

Abstract
Ferroportin 1 (FPN1) is an iron export protein found in mammals. FPN1 is important for the export of iron across the basolateral membrane of absorptive enterocytes and across the plasma membrane of macrophages. The expression of FPN1 is regulated by hepcidin, which binds to FPN1 and then induces its degradation. Previously, we demonstrated that divalent metal transporter 1 (DMT1) interacts with the intracellular iron chaperone protein poly(rC)-binding protein 2 (PCBP2). Subsequently, PCBP2 receives iron from DMT1 and then disengages from the transporter. In this study, we investigated the function of PCBP2 in iron export. Mammalian genomes encode four PCBPs (i.e. PCBP1–4). Here, for the first time, we demonstrated using both yeast and mammalian cells that PCBP2, but not PCBP1, PCBP3, or PCBP4, binds with FPN1. Importantly, iron-loaded, but not iron-depleted, PCBP2 interacts with FPN1. The PCBP2-binding domain of FPN1 was identified in its C-terminal cytoplasmic region. The silencing of PCBP2 expression suppressed FPN1-dependent iron export from cells. These results suggest that FPN1 exports iron received from the iron chaperone PCBP2. Therefore, it was found that PCBP2 modulates cellular iron export, which is an important physiological process.
Keywords: chaperone, iron, iron metabolism, protein-protein interaction, transporter, PCBP2, ferroportin1
Introduction
Iron is an essential but potentially hazardous bio-metal (1). Because of its ability to readily accept or donate electrons, iron is a valuable cofactor that is used in oxygen transport, electron transfer, and DNA synthesis (2, 3). However, iron is potentially toxic because it catalyzes the generation of reactive oxygen species. The ensuing oxidative stress is associated with damage to cellular molecules, injury to tissues, and disease via processes including hydroxyl radical formation, glutathione depletion, protein aggregation, lipid peroxidation, and nucleic acid modification (4, 5).
Despite its high abundance in nature, ferric iron is poorly bioavailable due to its exceedingly low solubility at physiological pH. Thus, the acquisition and usage of iron presents a considerable challenge to cells and organisms, which have evolved sophisticated mechanisms to satisfy their metabolic needs and concomitantly minimize the risk of toxicity.
It has long been known that there is very little, yet potentially toxic, low-molecular-weight iron in rapidly metabolizing cells (6). However, the molecular mechanisms involved in intracellular iron transport have remained elusive (7). Recently, it was revealed that one of the mechanisms of iron transport and metabolism involves intracellular chaperone proteins (8). It was reported that poly(rC)-binding protein 1 (PCBP1),3 also referred to as α-CP1 or hnRNP E1, is a cytosolic iron chaperone protein that delivers iron to ferritin (9). PCBP1 binds to iron with micromolar affinity at a molar ratio of 3:1 Fe:PCBP1. Notably, PCBP1 has been shown previously to function as an RNA- and DNA-binding protein and has been identified as a member of a family of four homologous proteins containing three heterogeneous nuclear ribonucleoprotein K-homology (KH) domains (10).
Mammalian genomes encode four PCBPs, and every PCBP has been found to exhibit iron chaperone activity (11). Interestingly, PCBP1 is an intronless gene (12) that likely arose from the retrotransposition of a splice variant of PCBP2 mRNA. This became fixed in the genome, probably because the associated gene product performed a unique function not shared by the other PCBPs (13). The other members of the PCBP gene family undergo alternative splicing that results in greater diversity in the expression of PCBPs in mammals (13). PCBP1 is specific to mammals, whereas PCBP2, PCBP3, and PCBP4 are present throughout vertebrates, flies, and worms (8).
Previously, we reported that PCBP2, but not PCBP1, binds to the transmembrane iron importer, divalent metal transporter 1 (DMT1) (14). DMT1 has four isoforms, each of which exhibits specific subcellular localizations, namely in the plasma membrane (DMT1A-I), recycling endosomes (DMT1A-II and DMT1B-II), and/or late endosomes/lysosomes (DMT1B-I) (15–18). PCBP2 receives iron directly from all four DMT1 isoforms (14). Iron-loaded DMT1 binds to iron-depleted PCBP2, whereas iron-chelated DMT1 cannot (14). Iron uptake into the cytoplasm in the form of either non-transferrin (Tf)-bound iron (NTBI) or Tf-bound iron (TBI) was dependent on both DMT1 and PCBP2 (14). Thus, it was suggested that PCBP2 might act as a “gateway keeper” for iron transport (19).
Ferroportin 1 (FPN1) is an important cellular iron exporter, which is found on the plasma membranes of certain types of cells, especially duodenal enterocytes and macrophages (20–22). One of the DMT1 isoforms is expressed specifically on the apical membrane of duodenal enterocytes (18), whereas FPN1 is expressed on the basolateral membrane of duodenal enterocytes (22). Hepcidin is a 25-amino acid peptide synthesized by hepatocytes (23–25). The major molecular target of hepcidin is FPN1, which transfers cellular iron to the plasma (26). Hepcidin binds to the extracellular loop of FPN1, and this binding induces the endocytosis and proteolysis of FPN1, leading to a decrease in the export of iron to the plasma (27). This hepcidin-FPN1 binding interaction is an essential mechanism for the regulation of body iron stores; it is not yet clear how FPN1 receives cytoplasmic iron to export it from cells. However, the process of iron release is crucial to the regulation of systemic iron metabolism (28, 29).
In the present study, we identified a novel function of PCBP2 as an iron chaperone. We demonstrated that the C-terminal cytoplasmic region of FPN1 interacts specifically with PCBP2 but not with PCBP1, PCBP3, or PCBP4. We also determined that iron-depleted FPN1 interacts with iron-loaded PCBP2 but not with iron-depleted PCBP2. Silencing PCBP2 expression suppressed the iron export activity of FPN1. In addition, we showed that DMT1 binds to PCBP2 but not PCBP1, PCBP3, or PCBP4. This observation indicates that the interaction of DMT1 with these candidate iron chaperone proteins is highly specific to PCBP2. All four PCBPs exhibit iron binding activity, and their target molecules could potentially be different. The results of our study suggest that PCBP2 has important functions in both iron import and export and serves as a gateway keeper for the safe transport of iron.
Results
FPN1 Interacts with PCBP2 but Not with PCBP1, PCBP3, or PCBP4 in Yeast Cells
Our previous study showed that PCBP2 was identified as a binding partner of DMT1 using the N-terminal cytoplasmic region of DMT1 as bait in yeast two-hybrid screening (18). FPN1 contains three major cytoplasmic regions (Fig. 1A): an N-terminal region (FPN1-N; amino acids 1–22), an intracellular loop between the sixth and seventh transmembrane regions (FPN1-M; amino acids 220–307) and a C-terminal region (FPN1-C; amino acids 534–572) (30). We hypothesized that one of these regions might function as a PCBP-binding domain. To assess this possibility, yeast two-hybrid assays were performed using these three regions of FPN1 as bait (Fig. 1).
FIGURE 1.

The C-terminal cytoplasmic region of FPN1 interacts with the KH2 domain of PCBP2 in yeast cells. A, schematic representation of FPN1 or the PCBPs showing the domains that were used as bait or prey in yeast cells. N, N-terminal cytoplasmic region; M, intracellular loop between the sixth and seventh transmembrane domains; C, C-terminal cytoplasmic region. FPN1 regions were cloned into a vector containing the GAL4 DNA-binding domain (bait vector). PCBP1 (amino acids 12–80 (I-1), 94–193 (I-2), 278–348 (I-3), or 1–356 (I-F)); PCBP2 (amino acids 12–80 (II-1), 94–192 (II-2), 285–357 (II-3), or 1–366 (II-F)); PCBP3 (amino acids 44–112 (III-1), 126–220 (III-2), 292–362 (III-3), or 1–371 (III-F)); and PCBP4 (amino acids 16–84 (IV-1), 98–192 (IV-2), 240–310 (IV-3) or 1–403 (IV-F)) were cloned into a vector containing the GAL4 DNA-activating domain (prey vector). B, confirmation of bait and prey expression. Yeast cells were harvested and extracted for whole protein using the method described under “Experimental Procedures.” Bait protein expression was detected by using the anti-Myc mAb, and prey was identified by anti-HA mAb. The arrowheads indicate the appropriate molecular weight of each recombinant protein. Results are typical experiments of at least three performed. C, analysis of the interaction between FPN1 and PCBPs. After mating yeast expressing the bait protein with yeast expressing the prey protein, yeast cells were grown on both SD/-Trp/-Leu and SD/-Trp/-Leu/-His/-Ade selection media and incubated for 48 at 30 °C. The vector (v) served as a negative control, and the well characterized interaction between p53 and SV40-T served as a positive control, as suggested by the manufacturer. Similar results were obtained from at least three independent experiments.
The PCBP family was reported to include four paralogues, PCBP1–4 (13). All members of the PCBP family contain three KH domains (i.e. KH1, KH2, and KH3 (Fig. 1A)) and exhibit iron chaperone activity by binding to iron (11). PCBP2 displays 83, 71, and 53% amino acid sequence similarity with PCBP1, PCBP3, and PCBP4, respectively (11, 12).
In this study, the full-length form of PCBP1 (amino acids 1–356, I-F) or PCBP2 (amino acids 1–366, II-F) was cloned into the prey vector and used for the yeast two-hybrid assay (Fig. 1A). We also cloned the full-length form of PCBP3 (amino acids 1–371, III-F) and PCBP4 (amino acids 1–403, IV-F) and made the recombinant yeast prey vectors in Escherichia coli (Fig. 1A). However, when we transformed the yeast with the prey vectors containing full-length PCBP3 or PCBP4, recombinant yeast clones could not be grown on selection plates. We repeatedly tried to isolate the recombinant yeast clones but were not successful. Hence, it was presumed that PCBP3 and PCBP4 in the prey vector were toxic for yeast cells.
To identify which domain of PCBPs are functional for binding to FPN1 in yeast cells, each KH domain was cloned into the prey vector (Fig. 1A). This included the KH1 domains of PCBP1 (amino acids 12–80, I-1), PCBP2 (amino acids 12–80, II-1), PCBP3 (amino acids 44–112, III-1), and PCBP4 (amino acids 16–84, IV-1); the KH2 domains of PCBP1 (amino acids 94–193, I-2), PCBP2 (amino acids 94–192, II-2), PCBP3 (amino acids 126–220, III-2), and PCBP4 (amino acids 98–192, IV-2); and the KH3 domains of PCBP1 (amino acids 278–348, I-3), PCBP2 (amino acids 285–357, II-3), PCBP3 (amino acids 292–362, III-3), and PCBP4 (amino acids 240–310, IV-3) (Fig. 1A). As an appropriate positive control, P53 was used as bait and SV40-T implemented as prey, as this protein-protein interaction is well characterized (31).
To examine the expression of recombinant proteins and assess whether each of them was expressed at their expected molecular weight in yeast cells, whole yeast protein was extracted and analyzed by immunoblotting (Fig. 1B). The expected molecular sizes of the bait fusion proteins were obtained (Fig. 1B, left panel): vector (v), 23 kDa; FPN1-N, 25 kDa; FPN1-M, 31 kDa; and FPN1-C, 26 kDa. Of note, when bait proteins were examined using the anti-Myc mAb, an extra band in the bait vector alone was evident (Fig. 1B, left panel). When the same bait vector and yeast strain were used in a previous report by others, a similar extra band was detected by the anti-Myc mAb (32).
The band in each lane indicated by an arrowhead (Fig. 1B, middle and right panels) was the expected molecular size of the prey fusion proteins, that is: vector (v), 23 kDa; I-F and II-F, 59 kDa (Fig. 1B, middle panel); I-1, II-1, III-1, and IV-1, 28 kDa; I-2, II-2, and III-2, 32 kDa; IV-2, 30 kDa; and I-3, II-3, III-3, and IV-3, 28 kDa (Fig. 1B, right panel). The KH2 domain is longer than KH1 or KH3, and thus the 28-kDa band was not observed in I-2, II-2, III-2, or IV-2.
When prey proteins were examined by anti-HA mAb, multiple bands on the blots were evident, even using the prey vector alone (Fig. 1B, middle and right panels). Considering this finding, two other sources of anti-HA mAbs (from Wako Chemicals (Osaka Japan) and Covance Laboratories (Madison, WI)) were utilized for detection, and again, a similar pattern of multiple bands was detected (data not shown). Of interest, when the same prey vector and yeast strain were used in previous reports by others, similar multiple bands were detected by the anti-HA mAb (32, 33).
In summary, the band in each lane indicated by an arrowhead (Fig. 1B) was the expected molecular size of the fusion proteins. Hence, we demonstrated that all bait and prey constructs worked appropriately in yeast cells generating recombinant proteins of the correct size. These were then used for the studies below in Fig. 1C.
When the N-terminal cytoplasmic region (FPN1-N) or the intracellular loop (FPN1-M) was expressed in yeast cells, these regions of FPN1 did not interact with PCBP1/PCBP2 or with any of the KH domains of PCBP1, PCBP2, PCBP3, and PCBP4 on SD-Leu-Trp-His-Ade-selective plates (Fig. 1C). In contrast, when the C-terminal cytoplasmic region of FPN1 (FPN1-C) was used as bait, we detected an interaction between FPN1 and the full-length form (II-F) or the KH2 domain (II-2) of PCBP2 (Fig. 1C). Importantly, the only other observed interaction under these conditions was the P53-SV40-T positive control. In summary, these findings suggest that the C-terminal cytoplasmic region of FPN1 interacts with PCBP2 and specifically the KH2 domain of PCBP2.
PCBP2 KH2 Domain Is a Crucial Region to Bind Not Only FPN1 but Also DMT1 in Yeast Cells
As shown in Fig. 1, the C-terminal cytoplasmic region of FPN1 as bait interacted with PCBP2 in yeast cells. Next, we investigated the interactions between DMT1 and the PCBP family (Fig. 2) and further confirmed the interaction between FPN1 and PCBP2 by means of exchanging the bait and prey inserts. We used the same region of the KH domains in prey (Fig. 1A) and bait (Fig. 2A) constructs. Using cDNA segments corresponding to the KH1, KH2, and KH3 domains of PCBP1, PCBP2, PCBP3, and PCBP4 or full-length forms of PCBP1, PCBP2, PCBP3, and PCBP4 as bait (Fig. 2A) and DMT1 or FPN1 as prey, we performed yeast two-hybrid assays (Fig. 2). In this case, the full-length forms of PCBP3 and PCBP4 cloned into bait vectors could be isolated in yeast cells.
FIGURE 2.

DMT1 and FPN1 can bind to PCBP2 but not PCBP1, PCBP3, or PCBP4 in yeast or mammalian cells. A, analysis of the interactions of DMT1 or FPN1 with the four PCBPs using a yeast two-hybrid assay. PCBP1 (amino acids 12–80 (I-1), 94–193 (I-2), 278–348 (I-3), or 1–356 (I-F)); PCBP2 (amino acids 12–80 (II-1), 94–192 (II-2), 285–357 (II-3), or 1–366 (II-F)); PCBP3 (amino acids 44–112 (III-1), 126–220 (III-2), 292–362 (III-3), or 1–371 (III-F)); and PCBP4 (amino acids 16–84 (IV-1), 98–192 (IV-2), 240–310 (IV-3), or (1–403 (IV-F)) were cloned into a vector containing the GAL4 DNA-binding domain (bait vector). Vectors containing the GAL4 DNA-activating domain fused to the N-terminal cytoplasmic region of DMT1 or the C-terminal cytoplasmic region of FPN1 were used as prey. B, confirmation of bait and prey expression. Yeast cells were harvested, and whole protein was prepared using the method described under “Experimental Procedures.” Bait protein expression was detected by using anti-Myc mAb, and prey was detected by anti-HA mAb. The arrowheads indicate the appropriate molecular weight of each recombinant protein. Results are typical experiments of at least three performed. v, vector. C, analysis of the interactions of DMT1 or FPN1 with PCBPs. After mating yeast expressing the bait protein with yeast expressing the prey protein, yeast cells were grown on both SD/-Trp/-Leu and SD/-Trp/-Leu/-His/-Ade selection media and incubated for 48 h at 30 °C. The vector alone served as a negative control, and the well characterized p53 and SV40-T interaction served as a positive control, as suggested by the manufacturer. Similar results were obtained from at least three independent experiments. D, analysis of the interactions of DMT1 or FPN1 with PCBP1–4 using a co-immunoprecipitation assay. HEp-2 cells were transfected with FPN1-GFP or DMT1A-I-GFP and incubated for 48 h at 37 °C. After incubation, total protein was extracted in TNE buffer and assessed by immunoblotting using the appropriate anti-PCBP1, -2, -3, or -4 or GFP Ab (left lane). The membrane fraction was prepared using the method described under “Experimental Procedures.” Using either the total protein extract or the membrane fraction, co-immunoprecipitation analyses (co-IP) were performed. The precipitates were analyzed by immunoblotting with anti-PCBP1 pAb, anti-PCBP2 mAb, anti-PCBP3 pAb, anti-PCBP4 pAb, and anti-GFP pAb (right lane). Similar results were obtained from at least three independent experiments.
To confirm the expression of recombinant proteins and ensure that each of them was expressed at its expected molecular weight in yeast cells, whole yeast protein was extracted and analyzed by immunoblotting (Fig. 2B). The expected molecular size of the bait fusion proteins was obtained: vector (v), 23 kDa; I-F and II-F, 59 kDa; III-F, 57 kDa; and IV-F, 61 kDa (Fig. 2B, top panel); I-1, II-1, III-1, and IV-1, 28 kDa; I-2, II-2, and III-2, 32 kDa; IV-2, 30 kDa; and I-3, II-3, III-3, and IV-3, 28 kDa (Fig. 2B, middle panel). The KH2 domain is longer than KH1 or KH3, and thus the 28-kDa band is not observed in I-2, II-2, III-2, or IV-2. Hence, the band in each lane was the expected molecular size (Fig. 2B).
The band in each lane indicated by an arrowhead (Fig. 2B, bottom panel) was the expected molecular size of the prey fusion proteins, that is: vector (v), 23 kDa; FPN1-C, 26 kDa; and DMT1-N, 40 kDa. When FPN1-C or DMT1-N was expressed in yeast cells and detected by anti-HA mAb, multiple bands were again detected as shown in Fig. 1B. However, the band in each lane indicated by an arrowhead was the expected molecular size (Fig. 2B, bottom panel).
Hence, we determined that all bait and prey constructs worked appropriately in yeast cells generating the correct size fusion proteins. These were then used for the further studies described below.
Notably, using SD-Leu-Trp-His-Ade-selective plates (Fig. 2C), the interactions of the KH2 domain and the full-length form of PCBP2 with the N-terminal cytoplasmic region of DMT1 and the C-terminal cytoplasmic region of FPN1 were detected in yeast cells along with the positive control (i.e. P53-SV40-T). However, other members of the PCBP family, or any of the KH domains of PCBP1, PCBP2, (except PCBP2 KH2), PCBP3, and PCBP4, were not demonstrated to interact with DMT1 or FPN1 (Fig. 2C).
FPN1/DMT1 Interacts with PCBP2 but Not with PCBP1, PCBP3, or PCBP4 in Mammalian Cells
Considering the results in yeast cells described above, we then examined the interactions of FPN1/DMT1 with all members of the PCBP family in mammalian cells (Fig. 2D). Expression of all four PCBPs was clearly detected in both the total protein extraction and the membrane fraction using immunoblotting (Fig. 2D). It should be noted that multiple PCBP2 bands on the immunoblot were observed throughout the present study. When the expression of PCBP2 was silenced using siRNA, the intensities of these multiple bands were reduced (as shown later in Figs. 6A and 7D). Previous investigations have reported that the multiple PCBP2 bands on the immunoblots may represent alternative PCBP2 gene splice forms (34–36).
FIGURE 6.

The NTBI-derived iron export activity of FPN1 was suppressed by PCBP2 silencing in mammalian cells. A, PCBP2 silencing decreases FPN1-dependent iron export. HEp-2 cells stably expressing DMT1A-I were cultured in serum-free DMEM and preloaded with 100 μm FAC or 100 μm Fe-NTA for 6 h at 37 °C. The cells were washed with PBS, transfected with FPN1-GFP expression plasmid or GFP and PCBP2 siRNA or control siRNA, and then incubated in serum-free RPMI 1640 medium for 18 h at 37 °C. The cells were then lysed in TNE buffer for immunoblotting analysis. The siRNA are denoted as: −, non-siRNA; N, negative control siRNA; P2, PCBP2 siRNA. Similar results were obtained from at least three independent experiments. B, the density of specific bands from immunoblotting was measured with a computer-assisted imaging analysis system (NIH ImageJ). The histogram shows the expression ratio of PCBP2, TfR1, tubulin, FPN1, GFP, and ferritin. Each column corresponds to a quantitative datum of band imaging appearing in A in order from left to right. Each column is indicated as follows: 0 h: post-transfection, 0 h siRNA(−), plasmid (−); FPN(−): post-transfection 18 h siRNA(−), plasmid (FPN1); FPN(N): post-transfection 18 h siRNA (negative control), plasmid (FPN1); FPN(P2): post-transfection 18 h siRNA(PCBP2), plasmid (FPN1); GFP(−): post-transfection 18 h siRNA(−), plasmid (GFP); GFP(N): post-transfection 18 h siRNA (negative control); plasmid (GFP) and GFP(P2): post-transfection 18 h siRNA(PCBP2), plasmid (GFP). Significance was determined by Student's t test: †, p < 0.05; §, p < 0.001. C, the histograms show the relative intracellular iron level. The cells were treated as described in A and lysed in 50 mm NaOH. The iron concentration was measured using a ferrozine-based iron assay. The detailed method is described under “Experimental Procedures.” The results in the histogram are presented as the means ± S.E. of three independent experiments. Significance was determined using Student's t test: †, p < 0.05; §, p < 0.001.
FIGURE 7.

The TBI-derived iron export activity of FPN1 was suppressed by PCBP2 silencing in mammalian cells. A and C, FPN1-GFP exports iron loaded as TBI. HEp-2 cells were cultured in serum-free DMEM and preloaded with 0.3 mg/ml holo-Tf for 12 h at 37 °C. The cells were washed with PBS, transfected with FPN1-GFP expression plasmid, and then incubated in serum-free RPMI 1640 medium for 18 h at 37 °C. The cells were lysed in TNE buffer for immunoblotting analysis (A) or in 50 mm NaOH (C) for a ferrozine-based iron assay. B, the density of specific bands from immunoblotting was measured with a computer-assisted imaging analysis system (NIH ImageJ), and normalization was to total protein loaded. The histogram shows the expression ratio of PCBP2, TfR1, tubulin, FPN1, GFP, and ferritin. Each column corresponds to an immunoblotting analysis lane in A in order from left to right. Each column is denoted in the following way: (−)12 h: holo-Tf(-)-incubation 12 h; (+)12 h: holo-Tf(+)-incubation 12 h; (+)18 h: holo-Tf(+)-incubation 12 and 18 h; (+)18 h FPN/GFP: holo-Tf(+) incubation 12 h; and 18 h FPN1 or GFP transfection. Significance was determined using Student's t test: †, p < 0.05; §, p < 0.001. C, the histogram shows the relative intracellular iron level. The detailed method is described under “Experimental Procedures.” The results in the histogram are presented as the means ± S.E. of experiments performed in triplicate. Significance was determined by Student's t test: ‡, p < 0.01; §, p < 0.001. D, PCBP2 silencing decreases FPN1-mediated iron export. HEp-2 cells were cultured in serum-free DMEM and preloaded with 0.3 mg/ml holo-Tf for 12 h at 37 °C. The cells were washed with PBS, transfected with FPN1-GFP expression plasmid and PCBP2 siRNA, and then incubated in serum-free RPMI 1640 medium for 18 h at 37 °C. The cells were lysed in TNE buffer for immunoblotting analysis. The symbols used to describe the siRNAs are as follows: −, non-siRNA; N, negative control siRNA; and P2, PCBP2 siRNA. Similar results were obtained from at least three independent experiments. E, the density of specific bands from immunoblotting was measured with a computer-assisted imaging analysis system (NIH ImageJ). The histogram shows the expression ratio of PCBP2, TfR1, tubulin, FPN1, GFP, and ferritin. Each column corresponds to an immunoblotting analysis lane in D in order from left to right. Each column is denoted in the following way in each band imaging of PCBP2, TfR1, Ferritin, Tubulin, FPN1 and GFP: 0 h: post-transfection 0 h siRNA (−), plasmid (−); FPN(−): post-transfection 18 h siRNA (−), plasmid (FPN1); FPN(N): post-transfection 18 h siRNA (negative control), plasmid (FPN1); FPN(P2): post-transfection 18 h siRNA(PCBP2), plasmid (FPN1); GFP(−): post-transfection 18 h siRNA(−), plasmid (GFP); GFP(N): post-transfection 18 h siRNA (negative control), plasmid (GFP); GFP(P2): post-transfection 18 h siRNA(PCBP2) and plasmid (GFP). Significance was determined using Student's t test: †, p < 0.05; §, p < 0.001. F, the histogram shows the relative intracellular iron level. Cells were treated as described in D and lysed in 50 mm NaOH. The iron concentration was measured using a ferrozine-based iron assay. The detailed method is described under “Experimental Procedures.” The results in the histogram are presented as the means ± S.E. of three independent experiments. Significance was determined by Student's t test: ‡, p < 0.01; §, p < 0.001.
We then performed co-immunoprecipitation assays in HEp-2 cells transfected with DMT1A-I-GFP or FPN1-GFP using anti-GFP (α-GFP) followed by immunoblotting with the relevant antibodies against the PCBP family (Fig. 2D). In these studies, PCBP2 co-immunoprecipitated with DMT1 or FPN1, whereas PCBP1, -3, and -4 did not (Fig. 2D). Thus the specific interaction between DMT1/FPN1 and PCBP2 was shown in mammalian cells.
The N-terminal Region of DMT1 and the C-terminal Region of FPN1 Are Crucial for Their Interactions with PCBP2 in Mammalian Cells
To investigate the importance of the cytoplasmic domains of both DMT1 and FPN1 in their interactions with PCBP2 in mammalian cells, two deletion mutants were constructed and expressed in HEp-2 cells (Fig. 3). These mutants were DMT1, lacking its N-terminal cytoplasmic region (DMT1ΔN), and FPN1, lacking its C-terminal cytoplasmic region (FPN1ΔC) (Fig. 3A). Mutants were prepared as described previously (18, 37).
FIGURE 3.

The N-terminal cytoplasmic region of DMT1 and the C-terminal cytoplasmic region of FPN1 are essential for their binding to PCBP2 in mammalian cells. A, the schematics show the full-length or deletion mutants of DMT1 or FPN1. The N-terminal cytoplasmic region of full-length DMT1A-I consists of 98 amino acids. DMT1ΔN is a deletion mutant of DMT1 that lacks amino acids 2–98. Amino acids 534–572 correspond to the C-terminal cytoplasmic region of FPN1. FPN1ΔC is a deletion mutant of FPN1 that lacks amino acids 535–572. B, the cellular localization of full-length or the deletion mutants of DMT1 or FPN1. The GFP-tagged deletion mutant forms of DMT1 and FPN1 were transiently transfected with mCherry-tagged full-length forms of DMT1, FPN1, and CPR. Cells were stained with anti-PDI mAb or anti-LAMP2 mAb. The arrowhead shows co-localization of full-length and deletion-mutant forms of DMT1 or FPN1 at the plasma membrane. Similar results were obtained from at least three experiments. C, GFP-tagged full-length and deletion-mutant forms of DMT1 and FPN1 were transiently transfected into HEp-2 cells. The cells were lysed in TNE buffer and analyzed by immunoblotting using α-PCBP2 Ab. Co-immunoprecipitation (IP) was also performed with anti-GFP pAb-conjugated protein A beads. These samples were analyzed by immunoblotting (WB) using an anti-PCBP2 mAb. Similar results were obtained from at least three independent experiments. D, iron transport activity of full-length and deletion-mutant forms of DMT1 or FPN1. Full-length or deletion-mutant forms of DMT1 were transfected into HEp-2 cells. Cells were loaded with 100 μm FAC for 6 h at 37 °C. The cells were lysed in 50 mm NaOH for the ferrozine-based iron assay. HEp-2 cells stably expressing DMT1A-I were cultured in serum-free DMEM and preloaded with 100 μm FAC for 6 h at 37 °C. The cells were washed with PBS, transfected with the full-length or deletion mutant of FPN1-GFP expression plasmid, and then incubated in serum-free RPMI 1640 medium for 18 h at 37 °C. The cells were then lysed in 50 mm NaOH for the ferrozine-based iron assay. The results shown on the histogram are presented as the means ± S.E. of three independent experiments. Significance was determined by Student's t test: ‡, p < 0.01; §, p < 0.001.
We then analyzed the subcellular localization of the deletion mutants, as this could affect their function (Fig. 3B). In these studies, confocal microscopy was used to examine the localization in HEp-2 cells of full-length (wild type (WT)) or deletion mutants of DMT1 or FPN1. The GFP-tagged deletion mutant forms of DMT1 or FPN1 (green) were transiently transfected with the mCherry-tagged full-length (WT) forms of DMT1, FPN1, or NADPH cytochrome P450 reductase (CPR) (red), and co-localization (yellow) in the electronic merge was then assessed (Fig. 3B). Cells transfected with the GFP-tagged deletion mutant forms of DMT1 or FPN1 (green) were also stained with the anti-protein disulfide isomerase (PDI) mAb (red) or anti-LAMP2 mAb (red) to examine co-localization in the endoplasmic reticulum (ER) lumen and lysosome, respectively (Fig. 3B).
The full-length forms of DMT1A-I and FPN1 were localized to the plasma membrane as described previously (18, 27). Importantly, both DMT1ΔN and FPN1ΔC were also expressed at the cell surface and intracellularly (Fig. 3B). Furthermore, they partially co-localized with the full-length forms of these molecules at the plasma membrane (Fig. 3B).
PDI is an ER protein that is localized to the ER lumen (38); it is notable that neither DMT1ΔN nor FPN1ΔC was strongly co-localized with PDI (Fig. 3B). In contrast, DMT1ΔN or FPN1ΔC demonstrated pronounced co-localization with CPR, which is known to be markedly expressed in the ER membrane (18). Neither DMT1ΔN nor FPN1ΔC was co-localized with LAMP2 (Fig. 3B), which is a marker of late endosomes/lysosomes (39).
DMT1 is known to exist in several isoforms (18, 40, 41). In fact, DMT1A-I has 98 amino acids in its N-terminal cytosolic region, and DMT1B-I lacks N-terminal amino acids 1–29 of DMT1A-I (18, 40, 41). DMT1A-I is localized to the plasma membrane, whereas DMT1B-I is localized to late endosomes/lysosomes (16, 18). As demonstrated from our studies examining the co-localization with LAMP2, the localization of DMT1ΔN was obviously different from late endosomes/lysosomes (shown in Fig. 3B). Indeed, DMT1ΔN and FPN1ΔC were localized to the plasma membrane and the ER membrane.
Co-immunoprecipitation was then performed to investigate the interaction of PCBP2 with DMT1 and FPN1 (Fig. 3C). In these studies, α-GFP was used to immunoprecipitate FPN1 and DMT1 from cells transfected with DMT1A-I-GFP, FPN1-GFP, and their deletion mutants. Immunoblotting was then performed using anti-PCBP2 (α-PCBP2). When the full-length form of DMT1 or FPN1 was expressed in HEp-2 cells, PCBP2 was co-immunoprecipitated with DMT1 or FPN1 (Fig. 3C).
In contrast, when DMT1ΔN or FPN1ΔC was expressed in HEp-2 cells, PCBP2 was not co-immunoprecipitated with DMT1ΔN or FPN1ΔC at all (Fig. 3C). This occurred even though deletion mutants were co-localized with the full-length forms of these proteins at the plasma membrane (Fig. 3B). Therefore, neither DMT1ΔN nor FPN1ΔC interacted with PCBP2 (Fig. 3). These results confirmed that the N-terminal cytoplasmic region of DMT1 is a domain that binds to PCBP2, as shown in our previous report (14). Moreover, these studies demonstrated that the C-terminal cytoplasmic region of FPN1 is responsible for its binding to PCBP2 in mammalian cells.
DMT1ΔN and FPN1ΔC Lose Iron Transport Activity
The cells expressing the full-length form of DMT1 incorporated iron very efficiently (Fig. 3D). When DMT1ΔN was expressed in cells, the cells did not incorporate iron from ferric ammonium citrate (FAC) (100 μm), and thus DMT1ΔN lost its iron import activity (p < 0.001). The cells expressing the full-length form of FPN1 exported iron efficiently (Fig. 3D). On the other hand, when FPN1ΔC was expressed, the cells did not export iron efficiently, indicating that FPN1ΔC lost its iron export activity (p < 0.01). Thus, these deletion mutants lost their iron transport activity in HEp-2 cells (Fig. 3D).
Taken together, the results shown in Figs. 1–3 confirmed the interactions between DMT1/FPN1 and PCBP2 in yeast and mammalian cells. In contrast, no interactions between DMT1/FPN1 and other members of the PCBP family were detected (Fig. 3). Therefore, we next investigated the function of PCBP2 in iron export.
FPN1 Interacts with Iron-loaded PCBP2
Previously, we described the iron-dependent DMT1-PCBP2 interaction (14). Iron-loaded, but not iron-depleted, DMT1 interacts with PCBP2 (14). However, on the other hand, the association of PCBP2 with iron does not alter its DMT1 binding activity (14). Therefore, in this study, we investigated the iron dependence of the FPN1-PCBP2 interaction and then examined whether FPN1 or PCBP2 needed to be in an iron-bound state to enable the FPN1-PCBP2 interaction (Fig. 4).
FIGURE 4.
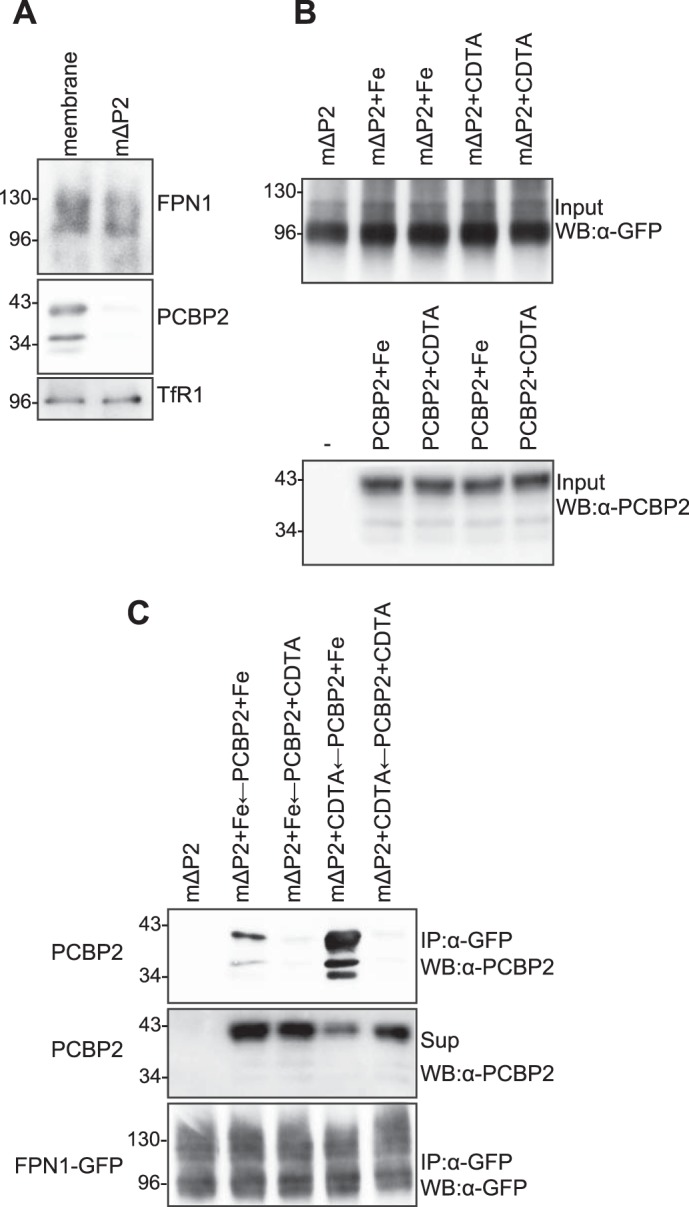
Iron-depleted FPN1 can bind to iron-loaded PCBP2 in mammalian cells. A, preparation of membrane and mΔP2 fractions. Membrane fractionation was performed on HEp-2 cells expressing FPN1-GFP as described under “Experimental Procedures.” The membrane fraction was pelleted via centrifugation, resuspended in homogenization buffer containing 0.1 m Na2CO3 (pH 11), and centrifuged again at 100,000 × g. The resultant pellet was collected as the PCBP2-depleted membrane (mΔP2) fraction and analyzed by immunoblotting using the appropriate FPN1, PCBP2, and TfR1 antibodies. B, analysis of the iron dependence of the FPN1-PCBP2 interaction. The mΔP2 fraction was incubated with iron (freshly prepared FeCl2) or the iron chelator CDTA for 4 h at 4 °C. The soluble fraction was preincubated with iron or CDTA for 4 h at 4 °C. Before immunoprecipitation, the mΔP2 fraction or soluble fraction was pretreated with FeCl2 or CDTA and analyzed by immunoblotting (WB) with anti-GFP pAb and anti-PCBP2 mAb. Similar results were obtained from at least three independent experiments. C, analysis of the iron dependence of the FPN1-PCBP2 interaction. The mΔP2 fraction was incubated with iron (freshly prepared FeCl2) or the iron chelator CDTA for 4 h at 4 °C and then immunoprecipitated (IP) with an anti-GFP pAb. The soluble fraction was preincubated with iron or CDTA for 4 h at 4 °C. The immunoprecipitated mΔP2 fraction and the preincubated soluble fraction were mixed for 2 h at 4 °C and then analyzed by immunoblotting with an anti-PCBP2 mAb. The supernatant (Sup) after mixing the mΔP2 fraction and the soluble fraction was collected and analyzed by immunoblotting with anti-PCBP2 mAb. The results are typical of at least three independent experiments.
It has been reported that PCBP2 is localized primarily to the nucleus and the cytoplasm (42, 43), and we discovered that some part of PCBP2 also resides within the cell membrane (14). Fig. 4A, using immunoblotting, demonstrates that FPN1 was not depleted from this fraction via Na2CO3 treatment, whereas membrane-bound PCBP2 was removed. This fraction is referred to as mΔP2 (Fig. 4A). Importantly, Na2CO3 treatment efficiently removes certain membrane-bound proteins that are not integrally associated with the membrane (14, 16, 44) Indeed, TfR1 is a transmembrane protein that resides in the membrane fraction (7), and its levels were not affected by Na2CO3 treatment (Fig. 4A). Thus, TfR1 acted as an appropriate control, which demonstrated that Na2CO3 did not lead to the extraction of integral membrane proteins.
To assess whether FPN1 can interact with iron-loaded PCBP2, studies were then performed to implement immunoprecipitation using α-GFP followed by immunoblotting using α-PCBP2 (Fig. 4B). These experiments utilized the mΔP2 fraction pretreated with either iron (i.e. FeCl2) or an iron chelator (i.e. 1,2-cyclohexylenedinitrilotetraacetic acid (CDTA) from Wako Chemicals) to deplete this fraction of iron (Fig. 4B). This step was performed to load or deplete the mΔP2 fraction, particularly FPN1, with iron. Iron or chelator pretreatment did not significantly (p > 0.05) alter the amount of FPN1 recovered in the mΔP2 fraction (Fig. 4B, upper panel). The free iron or chelator was then subsequently removed from the membrane by extensive washing with PBS as described previously (14).
The soluble cytosolic fraction containing PCBP2 was also premixed with FeCl2 or the CDTA iron chelator to load or deplete it of the metal ion, respectively (Fig. 4B, lower panel). Iron or chelator pretreatment did not alter the amount of PCBP2 in the soluble fraction. Then, the mΔP2 fraction and the soluble cytosolic fraction were mixed, immunoprecipitated with anti-GFP pAb-conjugated protein A beads and immunoblotted using an anti-PCBP2 mAb (Fig. 4C, top panel). The amount of FPN1 recovered by immunoprecipitation with anti-GFP pAb-conjugated protein A beads was not considerably changed among these conditions (Fig. 4C, bottom panel).
When the soluble cytosolic fraction was premixed with iron (PCBP2+Fe) and then mixed with the chelator-pretreated mΔP2 fraction (mΔP2 + CDTA), a substantial amount of PCBP2 was recovered on the immunoblot (Fig. 4C, top panel). However, after the soluble cytosolic fraction was pretreated with iron chelator (PCBP2 + CDTA) and mixed with the chelator-pretreated mΔP2 fraction (mΔP2 + CDTA), there was no appreciable co-immunoprecipitation of PCBP2 with FPN1 (Fig. 4C, top panel). This observation suggested that PCBP2 loaded with iron interacted preferably with immunoprecipitable FPN1-GFP in the mΔP2 membrane fraction relative to PCBP2 depleted of iron.
A markedly lower amount of PCBP2 was detected when the iron-premixed mΔP2 fraction (mΔP2 + Fe) was added to the iron-premixed soluble cytosolic fraction (PCBP2 + Fe; Fig. 4C, top panel). Hence, this observation suggested that potentially FPN1 with iron bound (or at least the mΔP2 + Fe fraction) inhibited the interaction with iron-loaded PCBP2 relative to when mΔP2 was depleted of iron (Fig. 4C, top panel).
When PCBP2 was depleted of iron using CDTA (PCBP2 + CDTA) and then mixed with the mΔP2 fraction loaded with iron (mΔP2 + Fe), no interaction was detected (Fig. 4C, top panel). Hence, unlike PCBP2 loaded with iron, these experiments suggested that when the PCBP2 fraction was depleted of iron, it could not interact at all with the iron-loaded immunoprecipitable FPN1 (mΔP2 + Fe).
Supernatants were analyzed after mixing the mΔP2 and the soluble fraction by immunoblotting using an anti-PCBP2 mAb (Fig. 4C, middle panel). When the soluble cytosolic fraction was premixed with iron (PCBP2 + Fe) and then added to the chelator-pretreated mΔP2 fraction (mΔP2 + CDTA), less PCBP2 was recovered in the supernatant fraction (Fig. 4C, middle panel). This observation suggests that iron-loaded PCBP2 in the soluble cytosolic fraction efficiently binds to iron-depleted FPN1, which is then immunoprecipitated with membrane-bound FPN1. Thus, the recovered PCBP2 in the supernatant fraction is reduced (Fig. 4C, middle panel).
Considering the results described above, and particularly the studies examining the iron loading of FPN1 and the decreased interaction with PCBP2 (i.e. mΔP2+Fe ← PCBP2 + Fe relative to mΔP2 + CDTA ← PCBP2 + Fe) (Fig. 4C, top panel), it is suggested that it is difficult to achieve saturation of the iron-binding site of FPN1 by applying iron under these conditions. This may result in some part of FPN1, which remains as an iron-lacking form, binding to iron-loaded PCBP2 (Fig. 4C, top panel). Such an interaction could explain the limited binding we observed. However, these data are consistent with the hypothesis that when FPN1 binds iron, there is a conformational change or alteration that prevents or diminishes the binding of iron-bound PCBP2.
Collectively, the results shown in Fig. 4 suggest that iron-depleted FPN1 interacts with iron-loaded PCBP2 but not with iron-depleted PCBP2. The depletion of iron from the PCBP2 fraction markedly decreased its FPN1 binding activity (Fig. 4C, top panel). Conversely, the association of iron with the PCBP2 fraction markedly increased its FPN1 binding activity. These observations agree with the hypothesis that the FPN1-PCBP2 interaction is iron-dependent and the transfer of iron from PCBP2 to FPN1 may occur via direct binding of FPN1 to PCBP2.
PCBP2 Directly Transfers Iron to FPN1
NTBI Iron-loading Studies
In general, there are two major pathways of iron uptake by mammalian cells: (i) the uptake of NTBI by DMT1A-I, which is localized to the plasma membrane; and (ii) the uptake of TBI via Tf-TfR1 endocytosis and subsequent transmembrane transport of Tf-derived iron by DMT1A-II/DMT1B-II, which are localized to recycling endosomes (7, 15, 16). In our previous report, we demonstrated that PCBP2 is indispensable for iron import through DMT1 via both pathways in HEp-2 cells (14). Therefore, we attempted to determine whether PCBP2 is involved in iron export through FPN1.
To investigate the function of PCBP2 in conjunction with FPN1, the ferritin and TfR1 expression levels and intracellular iron levels were analyzed (Figs. 5 and 6). It is well known that high levels of iron lead to increased ferritin and decreased TfR1 expression, whereas the opposite occurs under cellular iron depletion (7). Many cell types, including HEp-2 cells, internalize NTBI (e.g. FAC or ferric nitrilotriacetate (Fe-NTA)) resulting in iron loading (7, 44). In fact, their efficiency in incorporating NTBI is enhanced by the expression of DMT1A-I on the plasma membrane (45). It is notable that the expression level of endogenous FPN1 in HEp-2 cells was below the limit of detection of immunoblotting (Fig. 5A).
FIGURE 5.

FPN1 can export NTBI-derived iron. A and C, FPN1-GFP can export iron loaded as NTBI. HEp-2 cells stably expressing DMT1A-I were cultured in serum-free DMEM and preloaded with 100 μm FAC or 100 μm Fe-NTA for 6 h at 37 °C. The cells were washed with PBS, transfected with FPN1-GFP expression plasmid, and then incubated in serum-free RPMI 1640 medium for 18 h at 37 °C. The cells were lysed in TNE buffer for immunoblotting analysis (A) or in 50 mm NaOH for ferrozine-based iron assay (C). B, the density of specific bands from immunoblotting was measured with a computer-assisted imaging analysis system (NIH ImageJ), and normalization was to total protein loaded. The histogram shows the expression ratio of PCBP2, TfR1, tubulin, FPN1, GFP, and ferritin. Each column corresponds to the lane of immunoblotting analysis (A) in order from left to right. Each column is denoted as per the following labeling: (−)6 h, FAC/Fe-NTA(−) incubation 6 h; (+)6 h, FAC/Fe-NTA(+) incubation 6 h; (+)18 h, FAC/Fe-NTA(+) incubation 6 and 18 h; and (+)18 h FPN/GFP, FAC/Fe-NTA(+) incubation 6 h and FPN1 or GFP transfection 18 h. Significance was determined using Student's t test: †, p < 0.05; and §, p < 0.001. C, the histogram shows the relative intracellular iron level. The detailed method is described under “Experimental Procedures.” The results in the histogram are presented as the means ± S.E. of three independent experiments. Significance was determined using Student's t test: †, p < 0.05; §, p < 0.001.
Incubation of HEp-2 cells stably expressing DMT1A-I with NTBI (i.e. FAC or Fe-NTA) for 6 h/37 °C significantly increased the intracellular iron level (Fig. 5). This was demonstrated indirectly by the finding that TfR1 expression was significantly (p < 0.05) decreased and that ferritin expression was markedly and significantly (p < 0.001) increased under these conditions (Fig. 5, A and B). Furthermore, it was also assessed directly by quantitating the cellular iron levels, which were markedly and significantly (p < 0.001) increased after the loading period of 6 h at 37 °C (Fig. 5C).
Upon incubation of cells for an additional 18 h at 37 °C after NTBI had been withdrawn, TfR1 expression remained low and ferritin expression remained high (Fig. 5, A and B). In agreement with this observation, the intracellular iron level was not significantly (p > 0.05) decreased under these conditions (Fig. 5C). However, transfection of exogenous FPN1 into the NTBI-preloaded cells (confirmed by immunoblotting using an anti-FPN1 pAb (Fig. 5A)) markedly increased TfR1 (p < 0.05) and decreased ferritin (p < 0.001) (Fig. 5, A and B). When GFP as a control was transfected into the NTBI-preloaded cells (confirmed by immunoblotting using an anti-GFP pAb (Fig. 5A)), TfR1 expression remained low and ferritin expression remained high (Fig. 5, A and B). In agreement with these observations, the intracellular iron level remained high after GFP transfection (Fig. 5C). These changes in expression were consistent with the known ability of FPN1 to export iron out of cells to induce iron deficiency (29). This hypothesis was strongly supported by the significant (p < 0.05) decrease in the intracellular iron level after transfection of exogenous FPN1 (Fig. 5C). These results indicated that exogenously expressed FPN1 functioned properly as an iron exporter.
Considering these observations, to investigate the role of PCBP2 in iron export through FPN1, we then silenced PCBP2 expression using a specific siRNA (Fig. 6). The FPN1 expression plasmid and PCBP2 siRNA were transfected into NTBI-preloaded cells, and after incubation for 18 h at 37 °C in culture medium lacking iron, protein expression levels and intracellular iron levels were analyzed (Fig. 6). In the non-siRNA(−)/FPN1 or negative control-siRNA (N)/FPN1-transfected cells, the intracellular iron level was notably decreased, TfR1 expression was increased, and ferritin expression was decreased compared with the iron-replete, NTBI-preloaded cells incubated for 0 h (Fig. 6). These observations were considered to result from iron depletion in the cells by exogenously expressed FPN1.
When PCBP2 expression was silenced by specific PCBP2 siRNA (P2) in the FPN1-transfected cells, its levels were decreased by 80%, TfR1 was decreased by 70%, and ferritin was increased by 5-fold compared with their respective expression levels in non-siRNA(−)/FPN1 or negative control-siRNA (N)/FPN1-transfected cells (Fig. 6, A and B). The return of TfR1 and ferritin expression to almost the same levels as those observed in cells incubated for 0 h was attributed to PCBP2 silencing, which probably prevented iron export via the PCBP2-FPN1 interaction (Fig. 6, A and B). Importantly, the expression level of recombinant FPN1 was not altered by PCBP2 silencing (Fig. 6, A and B).
When GFP as a control was transfected into the NTBI-preloaded cells, TfR1 expression remained low and ferritin expression remained high in three conditions, namely non-siRNA(−)/GFP, negative control-siRNA(N)/GFP, and PCBP2-siRNA(P2)/GFP (Fig. 6, A and B). In agreement with these observations, the intracellular iron level remained high after GFP transfection (Fig. 6C).
The intracellular iron level in the PCBP2-siRNA/FPN1-transfected cells was 4-fold higher than that of the negative control-siRNA(N)/FPN1-transfected cells (Fig. 6C). Hence, the iron export activity of FPN1 was markedly reduced by PCBP2 silencing. These results suggest that PCBP2 functions to provide the iron supplied to the cells as NTBI to be exported out via FPN1.
Holo-Tf Iron-loading Studies
Next, we investigated the function of PCBP2 in conjunction with FPN1 when holo-Tf was used to load cells with iron (Fig. 7). Using a protocol similar to that implemented with NTBI loading (Figs. 5 and 6), very similar results were obtained utilizing holo-Tf as an iron donor (Fig. 7, A–C). That is, when cells were incubated without holo-Tf for 12 h at 37 °C, they expressed TfR1, whereas ferritin was not detected, suggesting a relative cellular iron deficit (Fig. 7A). In contrast, upon loading the cells with iron for 12 h at 37 °C with holo-Tf, there was a decrease in TfR1 expression and an increase in ferritin expression, denoting iron loading (Fig. 7A). This conclusion was confirmed by the direct determination of cellular iron levels (Fig. 7C).
Then, upon an additional incubation for 18 h at 37 °C, this iron-loading phenotype was maintained in the absence of FPN1 transfection, as shown by TfR1 and ferritin expression (Fig. 7, A and B). Again, the maintenance of iron loading was confirmed by the direct estimation of cellular iron levels (Fig. 7C). In contrast, when the cells were transfected with FPN1 and then incubated for 18 h at 37 °C, TfR1 increased (p < 0.05) and ferritin decreased (p < 0.001), suggesting cellular iron depletion via FPN1 (Fig. 7A). The decrease in the cellular iron levels was confirmed by direct estimation, showing a significant (p < 0.01) decrease in iron levels upon transfection with FPN1 (Fig. 7C).
When GFP as a control was transfected into the holo-Tf-preloaded cells, TfR1 expression remained low and ferritin expression remained high (Fig. 7, A and B). In agreement with this observation, the intracellular iron level remained high after GFP transfection (Fig. 7C). Collectively, these studies showed that holo-Tf loaded iron to the cells and that their transfection with FPN1 led to an iron deficiency.
Considering the results shown in Fig. 7, A–C, our studies then assessed the effect of silencing PCBP2 on cellular iron levels using this holo-Tf iron-loading system (Fig. 7, D–F). When PCBP2 expression was silenced using specific PCBP2 siRNA (P2) in FPN1-transfected cells, TfR1 was significantly (p < 0.05) decreased by 60%, whereas ferritin was significantly (p < 0.001) increased by 8-fold compared with their respective expression in non-siRNA(−)/FPN1 or negative control-siRNA(N)/FPN1-transfected cells (Fig. 7, C and D). In fact, both TfR1 and ferritin expression levels returned to nearly the same levels as those observed in the cells incubated for 0 h (Fig. 7D). Furthermore, the intracellular iron level in PCBP2-siRNA(P2)/FPN1-transfected cells was 3–4 times higher than that in non-siRNA(−)/FPN1 or negative control-siRNA(N)/FPN1-transfected cells (Fig. 7F). Hence, silencing PCBP2 resulted in the iron export activity of FPN1 being markedly reduced.
When GFP as a control was transfected into the holo-Tf-preloaded cells, TfR1 expression remained low and ferritin expression remained high under all three conditions, namely non-siRNA(−)/GFP, negative control-siRNA(N)/GFP, and PCBP2-siRNA(P2)/GFP (Fig. 7, D and E). In agreement with these observations, the intracellular iron level remained high after transfection with GFP (Fig. 7F).
Collectively, the results shown in Figs. 5–7 suggest that PCBP2 functions to provide iron to FPN1 prior to FPN1-dependent iron export. This occurs irrespective of whether cellular iron is initially supplied in the form of NTBI (Figs. 5 and 6) or holo-Tf (Fig. 7). Therefore, PCBP2 transfers iron to FPN1 to enable transmembrane export of iron.
Discussion
In this study, we investigated the interactions between FPN1 and the PCBP iron chaperones and found a specific interaction between FPN1 and PCBP2. It was reported that the KH domain of the PCBPs could interact with RNAs, DNAs, or proteins (12, 13). To analyze which domain in the PCBPs was important for the interaction between FPN1 and PCBPs, we focused on the three types of KH domains in these proteins using yeast two-hybrid assays. Bait and prey expression in yeast cells were confirmed by immunoblotting. The results obtained from the yeast two-hybrid assays showed that the C-terminal cytoplasmic region of FPN1 could bind to PCBP2, especially the KH2 domain of PCBP2 (Fig. 1).
Significantly, we did not detect any specific binding of FPN1 to members of the PCBP family aside from PCBP2. Hence, the specific membrane transporter binding activity of PCBP2 was revealed in this investigation. This specificity relative to the other members of the PCBP family could be due to differences in the amino acid sequences of the KH2 domains of the PCBPs (13). It is also possible that each member of the PCBP family may interact with its own specific target molecule in specific subcellular locations and/or cell types to perform specific functions.
To examine whether the N-terminal cytoplasmic region of DMT1 or the C-terminal cytoplasmic region of FPN1 was important for PCBP2 binding, we made deletion mutants (DMT1ΔN and FPN1ΔC) and then examined their intracellular distribution in mammalian cells. DMT1ΔN, lacking the N-terminal amino acids 1–98 of DMT1A-I, was localized to the plasma membrane and ER. DMT1B-I, lacking the N-terminal amino acids 1–29 of DMT1A-I, was localized to late endosomes/lysosomes (18, 40, 41). In a previous study, it was revealed that mutations in N-terminal cytoplasmic region of DMT1 caused mislocalization in the cells (37). Furthermore, FPN1ΔC was localized to the plasma membrane and also the ER. Therefore mislocalization of these proteins must be due to mutations in their cytoplasmic regions, which might induce mis-sorting of the molecules.
Notably, PCBP2 exists abundantly in the cytosol, and all four DMT1 isoforms can bind to PCBP2 regardless of the different distribution of the transporters (14). Therefore, the PCBP2-binding domains in the cytoplasmic regions could bind to PCBP2 even though they were localized to various intracellular membrane structures such as endosomes, lysosomes, and the ER.
In mammalian cells, both DMT1ΔN and FPN1ΔC completely lost their PCBP2 binding activity (Fig. 3C). Importantly, these deletion mutants remained localized to the plasma membrane (Fig. 3B). Irrespective of their plasma membrane location, and partial co-localization with the respective WT transporters (Fig. 3B), they still lost their PCBP2 binding activity (Fig. 3C) and iron transport activity (Fig. 3D). These data indicate that the reason for losing the iron transport activity in mammalian cells was the loss of the DMT1-PCBP2 or FPN1-PCBP2 interaction and not that these transporters became totally mislocalized.
We reported previously that mutants in the cytoplasmic regions of DMT1 still function as iron transporters in yeast cells (37), and the transmembrane-domain structure must be important for their iron transport activity itself (46). Thus, collectively, the specific PCBP2 binding activity of the cytoplasmic regions of each transmembrane iron transporter has been confirmed in both yeast and mammalian cells.
Using the UniProt Knowledgebase, we did not detect any amino acid sequence homology between the N-terminal cytoplasmic region of DMT1 and the C-terminal cytoplasmic region of FPN1. It remains unclear how PCBP2 binds to these specific regions of DMT1 or FPN1.
We investigated whether FPN1 or PCBP2 requires iron for the FPN1-PCBP2 interaction. After iron was removed from a PCBP2-containing fraction, neither iron-loaded FPN1 nor iron-depleted FPN1 could bind to PCBP2 (Fig. 4). Intriguingly, iron-depleted FPN1 strongly bound to iron-loaded PCBP2 (Fig. 4). In the case of DMT1, iron-depleted DMT1 did not bind to PCBP2 at all (14), but iron-loaded DMT1 did bind to PCBP2. Therefore, it is apparent that PCBP2 can bind to both DMT1 and FPN1 but that the effects and properties of each interaction are very different. The lack of similarity between the PCBP2-binding regions of these two transporters suggests a difference in their affinity for PCBP2. Hence, further structural analyses of iron-loaded and -depleted PCBP2 and these transporters are necessary but were beyond the scope of the current study.
We have shown that PCBP2 is essential for iron uptake via DMT1 (14). When PCBP2 expression was silenced, cells could not import iron. The process by which PCBP2 receives iron from DMT1 suggests a model in which PCBP2 disengages from DMT1 and subsequently transports iron to another acceptor. This could occur potentially through a subsequent interaction with a downstream acceptor such as ferritin or FPN1. Moreover, the direct delivery of iron from DMT1 to PCBP2 probably occurs while they are bound to each other. This integrated process could avoid the generation of hazardous free iron in the cytoplasm. Accordingly, we hypothesized that certain iron chaperones deliver iron directly to FPN1.
There are two forms of iron that can be imported by cells: NTBI and TBI (7). The NTBI is predominately relevant for iron-loading diseases such as hemochromatosis and β-thalassemia, whereas TBI is the physiologically important iron donor to tissues (7). The iron export activity of FPN1 was inhibited by PCBP2 silencing regardless of the pathway by which iron was supplied, suggesting that the pathways of iron uptake from NTBI or TBI merge intracellularly. As free iron is extremely hazardous to life because of its ability to induce reactive oxygen species production, it cannot be liberated freely into the cytoplasm. Thus, the task of each iron chaperone molecule is probably crucial to direct iron appropriately irrespective of whether it is supplied as NTBI or TBI. It can be further speculated that the role of each iron chaperone is likely to be somewhat unique in iron metabolism, as found for the metabolism of copper (47).
Considering this latter suggestion, emerging evidence indicates that PCBP1 and PCBP2 deliver iron not only to the iron storage protein, ferritin, but also directly to some enzymes that contain mono- and dinuclear iron centers (48). For instance, the prolyl and asparaginyl hydroxylases that modify the important transcriptional regulator hypoxia-inducible factor 1α (HIF-1α) accept iron from PCBPs (49). Similarly, deoxyhypusine hydroxylase, which is required for the modification of eukaryotic translation initiation factor 5A (eIF5A), is also a downstream target of these iron chaperones (48). These functions, together with our findings demonstrating that PCBP2 acts as a gateway keeper for cellular iron uptake (14) and also for cellular iron efflux via FPN1, should now focus attention on other possible molecular interactions of PCBPs. For instance, it is conceivable that PCBPs could interact with other iron-transporting transmembrane proteins such as ZIP14 (50) and mitoferrin (51). Hence, the participation of different PCBP family members in such processes needs to be clarified.
Another challenge in understanding the biology of the PCBPs will be to explain how functions in iron transport overlap with their described nuclear and cytoplasmic control over gene expression (52). For example, Pcbp2−/− mice demonstrate that PCBP2 has a non-redundant role in vivo during hematopoiesis, which is attributed to its post-transcriptional control over α-globin gene expression (53). However, another possibility regarding this latter finding emerges from our current and previous studies regarding PCBP2 function (14). That is, the potential ability of PCBP2 to control Tf iron uptake by DMT1 and also iron release by FPN1 in erythroid precursors may possibly affect erythropoiesis, and these results could explain the non-redundant role of PCBP2 in hematopoiesis.
In conclusion, PCBP2 acts as a recipient of iron from DMT1 and as a donor of iron to FPN1; both transmembrane transporters function as intracellular receptors for the iron chaperone PCBP2 but not PCBP1, PCBP3, or PCBP4. The role of PCBP2 as a crucial molecular partner in the release of iron from cells via FPN1 provides another link to understanding the complex protein network involved in cellular iron metabolism.
Experimental Procedures
Antibodies and Reagents
A mouse anti-human α-tubulin monoclonal antibody (mAb) was purchased from BD Transduction Laboratories (Franklin Lakes, NJ); a mouse anti-human PCBP2 mAb was purchased from Abnova (Taipei, Taiwan, catalog No. H00005094-M07); a goat anti-human PCBP1 polyclonal antibody (pAb) was purchased from Acris (Herford, Germany, catalog No. AP22434PU-N); a rabbit anti-human PCBP3 pAb was purchased from Atlas Antibodies (Stockholm, Sweden, catalog No. HPA030247); a rabbit anti-human PCBP4 pAb was purchased from Novus Biologicals (Minneapolis, MN, catalog No. NBP1-76729); a rabbit anti-human ferritin pAb was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, catalog No. sc-25617); a rabbit anti-human PDI mAb was purchased from Cell Signaling Technology (Boston MA, catalog No. 3501); an anti-human LAMP2 mAb (H4B4, developed by Drs. J. E. K. Hildreth and J. T. August) was obtained from the Developmental Studies Hybridoma Bank, University of Iowa; a mouse anti-human c-Myc mAb was purchased from Cell Signaling Technology (catalog No. 2276); a mouse anti-HA mAb was purchased from Cell Signaling Technology (catalog No. 2367). We previously raised a rabbit polyclonal antibody against GFP (16, 17). A mouse anti-TfR1 mAb (N-2) was prepared as described previously (54).
A rabbit anti-human FPN1 pAb was raised against amino acids 220–307 of FPN1 for the present study. Briefly, the affinity-purified anti-FPN1 pAb was purified from antiserum on a HiTrapTM N-hydroxysuccinimide-activated column (GE Healthcare Life Sciences) coupled with maltose-binding protein-tagged FPN1-(220–307), which was produced in E. coli strain JM109 and purified on a column of amylose resin. All animal experiments were approved by the Animal Ethics Committee of Kawasaki Medical School.
HRP-conjugated anti-goat IgG was purchased from Jackson ImmunoResearch (West Grove, PA, catalog No. 705-035-147), and HRP-conjugated anti-mouse (catalog No. 7076) and anti-rabbit IgG (catalog No. 7074) were purchased from Cell Signaling Technology. An Alexa 594-labeled goat anti-rabbit IgG (catalog No. A-11037) and an Alexa 594-labeled goat anti-mouse (catalog No. A-11005) IgG were purchased from Thermo Fisher Scientific. All antibodies were utilized in the range of 1/100–1/10,000. Most other general reagents were purchased from Wako Chemicals, Nacalai Tesque (Kyoto, Japan), or Sigma-Aldrich.
Cell Culture
HEp-2 cells were maintained in DMEM supplemented with 10% FBS. Cells were transfected with GFP-tagged full-length and deletion mutant forms of DMT1 and FPN1 or the mCherry-tagged full-length form of CPR by Lipofectamine® 3000 (Thermo Fisher Scientific).
Protein Extraction and Immunoblot Analysis
Whole cell lysates were extracted using TNE buffer (10 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 2% (w/v) Nonidet P-40) and immunoblotting performed by standard methods as described previously (55). Briefly, equal amounts of protein (15 μg), loaded and separated on 10 or 12.5% SDS-PAGE, were then transferred to PVDF membranes. The membranes were incubated with primary antibodies (see “Antibodies and Reagents” above) at 4 °C overnight and then incubated with HRP-conjugated secondary antibodies (as detailed above) at room temperature for 1 h.
Immunoprecipitation Protocol
Whole cell lysates or membrane fractions solubilized in TNE buffer were incubated with anti-GFP pAb conjugated to protein A-Sepharose beads (GE Healthcare Life Sciences) and incubated at 4 °C overnight. The beads were recovered by centrifugation and washed three times with TNE buffer. Proteins were extracted from the beads and then analyzed by immunoblotting (14).
Vectors and Plasmid Constructs
To express human DMT1A-I or FPN1, each cDNA was cloned into a pEGFP-N1 vector. The deletion mutant forms of DMT1 and FPN1 were generated via PCR-mediated mutagenesis using KOD-Plus DNA polymerase (Toyobo, Osaka, Japan). To express human CPR, the cDNA was cloned into the pRSET-B-mCherry vector (kindly provided by Dr. Roger Y. Tsien, University of California, San Diego). The amino acid coding regions of human PCBP1, PCBP2, and PCBP4 were amplified using total RNA from human HEp-2 cells, and the amino acid coding region of PCBP3 was amplified using total RNA from human placenta.
Yeast Two-hybrid Assay
The yeast two-hybrid assay was performed as described by the manufacturer (Clontech, Mountain View, CA). For bait plasmids, the N- or C-terminal cytoplasmic region or the intracellular loop between the sixth and seventh transmembrane domains of FPN1 (Fig. 1) was cloned into the GAL4 DNA-binding domain expression vector (Clontech). PCBP1 (amino acids 12–80 (I-1), 94–193 (I-2), 278–348 (I-3), and 1–356 (I-F)), PCBP2 (amino acids 12–80 (II-1), 94–192 (II-2), 285–357 (II-3), and 1–366 (II-F)), PCBP3 (amino acids 44–112 (III-1), 126–220 (III-2), 292–362 (III-3), and 1–371 (III-F)), or PCBP4 (amino acids 16–84 (IV-1), 98–192 (IV-2), 240–310 (IV-3), and 1–403 (IV-F)) was cloned into the GAL4 DNA-activating domain expression vector as prey (Fig. 1A).
To confirm the interaction between FPN1 and PCBP2, the C-terminal cytoplasmic region of FPN1 was cloned into the GAL4 activation domain expression vector as prey (Fig. 2A). PCBP1 (amino acids 12–80 (I-1), 94–193 (I-2), 278–348 (I-3), and 1–356 (I-F)), PCBP2 (amino acids 12–80 (II-1), 94–192 (II-2), 285–357 (II-3), and 1–366 (II-F)), PCBP3 (amino acids 44–112 (III-1), 126–220 (III-2), 292–362 (III-3), and 1–371 (III-F)), or PCBP4 (amino acids 16–84 (IV-1), 98–192 (IV-2), 240–310 (IV-3), and 1–403 (IV-F)) was cloned in to the GAL4 DNA-binding domain expression vector as bait (Fig. 2). Yeast whole protein was extracted using the lithium acetate/NaOH method as described previously (56).
Briefly, yeast cells were cultured in yeast nitrogen base with 2% glucose and the appropriate amino acids at 30 °C until A600 =1.0–3.0. Yeast cells were harvested and treated with 1.0 m lithium acetate for 10 min at room temperature and then treated for 5 min with 0.4 m NaOH on ice. These pretreated yeast cells were boiled in SDS-sample buffer for 3 min. The supernatant was collected and analyzed by immunoblotting.
Confocal Microscopy
Cells grown on glass coverslips were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 15 min. The coverslips were washed and blocked in 0.1% fish skin gelatin in PBS. Cells were incubated with primary antibodies for 60 min at room temperature. Coverslips were washed with 0.1% fish skin gelatin in PBS. Secondary antibody coupled to Alexa 594 was incubated with cells for 60 min at room temperature. Coverslips were washed and mounted on slides with Vectashield (catalog No. H-1000, Vector Laboratories, Burlingame, CA). The images were obtained by using a Zeiss LSM 700 confocal laser-scanning microscope system (Jena, Germany) (18).
Membrane Fractionation
HEp-2 cells were grown to confluence in 100-mm dishes. Cells were washed twice with PBS, removed from the plate in homogenization buffer (0.25 M sucrose, 10 mm Tris-HCl (pH 7.4), and protease inhibitor mixture), and homogenized in a Teflon-coated glass Potter homogenizer rotating at 1000 rpm for 15 strokes. The homogenate was then centrifuged at 1,000 × g for 10 min at 4 °C to obtain the postnuclear supernatant, which was centrifuged at 10,000 × g for 20 min/4 °C to obtain the postmitochondrial fraction. Then, the postmitochondrial fraction was centrifuged at 100,000 × g for 30 min at 4 °C to obtain the membrane fraction as described previously (14, 17). The membrane fraction was resolved in 250 μl of TNE buffer.
To remove PCBP2 from the membrane fraction, this fraction was resuspended in homogenization buffer containing 0.1 m Na2CO3 (pH 11.0). The suspension was incubated on ice for 30 min and then centrifuged at 100,000 × g for 30 min at 4 °C. The resulting precipitate, consisting of PCBP2-depleted cell membranes, is termed the “mΔP2 fraction” (14, 16). Protein concentrations were determined using Lowry's method (57).
In Vitro FPN1 and PCBP2-binding Assay: Loading or Depleting the mΔP2 Fraction of Iron
The mΔP2 fraction was solubilized in TNE buffer and mixed with the anti-GFP pAb conjugated to protein A-Sepharose beads as described previously (14). Then, 100 μm FeCl2 (Wako Chemicals; freshly prepared at time of use) or the chelator CDTA (100 μm, Wako Chemicals) was added to the mixture to load or deplete the fraction of iron, respectively. The beads were washed three times with PBS and then mixed with the soluble cytosolic fraction containing 100 μm FeCl2 or 100 μm CDTA for 2 h at 4 °C. The samples were washed again and analyzed by immunoblotting.
Stealth RNAi siRNA
Stealth RNAi siRNA targeting a 25-nucleotide sequence of PCBP2 was purchased from Thermo Fisher Scientific. HEp-2 cells were transfected with 10 nm Stealth RNAi using Lipofectamine® RNAiMAX (Thermo Fisher Scientific) and incubated for 18 h at 37 °C. The Lipofectamine® RNAiMAX:siRNA ratio was 1.0 μl:7.5 pmol. The sequence of PCBP2 Stealth RNAi was: 5′-CCGGUGUGAUUGAAGGUGGAUUAAA-3′. A Stealth RNAi with similar guanine-cytosine (GC) content (Stealth RNAiTM siRNA negative control medium-GC duplex No. 2) was purchased from Thermo Fisher Scientific (catalog No. 12935-112) and used as a control.
Iron Export Assay
Two procedures were used to load cellular iron; one method used NTBI and the other TBI. To load NTBI into cells, HEp-2 cells stably expressing DMT1A-I-GFP were cultured in serum-free DMEM supplemented with 100 μm FAC or 100 μm Fe-NTA (Fe:NTA ratio, 1:4) for 6 h at 37 °C.
To load TBI into cells, HEp-2 cells were cultured in serum-free DMEM supplemented with 0.3 mg/ml (3.75 μm) holo-Tf (Calbiochem) for 12 h at 37 °C. Cells were washed with PBS and then transfected with FPN1-GFP using Lipofectamine 3000 and with PCBP2 siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Cells were incubated in serum-free RPMI 1640 medium (Thermo Fisher Scientific) for 18 h at 37 °C. Then, the cells were washed with PBS containing 1 mm EDTA and lysed in TNE buffer for immunoblotting analysis or in 50 mm NaOH for the measurement of iron concentration.
Ferrozine-based Iron Assay
To directly quantify the intracellular iron concentration, we performed a ferrozine-based iron assay. The detailed method was described previously (58). Briefly, cells were lysed in 50 mm NaOH and then treated with acidic KMnO4 to release iron from proteins. The samples were then incubated in an iron detection agent (6.5 mm ferrozine, 6.5 mm neocuproine, 2.5 m ammonium acetate, and 1 m ascorbic acid dissolved in water). A colorimetric measurement was performed by calculating the absorbance of each sample relative to the absorbance of FeCl3 standards at a range of concentrations.
Statistics
The density of the specific bands from immunoblotting was measured with a computer-assisted imaging analysis system (NIH ImageJ) and normalization was to total protein loaded. All statistical analysis was performed using Student's t test and considered significant when p < 0.05. All results are typical of at least three experiments.
Author Contributions
I. Y. conducted most of the experiments, analyzed the results, and wrote the paper. D. R. R. suggested experiments and also was involved in analyzing results and writing the manuscript. K. I. conducted experiments for the binding assay in yeast. F. K. conceived the study and wrote the paper in association with I. Y.
Acknowledgments
We thank Dr. Darius Lane, Kevin Park, and Dr. Vera Richardson (Molecular Pharmacology and Pathology Program, University of Sydney) for suggestions, English proofreading, and careful editing of the manuscript.
This work was supported by a Grant-in-aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan (ID:22591052), the Research Program on Hepatitis from the Japan Agency for Medical Research and development (AMED), and research project grants from Kawasaki Medical School (ID:26-26, 27-50). The authors declare that they have no conflicts of interest related to the contents of this article.
- PCBP
- poly(rC)-binding protein
- KH
- K-homology
- DMT
- divalent metal transporter
- Tf
- transferrin
- TBI
- transferrin-bound iron
- NTBI
- non-Tf-bound iron
- FPN1
- ferroportin 1
- ER
- endoplasmic reticulum
- CPR
- cytochrome P450 reductase
- FAC
- ferric ammonium citrate
- CDTA
- 1,2-cyclohexylenedinitrilotetraacetic acid
- PDI
- protein disulfide isomerase
- pAb
- polyclonal antibody
- Fe-NTA
- ferric nitrilotriacetate.
References
- 1. Richardson D. R., Lane D. J., Becker E. M., Huang M. L., Whitnall M., Suryo Rahmanto Y., Sheftel A. D., and Ponka P. (2010) Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. U.S.A. 107, 10775–10782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Netz D. J., Stith C. M., Stümpfig M., Köpf G., Vogel D., Genau H. M., Stodola J. L., Lill R., Burgers P. M., and Pierik A. J. (2012) Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 8, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dunn L.L., Suryo Rahmanto Y., and Richardson D.R. (2007) Iron uptake and metabolism in the new millennium. Trends Cell Biol. 17, 93–100 [DOI] [PubMed] [Google Scholar]
- 4. Wolozin B., and Behl C. (2000) Mechanisms of neurodegenerative disorders: Part 1. Protein aggregates. Arch. Neurol. 57, 793–796 [DOI] [PubMed] [Google Scholar]
- 5. Fuhrman B., Oiknine J., and Aviram M. (1994) Iron induces lipid peroxidation in cultured macrophages, increases their ability to oxidatively modify LDL, and affects their secretory properties. Atherosclerosis 111, 65–78 [DOI] [PubMed] [Google Scholar]
- 6. Richardson D. R., Ponka P., and Vyoral D. (1996) Distribution of iron in reticulocytes after inhibition of heme synthesis with succinylacetone: examination of the intermediates involved in iron metabolism. Blood 87, 3477–3488 [PubMed] [Google Scholar]
- 7. Richardson D. R., and Ponka P. (1997) The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1331, 1–40 [DOI] [PubMed] [Google Scholar]
- 8. Philpott C. C. (2012) Coming into view: eukaryotic iron chaperones and intracellular iron delivery. J. Biol. Chem. 287, 13518–13523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi H., Bencze K. Z., Stemmler T. L., and Philpott C. C. (2008) A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leffers H., Dejgaard K., and Celis J. E. (1995) Characterisation of two major cellular poly(rC)-binding human proteins, each containing three K-homologous (KH) domains. Eur. J. Biochem. 230, 447–453 [PubMed] [Google Scholar]
- 11. Leidgens S., Bullough K. Z., Shi H., Li F., Shakoury-Elizeh M., Yabe T., Subramanian P., Hsu E., Natarajan N., Nandal A., Stemmler T. L., and Philpott C. C. (2013) Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 288, 17791–17802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Makeyev A. V., Chkheidze A. N., and Liebhaber S. A. (1999) A set of highly conserved RNA-binding proteins, αCP-1 and αCP-2, implicated in mRNA stabilization, are coexpressed from an intronless gene and its intron-containing paralog. J. Biol. Chem. 274, 24849–24857 [DOI] [PubMed] [Google Scholar]
- 13. Makeyev A. V., and Liebhaber S. A. (2002) The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8, 265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yanatori I., Yasui Y., Tabuchi M., and Kishi F. (2014) Chaperone protein involved in transmembrane transport of iron. Biochem. J. 462, 25–37 [DOI] [PubMed] [Google Scholar]
- 15. Canonne-Hergaux F., Fleming M. D., Levy J. E., Gauthier S., Ralph T., Picard V., Andrews N. C., and Gros P. (2000) The Nramp2/DMT1 iron transporter is induced in the duodenum of microcytic anemia mk mice but is not properly targeted to the intestinal brush border. Blood 96, 3964–3970 [PubMed] [Google Scholar]
- 16. Tabuchi M., Yoshimori T., Yamaguchi K., Yoshida T., and Kishi F. (2000) Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 275, 22220–22228 [DOI] [PubMed] [Google Scholar]
- 17. Tabuchi M., Tanaka N., Nishida-Kitayama J., Ohno H., and Kishi F. (2002) Alternative splicing regulates the subcellular localization of divalent metal transporter 1 isoforms. Mol. Biol. Cell 13, 4371–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yanatori I., Tabuchi M., Kawai Y., Yasui Y., Akagi R., and Kishi F. (2010) Heme and non-heme iron transporters in non-polarized and polarized cells. BMC Cell Biol. 11, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lane D. J., and Richardson D. R. (2014) Chaperone turns gatekeeper: PCBP2 and DMT1 form an iron-transport pipeline. Biochem. J. 462, e1–3 [DOI] [PubMed] [Google Scholar]
- 20. Abboud S., and Haile D. J. (2000) A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 275, 19906–19912 [DOI] [PubMed] [Google Scholar]
- 21. Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S. J., Moynihan J., Paw B. H., Drejer A., Barut B., Zapata A., Law T. C., Brugnara C., Lux S. E., Pinkus G. S., Pinkus J. L., et al. (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781 [DOI] [PubMed] [Google Scholar]
- 22. McKie A. T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D., Miret S., Bomford A., Peters T. J., Farzaneh F., Hediger M. A., Hentze M. W., and Simpson R. J. (2000) A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 5, 299–309 [DOI] [PubMed] [Google Scholar]
- 23. Nicolas G., Bennoun M., Devaux I., Beaumont C., Grandchamp B., Kahn A., and Vaulont S. (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 8780–8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park C. H., Valore E. V., Waring A. J., and Ganz T. (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 276, 7806–7810 [DOI] [PubMed] [Google Scholar]
- 25. Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P., and Loréal O. (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276, 7811–7819 [DOI] [PubMed] [Google Scholar]
- 26. Drakesmith H., Nemeth E., and Ganz T. (2015) Ironing out ferroportin. Cell Metab. 22, 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nemeth E., Tuttle M. S., Powelson J., Vaughn M. B., Donovan A., Ward D. M., Ganz T., and Kaplan J. (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 [DOI] [PubMed] [Google Scholar]
- 28. Ganz T. (2013) Systemic iron homeostasis. Physiol. Rev. 93, 1721–1741 [DOI] [PubMed] [Google Scholar]
- 29. Kalinowski D. S., Stefani C., Toyokuni S., Ganz T., Anderson G. J., Subramaniam N. V., Trinder D., Olynyk J. K., Chua A., Jansson P. J., Sahni S., Lane D. J., Merlot A. M., Kovacevic Z., Huang M. L., et al. (2016) Redox cycling metals: Pedaling their roles in metabolism and their use in the development of novel therapeutics. Biochim. Biophys. Acta 1863, 727–748 [DOI] [PubMed] [Google Scholar]
- 30. Wallace D. F., Harris J. M., and Subramaniam V. N. (2010) Functional analysis and theoretical modeling of ferroportin reveals clustering of mutations according to phenotype. Am. J. Physiol. Cell Physiol. 298, C75–C84 [DOI] [PubMed] [Google Scholar]
- 31. Chien C. T., Bartel P. L., Sternglanz R., and Fields S. (1991) The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. U.S.A. 88, 9578–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alver R. C., Zhang T., Josephrajan A., Fultz B. L., Hendrix C. J., Das-Bradoo S., and Bielinsky A. K. (2014) The N terminus of Mcm10 is important for interaction with the 9–1-1 clamp and in resistance to DNA damage. Nucleic Acids Res. 42, 8389–8404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu F. L., Liu P. H., Shao H. W., and Kung F. L. (2006) The intracellular domain of amyloid precursor protein interacts with FKBP12. Biochem. Biophys. Res. Commun. 350, 472–477 [DOI] [PubMed] [Google Scholar]
- 34. Toyoda H., Franco D., Fujita K., Paul A. V., and Wimmer E. (2007) Replication of poliovirus requires binding of the poly(rC) binding protein to the cloverleaf as well as to the adjacent C-rich spacer sequence between the cloverleaf and the internal ribosomal entry site. J. Virol. 81, 10017–10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walter B. L., Nguyen J. H., Ehrenfeld E., and Semler B. L. (1999) Differential utilization of poly(rC)-binding protein 2 in translation directed by picornavirus IRES elements. RNA 5, 1570–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walter B. L., Parsley T. B., Ehrenfeld E., and Semler B. L. (2002) Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J. Virol. 76, 12008–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tabuchi M., Yoshida T., Takegawa K., and Kishi F. (1999) Functional analysis of the human NRAMP family expressed in fission yeast. Biochem. J. 344, 211–219 [PMC free article] [PubMed] [Google Scholar]
- 38. Freedman R. B. (1989) Protein disulfide isomerase: multiple roles in the modification of nascent secretory proteins. Cell 57, 1069–1072 [DOI] [PubMed] [Google Scholar]
- 39. Carlsson S. R., Roth J., Piller F., and Fukuda M. (1988) Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2: major sialoglycoproteins carrying polylactosaminoglycan. J. Biol. Chem. 263, 18911–18919 [PubMed] [Google Scholar]
- 40. Kishi F., and Tabuchi M. (1997) Complete nucleotide sequence of human NRAMP2 cDNA. Mol. Immunol. 34, 839–842 [DOI] [PubMed] [Google Scholar]
- 41. Hubert N., and Hentze M. W. (2002) Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc. Natl. Acad. Sci. U.S.A. 99, 12345–12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghosh D., Srivastava G. P., Xu D., Schulz L. C., and Roberts R. M. (2008) A link between SIN1 (MAPKAP1) and poly(rC)-binding protein 2 (PCBP2) in counteracting environmental stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11673–11678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chkheidze A. N., and Liebhaber S. A. (2003) A novel set of nuclear localization signals determine distributions of the αCP RNA-binding proteins. Mol. Cell. Biol. 23, 8405–8415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujiki Y., Hubbard A. L., Fowler S., and Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yanatori I., Yasui Y., Noguchi Y., and Kishi F. (2015) Inhibition of iron uptake by ferristatin II is exerted through internalization of DMT1 at the plasma membrane. Cell Biol. Int. 39, 427–434 [DOI] [PubMed] [Google Scholar]
- 46. Rosakis A., and Köster W. (2005) Divalent metal transport in the green microalga Chlamydomonas reinhardtii is mediated by a protein similar to prokaryotic Nramp homologues. Biometals 18, 107–120 [DOI] [PubMed] [Google Scholar]
- 47. Palumaa P. (2013) Copper chaperones: the concept of conformational control in the metabolism of copper. FEBS Lett. 587, 1902–1910 [DOI] [PubMed] [Google Scholar]
- 48. Frey A. G., Nandal A., Park J. H., Smith P. M., Yabe T., Ryu M. S., Ghosh M. C., Lee J., Rouault T. A., Park M. H., and Philpott C. C. (2014) Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 111, 8031–8036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nandal A., Ruiz J. C., Subramanian P., Ghimire-Rijal S., Sinnamon R. A., Stemmler T. L., Bruick R. K., and Philpott C. C. (2011) Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 14, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liuzzi J. P., Aydemir F., Nam H., Knutson M. D., and Cousins R. J. (2006) Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. U.S.A. 103, 13612–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shaw G. C., Cope J. J., Li L., Corson K., Hersey C., Ackermann G. E., Gwynn B., Lambert A. J., Wingert R. A., Traver D., Trede N. S., Barut B. A., Zhou Y., Minet E., Donovan A., Brownlie A., et al. (2006) Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100 [DOI] [PubMed] [Google Scholar]
- 52. Ghanem L. R., Chatterji P., and Liebhaber S. A. (2014) Specific enrichment of the RNA-binding proteins PCBP1 and PCBP2 in chief cells of the murine gastric mucosa. Gene Expr. Patterns 14, 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghanem L. R., Kromer A., Silverman I. M., Chatterji P., Traxler E., Penzo-Mendez A., Weiss M. J., Stanger B. Z., and Liebhaber S. A. (2015) The poly(C)-binding protein pcbp2 and its retrotransposed derivative pcbp1 are independently essential to mouse development. Mol. Cell. Biol. 36, 304–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshimori T., Shimonishi Y., and Uchida T. (1988) Binding properties of monoclonal antibody to the cytoplasmic domain of transferrin receptor. Cell Struct. Funct. 13, 311–324 [DOI] [PubMed] [Google Scholar]
- 55. Kovacevic Z., Menezes S. V., Sahni S., Kalinowski D. S., Bae D. H., Lane D. J., and Richardson D. R. (2016) The metastasis suppressor, N-MYC downstream-regulated gene-1 (NDRG1), down-regulates the ErbB family of receptors to inhibit downstream oncogenic signaling pathways. J. Biol. Chem. 291, 1029–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang T., Lei J., Yang H., Xu K., Wang R., and Zhang Z. (2011) An improved method for whole protein extraction from yeast Saccharomyces cerevisiae. Yeast 28, 795–798 [DOI] [PubMed] [Google Scholar]
- 57. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 58. Riemer J., Hoepken H. H., Czerwinska H., Robinson S. R., and Dringen R. (2004) Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 331, 370–375 [DOI] [PubMed] [Google Scholar]