

Abstract
The Hedgehog (Hh) pathway is a highly conserved signaling cascade crucial for cell fate determination during embryogenesis. Response to the Hh ligands is mediated by the receptor Patched-1 (Ptch1), a 12-pass transmembrane glycoprotein. Despite its essential role in Hh signaling and its activity as a tumor suppressor, Ptch1 remains largely uncharacterized. We demonstrate here that Ptch1 binds to itself to form oligomeric structures. Oligomerization is mediated by two distinct, structurally disordered, intracellular domains spanning amino acids 584–734 (“middle loop”) and 1162–1432 (C terminus). However, oligomerization is not required for Ptch1-dependent regulation of the canonical Hh pathway operating through Smo. Expression of a mutant protein that deletes both regions represses the Hh pathway and responds to the addition of Hh ligand independent of its inability to bind other factors such as Smurf2. Additionally, deletion of the cytoplasmic middle loop domain generates a Ptch1 mutant that, despite binding to Hh ligand, constitutively suppresses Hh signaling and increases the length of primary cilia. Constitutive activity because of deletion of this region is reversed by further deletion of specific sequences in the cytoplasmic C-terminal domain. These data reveal an interaction between the cytoplasmic domains of Ptch1 and that these domains modulate Ptch1 activity but are not essential for regulation of the Hh pathway.
Keywords: cell signaling, cilia, Hedgehog signaling pathway, oligomerization, sonic hedgehog (SHH), Patched-1, Smoothened
Introduction
Central to the Hedgehog (Hh)2 signaling pathways is the receptor for the Hh ligands, Patched. Originally described in Drosophila (1–3), mammals harbor two variants of Patched, Patched-1 (Ptch1) and Patched-2 (Ptch2) (4–10). Although non-essential roles for Ptch2 during development have been recently elucidated (11–13), the expression and activities of Ptch1 are required for Hh ligand-dependent determination of cell fate during development (14). Ptch1 is a 12-pass, integral membrane glycoprotein for which the best-defined activity is the indirect repression of the activity of the G protein-coupled factor Smoothened (Smo) (15, 16). In the absence of Hh ligand, Ptch1-dependent repression of Smo promotes conversion of the transcriptional mediators of the Hh signaling pathway, the Gli family proteins Gli2 and Gli3, into transcriptional repressors (17). Hh ligand binding to Ptch1 inhibits its activity, activating Smo and thereby maintaining Gli2/3 in their transcriptionally active state. Referred to as the “canonical” pathway, this signaling cascade is typically organized by the primary cilium, where processing of specific components of the pathway is determined by their trafficking through this subcellular structure (18–21). In the absence of Hh ligand, Ptch1 localizes to the cilium, whereas Smo is excluded and is typically localized to the transition zone at its base. Activation of the canonical pathway by the Hh ligands results in the relocalization of Ptch1 to endosomes and Smo to the tip of the cilium. Defects in the molecular motors responsible for maintaining cilium structure also deleteriously affect Hh signaling (for a review, see Ref. 22).
Previously, we and others showed that Ptch1 complexes a number of proteins involved in signaling pathways distinct from those operating through Smo. For example, Ptch1 binds to the cyclin B1/cdk1 co-complex in an Hh-dependent manner (23, 24). Binding of the Hh ligands by Ptch1 liberates cyclin B1/cdk1, thereby promoting the transition through mitosis. Likewise, a point mutation in the C terminus of Ptch1 prevents binding of Tid1 (25). This mutation, present in FVB mice, segregates with their propensity to develop squamous cell carcinoma because of expression of activated Ras in a mouse model of skin cancer. We also reported that Ptch1 binds to factors containing SH3, SH2, and WW domains, including Smurf2, c-src, PIK3R2 (p85β), and Grb2 (26, 27). More detailed studies have defined two specific motifs in the cytoplasmic domains of Ptch1 that mediate binding to the HECT family of E3 ubiquitin ligases, such as Smurf2 and Itch1 (28–30).
Despite the centrality of Ptch1 to Hh signaling and its apparent role as a tumor suppressor (6, 31–34), the Ptch1 protein remains relatively uncharacterized. Furthermore, most mutations in Ptch1 found in human tumors result in substantial structural defects of this receptor, typically because of premature truncation of the protein. Among the informative variants of Ptch1 is one from the mesenchymal dysplasia (mes) mouse. This animal harbors a 32-base pair deletion in the second-last exon, causing truncation of the last 220 amino acids of the cytoplasmic C-terminal domain of Ptch1 and replacing it with an unrelated 68-amino acid peptide (35, 36). For some developing tissues, for example limb buds and adipose tissue, the mes variant of Ptch1 appears to behave as a hypomorphic allele of Ptch1 during embryogenesis. For these tissues, increased signaling through the canonical Hh pathway is observed, as evidenced by induction of the transcriptional targets of the Hh signaling pathway, Ptch1 and Gli1, as well as the resultant phenotypes that are consistent with constitutive canonical Hh signaling (37). However, in the skin of mes mice, where precocious hair follicle development occurs, altered canonical Hh signaling was not observed (38). Similarly, no increase in canonical Hh signaling was seen in epithelial cells of the mammary gland despite development of this skin appendage at puberty being blocked by the mes mutation (39, 40). Indeed, Hh signaling in mammary epithelial cells appears to require a cascade that involves estrogen receptor-α and c-src rather than the canonical signaling pathway operating through Smo (39, 40). Despite these apparent tissue-specific effects of truncating the C terminus of Ptch1, wild-type Ptch1 and mes repress Hh signaling similarly when introduced into Ptch1-deficient MEFs (27, 38). Furthermore, wild-type and mes Ptch1 bind an alkaline phosphatase-Shh (AP-Shh) fusion protein with indistinguishable avidities. In transient assays, Shh binding to either of these also inhibits their activity as a repressor of Smo (27). Thus, the structural basis in the mes mutant that gives rise to the non-lethal, tissue-specific effects of this variant of Ptch1 remains unclear.
To begin to define the structural aspects of Ptch1 that regulate its activities, the role of the two large intracellular domains (amino acids 584–734 and 1161–1434) that are highly conserved among vertebrates were characterized. Our analyses revealed that Ptch1-dependent repression of Smo does not require either of these two cytoplasmic domains. However, mutation of the cytoplasmic domain at the middle portion of Ptch1 generates a dominant variant that, despite binding to Shh ligand, constitutively represses Hedgehog signaling but does not prevent Smo localization to cilia. Furthermore, we show that, like dPtc (41), mPtch1 also forms multimers. However, two cytoplasmic domains facilitate Ptch1 binding to itself, and, in contrast to dPtc, multimerization activity is not required for mPtch1-dependent repression of Smo or its ability to respond to Hh ligand.
Results
Two Intracellular Regions of Murine Ptch1 Mediate Its Oligomerization
The 1434 a.a. mPtch1 protein is predicted to span the membrane 12 times, with an apparent gross structural symmetry between the first half and second half of the protein (Fig. 1A). Given the tissue-specific effects of the mes mutation and the highly conserved sequences between vertebrates of the two large cytoplasmic domains of Ptch1, the activities associated with these domains were characterized.
FIGURE 1.

Ptch1 structure and oligomerization. A, left panel, the predicted structure of the 12-pass transmembrane protein, Ptch1. Arrows indicate significant domains within the protein structure, and polyproline motifs are denoted in red. Right panel, the large deletion mutants of Ptch1 missing, either ΔML, ΔC, or ΔMC. Numbers refer to amino acids. B, left panel, the straight Western blot detecting HA-tagged full-length Ptch1, ΔC, and ΔMLΔC Ptch1 mutant proteins. Right panel, the ΔC mutant, but not the ΔMLΔC mutant, co-immunoprecipitates with full-length Ptch1. IB, immunoblot; IP, immunoprecipitation. C, GFP-tagged full-length Ptch1 transiently transfected with an HA-tagged ΔML mutant. Both α-GFP and reciprocal α-HA co-immunoprecipitations confirm that the ΔML deletion mutant of Ptch1 associates with full-length Ptch1. SSD, sterol sensing domain.
Oligomerization of dPtc is mediated by its C terminus (41). Thus, the ability of mPtch1 to form higher-order complexes was tested. Cells were co-transfected with HA-tagged full-length mPtch1 (mPtchHA) and mutants of mPtch1 that had gross deletions in the two large cytoplasmic domains of the protein: the C-terminal domain (ΔC, deletion of a.a. 1179–1434), the “middle loop” domain (ΔML, deletion of a.a. 614–709), or the two domains together (ΔMLΔC). As Fig. 1B illustrates, immunoprecipitation under stringent conditions of the full-length mPtch1HA using an α-HA antibody co-immunoprecipitated the ΔC mutant of mPtch1 but not the ΔMLΔC mutant that deleted both the C terminus and the middle loop domain. Because migration of the ΔML mutant overlaps that of full-length PtchHA in SDS-PAGE gels, GFP-tagged mPtch1 was co-transfected with HA-tagged ΔML, and co-immunoprecipitations were performed (Fig. 1C). The reciprocal immunoprecipitations in the Fig. 1C, center and right panels, showed that deletion of a.a. 614–709 in the ML region had no effect on mPtch1 binding to itself. Thus, oligomerization of mPtch1 was facilitated by two cytoplasmic domains, the C terminus, and the middle loop region. Either domain was sufficient to mediate oligomerization, and deletion of both domains was necessary to prevent mPtch1 from binding to itself.
Both the ML and C-terminal domains encode motifs that mediate binding to factors harboring SH2, SH3, and WW domains (26). However, only a random coil or disordered structure for these domains is predicted using several algorithms that predict secondary structure. Thus, the regions required for mPtch1 to bind to either the C-terminal or middle loop domains were determined. As summarized in Fig. 2A, a series of mutants of mPtch1 that harbored deletions in discrete portions of the C terminus and ML domains were co-transfected with constructs expressing an HA-tagged C-terminal protein (Fig. 2B) or a GFP-tagged ML fragment (Fig. 2C). Co-immunoprecipitation of the mPtch1 deletion mutants under high stringency conditions (i.e. radioimmune precipitation assay buffer) revealed that the individual domains readily associated with full-length mPtch1 that maintained either the ML or C-terminal portions of mPtch1. Simultaneous deletion of even small portions in the C terminus (e.g. ΔCX-A) in combination with the ML deletion prevented binding of either the isolated HAC-terminal or GFPML domains to mPtch1. In contrast, the small deletion (ΔMLB-X) near the end of the middle loop domain in combination with deletions in the C terminus retained binding activity to GFPML. Furthermore, we observed that point mutations in the polyproline motifs, determined previously to mediate binding to proteins with SH2, SH3, or WW domains (26, 28, 30), did not affect binding of Ptch1 to itself.
FIGURE 2.

Structural requirements for Ptch1 oligomerization. A, schematic illustrating additional mutants of Ptch1 containing discrete deletions in the middle loop and C-terminal domains. Numbers indicate boundaries of deleted amino acids. B, Ptch1 deletion mutants were co-transfected with HAC-term. Immunoprecipitation (IP) of Ptch1 shows binding of the isolated C-terminal domain of Ptch1 to the full-length, ΔML, and ΔC mutants but not to mutants with deletions simultaneously in the middle loop and C terminus. IB, immunoblot. C, co-expression and immunoprecipitation of the Ptch1 mutants with a GFP-tagged middle loop construct shows that the middle loop domain also binds to Ptch1 mutants with deletions in either the middle loop or C terminus but not when both deletions are present simultaneously. All blots are representative of at least two independent experiments. SSD, sterol sensing domain.
To verify that oligomerization activity was intrinsic to the ML and C-terminal domains, GFPML and HAC-term were co-transfected into HEK 293 cells, and coimmunoprecipitations were performed using an α-HA antibody (Fig. 3). GFPML co-immunoprecipitated with HAC-term (Fig. 3A). Furthermore, like its analogous region in dPtc, HAC-term co-immunoprecipitated the GFP-tagged version of this same domain (Fig. 3B). Thus, similar to dPtc, the C-terminal domain of mPtch1 facilitates oligomerization of this receptor. However, in the case of murine Ptch1, the cytoplasmic ML domain also exhibited this activity. Thus, the C terminus is able to form both homocomplexes and heterocomplexes with the middle loop.
FIGURE 3.

The cytoplasmic domains of Ptch1 are sufficient for oligomerization. A, GFP-tagged ML and HA-tagged C terminus constructs were co-transfected in HEK 293 cells. Immunoprecipitation of HAC-term co-immunoprecipitates GFPML. IB, immunoblot. B, co-expression of HA- and GFP-tagged C terminus constructs. α-HA immunoprecipitation pulls down the GFP-tagged variant of the ML domain. All blots are representative of at least two independent experiments.
The ML and C-terminal Domains of mPtch1 Are Not Required for Canonical Hh Signaling
Given the lack of similarity in the C-terminal and ML domains in dPtc versus mPtch1, it was predicted that these regions may be dispensable for control of Smo activity. Thus, specific mutants of mPtch1 were tested for their ability to repress canonical signaling and to respond to Hh ligand. mPtch1 mutants were co-transfected with the 8x-Gli1-Luc construct into Ptch1-deficient MEFs. Serum-starved cells were then stimulated with control medium or medium containing Shh ligand, and changes in canonical Hh signaling were determined (Fig. 4). All mutants with any deletion or alterations in either the ML or C terminus (Fig. 4A) strongly repressed the canonical pathway, as evidenced by the 60–80% reduction in luciferase activity relative to control cells. Mutants that deleted the ML domain in combination with various C-terminal deletions also strongly repressed canonical Hh signaling. Notably, this included the ΔMLΔC mutant that removed all but 12 amino acids in the C-terminal domain and almost 100 amino acids of the ML domain. These data reveal that the ML and C-terminal domains, individually or together, are not required for mPtch1-dependent repression of Smo activity in MEFs. Furthermore, combined with the data in Figs. 1–3, repression of canonical Hh signaling in mPtch1, unlike dPtc, does not depend on the ability of Ptch1 to form multimers.
FIGURE 4.

Activities of Ptch1 deletion mutants in the regulation of Hh-signaling. A, schematic of ΔML mPtch1 mutants. All mutants harbor deletion of a.a. 614 and 709 (ΔML) in conjunction with deletion of the indicated amino acids in the C-terminal domain. B and C, repression of canonical Hh signaling and response to Hh ligand of ΔMLΔC mutants. Deletions in the C terminus responsible for restoration of the Shh signaling response showed that loss of the sequence between 1179–1306 (ΔMLΔCP-A) reverses the constitutive repression of Smo by the ΔML mutant. Sequences down stream of 1274 (ΔMLΔCX-A and ΔMLΔCXcm) had no effect on the constitutive repression activity of the ΔML mutant. Data are displayed as mean ± S.E. (n = 5 for B and 2 for C). Independent experiments were done with biological duplicates. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, further refinement of the C-terminal deletions revealed that loss of a.a. 1179–1220 (ΔMLΔCP-S), which includes a polyproline motif (PP1) and a polyhydroxyl-rich region, or the loss of either motif individually (ΔMLΔCPP1, ΔMLΔCOH) restored the Shh signaling response of ΔML. Data are displayed as mean ± S.E. (n = 4). Independent experiments were done with biological duplicates. **, p < 0.01; ***, p < 0.001. E, Western blots were run using lysates taken from the same well as that used for the luciferase reporter assay. F, luciferase activity following titration of plasmids expressing either the full-length mPtch1 or the ΔMLΔC mutant. Data are displayed as mean ± S.E. (n = 3). G, Western blotting analysis showing the relative levels of Ptch1 expression in one set of lysates used for the luciferase experiments in F.
The ability of the mPtch1 deletion mutants to respond to Hh ligand was then determined. Full-length mPtch1 and variants that deleted sequences in the C terminus (e.g. ΔC) all responded to the addition of Hh ligand. So, although full-length mPtch1 or the ΔC truncation repressed canonical signaling by 60–80%, addition of medium containing N-Shh ligand reversed this repression, as was evident by the increased luciferase activity relative to the control medium (Fig. 4B). Interestingly, despite strong repression of the pathway by the ΔML mutant (deletion of a.a. 615–710), addition of N-Shh did not induce canonical Hh signaling. Thus, the deletion of 100 a.a. in the ML domain generated a constitutively active variant of mPtch1. The resistance to inhibition by Shh of the ΔML mutant lacking amino acids 615–710 was reversed, however, by simultaneous deletion of the C terminus of mPtch1, evident by the induction of luciferase activity of the ΔMLΔC mutant upon addition of N-Shh ligand. A representative Western blot for the core mutants (Fig. 4E) demonstrates that the ability to derepress in response to Hh ligand is not related to the expression level of the protein. The regions in the C terminus required for overcoming the constitutive activity of the deletion in the ML region were refined using small deletions of the C-terminal domain (Fig. 4, C and D). Deletions in the C terminus downstream from amino acid 1274 (ΔCX-A, ΔCXcm) failed to reverse constitutive repression of the canonical Hh pathway by ΔML. However, deletion of the sequence between amino acids 1179 and 1310 (ΔCP-A) restored the response to Shh ligand by ΔML. Fig. 4D illustrates refinement of this region where mutants that delete either the polyproline sequence (ΔMLΔCΔPP1) and/or the sequence with a high proportion of amino acids with hydroxyl side chains (ΔMLΔCΔOH) restored the sensitivity of mutants with the ΔML deletion to treatment with Shh. Thus, although deletion of specific portions of the C terminus, in combination with the middle loop, abrogates binding of these domains to Ptch1, deletion of the region between a.a.1174–1223 is sufficient to alter the constitutive repression activity imparted by the deletion of the Ptch1 ML domain.
The Western blot in Fig. 4E illustrates that the levels of ΔMLΔC protein are typically higher than those of other mutants, presumably because of its increased stability, a consequence of the loss of both binding sites for the E3 ubiquitin ligases Itch and Smurf1/2 (see below). To more carefully quantify its ability to regulate the Hh pathway, we titrated the amount of ΔMLΔC and measured its ability to modulate Hh signaling relative to full-length mPtch1. The relative levels of mPtch1 were then determined by Western blotting analysis using lysates derived from a portion of the same cells used for one of the luciferase experiments. The ΔMLΔC mutant still potently represses Hh signaling at 5 ng of added plasmid, and repression by ΔMLΔC remains sensitive to the addition of N-Shh ligand regardless of its level of expression. Thus, although the levels of ΔMLΔC at 5 ng of added plasmid are still slightly higher than the expression of full-length mPtch1 at 50 ng, the ability of the ΔMLΔC mutant remains indistinguishable from full-length mPtch1 in these transient assays.
E3 Ubiquitin Ligase Binding to mPtch1 Is Uncoupled from Oligomerization and Regulation of Hh Signaling
The refractory behavior of ΔML to inhibition by N-Shh was unrelated to its ability to complex factors known to bind to this region, namely HECT family E3 ubiquitin ligases (26, 28–30). Individual deletions of the ML or C-terminal domains of mPtch1 maintained the ability to complex Smurf2 and Itch (Fig. 5), consistent with previous observations (26, 28–30). However, the simultaneous deletion of both of these intracellular domains prevented the binding of these same factors to mPtch1. Thus, mPtch1 binding to members of the HECT family of E3 ubiquitin ligases can be uncoupled from its ability to regulate canonical Hh signaling
FIGURE 5.

Simultaneous deletion of the middle loop and C terminus is required to block binding of Smurf2 and Itch. Full-length mPtch1 as well as the ΔML, ΔC, and ΔMLΔC mutants were transiently transfected into HEK 293 cells with FLAG-tagged Itch (A) or FLAG-tagged Smurf2 (B). α-FLAG immunoprecipitations (IP) demonstrate that the ΔML and ΔC mutants are capable of binding to both Itch and Smurf2, whereas the ΔMLΔC mutant is unable to bind to these WW domain-containing ubiquitin ligases. IB, immunoblot.
mPtch1 Deletion Mutants Bind to N-Shh Ligand and Undergo Correct Posttranslational Processing
The inability of ΔML to respond to N-Shh was not due to its inability to bind to N-Shh ligand (Fig. 6). When expressed in HEK 293 cells, ΔML bound to an AP-N-Shh fusion protein with an avidity similar to that of wild-type mPtch1. These same mutants also appeared to be processed similarly in cells (Fig. 7). With the exception of the ΔMLΔCP-A mutant, which is fully cleaved by Endo H, all of the mutants were partially resistant to Endo H cleavage, indicating that they were processed and trafficked beyond the Golgi. Curiously, as Fig. 4C demonstrates, the ΔMLΔCP-A mutant behaves similar to wild-type Ptch1 in its ability to repress Smo and respond to Shh ligand, suggesting that posttranslational processing of Ptch1 may not be required for its canonical activities.
FIGURE 6.

The ΔML mutant of Ptch1 binds N-Shh-ligand. A, the ability of the ΔML mutant to bind to N-Shh ligand was determined by assaying alkaline phosphatase activity. The ΔML mutant binds to the AP-N-Shh protein with an avidity similar to wild-type Ptch1. AP-R154E, a mutant of Shh that does not bind to Ptch1, did not bind to the ΔML mutant, showing specificity of ligand binding. B, Western blot probed for N-Shh to show comparable levels of protein in AP-N-Shh and AP-R154E conditioned media. ***, p < 0.001.
FIGURE 7.

Posttranslational processing occurs normally in mPtch1 mutants. Ptch1 deletion mutants exhibit correct secretory pathway trafficking in HEK 293 cells. Equal amounts of Ptch1FL, ΔML, ΔC, ΔMLΔC, ΔMLΔCX-A, ΔMLΔCXcm, and ΔMLΔCP-A were transfected into HEK 293 cells. Cell lysates were taken and subjected to Endo H or PNGase F treatment for 1 h. All Ptch1 constructs tested, except ΔMLΔCP-A, are partially resistant to Endo H cleavage, indicating correct posttranslational processing and secretory pathway trafficking.
Smo Localization to the Cilia in Response to Hh Ligand
To ensure that the activity of ΔML was not due to a defect in its ability to localize to the cilia, the localization of transiently expressed ΔML in Ptch1−/− MEFs was determined (Fig. 8). A very strong signal for ΔML appears coincident with the marker of cilia acetylated tubulin. It was noted further that expression of the ΔML mutant significantly lengthened cilia relative to MEFs expressing wild-type mPtch1 (Fig. 8B).
FIGURE 8.

Expression of ΔML causes an increase in primary cilium length. A, Ptch1FL, ΔML, ΔMLΔC, and ΔsML readily locate to cilia in Ptch−/− MEFs. FL, full-length. B, ΔML and ΔsML caused a significant increase in the length of cilia. A minimum of six cilia were analyzed per independent experiment for a total of at least 20 cilia/treatment (20 for pcDNA3, 25 for mPtch1, 62 for ΔML, and 31 for ΔsML). Data were analyzed by Student's t test. Data are displayed as mean ± S.E. (n = 3). *, p < 0.05. px, pixels.
To test whether the observed constitutive repression by the ΔML mutant was due to the exclusion of Smo from the cilia, the ability of Smo to enter the cilia in response to Hh ligand was tested. Fig. 9A shows that Smo is localized to cilia in serum-starved Ptch1−/− MEFs regardless of the presence of Shh. Localization of Smo to this structure could be blocked, however, following treatment of cells with the small molecule Smo inhibitor smoothened antagonist-1 (Fig. 9B). Probing for Smo in Smo−/− MEFs in the absence or presence of smoothened agonist (Fig. 9, C and D) confirms the specificity of the α-Smo antibody for Smo in cilia (staining in nuclei is nonspecific).
FIGURE 9.
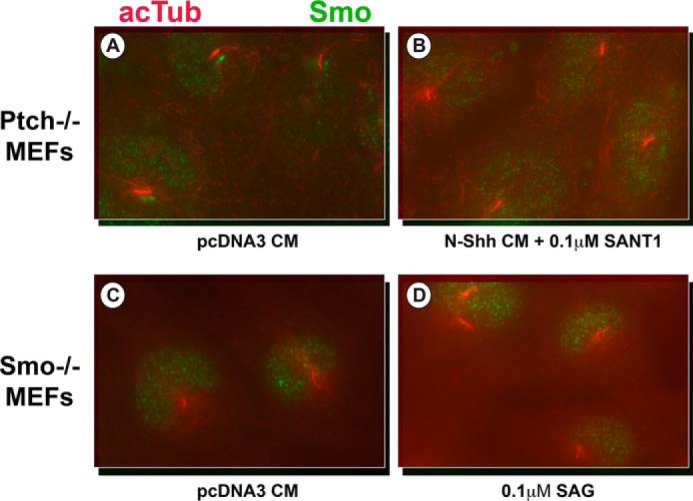
Smo is present in the cilia of Ptch−/− MEFs in the absence of Shh ligand. A–B, Ptch−/− MEFs were serum-starved for 48 h and then treated with either control medium (A) or N-Shh conditioned medium with 0.1 μm smoothened antagonist-1 (B) for 8 h. Cells were then fixed with paraformaldehyde and probed for acetylated tubulin (acTub, red) and Smo (green). Smo localizes to the cilia in the absence of Shh ligand, and this localization can be reversed by the Smo inhibitor smoothened antagonist-1. C–D, Smo−/− MEFs were serum-starved for 48 h and then treated with either control medium (C) or 0.1 μm smoothened agonist (D) for 8 h. Cells were then fixed with paraformaldehyde and probed for acetylated tubulin (red) and Smo (green). Smo is not detectable in this cell line, even in the presence of smoothened agonist.
To assess the effect of the ΔML mutation on Smo localization, Ptch1-deficient MEFs were transiently transfected with mPtch1FL or the ΔML mutant. In cells expressing visible Ptch1FL or ΔML in the primary cilia, Smo was excluded from the cilia when treated with control medium (Fig. 10). However, treatment with Hh ligand resulted in relocalization of Smo to cilia in the majority of cells (Fig. 10). Curiously, both PtchFL and the ΔML mutant were still present in cilia in the presence of Hh ligand in these transient assays. However, this constitutive localization in the cilia of MEFs did not prevent the localization of Smo to this structure.
FIGURE 10.

Smo localizes to the cilia in response to Hh ligand in the ΔML mutant of Ptch1. Ptch−/− MEFs transiently transfected with Ptch1FL (A) or ΔML (B) were serum-starved for 48 h prior to treatment with either control or N-Shh conditioned medium for 8 h. Cells were fixed in 4% paraformaldehyde and probed for acetylated tubulin (acTub, blue), Ptch1 (red), and Smo (green). Smo enters the cilia in response to Shh ligand with either Ptch1FL or ΔML. C, straight Western blots were run using lysates taken from identical wells as the coverslips used for immunofluorescence staining. Staining and Western blots are representative of three independent experiments.
Taken together, these data demonstrate that the two cytoplasmic domains of Ptch1, between a.a. 584–734 and 1162–1432, mediate oligomerization of this receptor. This activity can be uncoupled from Ptch1-dependent repression of Smo activity and its ability to respond to Hh ligand. However, these same two interacting domains are involved in the regulation of canonical Hh signaling, as evidenced by the constitutive repression observed for the ΔML mutant and the reversal of this constitutive activity by deletion of a specific region of the C terminus of Ptch1.
Discussion
The Ptch1 protein plays a central and essential role in the Hedgehog signaling pathway. Deletion of mPtch1 results in severe defects in the mouse embryo, particularly evident during early organogenesis (14). Genetic evidence from mice and humans further revealed the importance of Hh signaling in tumors. Indeed, loss of Ptch1 activity and subsequent transformation of specific tissues is consistent with Ptch1 acting as a tumor suppressor (6, 31–34).
Given the distinct activities for Ptch1 revealed by the mes mouse and the relative paucity of data detailing the functional domains of Ptch1, we have begun to characterize the structural aspects of Ptch1 that determine the distinct activities of this receptor. A comparison of the sequence of Ptch1 with its vertebrate counterparts reveals a significant similarity. This high similarity includes sequences in the cytoplasmic domains. In contrast, little similarity exists in these regions compared with its Drosophila homologue, dPtc, or with analogous regions in the related vertebrate protein, Ptch2, which is otherwise 57% identical with Ptch1 (7, 8). Given the high degree of similarity between the cytoplasmic C terminus and middle loop regions among vertebrates but their divergence from dPtc and Ptch2, we hypothesized that these regions might not be essential for the Ptch1-dependent regulation of canonical Hh signaling. As shown by the ΔMLΔC mutant, which deletes 100 a.a. in the ML domain and all but 12 a.a. in the C terminus, these domains, individually or together, were not essential for Ptch1-dependent repression of canonical Hh signaling in MEFs. Furthermore, these data demonstrated that simultaneous loss of both of these regions in the ΔMLΔC mutant did not affect the apparent ability of N-Shh ligand to inhibit Ptch1-dependent repression of Smo. These activities were shown by transient expression of Ptch1 in Ptch1-deficient MEFs, where the constitutively high level of canonical Hh signaling was repressed, and this repression was reversed upon stimulation with N-Shh ligand.
Thus, the essential activity of Ptch1, specifically the Hh-ligand-dependent modulation of Smo activity, does not require the activities of the two large cytoplasmic domains of Ptch1. However, data from observations using adipocytes or mammary mesenchymal cells from mes mice demonstrated that the C-terminal region did play a role in the regulation of canonical Hh signaling. For both cell types, constitutively higher levels of canonical Hh signaling were apparent in cells from mes mice, as evidenced by the increased expression of transcriptional targets of the Hh pathway, Gli1 and Ptch1. The basis for these cell-specific effects observed when the C-terminal domain of Ptch1 is deleted is not apparent. It is clear, however, that the activities of these two regions can be uncoupled from the Ptch1-dependent modulation of Smo activity.
Our data also revealed that the C-terminal domain and the cytoplasmic ML region interacted and participated in a complex manner to regulate the activities of Ptch1. Although it has been demonstrated previously for dPtc that its C terminus mediated trimerization of this receptor and that this activity was required for canonical Hh signaling (41), we showed that multimerization of mPtch1 was facilitated by two distinct interacting cytoplasmic domains. This activity was not required for mPtch1-dependent regulation of canonical Hh signaling. In transient assays, the simultaneous loss of both of these domains prevented Ptch1 from associating with wild-type Ptch1 or the isolated ML and C-terminal domains. Interestingly, the coincident deletion of these domains failed to have any discernible effects on canonical Hh signaling. The lack of a requirement for these domains in the regulation of canonical Hh signaling is consistent with the overall lack of sequence similarity for these regions between invertebrate and vertebrate Patched.
Loss of the C terminus of Ptch1 was shown recently to prevent repression of canonical Hh signaling and localization of Ptch1 to the primary cilia (42). In contrast, we observed here and in previous experiments (27) that loss of the C terminus did not prevent Ptch1 from repressing Smo in Ptch1-deficient MEFs or responding to N-Shh ligand. Our data were also in agreement with those reported for the mes variant of Ptch1 (38). The basis for these differences is not apparent, particularly given that Ptch1-deficient MEFs were used in both studies. That the lines used in these various studies might have a number of differences is supported by our observation and those of others (43) that Ptch2 is expressed in Ptch1-deficient MEFs, whereas another group (42) stated that Ptch2 was not expressed in their MEF line.
It has also been shown previously that binding of E3 ubiquitin ligases, specifically Smurf2 and Itch1, was required for repression of Ptch1 activity (28, 30, 44). In contrast, in similar transient assays, we determined that Shh ligand inhibited the repression of Smo by Ptch1 regardless of whether the domains required for Ptch1 binding to Smurf2 and Itch1 were present.
Despite the lack of sequence similarity with dPtc, the very high sequence similarity between vertebrates for the ML and C-terminal domains of Ptch1 suggested that these regions mediated important activities of Ptch1. For example, a complex interaction between the ML and C-terminal domains is evident. Deletion of the ML region between a.a. 614 and 709 resulted in a protein that strongly repressed canonical Hh signaling but was refractory to the reversal of this repression upon addition of Shh ligand. Indeed, refinement of this region by deleting a.a. 630–673 (ΔsML) resulted in a mutant protein with the same qualities as ΔML. The failure to respond to Shh ligand was not due to a gross overexpression of the protein or an inability of the ΔML mutant to bind the ligand because it bound to the AP-Shh fusion protein with the same avidity as wild-type Ptch1. Interestingly, the refractory nature of mutants that deleted portions of the ML was overcome by deletion of a specific portion of the C-terminal domain. Using more discrete mutations, the region in the C terminus that was defined as being required to reverse the constitutive activity of ΔML harbored a polyproline motif (a.a. 1180–1188) and a stretch of amino acids with a high proportion (11 of 18 a.a.) of side chains containing hydroxyl groups (a.a. 1204–1221). Deletion of this region overcame the constitutive repression of canonical Hh signaling because of deletion of sequences in the ML region.
We suggest that a potential mechanism exists where the C terminus mediates a secondary regulatory mechanism that modifies mPtch1 activity. This activity is not required for canonical signaling but can contribute to the active “repressor state” of Ptch1. This activity of the C terminus can be repressed in response to Hh ligand binding by a signal mediated through the ML region. Thus, when ML is deleted, inhibition of repressor activity imparted by the C terminus is abrogated, thereby maintaining Ptch1 in a constitutively active repressor state. However, when the C terminus is also removed, this secondary regulatory mechanism is lost so that the effect of the ΔML deletion is irrelevant, the remaining sequences in Ptch1 being sufficient to regulate Hh ligand-dependent activity of Smo. It was also noted that the Smo agonist smoothened agonist, which bypasses Ptch1 activity to directly activate Smo, also activated Smo in the presence of the ΔML mutant. This observation is consistent with a model reported previously (45) that suggests that, following Smo localization to the cilia, Ptch1 regulates a second process that is required for the activation of Smo in this organelle. We propose that the ΔML mutant constitutively blocks this activation step.
It has been proposed previously that translocation of Smo to cilia is insufficient for its activation and that an additional activation step is required (45, 46). Thus, although the ΔML mutant does not prevent Smo from entering cilia in response to Hh ligand, it is possible that ΔML may interfere with this secondary activation. That expression of ΔML causes a significant increase in cilia length may suggest further that alterations in cilia trafficking may play a role in this process. Alterations in cilia function have been shown previously to effect the output of the canonical Hh signaling cascade (for a review, see Ref. 42). Thus, we propose that constitutive repression by the ΔML mutant of Hh signaling alters normal cilium function, thereby preventing a process required for normal activation of Smo in the cilia.
In conclusion, the ML and C-terminal domains play important roles in the regulation of Ptch1 activities during Hh signaling. However, these domains are not essential for the central activity of Ptch1; specifically, the Hh ligand-dependent regulation of Smo trafficking and activity. Rather, these cytoplasmic domains of Ptch1 mediate a second tier of control that influences the signal imparted by Hh ligand binding to this essential receptor.
Materials and Methods
Cell Culture
HEK 293 cells (a gift from S. Girardin, University of Toronto), Ptch1−/− MEFs (a gift from C. C. Hui, Hospital for Sick Children Research Institute), Smo−/− MEFs (kindly provided by M. Lewis, Baylor College of Medicine), and MDA-MB-231 cells (ATCC HTB-26) were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Shh Light II fibroblasts (47) were cultured in the same medium with the addition of 400 μg/ml G418 and 150 μg/ml zeocin.
Transfections, Western Blotting, and Co-immunoprecipitations
HEK293 cells were grown to 60% confluence in 60-mm plates and transfected with 1 μg of the various Ptch1 mutants using 2 mg/ml PEI at a 2:1 ratio. Cell lysates in 0.5% Nonidet P-40 were prepared as described previously (26). For straight Western blots, 50 μg of lysate was mixed with 4× SDS loading buffer (50 mm Tris (pH 6.8), 100 mm DTT, 2% SDS, 0.1% bromphenol blue, and 10% glycerol) and incubated for at least 20 min at 37 °C. Samples were separated using 10% SDS-PAGE and blotted onto a nitrocellulose membrane. The blots were blocked in 5% skim milk and probed overnight with primary antibodies in a 3% BSA solution. Antibodies used were as follows: 1:1000 goat α-Ptch1 (Santa Cruz Biotechnology, sc-6149), 1:1000 mouse α-Ptch1 (5c7, Genetex, 83771), 1:1000 mouse α-FLAG (ABM, G191), 1:1000 rabbit α-HA (ABM, G166), 1:500 12CA5 mouse α-HA, 1:200 5E1 mouse α-Shh, 1:1000 goat α-GFP (ABM, G095), and 1:1000 mouse α-GFP (ABM, G096).
For immunoprecipitations, primary antibody was added to 250 μg of total cell lysate and incubated overnight at 4 °C in 0.5% Nonidet P-40 lysis buffer or radioimmune precipitation assay buffer (25 mm Tris (pH 7.6), 150 mm NaCl, 1% Nonidet P-40, 1% deoxycholic acid, and 0.1% SDS). The following day, 20 μl of protein G-agarose or protein A-agarose beads were added at 4 °C for 2 h. The beads were then spun down and washed five times in 0.5% Nonidet P-40 or in radioimmune precipitation assay buffer, respectively, and then resuspended in 25 μl of SDS loading buffer. Proteins were separated on 10% SDS-PAGE gels, and Western blotting analyses were performed as described above.
Luciferase Reporter Assays
Shh-conditioned and control media were prepared by transiently transfecting pcDNA3.1-N-Shh or empty pcDNA3.1 into HEK 293 cells. After 24 h, cells were serum-starved using medium with 0.5% FBS for 48–72 h. The medium was recovered and filter-sterilized using a 0.22-μm syringe filter. Shh ligand-activity was verified using Shh Light II fibroblasts, which harbor an intrinsic 8X-Gli-Luciferase promoter as described previously (47). To assay the ability of Ptch1 mutants to regulate canonical Hh signaling in transient assays, Ptch1-deficient MEFs were transfected with 400 ng of the 8X-Gli-Luciferase firefly reporter transgene, 40 ng of a constitutive Renilla luciferase transgene, and 25 ng of a pcDNA3 expression vector encoding various Ptch1 mutants using FuGENE 6 (4:1 ratio, Promega) according to the instructions of the manufacturer. After 24 h, cells were switched to 0.5% FBS. Twenty-four hours later, cells were treated with Shh-conditioned or control medium, and cell lysates were taken after another 24 h. Firefly and Renilla luciferase activities were determined using the Dual-Luciferase reporter assay system (Promega) according to the instructions of the manufacturer. Data were analyzed by two-way analysis of variance followed by pairwise comparison of means using a Student's t test.
Immunofluorescence
Ptch1-deficient MEFs were grown in 100-mm dishes until confluent and then trypsinized, and 1.2 × 106 cells were transfected with 2 μg of plasmid using a 3:1 ratio of GenJet Reagent II (SignaGen Laboratories) according to the instructions of the manufacturer. Transfected cells were seeded onto a 12-mm coverslip in a 6-well plate. Cells were grown for 24–48 h and serum-starved for 24 h in DMEM containing 0.5% FBS. Cells were washed twice in ice-cold PBS and fixed in 4% paraformaldehyde in PBS for 10 min at room temperature. Samples were blocked and permeabilized using 0.2% Triton X-100 and 3% BSA in PBS for 10 min at room temperature. Primary antibodies (1:400 goat α-Ptch1 (Santa Cruz Biotechnology), 1:400 mouse α-acetylated tubulin (Sigma, T6793), and 1:200 rabbit α-Smoothened (Abcam, ab38686)) were diluted in 3% BSA in PBS and incubated for 4 h at room temperature or overnight at 4 °C in a humidified chamber. After three washes in PBS, coverslips were incubated in the dark with secondary antibodies (1:400 FITC-conjugated donkey α-goat IgG (Jackson ImmunoResearch Laboratories), 1:100 TRITC-conjugated donkey α-goat IgG (Jackson ImmunoResearch Laboratories), 1:200 Alexa Fluor 568 donkey α-mouse IgG (Invitrogen), 1:200 Alexa Fluor 488 donkey α-rabbit IgG (Invitrogen), or 1:200 AMCA AffiniPure donkey α-mouse (Jackson ImmunoResearch Laboratories)) for 1 h at room temperature in 3% BSA in PBS. Coverslips were washed three times in PBS and then mounted on slides with Vectashield mounting medium with or without DAPI (Vector Laboratories). Slides were viewed on a Nikon Eclipse 80i, and images were taken with a QICAM FAST 1394 camera using QCapture PLUS software (QImaging).
Hh Ligand Binding Assay
Conditioned medium containing an AP-Shh fusion protein or a mutant construct, AP-R154E, which does not bind to Ptch1, was prepared by transiently transfecting the respective construct into HEK 293 cells as described above for the Shh conditioned medium (48, 49). HEK 293 cells were transiently transfected with the indicated Ptch1 expression constructs and then lysed after 48–72 h in 0.5% Nonidet P-40 lysis buffer. An agarose bead-conjugated antibody against Ptch1 (Santa Cruz Biotechnology, sc-6149 AC) was used to isolate Ptch1 from 400 μg of lysate. Immunoprecipitated Ptch1 proteins were then washed twice with cold HBAH (0.5 mg/ml BSA, 0.1% sodium azide, and 20 mm HEPES (pH 7.0)) and then incubated with 600 μl of AP-Shh-conditioned medium for 2 h at room temperature. Complexes were then washed five times with HBAH, once with HEPES buffered saline (150 mm NaCl and 20 mm HEPES (pH 7.0)), resuspended in 100 μl of HEPES buffered saline, and heated at 65 °C for 10 min. The samples were then transferred to ice and mixed with 100 μl of 2× AP substrate buffer (31 mm p-nitrophenyl phosphate, 1 mm MgCl2, and 2 m diethanolamine (pH 9.8)) for measurement of alkaline phosphatase activity via absorbance at 412 nm.
Glycosidase Assay
Transiently transfected HEK 293 cells were lysed in Nonidet P-40 lysis buffer. 40 μg of lysates was incubated for 1 h at 37 °C with 250 units of Endo H (New England Biolabs) or 500 units of PNGase F (New England Biolabs). Lysates were then run on Western blot as described above.
Quantification of Cilium Length
Immunofluorescent images were taken by a QICAM FAST 1394 camera using QCapture PLUS software at ×60 objective magnification. Images were opened with Inkscape graphics editor software, and cilia were traced using the draw tool and measured using the Measure Path function.
Author Contributions
A. F. wrote the paper and designed, performed, and analyzed the experiments shown in Figs. 2, 3, 9, and 10. J. P. Y. L. designed, performed, and analyzed the experiments shown in Figs. 4, 7, and 8. A. T. designed, performed, and analyzed the experiments shown in Figs. 1 and 5. I. J. performed and analyzed the experiment shown in Fig. 6. P. A. H. conceived and coordinated the study and wrote the paper. A. F., J. P. Y. L., and P. A. H. reviewed the results, and all authors approved the final version of the manuscript.
This work was supported by Canadian Institutes of Health Research Grant MOP-97929 (to P. A. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- Hh
- Hedgehog
- SH
- Src homology
- MEF
- mouse embryonic fibroblast
- AP
- alkaline phosphatase
- a.a.
- amino acid(s)
- C-term
- C-terminal
- Endo H
- endo-β-N-acetylglucosaminidase H
- TRITC
- tetramethylrhodamine isothiocyanate
- HECT
- homologous to the E6-AP carboxyl terminus.
References
- 1. Nakano Y., Guerrero I., Hidalgo A., Taylor A., Whittle J. R., and Ingham P. W. (1989) A protein with several possible membrane-spanning domains encoded by the Drosophila segment polarity gene patched. Nature 341, 508–513 [DOI] [PubMed] [Google Scholar]
- 2. Hidalgo A., and Ingham P. (1990) Cell patterning in the Drosophila segment: spatial regulation of the segment polarity gene patched. Development 110, 291–301 [DOI] [PubMed] [Google Scholar]
- 3. Chen Y., and Struhl G. (1996) Dual roles for patched in sequestering and transducing Hedgehog. Cell 87, 553–563 [DOI] [PubMed] [Google Scholar]
- 4. Goodrich L. V., Johnson R. L., Milenkovic L., McMahon J. A., and Scott M. P. (1996) Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 10, 301–312 [DOI] [PubMed] [Google Scholar]
- 5. Hahn H., Christiansen J., Wicking C., Zaphiropoulos P. G., Chidambaram A., Gerrard B., Vorechovsky I., Bale A. E., Toftgard R., Dean M., and Wainwright B. (1996) A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J. Biol. Chem. 271, 12125–12128 [DOI] [PubMed] [Google Scholar]
- 6. Johnson R. L., Rothman A. L., Xie J., Goodrich L. V., Bare J. W., Bonifas J. M., Quinn A. G., Myers R. M., Cox D. R., Epstein E. H. Jr., and Scott M. P. (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671 [DOI] [PubMed] [Google Scholar]
- 7. Motoyama J., Takabatake T., Takeshima K., and Hui C. (1998) Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat. Genet. 18, 104–106 [DOI] [PubMed] [Google Scholar]
- 8. Smyth I., Narang M. A., Evans T., Heimann C., Nakamura Y., Chenevix-Trench G., Pietsch T., Wicking C., and Wainwright B. J. (1999) Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene in basal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 8, 291–297 [DOI] [PubMed] [Google Scholar]
- 9. Zaphiropoulos P. G., Undén A. B., Rahnama F., Hollingsworth R. E., and Toftgård R. (1999) PTCH2, a novel human patched gene, undergoing alternative splicing and up-regulated in basal cell carcinomas. Cancer Res. 59, 787–792 [PubMed] [Google Scholar]
- 10. Carpenter D., Stone D. M., Brush J., Ryan A., Armanini M., Frantz G., Rosenthal A., and de Sauvage F. J. (1998) Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc. Natl. Acad. Sci. U.S.A. 95, 13630–13634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holtz A. M., Peterson K. A., Nishi Y., Morin S., Song J. Y., Charron F., McMahon A. P., and Allen B. L. (2013) Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development 140, 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adolphe C., Nieuwenhuis E., Villani R., Li Z. J., Kaur P., Hui C.-C., and Wainwright B. J. (2014) Patched 1 and patched 2 redundancy has a key role in regulating epidermal differentiation. J. Invest. Dermatol. 134, 1981–1990 [DOI] [PubMed] [Google Scholar]
- 13. Alfaro A. C., Roberts B., Kwong L., Bijlsma M. F., and Roelink H. (2014) Ptch2 mediates the Shh response in Ptch1−/− cells. Development 141, 3331–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milenkovic L., Goodrich L. V., Higgins K. M., and Scott M. P. (1999) Mouse patched1 controls body size determination and limb patterning. Development 126, 4431–4440 [DOI] [PubMed] [Google Scholar]
- 15. Chen Y., and Struhl G. (1998) In vivo evidence that Patched and Smoothened constitute distinct binding and transducing components of a Hedgehog receptor complex. Development 125, 4943–4948 [DOI] [PubMed] [Google Scholar]
- 16. Ingham P. W., Nystedt S., Nakano Y., Brown W., Stark D., van den Heuvel M., and Taylor A. M. (2000) Patched represses the Hedgehog signalling pathway by promoting modification of the Smoothened protein. Curr. Biol. 10, 1315–1318 [DOI] [PubMed] [Google Scholar]
- 17. Sasaki H., Nishizaki Y., Hui C., Nakafuku M., and Kondoh H. (1999) Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915–3924 [DOI] [PubMed] [Google Scholar]
- 18. Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., and Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 19. Liu A., Wang B., and Niswander L. A. (2005) Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132, 3103–3111 [DOI] [PubMed] [Google Scholar]
- 20. Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J., and Yoder B. K. (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rohatgi R., Milenkovic L., and Scott M. P. (2007) Patched1 regulates Hedgehog signaling at the primary cilium. Science 317, 372–376 [DOI] [PubMed] [Google Scholar]
- 22. Wilson C. W., and Stainier D. Y. (2010) Vertebrate Hedgehog signaling: cilia rule. BMC Biol. 8, 102–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barnes E. A., Kong M., Ollendorff V., and Donoghue D. J. (2001) Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 20, 2214–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adolphe C., Hetherington R., Ellis T., and Wainwright B. (2006) Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 66, 2081–2088 [DOI] [PubMed] [Google Scholar]
- 25. Wakabayashi Y., Mao J.-H., Brown K., Girardi M., and Balmain A. (2007) Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature 445, 761–765 [DOI] [PubMed] [Google Scholar]
- 26. Chang H., Li Q., Moraes R. C., Lewis M. T., and Hamel P. A. (2010) Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int. J. Biochem. Cell Biol. 42, 1462–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harvey M. C., Fleet A., Okolowsky N., and Hamel P. A. (2014) Distinct effects of the mesenchymal dysplasia gene variant of murine Patched-1 protein on canonical and non-canonical Hedgehog signaling pathways. J. Biol. Chem. 289, 10939–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yue S., Tang L.-Y., Tang Y., Tang Y., Shen Q.-H., Ding J., Chen Y., Zhang Z., Yu T.-T., Zhang Y. E., and Cheng S. Y. (2014) Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. eLife 3, e02555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen X. L., Chinchilla P., Fombonne J., Ho L., Guix C., Keen J. H., Mehlen P., and Riobo N. A. (2014) Patched-1 pro-apoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell Biol. 10.1128/MCB.00960-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang S., Zhang Z., Zhang C., Lv X., Zheng X., Chen Z., Sun L., Wang H., Zhu Y., Zhang J., Yang S., Lu Y., Sun Q., Tao Y., Liu F., Zhao Y., and Chen D. (2013) Activation of Smurf E3 ligase promoted by smoothened regulates hedgehog signaling through targeting patched turnover. PLoS Biol. 11, e1001721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stone D. M., Hynes M., Armanini M., Swanson T. A., Gu Q., Johnson R. L., Scott M. P., Pennica D., Goddard A., Phillips H., Noll M., Hooper J. E., de Sauvage F., and Rosenthal A. (1996) The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134 [DOI] [PubMed] [Google Scholar]
- 32. Wolter M., Reifenberger J., Sommer C., Ruzicka T., and Reifenberger G. (1997) Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 57, 2581–2585 [PubMed] [Google Scholar]
- 33. Braun S., Oppermann H., Mueller A., Renner C., Hovhannisyan A., Baran-Schmidt R., Gebhardt R., Hipkiss A., Thiery J., Meixensberger J., and Gaunitz F. (2012) Hedgehog signaling in glioblastoma multiforme. Cancer Biol. Ther. 13, 487–495 [DOI] [PubMed] [Google Scholar]
- 34. Bhattacharya R., Kwon J., Ali B., Wang E., Patra S., Shridhar V., and Mukherjee P. (2008) Role of hedgehog signaling in ovarian cancer. Clin. Cancer Res. 14, 7659–7666 [DOI] [PubMed] [Google Scholar]
- 35. Sweet H. O., Bronson R. T., Donahue L. R., and Davisson M. T. (1996) Mesenchymal dysplasia: a recessive mutation on chromosome 13 of the mouse. J. Hered. 87, 87–95 [DOI] [PubMed] [Google Scholar]
- 36. Makino S., Masuya H., Ishijima J., Yada Y., and Shiroishi T. (2001) A Spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of patched (ptc). Dev. Biol. 239, 95–106 [DOI] [PubMed] [Google Scholar]
- 37. Li Z., Zhang H., Denhard L. A., Liu L.-H., Zhou H., and Lan Z.-J. (2008) Reduced white fat mass in adult mice bearing a truncated Patched 1. Int. J. Biol. Sci. 4, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nieuwenhuis E., Barnfield P. C., Makino S., and Hui C.-C. (2007) Epidermal hyperplasia and expansion of the interfollicular stem cell compartment in mutant mice with a C-terminal truncation of Patched1. Dev. Biol. 308, 547–560 [DOI] [PubMed] [Google Scholar]
- 39. Okolowsky N., Furth P. A., and Hamel P. A. (2014) Oestrogen receptor-α regulates non-canonical Hedgehog-signalling in the mammary gland. Dev. Biol. 391, 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang H., Balenci L., Okolowsky N., Muller W. J., and Hamel P. A. (2012) Mammary epithelial-restricted expression of activated c-src rescues the block to mammary gland morphogenesis due to the deletion of the C-terminus of Patched-1. Dev. Biol. 370, 187–197 [DOI] [PubMed] [Google Scholar]
- 41. Lu X., Liu S., and Kornberg T. B. (2006) The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev. 20, 2539–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim J., Hsia E. Y., Brigui A., Plessis A., Beachy P. A., and Zheng X. (2015) The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal. 8, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhulyn O., Nieuwenhuis E., Liu Y. C., Angers S., and Hui C. (2015) Ptch2 shares overlapping functions with Ptch1 in Smo regulation and limb development. Dev. Biol. 397, 191–202 [DOI] [PubMed] [Google Scholar]
- 44. Chen X. L., Chinchilla P., Fombonne J., Ho L., Guix C., Keen J. H., Mehlen P., and Riobo N. A. (2014) Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell Biol. 34, 3855–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rohatgi R., Milenkovic L., Corcoran R. B., and Scott M. P. (2009) Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. U.S.A. 106, 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilson C. W., Chen M.-H., and Chuang P.-T. (2009) Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS ONE 4, e5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sasaki H., Hui C., Nakafuku M., and Kondoh H. (1997) A binding site for Gli proteins is essential for HNF-3β floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313–1322 [DOI] [PubMed] [Google Scholar]
- 48. Chuang P.-T., and McMahon A. P. (1999) Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 397, 617–621 [DOI] [PubMed] [Google Scholar]
- 49. Okada A., Charron F., Morin S., Shin D. S., Wong K., Fabre P. J., Tessier-Lavigne M., and McConnell S. K. (2006) Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature 444, 369–373 [DOI] [PubMed] [Google Scholar]