

Abstract
A variant of the cation channel channelrhodopsin-2 from Chlamydomonas reinhardtii (CrChR2) was selectively labeled at position Cys-79 at the end of the first cytoplasmic loop and the beginning of transmembrane helix B with the fluorescent dye fluorescein (acetamidofluorescein). We utilized (i) time-resolved fluorescence anisotropy experiments to monitor the structural dynamics at the cytoplasmic surface close to the inner gate in the dark and after illumination in the open channel state and (ii) time-resolved fluorescence quenching experiments to observe the solvent accessibility of helix B at pH 6.0 and 7.4. The light-induced increase in final anisotropy for acetamidofluorescein bound to the channel variant with a prolonged conducting state clearly shows that the formation of the open channel state is associated with a large conformational change at the cytoplasmic surface, consistent with an outward tilt of helix B. Furthermore, results from solute accessibility studies of the cytoplasmic end of helix B suggest a pH-dependent structural heterogeneity that appears below pH 7. At pH 7.4 conformational homogeneity was observed, whereas at pH 6.0 two protein fractions exist, including one in which residue 79 is buried. This inaccessible fraction amounts to 66% in nanodiscs and 82% in micelles. Knowledge about pH-dependent structural heterogeneity may be important for CrChR2 applications in optogenetics.
Keywords: fluorescence, fluorescence anisotropy, membrane protein, optogenetics, protein conformation, channelrhodopsin, fluorescence quenching
Introduction
Channelrhodopsins are involved in phototaxis and photophobia of unicellular green algae (1). They constitute a new class of light-gated ion channels containing the seven-transmembrane helix motif and the chromophore retinal as the light-sensitive cofactor, which are both common to the other retinal-containing proteins such as bacteriorhodopsin (bR)4 or visual rhodopsin (2). In CrChR2 the chromophore retinal is bound to Lys-257 (3). Channelrhodopsin activation via light-induced isomerization of retinal from all-trans to 13-cis is coupled to a transient cofactor deprotonation and a functional protein structural change. In channelrhodopsin, as well as in other retinal-containing photoreceptors, subtle changes in the chromophore vicinity trigger large scale protein conformational changes remote from the chromophore binding pocket. Rearrangement of helix B is hypothesized to constitute a key element in channel opening by allowing the entry of water molecules (4–6). A continuous water wire is a prerequisite for the formation of the ion-conducting pathway. A hydrophilic pore between helices A, B, C, and G was suggested to serve as the ion permeation pathway based on the high resolution crystal structure of the C1C2 chimera (3). Polar and charged residues of transmembrane helix B line this pore. Important constituents of the pathway are the inner gate (intracellular (cytoplasmic) side) and the access channel (extracellular side), as well as the central gate with critical determinants to regulate cation selectivity in CrChR2 (for a review see Ref. 5). Residue Cys-128, close to the retinal binding pocket, constitutes the so-called DC gate together with Asp-156 (7). Mutants of Cys-128, e.g. C128T, show a prolonged lifetime of the conducting state (8). These mutants are thus very well suited for trapping the P3520 state, the conducting open channel state (also named P520), in spectroscopic measurements, because no other effect except a prolonged open state was found upon mutation of Cys-128 (9, 10).
The inner gate was hypothesized to be involved in CrChR2 activation (formation of the open conducting channel state) together with the tilt of helix B (5). Evidence for light-induced movements of helix B comes from structural studies using electron crystallography and EPR spectroscopy (double electron-electron resonance) (11–13). Together these data suggest that in contrast to bR and sensory rhodopsin, where large helix movements were observed for helix F upon reprotonation of the retinal Schiff base (14–18), helix B in CrChR2 undergoes prominent light-induced rearrangements. However, the EPR distance measurements are not able to discriminate between conformational changes of the spin label and structural changes of the protein moiety. Therefore, it is not entirely clear whether the light-induced changes (11, 13) originate from helix B displacements or local structural changes of the loop or label conformation.
To investigate the conformational dynamics of the cytoplasmic end of helix B in real time, i.e. on the picosecond and nanosecond time scale, we selectively labeled position Cys-79 in a C128T variant of CrChR2 (11) with a fluorescent dye. Then we followed the conformational changes of the channel in detergent (CrChR2 micelles) and lipid environment (CrChR2 nanodiscs) by time-resolved fluorescence depolarization. This technique is very well suited to obtain information on local and global protein dynamics on the nanosecond time scale, because the dynamics of the covalently bound dye is affected by the motion of the protein segment to which it is covalently attached. The different modes of motion can be separated in the time domain (16, 19–21). We measured the time-resolved anisotropy curves in the dark state of the channel and after light activation at pH 6.0 and pH 7.4. The increase in final anisotropy at both pH values in nanodiscs clearly showed that the light-induced formation of the open channel state is associated with a strong increase in sterical hindrance of helix B motion, suggesting a large conformational change at the cytoplasmic surface. Accessibility studies by means of fluorescence quenching with iodide, a polar quencher, support these interpretations. Moreover, the latter experiments revealed a pH-dependent structural heterogeneity of helix B in close vicinity to the inner gate.
Results
Ion Channel Functionality
ChR2CA/C128T-C79/208 (11), a CrChR2 variant with a prolonged open channel state containing Cys-79 and Cys-208, was used in our studies. To show ion channel functionality ChR2-C128T and ChR2CA/C128T-C79/208 were expressed in HEK-293T cells, and photocurrents were measured using whole cell patch clamp recordings. At −60 mV blue light (460 nm) evoked inward-directed currents of similar shape and amplitude, but with different kinetics in ChR2-C128T and the Cys mutant (Fig. 1, A, B, and E–G). Notably, the current decay following a 100-ms light pulse was more than two times slower in ChR2CA/C128T-C79/208 (4890 ± 390 ms) compared with ChR2-C128T (2300 ± 170 ms, p < 0.0001 (Fig. 1G). ChR2CA/C128T-C79/208 showed the typical inward rectifying current-voltage relationship with a similar reversal potential Erev as in ChR2-C128T (Fig. 1C). Finally, light responsiveness as quantified by the velocity of initial current rise was not different in-between both variants (Fig. 1D); thus ChR2CA/C128T-C79/208 is a functional light-activated channel with similar photocurrent properties as found in the step-function-rhodopsin ChR2-C128T.
FIGURE 1.

Photocurrent properties of ChR2-C128T and ChR2CA/C128T-C79/208. A and B, typical photocurrent traces of HEK293T cells expressing ChR2-C128T-mCherry (shown in red) and ChR2CA/C128T-C79/208-mCherry (shown in blue) at −60 mV following blue light activation (460 nm, 11.9 milliwatt·mm−2) for 100 ms (upper trace) and for 10 s (lower trace). Please note the slower current decay of ChR2CA/C128T-C79/208. C, current-voltage relationship of stationary photocurrent Is. Arrows indicate the reversal potentials Erev. D, normalized velocity of the initial current rise ΔI/Δt at different light intensities (−60 mV, 460 nm). E–G, comparison of peak current amplitude Ip after 100-ms light pulse, stationary current amplitude Is after illumination for 10 s, and current decay time τoff after 100-ms light pulse (all at −60 mV, 460 nm, 11.9 milliwatt·mm−2). n.s., not significant; ***, p < 0.0001, Student's t test. Error bars reflect S.E.; number of cells (n) is indicated in each figure panel.
Selective Labeling of Cys-79 with Fluorescein and Spectroscopic Characterization of ChR2CA/C128T-C79-AF/C208
Under the conditions described under “Experimental Procedures” 0.95 mol of 5-iodo-acetamidofluorescein (IAF) per mol ChR2CA/C128T-C79/208 was selectively bound to the cysteine in position 79 at the cytoplasmic end of helix B, as calculated according to Equation 1. Fig. 2A shows the absorption spectra of the channel before and after labeling (dashed and solid lines, respectively) with IAF. When fluorescein is bound to the protein, the absorption band has its maximum at λmax = 502 ± 1 nm in the alkaline form (pH 9) and thus is red-shifted compared with that of IAF (λmax = 490 nm), indicating a rather apolar environment of the fluorescein label (22, 23) at position 79 of the channel in n-decyl-β-d-maltopyranoside (DM) micelles. The emission spectrum of bound fluorescein is shown in Fig. 2B. The λmax value of fluorescence emission is 523 ± 0.5 nm.
FIGURE 2.

Absorption and emission spectra of ChR2CA/C128T-C79-AF/C208. A, absorption spectra before (dashed line) and after (solid line) labeling with IAF at alkaline pH (pH 8), yielding ChR2CA/C128T-C79-AF/C208 (in short ChR2-C79-AF). The arrow indicates the absorption maximum of deprotonated bound fluorescein. The inset shows the absorption of fluorescein derived from the difference spectrum between labeled and unlabeled channel. B, emission spectrum of ChR2CA/C128T-C79-AF/C208. The conditions were 0.2% DM, 100 mm NaCl at 20 °C.
The specific labeling of position 79 by IAF was verified by proteolytic digestion with the protease AspN and visualized by gel electrophoresis of the proteolytically cleaved fragments (Fig. 3). AspN selectively and exclusively cleaves peptide bonds N-terminal to aspartic acid residues under our experimental conditions. Three of seven restriction sites are located in the transmembrane part of the protein (Fig. 3A). The proteolytic fragments containing transmembrane parts of the channel are designated with “Fm.” The cysteines in positions 79 and 208 of the channel variant (36.4 kDa) are located between Asp-32–Asp-144 (fragment size 12.6 kDa, Fm1) and Asp-156–Asp-256 (fragment size, 10.5 kDa; Fm3), respectively (Fig. 3A). The digest was performed with three enzyme:sample-protein ratios (w/w) of 1:25, 1:30, and 1:250 at varying incubation times to follow the digestion at the different stages. With the lowest ratio (1:250) and a short incubation time of 0.5 h, a fluorescent fragment with a size of ∼30 kDa appears first (Fig. 3B, lane 3), corresponding to protein fragments Asp-2–Asp-256 or Asp-32–Asp-302 (Fm+), that is followed by a smaller fragment above 22 kDa. This smaller fragment above 22 kDa, when derived from Fm+, can only correspond to the protein fragment located between Asp-32–Asp-256 (Fm1–3) with a calculated size of 24.5 kDa. This pattern indicates that limited proteolysis starts from both the N- and C-terminal ends and results in fragments containing the membrane portion of the protein. Further digestion of Fm1–3 for 3 h (Fig. 3B, lane 4) yields additional fluorescent fragments with sizes of ∼14 and 12 kDa, corresponding to the Fm1–2 fragment (14.0 kDa) and the Fm1 fragment (12.6 kDa) or the Fm2–3 fragment (11.9 kDa). To test whether the latter fluorescent band belongs to Fm1 (labeling position 79) or Fm3 (labeling position 208), a digest with an enzyme:sample-protein ratio (w/w) of 1:30 for a prolonged incubation time of 18 h was performed (Fig. 3B, lane 5). At similar conditions a protein sample labeled with fluorescein at pH 10 (to access both position 79 and 208) was digested (Fig. 3B, lane 9). Whereas in lane 5 even after prolonged incubation time with the protease AspN, no further band appears that indicates the labeling of position 208 in Fm3 (10.5 kDa), this band is clearly visible in lane 9, indicating labeling of both positions 79 and 208 at pH 10. Thus, the proteolytic digestion reveals that the sample derivatized with IAF under our labeling conditions (pH 7.4, 1 h of incubation) is labeled at position 79.
FIGURE 3.

Specific fluorescence labeling at position 79 of ChR2CA/C128T-C79/208 with IAF. A, secondary structural model of the protein. α-Helices are depicted as gray boxes. The restriction sites of endoprotease AspN are indicated. The membrane fragments Fm1, Fm2, and Fm3 are highlighted (Fm1, light orange; Fm2, light yellow; and Fm3, light blue). B, SDS-PAGE gel of samples specifically labeled with fluorescein at Cys-79 before and after proteolytic digestion by AspN. Lane 1, molecular weight marker; lane 2, undigested sample; lanes 3 and 4, proteolytic fragments of a sample labeled at pH 7.4 for 1 h after digestion with enzyme:sample protein ratios of 1:25 for 0.5 h (lane 3) and 3 h (lane 4); lane 5, proteolytic fragments of a sample labeled at pH 7.4 for 1 h after digestion with enzyme:sample protein ratio of 1:30 for 18 h; lane 6, proteolytic fragments of a sample labeled at pH 7.4 for 12 h after digestion with enzyme:sample protein ratio of 1:30 for 18 h; lane 7, undigestedy sample; lane 8, molecular weight marker; lane 9, proteolytic fragments of a sample labeled at pH 10 for 1 h after digestion with enzyme:sample protein ratio of 1:25 for 20 h; lane 10, molecular weight marker (BenchMarkTM fluorescent protein standard; Life Technologies). ChR2, Fm+, Fm*, Fm1–3, Fm1–2, and Fm1 mark the positions of the undigested channelrhodopsin-2 variant monomer (36.4 kDa) and the respective proteolytic fragments (see text for further explanation) in the gel. Fm* is proteolytic fragment that appears only at longer incubations time with AspN and probably results from Fm+. Specific labeling of Cys-79 is shown by the fluorescence of the band corresponding to fragment Fm1, which contains Cys-79. C, structural model of the ChR2 dimer based on the crystal structure of the dark-adapted C1C2 chimera by Ref. 3 (Protein Data Bank code 3ug9). The transmembrane helices are labeled (A–G) in one of the monomers. Amino acids belonging to the inner gate (conducting pore entrance at the intracellular side) are highlighted in yellow, and their position in ChR2 (C1C2 numbering in brackets) is given. Cys-79 is highlighted in red. The green- (gray) and red-colored areas around position Cys-79 indicate the conformational space of the bound fluorescein as determined from time-resolved anisotropy experiments in the dark state (green and gray) and after illumination (red).
The exclusive labeling of position 79 with fluorescein under our labeling conditions (pH 7.4, 1 h of incubation; Fig. 3B, lane 5) was further confirmed by a sample that was labeled at pH 7.4 but for 12 h at room temperature. The digest of this sample with the protease AspN is shown in Fig. 3B (lane 6), revealing that even after prolonged labeling times, the band at 10.5 kDa (Fm3) is absent, i.e. labeling in position 208 is absent under these conditions in contrast to labeling at pH 10 (Fig. 3B, s). We note that an additional proteolytic fragment at ∼20 kDa (Fm*) appears at longer incubation times with AspN and probably results from Fm+.
Time-resolved Fluorescence Anisotropy for Detection of Dynamic Helix B Conformations
We investigated the dynamics and conformational changes of helix B by time-resolved fluorescence depolarization in the dark state and after illumination with blue light (465 nm) in samples consisting of ChR2CA/C128T-C79-AF/C208 in DM micelles or reconstituted in nanodiscs. These measurements allow for the analysis of the diffusional dynamics of protein segments directly on the nanosecond time scale (16, 19). Position Cys-79 is located at the end of the first cytoplasmic loop and the beginning of helix B facing toward the lipid shell and the putative CrChR2 dimer interface (Fig. 3C). The time-resolved anisotropy curves were fitted with a model function (Equations 2 and 3) as shown in Figs. 4 and 5 and summarized in Table 1. Because the rotational diffusion of the dye is affected by the motion of the protein segment to which it is covalently attached, the anisotropy decay curve yields information on global and local protein dynamics, as well as on the protein structure and conformational changes. The analysis of the anisotropy decay curves (Figs. 4 and 5) was performed as described previously (16, 19, 20, 24–26). The main decay components were assigned to the dynamics of (i) the dye itself with a rotational correlation time in the 150–350-ps range (φ1, β1), (ii) the cytoplasmic end of helix B to which the dye is bound to (φ2, β2), and (iii) a constant end value of the anisotropy r∞ (or the amplitude of the slowest decay component (β3)), which represents a measure of steric hindrance of helix B motion imposed by the constituents of the surrounding surface. The rotational correlation time φ3 of ∼22 ns (Table 1) describes the tumbling of the channel micelle.
FIGURE 4.

Decay of the time-resolved fluorescence anisotropy r(t) of ChR2CA/C128T-C79/208 labeled with fluorescein at cysteine 79. A, ChR2CA/C128T-C79-AF/C208 (in short ChR2-C79-AF) micelles, pH 7.4 (0.2% DM). B, ChR2-C79-AF nanodiscs pH 6.0. C, ChR2-C79-AF nanodiscs, pH 7.4. The conditions were 20 mm MES, 100 mm NaCl, pH 6.0, or 20 mm HEPES, 100 mm NaCl, pH 7.4, at 4 °C. The fitted curves are least squares fits to Equation 2 for A and to Equation 3 for B and C. The weighted residuals are shown below each panel for parallel (‖) and perpendicular (⊥) intensities I(t). The fit parameters are collected in Table 1.
FIGURE 5.
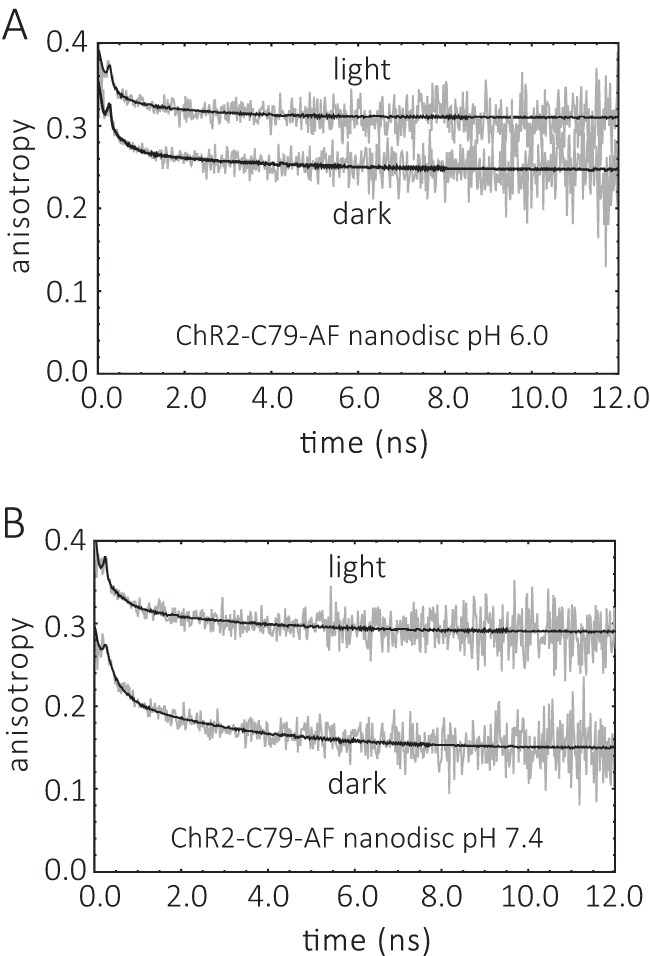
Comparison of the time-resolved fluorescence anisotropy r(t) decays of ChR2CA/C128T-C79-AF/C208 in the dark and after light-illumination. A, ChR2CA/C128T-C79-AF/C208 (in short ChR2-C79-AF) nanodiscs, pH 6.0. B, ChR2-C79-AF nanodiscs, pH 7.4. The conditions were as described in the legend to Fig. 4. The fit parameters are collected in Table 1.
TABLE 1.
Fluorescence anisotropy decay parameters of fluorescein bound to cysteine 79 of ChR2CA/C128T-C79/208
The conditions were 20 mm MES, pH 6.0, or HEPES, pH 7.4, 100 mm NaCl at 4 °C. The model functions (Equations 2 and 3) were used for obtaining the fit parameters. In the case of channelrhodopsin micelles (dark, pH 7.4), Equation 2 with a sum of two exponentials was sufficient to fit the data. The goodness of the fit was judged by the residuals (Fig. 4) and the xred2.
| System | State | pH | φ1 (dye) | φ2 (segment) | φ3 (micelles) | β1 | β2 | β3 | r∞ | xred2 |
|---|---|---|---|---|---|---|---|---|---|---|
| ns | ns | ns | ||||||||
| Nanodisc | Dark | 6.0 | 0.26 | 3.28 | 0.04 | 0.03 | 0.25 | 1.2 | ||
| Nanodisc | Light | 6.0 | 0.15 | 1.83 | 0.04 | 0.02 | 0.31 | 1.2 | ||
| Nanodisc | Dark | 7.4 | 0.24 | 3.00 | 0.07 | 0.07 | 0.14 | 1.1 | ||
| Nanodisc | Light | 7.4 | 0.31 | 3.05 | 0.04 | 0.03 | 0.29 | 1.2 | ||
| Micelle | Dark | 7.4 | 0.35 | 21.61 | 0.07 | 0.19 | 1.1 | |||
| Micelle | Light | 7.4 | 0.35 | 1.32 | 21.26 | 0.05 | 0.02 | 0.16 | 1.1 |
Dark State of the CrChR2 Variant
Fig. 4 shows the anisotropy decay curves for the channel in micelles at pH 7.4, and in nanodiscs at pH 6.0 and 7.4. The anisotropy decay parameters are presented in Table 1 and Fig. 6. Fig. 6C compares the steric hindrance of the motion of the dye-tagged cytoplasmic end of helix B. Both in micelles and in nanodiscs a steric restriction with anisotropy values (r∞ and β3) between 0.14 and 0.25 was observed. With r∞ and β3 values of 0.19 and 0.14, respectively, the protein surface imposes less steric restriction at pH 7.4 compared with pH 6.0 (r∞ = 0.25) for the cytoplasmic end of helix B. Interestingly, the flexibility and mobility of the cytoplasmic part of transmembrane helix B depends both on pH and the lipid environment, respectively, the detergent, as judged from the rotational correlation time φ2 and the corresponding amplitude β2 (Fig. 6, A and B). In micelles no mobility of the cytoplasmic part of helix B, based on the absence of a rotational correlation time in the 1–5-ns time range, was observed. In contrast, the cytoplasmic part of helix B is mobile in nanodiscs, and the mobility depends on pH (Fig. 6B). At pH 6.0 the conformational space for helix B movement at the cytoplasmic surface is ∼3-fold lower than at pH 7.4. This is in agreement with the higher steric restriction by the cytoplasmic protein surface at pH 6.0 (Fig. 6C). The rotational correlation time of the flexible end of helix B is ∼3 ns at both pH 6.0 and 7.4 (Fig. 6A).
FIGURE 6.
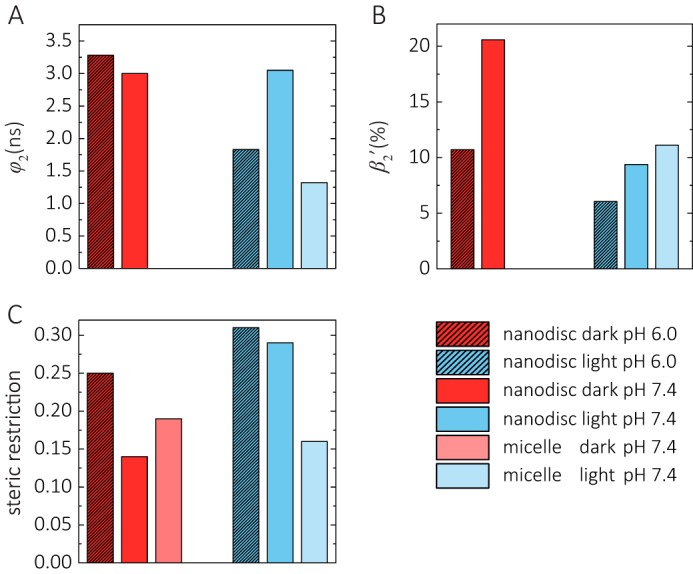
Helix B dynamics in the dark and after illumination. The fluorescence anisotropy decay parameters of the curves shown in Figs. 4 and 5 are graphed. A, rotational correlation time of cytoplasmic helix B segment (φ2). B, conformational space of helix B expressed as relative mobility β2′ in percentage (β2′ = β2/(β2 + r∞) for nanodiscs and β2′ = β2/(β2 + β3) for micelles). C, steric restriction of helix B (r∞ or β3).
The CrChR2 Variant after Illumination
In micelles only minor conformational changes of the channel were observed after blue light illumination. Fig. 6A shows that the most prominent change is the increase in flexibility of the cytoplasmic end of helix B with a rotational correlation time of 1.3 ns.
The anisotropy decay curves of ChR2CA/C128T-C79-AF/C208 in nanodiscs before and after illumination are presented in Fig. 5. Large conformational changes upon illumination with blue light, i.e. upon formation of the open conducting state of the channel, are apparent from the change in the anisotropy decay curves. These light-induced conformational changes occur at both pH 7.4 and 6.0, as monitored by the change in final anisotropy r∞ and the dynamics of helix B movement (Fig. 6).
At pH 6.0 the rotational correlation time of the flexible end of helix B becomes faster with 1.8 ns compared with 3.3 ns in the dark state (Fig. 6A). The already reduced conformational space for helix B movement at the cytoplasmic interface becomes even smaller after illumination (Fig. 6B), whereas the steric restriction imposed by the cytoplasmic surface becomes larger with r∞ = 0.31 after illumination compared with r∞ = 0.25 in the dark state (Fig. 6C).
At pH 7.4 the most prominent change is the reduction in conformational space for helix B movement at the cytoplasmic interface and the corresponding increase in steric restriction imposed by the cytoplasmic surface from r∞ = 0.14 to r∞ = 0.29 after illumination (Fig. 6, B and C). Thus, this increase in final anisotropy at both pH values clearly shows that the light-induced formation of the open channel state in nanodiscs (i.e. in a lipid membrane environment) is associated with a large change at the cytoplasmic surface affecting helix B mobility.
Accessibility of Helix B by Collisional Quenching Experiments
The charged collisional quencher iodide (I−) was used to investigate the accessibility of the label at position Cys-79. The experiments were carried out with DM micelles and nanodiscs at pH 6.0 and 7.4. Typical fluorescence decay traces for all four samples in the dark-adapted state are shown in Fig. 7. The highest concentration of the quencher iodide was 400 mm KI (Fig. 7, blue traces). Clear iodide quenching effects were observed in nanodiscs for pH 6.0 and 7.4 (Fig. 7, C and D). In micelles a similar clear quenching was only observed at pH 7.4 (Fig. 7B). Control experiments with and without KCl showed no effect on the fluorescence decay curves after addition of 400 mm KCl (Fig. 8).
FIGURE 7.
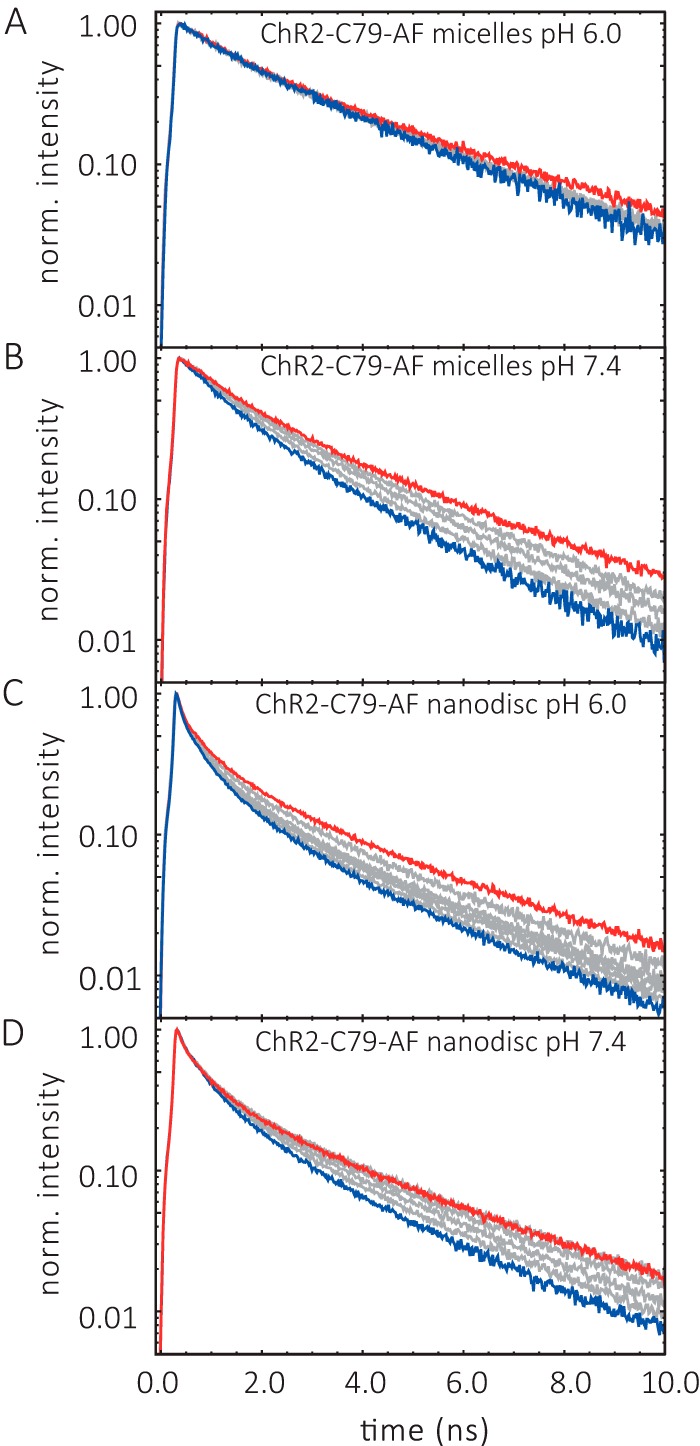
Iodide quenching of ChR2CA/C128T-C79-AF/C208 fluorescence lifetime. A, ChR2CA/C128T-C79-AF/C208 (in short ChR2-C79-AF) micelles, pH 6.0. B, micelles, pH 7.4. C, nanodiscs, pH 6.0. D, nanodiscs, pH 7.4. The conditions were 20 mm buffer, 100 mm NaCl at 4 °C. Varying I- concentrations are indicated by the different colors of the fluorescence decay curves. Red and blue curves refer to the lifetime curve in the absence and presence of 400 mm KI, respectively, and gray curves indicate 100, 200, and 300 mm KI.
FIGURE 8.
Fluorescence decay curves of ChR2CA/C128T-C79-AF/C208 in the absence and presence of KCl. The conditions were 0.2% DM, 20 mm HEPES, 100 mm NaCl, pH 7.4, at 4 °C. The concentration of KCl is indicated.
Light- and Environment-induced Changes in Helix B Accessibility at pH 7.4
Linear Stern-Volmer (SV) plots (Equation 6 and Fig. 9) were obtained over a wide range of iodide quencher concentrations for ChR2CA/C128T-C79-AF/C208 in micelles and nanodiscs. The good linearity of the SV plots both in the dark and in the open state of the channel suggests the presence of a single population of fluorophores, i.e. conformational homogeneity at pH 7.4. The slope of the SV plot in Fig. 9 is clearly smaller for samples in nanodiscs than in DM micelles. Illumination with blue light results only in minor changes. Thus, quenching as deduced from the SV quenching constant KD is always less in nanodiscs. For dynamic quenching the slope of the SV plot is equal to the product of the quenching rate kq and the lifetime in the absence of quencher (Equation 6). The respective iodide quenching values are summarized in Table 2. When compared with the collisional quenching with iodide of unbound IAF in aqueous solution (KD = 12 m−1 and kq = 3 × 109 m−1 s−1), both the SV quenching constant and the bimolecular quenching rate values are reduced to KD = ∼1 m−1 and kq = ∼0.4–0.5 × 109 m−1 s−1. Similar reduced iodide quenching values were obtained for fluorescein-labeled α-toxin bound to the surface of the acetylcholine receptor, and the reduction in quenching constant and rate can be explained by the geometric constraint for collisional quenching at the surface of proteins (27, 28).
FIGURE 9.
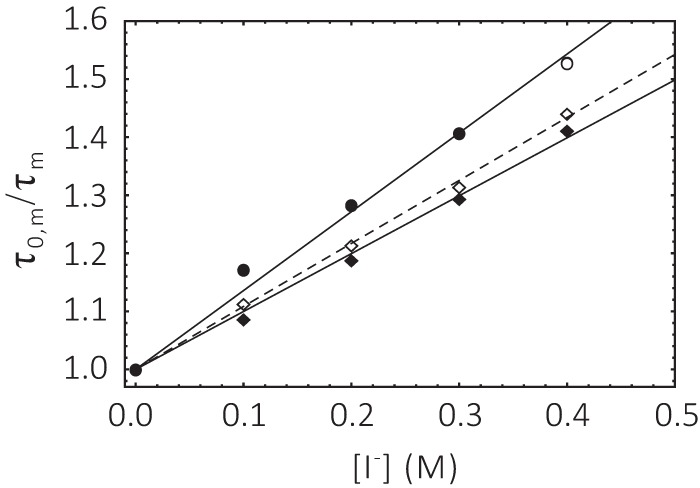
Stern-Volmer plot of ChR2CA/C128T-C79-AF/C208 at pH 7. 4. Circle, micelle; square, nanodiscs. Filled symbols, dark; open symbols, after blue light illumination. The conditions as described in the legend to Fig. 7.
TABLE 2.
Collisional quenching parameters of fluorescein bound to cysteine 79 of ChR2CA/C128T-C79/208 by I− at pH 7.4
The mean lifetime of bound fluorescein in the absence of the quencher τ0,m, the SV quenching constant KD, and the quenching rate kq are given. Fit is according to Equation 6 of the data shown in Fig. 8. The errors in kq take into account the uncertainty in the slope of the linear fit and the error in the lifetime. The conditions were 20 mm HEPES buffer, pH 7.4, 100 mm NaCl at 4 °C.
| Micelle |
Nanodisc |
IAF | |||
|---|---|---|---|---|---|
| Dark | Light | Dark | Light | ||
| τ0,m (ns) | 2.88 ± 0.07 | 2.88 ± 0.08 | 2.59 ± 0.02 | 2.84 ± 0.04 | 3.90 ± 0.01 |
| KD (m−1) | 1.4 ± 0.04 | 1.3 ± 0.04a | 1.00 ± 0.02 | 1.10 ± 0.02 | 12.1 ± 0.3 |
| kq (109 m−1 s−1) | 0.47 ± 0.02 | 0.46 ± 0.02a | 0.389 ± 0.008 | 0.384 ± 0.006 | 3.1 ± 0.8 |
| fa (%) | 100 | 100a | 100 | 100 | 100 |
a As estimated from the fluorescence lifetime at the highest KI concentration.
Light- and Environment-induced Changes in Helix B Accessibility at pH 6.0
Iodide quenching of fluorescein bound to helix B in position 79 at pH 6.0, both in micelles and nanodiscs, appears to result in quenching data that deviate from the SV equation (Equation 6), as shown by the downward curvature in the SV plot in Fig. 10A. This nonlinearity suggests the existence of multiple fluorophore populations with differing accessibilities. In the simplest case an accessible and a buried (inaccessible) population of fluorophores can be assumed (Equations 7–9 and Fig. 10). Because a unique fluorophore attachment site (Cys-79) was established, this quenching heterogeneity reflects different protein conformations at pH 6.0 (Equations 8 and 9). In nanodiscs ∼66% of the cytoplasmic end of helix B was not accessible to the polar quencher in the dark state. Upon illumination only minor changes in accessibility were detectable. The inaccessible fraction is reduced to ∼62%, i.e. the major fraction of the cytoplasmic end of helix B is still inaccessible. The accessible protein fraction, however, displayed a high quenching rate (4.9 × 109 m−1 s−1) that decreases to 3.9 × 109 m−1 s−1 upon blue light illumination. Both quenching rates are much higher than those observed for protein-bound fluorescein at pH 7.4 (Table 2) and are close to the values obtained for free fluorescein in solution (Table 3) (27–29). In micelles the accessible fraction amounts only to 18% and does not change upon blue light illumination. The quenching rate of 2.7 × 109 m−1 s−1 for the accessible fraction is slightly lower than in nanodiscs (Table 3).
FIGURE 10.

Stern-Volmer plot of ChR2CA/C128T-C79-AF/C208 at pH 6.0. Circle, micelle; square, nanodiscs. Filled symbols, dark; open symbols, after blue light illumination. A, conventional SV plot and fit according to Equation 8. B, linearized modified SV plot according to Equation 9. Fit parameters are summarized in Table 3. The conditions as described in the legend to Fig. 7.
TABLE 3.
Collisional quenching parameters of fluorescein bound to cysteine 79 of ChR2CA/C128T-C79/208 by I− at pH 6.0
The mean lifetime of bound fluorescein in the absence of the quencher τ0,m, the SV quenching constant KD, and the quenching rate kq are given. Fit is according to Equations 8 and 9. The errors in kq take into account the uncertainty in the slope of the linear fit and the error in the lifetime. The conditions were 20 mm MES buffer, pH 6.0, 100 mm NaCl at 4 °C.
| Micelle |
Nanodisc |
IAF | |||
|---|---|---|---|---|---|
| Dark | Light | Dark | Light | ||
| τ0,m (ns) | 3.30 ± 0.07 | 3.28 ± 0.06 | 2.58 ± 0.03 | 2.74 ± 0.04 | 3.59 ± 0.02 |
| KD (m−1) | 9 ± 2 | 9a | 13 ± 1 | 11.0 ± 0.4 | 11.8 ± 0.3 |
| kq (109 m−1 s−1) | 2.7 ± 0.6 | 2.7a | 4.9 ± 0.4 | 3.9 ± 0.1 | 3.2 ± 0.8 |
| fa (%) | 18 ± 1 | 18a | 34 ± 1 | 38.3 ± 0.6 | 100 |
a As estimated from the fluorescence lifetime at the highest KI concentration.
Discussion
Our goal was to gain new insights from structural dynamics into the mechanism underlying the formation of the open channel state in CrChR2. In particular, we focused on the cytoplasmic part of helix B. A key role for helix B in light-induced channel opening and closing has been suggested. Evidence for light-induced movements of helix B is based on structural studies using electron crystallography and EPR spectroscopy (double electron-electron resonance) (11–13). Although double electron-electron resonance measurements suggest a light-induced displacement/outward tilt of helix B (11, 13), electron crystallographic studies found additional evidence for a loss of order in that helix (12).
The prominent light-induced movement/structural changes of helix B in CrChR2 are unique, because for other microbial rhodopsin, such as sensory rhodopsin and bR, major helix displacements occur upon light activation in helix F (14–18). In the latter case, the light-induced helix F movements are connected to the reprotonation of the retinal Schiff base via the internal proton donor Asp-96 in the proton pump cycle (30, 31). Because CrChR2 is a light-triggered cation channel, it was suggested that the changes around helix B seem to be involved in cation permeation by creating a water-filled pore (12).
To detect dynamic structures and conformational changes of helix B via time-resolved fluorescence depolarization (16, 21), we selectively labeled position Cys-79 in a CrChR2 variant, in which the native cysteines, except for Cys-79 and Cys-208, were exchanged to alanine (11). Because the two remaining cysteines exhibit differential reactivity toward IAF, we were able to exclusively label position 79 at helix B (Fig. 3). The cysteine mutations were generated in the slowly cycling variant C128T (8), which accumulates the conducting state by slowing down the respective time constants (Fig. 1). This allowed us to specifically address the conducting state after blue light illumination. In addition, these so-called step function mutants of CrChR2 are particularly interesting in optogenetics because they offer the opportunity to generate a more permanent cell depolarization (8).
Time-resolved fluorescence depolarization experiments allowed us to monitor the conformational dynamics of the cytoplasmic end of helix B in the dark state and the changes upon blue light illumination via the analysis of the rotational correlation time and the amplitude assigned to the anisotropy decay component (second decay component in Table 1), which reflects the motion of helix B. Moreover, the final anisotropy is a measure for the steric hindrance of helix B motion. The increase in final anisotropy r∞ at both pH values in nanodiscs upon light activation (Fig. 5) clearly shows that the light-induced formation of the open channel state is associated with an increased steric hindrance of helix B motion at the inner gate because of a large conformational change at the cytoplasmic surface. These changes are schematically indicated in Fig. 3C, which shows the structural model of dark-adapted CrChR2 and thus a closed channel conformation. The red-colored area around position Cys-79 indicates the reduction of conformational space of helix B/dye in the open channel conformation compared with dark CrChR2. In addition to changes in conformational space for helix B motion, the rotational correlation times of the cytoplasmic end of helix B in the dark and in the open conducting P3520 state indicate changes in helix B flexibility. In CrChR2 micelles the cytoplasmic end of helix B is immobile in the dark state and becomes flexible in the P3520 state with a rotational correlation time ϕ2 of 1.3 ns. Similarly, a faster motion of helix B was observed in nanodiscs at pH 6.0 in the P3520 state with a rotational correlation time φ2 of 1.8 ns compared with 3.3 ns in the dark state. Because only in CrChR2 micelles was immobility of the cytoplasmic end of helix B in the dark state observed, we speculate that the detergent molecules restrict the mobility of the first intracellular loop. It is known from the literature (32) that detergent molecules may interact with solvent-exposed surface regions of membrane proteins.
As in channelrhodopsin, light-induced conformational changes were observed at the cytoplasmic surface of the retinal proteins bR and visual rhodopsin (24, 26). A general increase in protein flexibility/protein softening was assumed to be a prerequisite to overcome potential barriers in the large scale structural changes during the M-intermediate of bR (33). The latter were correlated with anisotropy changes in the M-intermediate and the Meta-II state of bR and visual rhodopsin, respectively (24, 26). The clear reduction in conformational space of helix B movement in nanodiscs upon light activation together with a shortening of the rotational correlation time at pH 6 indicates that changes in the dynamics of the cytoplasmic part of helix B are involved in the formation of the open conducting state. Moreover, a higher steric restriction of the fluorescent label in position 79 pointing toward the putative dimer interface (Fig. 3C) would agree with the structural model of an open conductive state with changes at the dimer interface (11) and the tilt of helix B observed in MD simulations (6). It seems that these conformational changes on the cytoplasmic surface induced by retinal isomerization led to the required structural rearrangements allowing transient water influx necessary for either proton uptake (e.g. in bR) or cation permeation in CrChR2.
As indicated in Fig. 3C, position 79 at the cytoplasmic end of helix B is located at the entrance of the conducting pore (for a review see Ref. 5) in close proximity to the inner gate, a hydrophilic amino acid cluster that blocks the cation permeation pathway at the cytoplasmic (intracellular) side. The amino acids belonging to the inner gate are Glu-82, Glu-83, Tyr-70, His-134, His-265, and Arg-268 (Fig. 3C). It was hypothesized that a reorientation of this cluster might be the final step in CrChR2 gating (5). To detect possible changes at the inner gate, we investigated the accessibility of the bound fluorophore in position 79 to the polar quencher I− in the dark and after light illumination and in dependence of pH. When bound to the surface of the channel, fluorescein exhibits a fluorescence lifetime of ∼3 ns, clearly smaller than for free fluorescein (∼4 ns). This faster fluorescence lifetime of fluorescein when bound to the channel surface can be explained mainly by quenching effects from the protein surface. A similar effect on the fluorescence lifetime was found when fluorescein was covalently bound to the surface of bR (34) and visual rhodopsin (16, 35). At pH 7.4 we observed reduced quenching rates for the collisional polar quencher iodide in both micelles and nanodiscs compared with free fluorescein in solution. No changes were observed after blue light illumination. Linear SV plots, both in the dark state and after illumination (Fig. 9), suggest the presence of a single population of fluorophores, i.e. a homogeneous protein conformation. In contrast, at pH 6.0 structural heterogeneity with two different protein conformations was observed as deduced from the analysis of the modified SV plots (Fig. 10). Thus, a pH-dependent conformational change takes place at the cytoplasmic surface of the channel, close to the inner gate. Analysis of this structural heterogeneity (36, 37) reveals a protein population in which position 79, i.e. the side of helix B pointing toward the putative dimer interface (Fig. 3C), is not solute accessible from the aqueous solution. This protein population constitutes the majority of the sample with ∼82% (micelles) and 66% (nanodiscs). The higher final anisotropy r∞ observed in nanodiscs at pH 6.0 compared with pH 7.4 in the dark state (Figs. 4 and 6C) can be explained by the large buried fraction of bound fluorescein at pH 6.0 that is expected to experience a higher degree of steric restriction. Note that this structural heterogeneity does not affect the overall change observed in the time-resolved anisotropy signal after illumination. However, only minor changes were detected in the fractions after illumination, indicating that the conformational heterogeneity is rather pH-dependent than light activation-dependent. The protein fraction that was accessible, however, showed an unusual high accessibility for a fluorescein bound to a protein surface, because quenching values on the order of free fluorescein were observed. We speculate that in this fraction fluorescein bound in position 79 is oriented toward the hydrophilic cluster of the inner gate attracting water molecules to the cytoplasmic surface (6) and that positively charged residues, such as Arg-268, locally increase the iodide concentration, leading to an apparent higher quenching compared with the lower bulk I− concentrations. This effect may also be transient, because transient surface changes (in addition to the discussed helix tilt) are known to occur at the cytoplasmic surface of both bR (38) and visual rhodopsin (39) upon light activation. For instance, a conformational change of the EF-Loop at the cytoplasmic surface of bR was correlated with surface charge changes (16, 38).
Because conformational heterogeneity was not observed at pH 7.4, a structural pH-dependent rearrangement of helix B below pH 7 must occur that affects the region of the inner gate. Support for this conclusion comes from recent x-ray crystallographic studies of channelrhodopsin, because the available C1C2 x-ray crystal structures (3, 40) suggest pH-dependent changes at the cytosolic side of helix B. Although the crystal structure of a C1C2 chimera was solved at pH 6.0 (7), another C1C2 variant was crystallized at pH 7.0 (40). The latter C1C2 variant contains two amino acid substitutions (corresponding amino acids in CrChR2: T159G/G163A) in close proximity to the β-ionone ring of the retinal. When comparing these C1C2 structures at the two pH values of 6.0 and 7.0, different protonation states for four amino acids were resolved; three of them are located in the N-terminal region. The only structural difference observed in the transmembrane part (apart from the amino acid substitutions) is at position Glu-83 (Fig. 3, A and C), which is part of the inner gate and located in helix B close to residue Cys-79 (Fig. 3C). In its protonated state at pH 6.0, Glu-83 is hydrogen-bonded to His-134 (helix C), whereas at pH 7.0, it forms a salt bridge with Arg-268 (helix G). Thus, protonation of Glu-83 at pH 6.0 in CrChR2 and consequently hydrogen bonding to His-134 might lead to the observed structural pH-dependent rearrangement of the cytoplasmic end of helix B below pH 7 in our experiments. Because Glu-83 belongs to the 12 polar residues along the channel pore, forming a hydrophilic and strongly electronegative surface, structural changes close to the cytoplasmic end of this pore (i.e. close to the inner gate) may affect gating. This will be the subject of future studies.
Moreover, pH-dependent structural heterogeneity of helix B in the C128T variant would add a new perspective to the finding of multiple dark-adapted states in C128T (41). Hence, our data not only show the nature of the conformational change of the cytoplasmic end of helix B in channel opening but also highlight the pH-dependent conformational heterogeneity of a CrChR2 step function mutant that belongs to the neurophysiological optogenetics tool kit.
By a combination of different time-resolved fluorescence techniques, we have presented results that lend support to the helix tilt model (5, 11–13) of helix B upon formation of the open channel state and gave insight in the possible mechanism by comparing the CrChR2 data with time-resolved anisotropy data from other retinal proteins. Accessibility studies revealed a pH-dependent structural heterogeneity close to the inner gate. This kind of information is important when comparing and interpreting crystallographic and spectroscopic results for CrChR2 photocycle or electrophysiological measurements, because crystal structures were obtained at pH 6 and 7 (discussed above), and spectroscopic as well as electrophysiology data were measured at a variety of pH values (9, 40–42). Structural heterogeneity might also contribute to the observed multiple dark states of the ChR2-C128T variant (41). Moreover, structural heterogeneity may affect the photocurrent, open state time of the channel, and degree of inactivation, important parameters when employing step function mutants of CrChR2 in optogenetics experiments.
Experimental Procedures
Expression of a Recombinant CrChR Variant
A CrChR variant (amino acid residues 1–307 of CrChR2, a linker of two residues, alanine and serine, and a 10× His tag) was constructed that still contains Cys-79 and Cys-208 as in wild type, but where all other cysteines were substituted by alanine (C34A,C36A,C87A,C179A,C183A,C259A) or threonine (C128T) (Fig. 3A). This variant was first described in Ref. 11 and here renamed as ChR2CA/C128T-C79/208 (formerly, ChR2-C79/208). The C128T substitution is known to prolong the conducting state and thus to enable the accumulation of P3520 under continuous illumination (9, 10).
The variant with two remaining cysteines, ChR2CA/C128T-C79/208, has been used in this study for two reasons. First of all, ChR2CA/C128T-C79/208 is a functional light-activated ion channel with similar photocurrent properties as the step-function-rhodopsin ChR2-C128T (Fig. 1). Second, the further exchange of cysteine in position 208 to alanine in the cysteine-conversion mutant leads to a drastic decrease in protein expression. This makes it difficult to prepare high quality samples that are needed for the fluorescence experiments. Further, Cys-79 can be selectively labeled with 5-iodoacetamido-fluorescein in the presence of Cys-208, as shown under “Results.”
ChR2CA/C128T-C79/208 was purified as described (11). The solubilized purified protein was kept in 0.2% DM, 100 mm NaCl, and 20 mm HEPES buffer, pH 7.4, at −80 °C prior to fluorescence labeling and nanodisc preparation.
Electrophysiological Measurements
HEK293T cells were seeded on glass coverslips (16 mm, thickness 0) at 1 × 105 cells ml−1 and were grown in Dulbecco's modified medium supplemented with 10% FCS and 100 μg·ml−1 penicillin/streptomycin at 37 °C and 5% CO2. HEK cells were transfected with ChR2-C128T-mCherry and ChR2CA/C128T-C79/208-mCherry using jetPEI (PolyPlus Transfection) at between 48 and 60 h prior to patch clamp measurements. Whole cell patch clamp recordings were performed using an inverted DMI 4000B microscope (Leica), an Axopatch 200B amplifier, and an Axon Digidata 1550A (both Molecular Devices). Activation light was delivered by a 460-nm LED (maximal intensity 11.9 milliwatt·mm−2 in the object plane) and controlled via a custom-built control unit (Essel Research and Development). The pipette solution contained 110 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm EGTA, and 10 mm HEPES, and the bath solution contained 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, and 10 mm HEPES. The pH was titrated to 7.2 with N-methyl-d-glucamin, and the osmolarity was adjusted to 290 and 320 mosm with d-glucose for pipette and bath solution, respectively. The data were recorded with pClamp 10 and analyzed using Microsoft Excel 2013 and Sigma Plot 11.0.
Site-specific Labeling of ChR2CA/C128T-C79/208 at Position Cys-79
The variant ChR2CA/C128T-C79/208 was labeled with a 10-fold excess of 5-iodoacetamidofluorescein (Invitrogen) in 20 mm HEPES buffer, pH 7.4, for 1 h at room temperature, essentially as described for bR (43). Unbound dye was removed by gel filtration using Sephadex G-25 fine (GE Healthcare). The labeling stoichiometry was calculated using Equation 1,
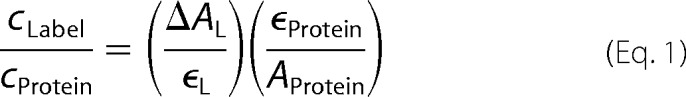 |
where AProtein is the absorbance of ChR2CA/C128T-C79/208 at 500 nm, and ϵProtein is 45,000 m−1 cm−1, the corresponding extinction coefficient at 500 nm. AL is the absorbance of the label fluorescein. The extinction coefficient of fluorescein at 492 nm is ϵL = 77000 m−1 cm−1 (20). The covalent binding of the fluorescent label and the removal of excess label were verified by SDS-PAGE.
Digestion with AspN
The labeling position was determined by limited enzymatic proteolysis of the fluorescein-labeled channel variant with the metalloendopeptidase AspN (Sigma). The digest was performed at different enzyme:sample-protein ratios (1:25, 1:30, and 1:250 (w/w)) and incubation times (between 0.5 and 20 h) at 37 °C. The peptide fragments were separated by SDS-PAGE using a 15% gel.
Preparation of CrChR2-containing Nanodiscs
The reconstitution procedure was adopted from Ref. 44. In brief, fluorescein-labeled ChR2CA/C128T-C79/208 (typically 100–200 μm) in DM was mixed with MSP1D1 (membrane scaffold protein 1D1) (typically 200 μm) and a dimyristoylphophatidylcholine/cholate solution (50 mm/100 mm). The molar ratio of the channel to MSP1D1 to dimyristoylphophatidylcholine was 0.5 to 1 to 55. Additional cholate was added to adjust the concentration of the mixture to 20 mm. The solution was incubated for 1 h at 25 °C. Subsequently biobeads (1.5 g of wet weight/ml of solution) were added and incubated for 2 h at 25 °C to remove the detergent. The biobeads were discarded, and the reconstitution mixture was centrifuged for 20 min at 21,000 × g to remove larger aggregates. The supernatant was run on a size exclusion column (Superdex 200 10/300, GE Healthcare; buffer: 20 mm HEPES, 100 mm NaCl, pH 7.4) at 0.5 ml/min, and the peak fractions containing the self-assembled ChR2 channel nanodiscs were pooled.
Optical Spectra
Absorption spectra were measured with a Shimadzu UV2450 UV-visible spectrophotometer. Fluorescence spectra were recorded with a Spex Fluoromax. Excitation wavelength was 488 nm. The bandwidth was 1 nm.
Time-resolved Fluorescence and Fluorescence Depolarization
ChR2CA/C128T-C79-AF/C208 samples were measured with a tunable picosecond laser in a time-correlated single photon counting setup, which is based on a microchannel plate detector (16, 34). Sample fluorescence was excited by a white light laser system together with an acousto-optical tunable filter (SuperK Extreme EUV3 and SELECT UV-visible, NKT) (45). The output power was 140 μW (488 nm) at 19.5 MHz repetition rate. Emission was spectrally selected with a long pass filter at ≥515 nm (OG515).
Fluorescence decay traces were collected in 1024 time channels with a channel width of 19.5 ps. The measurements of samples containing 100 mm NaCl were performed at 4 °C, at pH 6.0 (20 mm MES, 100 mm NaCl) and pH 7.4 (20 mm HEPES, 100 mm NaCl). Light activation of the channel variant was achieved by illuminating the sample for 2 or 5 min with a light emitting diode of λ = 465 nm (0.17 lumen; L-7113QBC-D Blue, Knightbright). Formation of the light-activated state and its decay was checked by UV-visible spectroscopy.
The instrumental response function of the time-correlated single photon counting setup was determined at the corresponding wavelengths with a colloidal silica solution as scattering material (LUDOX, Grace). The instrumental response function of the system had a full width at half-maximum of 60–70 ps. After deconvolution with the instrumental response function the fluorescence decay traces were fitted with a sum of exponentials (16, 19). Fluorescence anisotropy data of micelles were fitted to the model function shown in Equation 2.
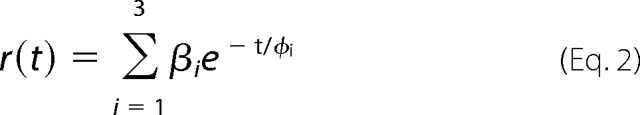 |
For nanodiscs, which are too large to observe the rotational diffusion of the whole system on the time scale of the fluorescence lifetime of fluorescein, anisotropy data were fitted to the following model function.
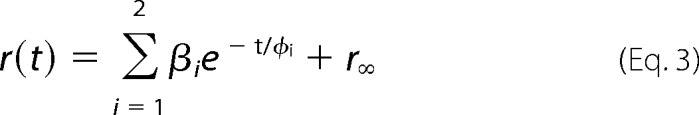 |
The rotational correlation times φ1 and φ2 describe the rotational motion of the label and the segment the label is attached to, respectively. φ3 describes the rotational motion of the whole system. The final anisotropy r∞ in the case of nanodiscs, and β3 in the case of micelles, is a measure for the degree of constraint (sterical hindrance) imposed by the constituents of the protein surface and/or lipid environment.
Fluorescence Quenching Experiments
Time-resolved fluorescence quenching experiments were performed with the time-correlated single photon counting setup. As a polar quencher, iodide (I−) was used. The KI concentration was varied between 0 and 400 mm KI. Fluorescence decay curves of the KI-dependent quenching measurements were analyzed using the software package Globals Unlimited V2.2 (Laboratory for Fluorescence Dynamics). Fluorescence time traces were fitted by an iterative nonlinear least squares analysis using a sum of exponentials,
where n is the total number of decay components, αi is the amplitude, and τi is the fluorescence lifetime of the ith component. The analysis of all time-resolved quenching data is based on the mean fluorescence lifetime τm, is given by the following equation.
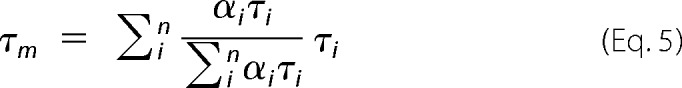 |
The Stern-Volmer (SV) equation (Equation 6) was used to obtain both the bimolecular quenching constant kq and the SV quenching constant KD,
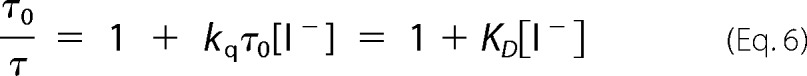 |
where τ and τ0 are the (mean) fluorescence lifetimes in the presence and absence of iodide, respectively, and [I−] is the quencher concentration. In the case of quenching heterogeneity, i.e. two protein conformations with an accessible (a) and a buried (unquenchable) population (b) of bound fluorophores, we used the modified SV equation (37),
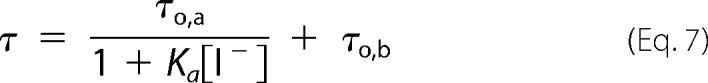 |
allowing the determination of the accessible fraction fa and the SV quenching constant of the accessible fraction Ka in the form of
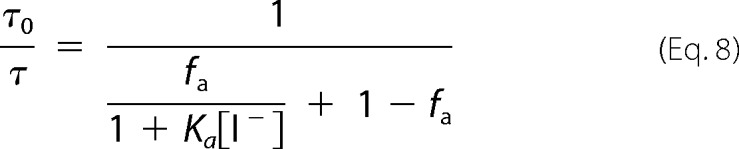 |
Linearization of the modified SV equation yields Equation 9.
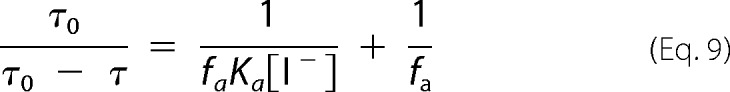 |
From Equation 9, the accessible fraction fa and the corresponding quenching constant Ka can be determined directly from the intercept and the slope of the ordinate, respectively. To obtain reliable fit results the quenching data were fitted simultaneously to Equations 8 and 9, yielding fa, Ka, and the accessible fraction. Fitting was performed with Mathematica 10 (Wolfram Research).
Author Contributions
P. V. performed time-resolved fluorescence experiments, analyzed and interpreted the data, and contributed to the writing of the manuscript; N. K. generated the mutant protein, developed the preparation in nanodiscs, and contributed to the writing of the manuscript; J. B. contributed to the time-resolved fluorescence experiments and sample preparation; C. S. characterized the fluorescently labeled sample and performed experiments shown in Figs. 1 and 2; M. W. prepared the protein sample; F. S. performed the electrophysiology experiments and wrote the corresponding parts of the manuscript; R. S. conceived and coordinated the study, contributed to the design of the experiments, and contributed to the writing of the manuscript; U. A. conceived and coordinated the study, designed the experiments, analyzed and interpreted the data, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Peter Hegemann (Humboldt-Universität zu Berlin) for support.
This work was supported by grants from the Deutsche Forschungsgemeinschaft: Sfb1078, project A2 (to U. A.), project B4 (to R. S.), project B2 (to P. Hegemann), and Sfb1112, project B03 to (U. A.). The authors declare that they have no conflicts of interest with the contents of this article.
- bR
- bacteriorhodopsin
- AF
- acetamidofluorescein
- CrChR2
- Chlamydomonas reinhardtii channelrhodopsin-2
- DM
- n-decyl-β-d-maltopyranoside
- Fm
- proteolytic fragments containing transmembrane parts of CrChR2
- IAF
- 5-iodoacetamidofluorescein
- SV
- Stern-Volmer.
References
- 1. Sineshchekov O. A., Govorunova E. G., and Spudich J. L. (2009) Photosensory functions of channelrhodopsins in native algal cells. Photochem. Photobiol. 85, 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nagel G., Ollig D., Fuhrmann M., Kateriya S., Musti A. M., Bamberg E., and Hegemann P. (2002) Channelrhodopsin-1: a light-gated proton channel in green algae. Science 296, 2395–2398 [DOI] [PubMed] [Google Scholar]
- 3. Kato H. E., Zhang F., Yizhar O., Ramakrishnan C., Nishizawa T., Hirata K., Ito J., Aita Y., Tsukazaki T., Hayashi S., Hegemann P., Maturana A. D., Ishitani R., Deisseroth K., and Nureki O. (2012) Crystal structure of the channelrhodopsin light-gated cation channel. Nature 482, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lórenz-Fonfría V. A., Bamann C., Resler T., Schlesinger R., Bamberg E., and Heberle J. (2015) Temporal evolution of helix hydration in a light-gated ion channel correlates with ion conductance. Proc. Natl. Acad. Sci. U.S.A. 112, E5796–E5804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schneider F., Grimm C., and Hegemann P. (2015) Biophysics of channelrhodopsin. Annu. Rev. Biophys. 44, 167–186 [DOI] [PubMed] [Google Scholar]
- 6. Watanabe H. C., Welke K., Sindhikara D. J., Hegemann P., and Elstner M. (2013) Towards an understanding of channelrhodopsin function: simulations lead to novel insights of the channel mechanism. J. Mol. Biol. 425, 1795–1814 [DOI] [PubMed] [Google Scholar]
- 7. Nack M., Radu I., Gossing M., Bamann C., Bamberg E., von Mollard G. F., and Heberle J. (2010) The DC gate in channelrhodopsin-2: crucial hydrogen bonding interaction between C128 and D156. Photochem. Photobiol. Sci. 9, 194–198 [DOI] [PubMed] [Google Scholar]
- 8. Berndt A., Yizhar O., Gunaydin L. A., Hegemann P., and Deisseroth K. (2009) Bi-stable neural state switches. Nat. Neurosci. 12, 229–234 [DOI] [PubMed] [Google Scholar]
- 9. Stehfest K., Ritter E., Berndt A., Bartl F., and Hegemann P. (2010) The branched photocycle of the slow-cycling channelrhodopsin-2 mutant C128T. J. Mol. Biol. 398, 690–702 [DOI] [PubMed] [Google Scholar]
- 10. Bamann C., Gueta R., Kleinlogel S., Nagel G., and Bamberg E. (2010) Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry 49, 267–278 [DOI] [PubMed] [Google Scholar]
- 11. Krause N., Engelhard C., Heberle J., Schlesinger R., and Bittl R. (2013) Structural differences between the closed and open states of channelrhodopsin-2 as observed by EPR spectroscopy. FEBS Lett. 587, 3309–3313 [DOI] [PubMed] [Google Scholar]
- 12. Müller M., Bamann C., Bamberg E., and Kühlbrandt W. (2015) Light-induced helix movements in channelrhodopsin-2. J. Mol. Biol. 427, 341–349 [DOI] [PubMed] [Google Scholar]
- 13. Sattig T., Rickert C., Bamberg E., Steinhoff H. J., and Bamann C. (2013) Light-induced movement of the transmembrane helix B in channelrhodopsin-2. Angew. Chem. Int. Ed. Engl. 52, 9705–9708 [DOI] [PubMed] [Google Scholar]
- 14. Sass H. J., Büldt G., Gessenich R., Hehn D., Neff D., Schlesinger R., Berendzen J., and Ormos P. (2000) Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature 406, 649–653 [DOI] [PubMed] [Google Scholar]
- 15. Radzwill N., Gerwert K., and Steinhoff H. J. (2001) Time-resolved detection of transient movement of helices F and G in doubly spin-labeled bacteriorhodopsin. Biophys. J. 80, 2856–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexiev U., Rimke I., and Pöhlmann T. (2003) Elucidation of the nature of the conformational changes of the EF-interhelical loop in bacteriorhodopsin and of the helix VIII on the cytoplasmic surface of bovine rhodopsin: a time-resolved fluorescence depolarization study. J. Mol. Biol. 328, 705–719 [DOI] [PubMed] [Google Scholar]
- 17. Moukhametzianov R., Klare J. P., Efremov R., Baeken C., Göppner A., Labahn J., Engelhard M., Büldt G., and Gordeliy V. I. (2006) Development of the signal in sensory rhodopsin and its transfer to the cognate transducer. Nature 440, 115–119 [DOI] [PubMed] [Google Scholar]
- 18. Wegener A. A., Chizhov I., Engelhard M., and Steinhoff H. J. (2000) Time-resolved detection of transient movement of helix F in spin-labelled pharaonis sensory rhodopsin II. J. Mol. Biol. 301, 881–891 [DOI] [PubMed] [Google Scholar]
- 19. Alexiev U., and Farrens D. L. (2014) Fluorescence spectroscopy of rhodopsins: insights and approaches. Biochim. Biophys. Acta 1837, 694–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirchberg K., Kim T. Y., Möller M., Skegro D., Dasara Raju G., Granzin J., Büldt G., Schlesinger R., and Alexiev U. (2011) Conformational dynamics of helix 8 in the GPCR rhodopsin controls arrestin activation in the desensitization process. Proc. Natl. Acad. Sci. U.S.A. 108, 18690–18695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schröder G. F., Alexiev U., and Grubmüller H. (2005) Simulation of fluorescence anisotropy experiments: probing protein dynamics. Biophys. J. 89, 3757–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kirchberg K., Michel H., and Alexiev U. (2013) Exploring the entrance of proton pathways in cytochrome c oxidase from Paracoccus denitrificans: surface charge, buffer capacity and redox-dependent polarity changes at the internal surface. Biochim. Biophys. Acta 1827, 276–284 [DOI] [PubMed] [Google Scholar]
- 23. Alexiev U., Marti T., Heyn M. P., Khorana H. G., and Scherrer P. (1994) Covalently bound pH-indicator dyes at selected extracellular or cytoplasmic sites in bacteriorhodopsin. 2. Rotational orientation of helices D and E and kinetic correlation between M formation and proton release in bacteriorhodopsin micelles. Biochemistry 33, 13693–13699 [DOI] [PubMed] [Google Scholar]
- 24. Kim T. Y., Schlieter T., Haase S., and Alexiev U. (2012) Activation and molecular recognition of the GPCR rhodopsin: insights from time-resolved fluorescence depolarisation and single molecule experiments. Eur. J. Cell Biol. 91, 300–310 [DOI] [PubMed] [Google Scholar]
- 25. Dutta A., Kim T. Y., Möller M., Wu J., Alexiev U., and Klein-Seetharaman J. (2010) Characterization of membrane protein non-native states: 2. The SDS-unfolded states of rhodopsin. Biochemistry 49, 6329–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim T. Y., Möller M., Winkler K., Kirchberg K., and Alexiev U. (2009) Dissection of environmental changes at the cytoplasmic surface of light-activated bacteriorhodopsin and visual rhodopsin: sequence of spectrally silent steps. Photochem. Photobiol. 85, 570–577 [DOI] [PubMed] [Google Scholar]
- 27. Johnson D. A., and Yguerabide J. (1985) Solute accessibility to N-epsilon-fluorescein isothiocyanate-lysine-23 cobra α-toxin bound to the acetylcholine-Receptor: a consideration of the effect of rotational diffusion and orientation constraints on fluorescence quenching. Biophys. J. 48, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson D. A., Cushman R., and Malekzadeh R. (1990) Orientation of cobra α-toxin on the nicotinic acetylcholine-receptor: fluorescence studies. J. Biol. Chem. 265, 7360–7368 [PubMed] [Google Scholar]
- 29. Watt R. M., and Voss E. W. (1979) Solvent perturbation of the fluorescence of fluorescein bound to specific antibody: fluorescence quenching of the bound fluorophore by iodide. J. Biol. Chem. 254, 1684–1690 [PubMed] [Google Scholar]
- 30. Dickopf S., Alexiev U., Krebs M. P., Otto H., Mollaaghababa R., Khorana H. G., and Heyn M. P. (1995) Proton transport by a bacteriorhodopsin mutant aspartic acid-85 → asparagine, initiated in the unprotonated Schiff-base state. Proc. Natl. Acad. Sci. U.S.A. 92, 11519–11523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexiev U., Mollaaghababa R., Scherrer P., Khorana H. G., and Heyn M. P. (1995) Rapid long-range proton diffusion along the surface of the purple membrane and delayed proton-transfer into the bulk. Proc. Natl. Acad. Sci. U.S.A. 92, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Böckmann R. A., and Caflisch A. (2005) Spontaneous formation of detergent micelles around the outer membrane protein OmpX. Biophys. J. 88, 3191–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pieper J., Buchsteiner A., Dencher N. A., Lechner R. E., and Hauss T. (2008) Transient protein softening during the working cycle of a molecular machine. Phys. Rev. Lett. 100, 228103. [DOI] [PubMed] [Google Scholar]
- 34. Kim T. Y., Winkler K., and Alexiev U. (2007) Picosecond multidimensional fluorescence spectroscopy: a tool to measure real-time protein dynamics during function. Photochem. Photobiol. 83, 378–384 [DOI] [PubMed] [Google Scholar]
- 35. Boreham A., Kim T. Y., Spahn V., Stein C., Mundhenk L., Gruber A. D., Haag R., Welker P., Licha K., and Alexiev U. (2011) Exploiting fluorescence lifetime plasticity in FLIM: target molecule localization in cells and tissues. ACS Med. Chem. Lett. 2, 724–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eftink M. R., and Ghiron C. A. (1981) Fluorescence quenching studies with proteins. Anal. Biochem. 114, 199–227 [DOI] [PubMed] [Google Scholar]
- 37. Lehrer S. S. (1971) Solute perturbation of protein fluorescence: the quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10, 3254–3263 [DOI] [PubMed] [Google Scholar]
- 38. Alexiev U., Scherrer P., Marti T., Khorana H. G., and Heyn M. P. (1995) Time-resolved surface-charge change on the cytoplasmic side of bacteriorhodopsin. FEBS Lett. 373, 81–84 [DOI] [PubMed] [Google Scholar]
- 39. Möller M., and Alexiev U. (2009) surface charge changes upon formation of the signaling state in visual rhodopsin. Photochem. Photobiol. 85, 501–508 [DOI] [PubMed] [Google Scholar]
- 40. Kato H. E., Kamiya M., Sugo S., Ito J., Taniguchi R., Orito A., Hirata K., Inutsuka A., Yamanaka A., Maturana A. D., Ishitani R., Sudo Y., Hayashi S., and Nureki O. (2015) Atomistic design of microbial opsin-based blue-shifted optogenetics tools. Nat. Commun. 6, 7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ritter E., Piwowarski P., Hegemann P., and Bartl F. J. (2013) Light-dark adaptation of channelrhodopsin C128T mutant. J. Biol. Chem. 288, 10451–10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lórenz-Fonfría V. A., and Heberle J. (2014) Channelrhodopsin unchained: structure and mechanism of a light-gated cation channel. Biochim. Biophys. Acta 1837, 626–642 [DOI] [PubMed] [Google Scholar]
- 43. Alexiev U., Mollaaghababa R., Khorana H. G., and Heyn M. P. (2000) Evidence for long range allosteric interactions between the extracellular and cytoplasmic parts of bacteriorhodopsin from the mutant R82A and its second site revertant R82A/G231C. J. Biol. Chem. 275, 13431–13440 [DOI] [PubMed] [Google Scholar]
- 44. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Chapter eleven-reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Richter C., Schneider C., Quick M. T., Volz P., Mahrwald R., Hughes J., Dick B., Alexiev U., and Ernsting N. P. (2015) Dual-fluorescence pH probe for bio-labelling. Phys. Chem. Chem. Phys. 17, 30590–30597 [DOI] [PubMed] [Google Scholar]