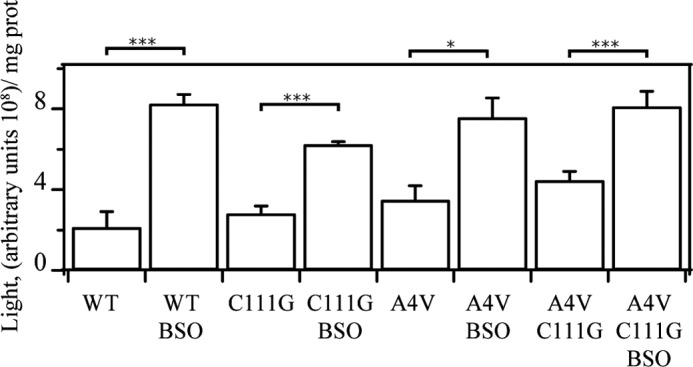
ROS level detection in cell lysates. Cells (1 × 106) were treated with BSO for 24 h and then lysed in PBS buffer. ROS levels in the lysates were measured by a luminol-dependent chemiluminescence method. Data were obtained from n = 6. *, p < 0.05; ***, p < 0.001.
SOD1 Redox State in Cells
In addition to studying the effects of treatment with ATG and BSO on cell morphology, we analyzed the redox state of hSOD1-GFP molecules in cells harboring single- and double-mutant hSOD1 by redox Western blotting (Fig. 7). In cells expressing WT-SOD1-GFP, there were no changes in the ratio of the reduced and oxidized form of SOD1 upon treatment with BSO or ATG. In cells expressing G93A SOD1-GFP, treatment with either BSO or ATG increased the ratio of oxidized forms (Fig. 7). In cells expressing A4V SOD1-GFP, the oxidized forms were increased significantly under both treatment conditions compared with untreated cells. We also found that mutation at Cys111 made the redox state of G93A/C111G and A4V/C111G SOD1 independent of cellular redox environment change caused by treatment with ATG or BSO (Fig. 7).
FIGURE 7.
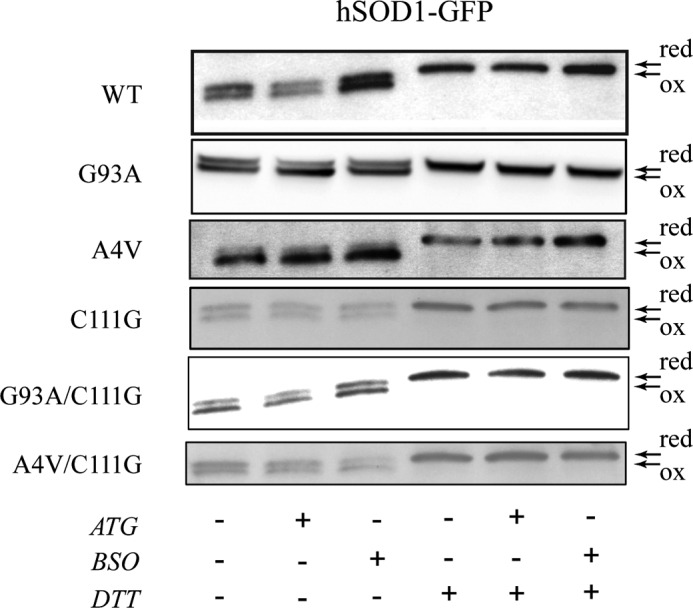
Analysis of the redox state of SOD1 in cell lysates. After RT4-D6P2T cells expressing WT and mutant hSOD1 were treated with BSO and ATG, the cells were lysed in buffer containing 20 mm iodoacetamide to block the redox state of the proteins. The protein were separated under non-reducing SDS-PAGE (left lanes) or in reducing condition (right lanes, DTT) and detected by Western blotting. red, reduced; ox, oxidized.
MS/MS Analysis for the Disulfide Formation in Recombinant Proteins
To investigate whether the role of Cys111 in the hSOD1 aggregation involves the formation of disulfides, we used MS/MS to detect the disulfide formation in recombinant A4V SOD1 proteins. The protein was analyzed as purified. Besides the structural disulfide between Cys57 and Cys146, the peptide V4VCVLK9-H80VGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGR114 was identified with high confidence with a 5+ precursor with a p value of 1.68e−6 and with a 6+ precursor with a p value of 8.9e−8 (Fig. 8, A and B), indicating that the disulfide of Cys6-Cys111 was formed. Fig. 8, C and D, shows the extracted ion chromatography of peptide L144ACGVIGIAQ153-H80VGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGR114 and MS/MS fragmentation annotation of the higher energy collisional dissociation spectrum of the 5+ peptide, indicating that a disulfide between Cys111 and Cys146 (p = 5.25e−4) was present. In contrast, a disulfide between Cys57 and Cys146 was detected, but disulfides of Cys6-Cys111 and Cys111-Cys146 were not detected in purified recombinant WT hSOD1 protein.
FIGURE 8.

MS/MS analysis of purified recombinant A4V hSOD1 protein. A, extracted ion chromatography of peptide V4VCVLK9-H80VGDLGNVTADKDGVADVSIEDSVISLSG DHCIIGR114 and the isotopic pattern of the 6+ ions. B, MS/MS fragmentation annotation of the HCD spectrum of the 6+ peptide, indicating the disulfide bond between Cys6 and Cys111. C, extracted ion chromatography of peptide L144ACGVIGIAQ153-H80VGDLG NVTADKDGVADVSIEDSVISLSGDHCIIGR114 and the isotopic pattern of the 5+ ions. D, MS/MS fragmentation annotation of the HCD spectrum of the 5+ peptide, indicating the disulfide bond between Cys111 and Cys146. Upper fragments are counted from peptide 144LACGVIGIAQ153, whereas the bottom fragments are counted from the other peptide.
Discussion
ALS is a complex disease in which the main targets are the cortical, brain stem, or spinal cord motoneurons. The intracellular protein aggregate of SOD1 is a hallmark character in nervous tissue of fALS patients (32). It has been described that cysteines play an important role in ALS-linked SOD1 misfolding (14, 33–40). The Trx and Grx systems are the two major cellular disulfide reductase systems. Trx family proteins control protein folding (41), regulating protein disulfide formation. We show here that both the Trx and Grx systems can reduce the oxidized forms of ALS-linked A4V and G93A hSOD1 mutants (Fig. 2A), which is consistent with previous reports (25–27). WT hSOD1 is much more resistant to reduction by both systems. Disulfide reduction and metal ion removal may trigger SOD1 aggregation (42), but, in fact, disulfide bonds cause the initiation of its fibrillation (43). In our study, the reduced mutant hSOD1 was detected to be catalytically active.
Oxidative stress has been indicated to be a key factor for neuron cell injury during ALS progression (11). The ROS level is reported to be increased in ALS patients because antioxidant enzymes, including glutathione S-transferase, peroxiredoxins, and the redox-sensitive transcription factor Nrf2 are down-regulated, and NADPH oxidase activity is elevated, in motor neuronal cells (32). Here we found that interruption of the Trx system by treatment with ATG or depletion of GSH by BSO treatment caused an increase in ROS level and aggregation of mutant SOD1 but not WT SOD1 (Fig. 3). This is consistent with the result that the redox state of WT SOD1 was hardly affected by treatment with ATG and BSO (Fig. 7), although the ROS level was increased in all tested cells, including those expressing WT SOD1, following treatment with BSO (Fig. 6). Trx-dependent peroxidase 2 (Prx2), and glutathione peroxidase 1 (GPx1) are colocalized with SOD1 in neuronal Lewy body-like hyaline inclusions in the spinal cords of fALS patients (44).
An important observation is that Cys111 is critical for the formation of hSOD1 aggregates (Fig. 5) (45). Although Cys111 is not the only residue to mediate misfolding/aggregation, it is the only Cys residue present in SOD1 from primates, and its role in ALS pathophysiology has been considered (28, 45). Because of its location in the surface of the protein, Cys111 may interact with Cys57 or Cys146 and reorganize the intramolecular disulfide bond (20) and would reorient the Cu2+. As it has been described, glutathionylation of Cys111 increases the proportion of highly fibrillogenic hSOD1 monomers (46, 47), interrupting the dimer contact at the interface stereochemically and causing the dissociation. Indeed, the point mutation of Cys111 in A4V and G93A mutant SOD1 proteins reduces the amount of aggregates (Fig. 5) (30, 48, 49).
A potential mechanism involving the role of Cys111 in hSOD1 mutant protein aggregation is proposed, as shown in Fig. 9, which also shows how the Trx and GSH-Grx systems participate in this process. Mutation in hSOD1 dimers exposes its conserved Cys57-Cys146 disulfide on the protein surface in either one or both subunits (28, 29), where it is easy to be reduced by the Trx and Grx systems into dithiol form, followed by the dimer dissociation. We can also find non-conserved disulfide bond forms, as Cys6-Cys111 and Cys146-Cys111 in SOD1 dimers, that are in a balance because of the Trx-GSH/Grx redox systems. High accessibility of the exposure to the Trx and Grx systems may lead to intermolecular thiol/disulfide exchange reactions, the dissociation of the dimers, and misfolding when they are not in an equilibrium (29, 37, 50). The monomers, which are released by the protein reduction, could promote the non-native and less stable disulfides. Then, under the oxidation state, including treatment with BSO or ATG, mainly Cys111 can intermolecularly attack a cysteine from another SOD1 molecule, particularly Cys146, which is exposed to form an intermolecular disulfide. Cys146 was indicated previously to be involved in the formation of SOD1 insoluble multimers and aggregates (37, 51), and it is in agreement with our results, as disulfide bonds between Cys111 and Cys146 were identified in MS experiment. These intermolecular disulfide bonds will induce the formation of insoluble high molecular weight aggregates, and it is not necessary for the proteins to be fully reduced to start polymerization (Fig. 9) (52, 53). Therefore, the formation of insoluble SOD1 oligomeric inclusions, which is commonly described in fALS cases, is promoted by the modification or destabilization of the redox cytosolic environment.
FIGURE 9.

A potential mechanism for mutant hSOD1 aggregation. The Trx and GSH-Grx systems may play dual roles in hSOD1 mutant protein aggregation. A, WT SOD1 is highly resistant to GSH-Grx reducing conditions because the disulfide bond is located intramolecularly. B, on the contrary, mutant hSOD1 protein is easier to be reduced by the Trx and GSH-Grx systems, reflecting that disulfide bonds in the molecule are mostly exposed to the external environment compared with hSOD1 WT protein. A balance stabilized by redox systems will induce the formation of three disulfide bond patterns that can be reduced by the destabilization of the systems. C, the hSOD1 monomer is prone to form aggregates when the redox balance is disturbed by ATG or BSO and the Trx and GSH-Grx systems are not in equilibrium. Some monomers could return to their original form, and some others could enhance an intermolecular disulfide bond formation. This panel shows an example of how the aggregates can be organized, but other combinations would be possible. The Trx and GSH-Grx systems are quite susceptible to be interrupt by environmental factors and others, like aging, which can result in mild oxidative stress. Under such conditions, the balance between the thiol-dependent antioxidant system and reactive oxygen species will be altered, which may cause the formation of intermolecular disulfide and further insoluble high molecular weight aggregates.
Treatment with BSO and ATG interrupts the GSH-Grx and Trx systems, causing mild oxidative stress and a subsequent cellular response, including Nrf-2 regulated antioxidant expression, for compensation. For example, treatment with ATG can specifically inhibit TrxR1 because Trx1 may get the electrons from the GSH-Grx system, a less efficient electron donor for Trx1, and it is kept in the reduced form (54). However, the change in the Trx system causes mutant SOD1 aggregation, indicating that the mutant SOD1 is susceptible to cytoplasmic oxidative stress. Many environmental factors, including some toxic heavy metal ions and some pharmaceutical compounds, can cause inhibition of TrxR1, inducing oxidative stress (55). Aging is also a factor that can cause alteration of the Trx and GSH-Grx systems. All of these factors may thus contribute to the pathophysiology via the change of the Trx and GSH-Grx systems. In fact, TrxR1 haplotypes affect the onset of fALS (56). In this regard, a role of the Trx system in ALS was also shown to be involved in the cell apoptosis process (57). G93A mutant SOD1 can induce dissociation of mammalian sterile 20-like kinase 1 (MST1) from Trx1. This results in the activation, in a ROS-dependent manner, of p38 mitogen protein kinase in SOD1 (G93A) mice spinal cord motoneurons (57).
In summary, we showed here that mutations of SOD1 affect susceptibility toward reduction by the Trx and GSH-Grx systems. Inhibitors of TrxR and GSH can trigger mutant SOD aggregation formation in cells. These results strongly suggest that the Trx and Grx systems are key players in SOD1 progression and ALS pathogenesis.
Experimental Procedures
Chemicals and Proteins
Human Trx1, recombinant rat TrxR1, Grx1, rabbit anti-human Trx1, and anti-rat TrxR1 antibodies were from IMCO Ltd. (Stockholm, Sweden). GR and GAPDH antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Other chemicals were from Sigma.
Human SOD1 Expression and Purification
The plasmids to express human SOD1 were constructed by insertion of the human SOD1 gene in the PGEX 4T-1 plasmid. The human SOD1 mutant proteins were obtained by site-directed mutagenesis kit (QuikChange kit, Stratagene). The expression of either the WT or mutations G93A and A4V was induced in bacteria overnight by 0.4 mm isopropyl β-d-thiogalactopyranoside at room temperature, with 250-ml Luria Bertani medium cultures. Cells were harvested by centrifugation and resuspended in ice-cold 250 mm NaCl and 20 mm sodium phosphate (pH 7.5), sonicated, and centrifuged at 15,000 × g for 20 min. The resultant supernatant was incubated with 0.7 ml of glutathione-SepharoseTM 4B beads (Amersham Biosciences, Freiburg, Germany) for 1 h at 4 °C to isolate the recombinant proteins. Finally, the recombinant proteins were eluted by thrombin cleavage in 250 mm NaCl, 2.5 mm CaCl2, and 20 mm sodium phosphate (pH 7). The concentration of proteins was determined by the Lowry or Bradford methods using BSA as the standard.
In Vitro hSOD1 Reduction Assays
The oxidized human SOD1 proteins (0.5 μm) were incubated with the GSH-Grx system (including 1 mm GSH, 100 nm GR, 2 μm Grx1, and 200 μm NADPH) or Trx system (including 100 nm TrxR1, 200 μm NADPH, and 10–40 μm Trx1) in 1.0 m PBS buffer for 1 h at room temperature. To prepare fully reduced proteins, 10 mm DTT was used in a separate experiment. Afterward, the resulting free cysteines of proteins were alkylated with 30 mm iodoacetamide at 37 °C for 30 min. After these treatments, proteins were separated on a 12% non-reducing SDS-PAGE. The proteins were then transferred to a nitrocellulose membrane to perform Western blotting analysis and detected using a sheep polyclonal antibody against human Cu,Zn-SOD1 (1:2500 dilution) (Calbiochem, Darmstadt, Germany). Western blotting bands were quantified using ImageJ software.
SOD1 Activity Assay
SOD1 activities were measured according to the SOD1 activity assay kit from Enzo Life Sciences. Briefly, in this colorimetric assay, superoxide ions were generated from the conversion of xanthine and oxygen to uric acid by xanthine oxidase. The superoxide anion then converted WST-1 to WST-1 formazan, a colored product with absorbance at 450 nm. SOD1 reacted and reduced the superoxide ion concentration and thereby decreased the rate of WST-1-formazan production. Reduction in the production of WST-1-formazan was used to represent SOD1 activity in experimental samples. In this experiment, 0.1, 1, and 10 units of SOD1 from human erythrocytes were used as standards.
Construction of the Stably hSOD1-transfected Cell Lines
Fluorescent hSOD1-GFP protein was generated in plasmid pEGFP-N1 by inserting human SOD1 DNA sequences. Mutations were performed as described above in section human SOD1 expression and purification. The schwannoma cell line RT4-D6P2T SCs (ATCC) was transfected with these plasmids using Lipofectamine 2000 (Invitrogen) according to instructions of the manufacturer. The cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2.
To screen hSOD1-GFP-positive transfected cell lines, cells were washed in PBS, dissociated by trypsin treatment, diluted 10-fold in cell sorter buffer, and filtered to prevent cell aggregation. GFP-positive cells were selected in a FACSCalibur and BD CellQuest Pro software (BD Biosciences) in which the excitation wavelength was set at 488 nm and the fluorescence emission was detected at 585 nm. Isolated fluorescent cells were grown and subsequently submitted to a process of selection. Cells were routinely checked for the purity to get cell lines containing the following constructs: WT hSOD1-GFP, C111G-GFP, G93A hSOD1-GFP, G93A/C111G, A4V hSOD1-GFP, and A4V/C111G hSOD1-GFP.
Detection of Protein Aggregation in Cells
The SOD1-GFP stably transfected cell lines were grown at a confluence of 80% and then treated with 100 μm ATG or 100 μm BSO for 24 h. The cells were fixed with paraformaldehyde (4%) and prepared for the detection of GFP aggregation with a confocal microscope (Zeiss Axioplan 2). Nuclei were stained with ToPro (1:1000). Digital images were taken by an AxioCamHRm camera and AxioVision 4.2 software. The size of aggregates and distribution of aggregates per cell were determined using ImageJ software.
Detection of the Redox State of SOD1 in Cells
Cells treated with 100 μm ATG or BSO were washed with 1 ml of PBS, scratched, and centrifuged at 1600 rpm for 5 min. Cells were resuspended in 100 μl of lysis buffer (100 mm NaCl, 1 mm EDTA, 20 mm iodoacetamide, and 10 mm Tris (pH 7.4)) containing 1% Nonidet P40 and protease inhibitor mixture (1:100 dilution) (Roche Diagnostics) and sonicated at 50 W for 10–15 s. The resulting lysates were centrifuged at 50,000 × g for 10 min in a Beckman Airfuge to remove the insoluble pellet. The supernatants were saved for analysis and mixed with loading buffer with or without 20 mm DTT. The SDS-PAGE and Western blotting experiments were performed as indicated above.
Intracellular ROS Production
To detect ROS production, a luminol-dependent chemiluminescence method was utilized (31). Briefly, cells (1 × 106) were treated with BSO for 24 h and then harvested by centrifugation. The cells were washed twice in 4 ml of 1× PBS and resuspended in 100 μl of 1× PBS (pH 7.2). Subsequently the suspension was homogenized by sonication. The resulting homogenates were used for the measurement of ROS. A stock solution of 2 mm luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) was prepared by dissolving it in a drop of 10 m NaOH diluted in 10 mm Tris-HCl (pH 8.0) and stored in the dark. An aliquot of cell homogenate (20 μl) was added with 40 μl of luminol stock solution to a hemolysis tube containing 500 μl of 10 mm Tris-HCl (pH 8.0). When ROS reacted with luminol, emitted light was captured by a photomultiplier (R374, Hamamatsu) fed at high voltage (700–800 V, high power supply, C-9525-01, Hamamatsu). The resulting signal was amplified in a low-noise amplifier (P16, Grass), filtered at 0.5 Hz in a Bessel filter (Frequency Devices), and digitized at 25 Hz using WinWCP (v3.3.3) software (Prof. John Dempster, Strathclyde University). Results are expressed as arbitrary units, obtained by quantification of areas under the curve of emission as a function of time and amount of protein.
Mass Spectrometry Analysis
Recombinant SOD1 A4V mutant protein was dissolved in 2 m urea and 50 mm ammonia bicarbonate buffer and digested overnight at 37 °C with sequencing grade-modified trypsin (Promega, 5% weight of SOD1 A4V). The digests were then desalted by stage tips (Thermo Fisher Scientific), dried by speedvac, and stored at −20 °C until further analysis.
The digests were dissolved in 0.1% formic acid and injected into a C18 EASY-Spray column (Thermo Fisher Scientific), 50 cm × 75 μm inner diameter, with a 2-h gradient coupled with an Orbitrap FusionTM TribridTM mass spectrometer (Thermo Fisher Scientific). The mass spectra were obtained with a full scan at 120,000 resolution in the Orbitrap and MS/MS with the top speed method at 3 s with 15,000 resolution in the Orbitrap using HCD energy at 35%. The resulting raw files were analyzed by Pepfinder 2.0 (Thermo Fisher Scientific) with a mass accuracy of 5 ppm.
Statistic Analysis
Mean, standard error of the mean, and t test significances were calculated in IgorPro (Wavemetrics) and GraphPad software.
Author Contributions
C. A. Z., J. L., C. S., and A. H. conceived the study, designed the experiments, and wrote the paper. C. A., J. L., Y. Z., H. Y., and J. B. performed the experiments. All authors analyzed the results, and approved the final version of the manuscript.
Acknowledgments
We thank the Scientific Facilities of the University of Barcelona. We also thank Prof. Mikael Oliveberg (Stockholm University) for kindly providing SOD1 proteins to perform the MS/MS experiment.
This work was supported by the Swedish Cancer Society (961), the Swedish Research Council Medicine (13X-3529), the K&A Wallenberg Foundation, grants from Karolinska Institutet and Boehringer Ingelheim Fonds, and Grant SAF2014-56811R from the Spanish Ministry of Economy and Competitiveness. The authors declare that they have no conflicts of interest with the contents of this article.
- fALS
- familial ALS
- SOD
- superoxide dismutase
- redox
- reduction-oxidation
- Trx
- thioredoxin
- Grx
- glutaredoxin
- TrxR
- thioredoxin reductase
- GR
- glutathione reductase
- hSOD
- human superoxide dismutase
- ATG
- aurothioglucose
- BSO
- buthionine sulfoximine
- ROS
- reactive oxygen species.
References
- 1. Seetharaman S. V., Prudencio M., Karch C., Holloway S. P., Borchelt D. R., and Hart P. J. (2009) Immature copper-zinc superoxide dismutase and familial amyotrophic lateral sclerosis. Exp. Biol. Med. 234, 1140–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kiernan M. C., Vucic S., Cheah B. C., Turner M. R., Eisen A., Hardiman O., Burrell J. R., and Zoing M. C. (2011) Amyotrophic lateral sclerosis. Lancet 377, 942–955 [DOI] [PubMed] [Google Scholar]
- 3. Turner M. R., Hardiman O., Benatar M., Brooks B. R., Chio A., de Carvalho M., Ince P. G., Lin C., Miller R. G., Mitsumoto H., Nicholson G., Ravits J., Shaw P. J., Swash M., Talbot K., et al. (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., and Deng H. X. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 5. Valentine J. S., Doucette P. A., and Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 6. Chattopadhyay M., and Valentine J. S. (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox Signal. 11, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cudkowicz M. E., McKenna-Yasek D., Sapp P. E., Chin W., Geller B., Hayden D. L., Schoenfeld D. A., Hosler B. A., Horvitz H. R., and Brown R. H. (1997) Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 41, 210–221 [DOI] [PubMed] [Google Scholar]
- 8. Wang J., Farr G. W., Zeiss C. J., Rodriguez-Gil D. J., Wilson J. H., Furtak K., Rutkowski D. T., Kaufman R. J., Ruse C. I., Yates J. R. 3rd, Perrin S., Feany M. B., and Horwich A. L. (2009) Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., and Deng H. X. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 10. Ilieva H., Polymenidou M., and Cleveland D. W. (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barber S. C., and Shaw P. J. (2010) Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 48, 629–641 [DOI] [PubMed] [Google Scholar]
- 12. Goodall E. F., and Morrison K. E. (2006) Amyotrophic lateral sclerosis (motor neuron disease): proposed mechanisms and pathways to treatment. Expert Rev. Mol. Med. 8, 1–22 [DOI] [PubMed] [Google Scholar]
- 13. Tiwari A., Liba A., Sohn S. H., Seetharaman S. V., Bilsel O., Matthews C. R., Hart P. J., Valentine J. S., and Hayward L. J. (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 284, 27746–27758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnston J. A., Dalton M. J., Gurney M. E., and Kopito R. R. (2000) Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 97, 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shinder G. A., Lacourse M. C., Minotti S., and Durham H. D. (2001) Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J. Biol. Chem. 276, 12791–12796 [DOI] [PubMed] [Google Scholar]
- 16. Elam J. S., Malek K., Rodriguez J. A., Doucette P. A., Taylor A. B., Hayward L. J., Cabelli D. E., Valentine J. S., and Hart P. J. (2003) An alternative mechanism of bicarbonate-mediated peroxidation by copper-zinc superoxide dismutase: rates enhanced via proposed enzyme-associated peroxycarbonate intermediate. J. Biol. Chem. 278, 21032–21039 [DOI] [PubMed] [Google Scholar]
- 17. Elam J. S., Taylor A. B., Strange R., Antonyuk S., Doucette P. A., Rodriguez J. A., Hasnain S. S., Hayward L. J., Valentine J. S., Yeates T. O., and Hart P. J. (2003) Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 10, 461–467 [DOI] [PubMed] [Google Scholar]
- 18. Tiwari A., and Hayward L. J. (2005) Mutant SOD1 instability: implications for toxicity in amyotrophic lateral sclerosis. Neurodegener. Dis. 2, 115–127 [DOI] [PubMed] [Google Scholar]
- 19. Tiwari A., Xu Z., and Hayward L. J. (2005) Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J. Biol. Chem. 280, 29771–29779 [DOI] [PubMed] [Google Scholar]
- 20. Ogawa M., Shidara H., Oka K., Kurosawa M., Nukina N., and Furukawa Y. (2015) Cysteine residues in Cu,Zn-superoxide dismutase are essential to toxicity in Caenorhabditis elegans model of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 463, 1196–1202 [DOI] [PubMed] [Google Scholar]
- 21. Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J. P., Brady S. T., and Brown R. H. Jr. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu H., Zhu H., Eggers D. K., Nersissian A. M., Faull K. F., Goto J. J., Ai J., Sanders-Loehr J., Gralla E. B., and Valentine J. S. (2000) Copper2+ binding to the surface residue cysteine 111 of His46Arg human copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant. Biochemistry 39, 8125–8132 [DOI] [PubMed] [Google Scholar]
- 23. Holmgren A. (1989) Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264, 13963–13966 [PubMed] [Google Scholar]
- 24. Lu J., and Holmgren A. (2014) The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 [DOI] [PubMed] [Google Scholar]
- 25. Bouldin S. D., Darch M. A., Hart P. J., and Outten C. E. (2012) Redox properties of the disulfide bond of human Cu,Zn superoxide dismutase and the effects of human glutaredoxin 1. Biochem. J. 446, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carroll M. C., Outten C. E., Proescher J. B., Rosenfeld L., Watson W. H., Whitson L. J., Hart P. J., Jensen L. T., and Cizewski Culotta V. (2006) The effects of glutaredoxin and copper activation pathways on the disulfide and stability of Cu,Zn superoxide dismutase. J. Biol. Chem. 281, 28648–28656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferri A., Fiorenzo P., Nencini M., Cozzolino M., Pesaresi M. G., Valle C., Sepe S., Moreno S., and Carrì M. T. (2010) Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 19, 4529–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Solsona C., Kahn T. B., Badilla C. L., Álvarez-Zaldiernas C., Blasi J., Fernandez J. M., and Alegre-Cebollada J. (2014) Altered thiol chemistry in human amyotrophic lateral sclerosis-linked mutants of superoxide dismutase 1. J. Biol. Chem. 289, 26722–26732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roberts B. L., Patel K., Brown H. H., and Borchelt D. R. (2012) Role of disulfide cross-linking of mutant SOD1 in the formation of inclusion-body-like structures. PLoS ONE 7, e47838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cozzolino M., Amori I., Pesaresi M. G., Ferri A., Nencini M., and Carrì M. T. (2008) Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Catz S. D., Carreras M. C., and Poderoso J. J. (1995) Nitric oxide synthase inhibitors decrease human polymorphonuclear leukocyte luminol-dependent chemiluminescence. Free Radic. Biol. Med. 19, 741–748 [DOI] [PubMed] [Google Scholar]
- 32. Ferraiuolo L., Kirby J., Grierson A. J., Sendtner M., and Shaw P. J. (2011) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630 [DOI] [PubMed] [Google Scholar]
- 33. Furukawa Y., Fu R., Deng H. X., Siddique T., and O'Halloran T. V. (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu,Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7148–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chattopadhyay M., Durazo A., Sohn S. H., Strong C. D., Gralla E. B., Whitelegge J. P., and Valentine J. S. (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 18663–18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Lindberg M., Oliveberg M., and Marklund S. L. (2006) Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129, 451–464 [DOI] [PubMed] [Google Scholar]
- 36. Niwa J., Yamada S., Ishigaki S., Sone J., Takahashi M., Katsuno M., Tanaka F., Doyu M., and Sobue G. (2007) Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J. Biol. Chem. 282, 28087–28095 [DOI] [PubMed] [Google Scholar]
- 37. Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., and Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J., Slunt H., Gonzales V., Fromholt D., Coonfield M., Copeland N. G., Jenkins N. A., and Borchelt D. R. (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum. Mol. Genet. 12, 2753–2764 [DOI] [PubMed] [Google Scholar]
- 39. Furukawa Y., Torres A. S., and O'Halloran T. V. (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tiwari A., and Hayward L. J. (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 278, 5984–5992 [DOI] [PubMed] [Google Scholar]
- 41. Lu J., and Holmgren A. (2014) The thioredoxin superfamily in oxidative protein folding. Antioxid. Redox Signal. 21, 457–470 [DOI] [PubMed] [Google Scholar]
- 42. Furukawa Y., Kaneko K., Yamanaka K., O'Halloran T. V., and Nukina N. (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 283, 24167–24176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chattopadhyay M., Nwadibia E., Strong C. D., Gralla E. B., Valentine J. S., and Whitelegge J. P. (2015) The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu,Zn superoxide dismutase (SOD1) fibrillation. J. Biol. Chem. 290, 30624–30636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kato S., Saeki Y., Aoki M., Nagai M., Ishigaki A., Itoyama Y., Kato M., Asayama K., Awaya A., Hirano A., and Ohama E. (2004) Histological evidence of redox system breakdown caused by superoxide dismutase 1 (SOD1) aggregation is common to SOD1-mutated motor neurons in humans and animal models. Acta Neuropathol. 107, 149–158 [DOI] [PubMed] [Google Scholar]
- 45. Nagano S., Takahashi Y., Yamamoto K., Masutani H., Fujiwara N., Urushitani M., and Araki T. (2015) A cysteine residue affects the conformational state and neuronal toxicity of mutant SOD1 in mice: relevance to the pathogenesis of ALS. Hum. Mol. Genet. 24, 3427–3439 [DOI] [PubMed] [Google Scholar]
- 46. Wilcox K. C., Zhou L., Jordon J. K., Huang Y., Yu Y., Redler R. L., Chen X., Caplow M., and Dokholyan N. V. (2009) Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 284, 13940–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Redler R. L., Wilcox K. C., Proctor E. A., Fee L., Caplow M., and Dokholyan N. V. (2011) Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry 50, 7057–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prudencio M., Lelie H., Brown H. H., Whitelegge J. P., Valentine J. S., and Borchelt D. R. (2012) A novel variant of human superoxide dismutase 1 harboring amyotrophic lateral sclerosis-associated and experimental mutations in metal-binding residues and free cysteines lacks toxicity in vivo. J. Neurochem. 121, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karch C. M., and Borchelt D. R. (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 13528–13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang J., Xu G., and Borchelt D. R. (2006) Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J. Neurochem. 96, 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toichi K., Yamanaka K., and Furukawa Y. (2013) Disulfide scrambling describes the oligomer formation of superoxide dismutase (SOD1) proteins in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 288, 4970–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cardoso R. M., Thayer M. M., DiDonato M., Lo T. P., Bruns C. K., Getzoff E. D., and Tainer J. A. (2002) Insights into Lou Gehrig's disease from the structure and instability of the A4V mutant of human Cu,Zn superoxide dismutase. J. Mol. Biol. 324, 247–256 [DOI] [PubMed] [Google Scholar]
- 53. Hough M. A., Grossmann J. G., Antonyuk S. V., Strange R. W., Doucette P. A., Rodriguez J. A., Whitson L. J., Hart P. J., Hayward L. J., Valentine J. S., and Hasnain S. S. (2004) Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc. Natl. Acad. Sci. U.S.A. 101, 5976–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Du Y., Zhang H., Lu J., and Holmgren A. (2012) Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 287, 38210–38219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu J., and Holmgren A. (2012) Thioredoxin system in cell death progression. Antioxid. Redox. Signal. 17, 1738–1747 [DOI] [PubMed] [Google Scholar]
- 56. Mitchell J., Morris A., and de Belleroche J. (2009) Thioredoxin reductase 1 haplotypes modify familial amyotrophic lateral sclerosis onset. Free Radic. Biol. Med. 46, 202–211 [DOI] [PubMed] [Google Scholar]
- 57. Lee J. K., Shin J. H., Hwang S. G., Gwag B. J., McKee A. C., Lee J., Kowall N. W., Ryu H., Lim D. S., and Choi E. J. (2013) MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc. Natl. Acad. Sci. U.S.A. 110, 12066–12071 [DOI] [PMC free article] [PubMed] [Google Scholar]