

Abstract
Contact-dependent growth inhibition (CDI) is a widespread mechanism of inter-bacterial competition. CDI+ bacteria deploy large CdiA effector proteins, which carry variable C-terminal toxin domains (CdiA-CT). CDI+ cells also produce CdiI immunity proteins that specifically neutralize cognate CdiA-CT toxins to prevent auto-inhibition. Here, we present the crystal structure of the CdiA-CT/CdiIE479 toxin/immunity protein complex from Burkholderia pseudomallei isolate E479. The CdiA-CTE479 tRNase domain contains a core α/β-fold that is characteristic of PD(D/E)XK superfamily nucleases. Unexpectedly, the closest structural homolog of CdiA-CTE479 is another CDI toxin domain from B. pseudomallei 1026b. Although unrelated in sequence, the two B. pseudomallei nuclease domains share similar folds and active-site architectures. By contrast, the CdiIE479 and CdiI1026b immunity proteins share no significant sequence or structural homology. CdiA-CTE479 and CdiA-CT1026b are both tRNases; however, each nuclease cleaves tRNA at a distinct position. We used a molecular docking approach to model each toxin bound to tRNA substrate. The resulting models fit into electron density envelopes generated by small-angle x-ray scattering analysis of catalytically inactive toxin domains bound stably to tRNA. CdiA-CTE479 is the third CDI toxin found to have structural homology to the PD(D/E)XK superfamily. We propose that CDI systems exploit the inherent sequence variability and active-site plasticity of PD(D/E)XK nucleases to generate toxin diversity. These findings raise the possibility that many other uncharacterized CDI toxins may belong to the PD(D/E)XK superfamily.
Keywords: crystal structure, protein complex, ribonuclease, small-angle x-ray scattering (SAXS), toxin, Burkholderia pseudomallei, toxin/immunity complexes, tRNase
Introduction
Bacterial contact-dependent growth inhibition (CDI) 4 is an important mechanism of intercellular competition in which Gram-negative bacteria intoxicate neighboring cells upon direct contact. Genes encoding CDI systems are distributed throughout α-, β-, and γ-proteobacteria and are commonly found in human pathogens such as enterohemorrhagic Escherichia coli, Neisseria meningitidis, Pseudomonas aeruginosa, and Burkholderia pseudomallei (1–3). CDI is a function of the CdiB/CdiA family of two-partner secretion systems. CdiB is an Omp85 β-barrel protein required for the export and display of CdiA effectors on the cell surface. Based on its homology to filamentous hemagglutinin from Bordetella species, CdiA proteins are predicted to form filaments that project several hundred Å from the cell surface (3–5). Upon binding to specific receptors on susceptible bacteria, CdiA delivers its C-terminal toxin domain (CdiA-CT) into the target cell to inhibit growth (6–8). CDI+ bacteria are protected from toxicity by CdiI immunity proteins, which bind to the CdiA-CT toxin domain and neutralize its activity. Although the general architecture of CdiA proteins is conserved across bacteria, the effectors vary considerably in size (180–640 kDa), and CdiA-CT toxin sequences are remarkably diverse (1, 2, 9–13). Thus, CdiA effectors collectively deploy a variety of toxin domains with distinct biochemical activities (1, 3, 14, 15). Moreover, CdiI immunity proteins are also highly variable in sequence and only protect cells against cognate CdiA-CT toxins. The polymorphic nature of CDI toxin/immunity protein pairs and the specificity of their binding interactions suggest that the systems mediate inter-strain competition and self/nonself recognition.
We previously characterized CdiA-CT/CdiI pairs from environmental isolates of Burkholderia pseudomallei, using these systems as a model to explore toxin/immunity protein structure and evolution (16, 17). B. pseudomallei is a category B pathogen and the causative agent of melioidosis, a serious human disease endemic to southeast Asia and northern Australia (18, 19). B. pseudomallei isolates are genetically heterogeneous, and different strains are thought to compete with one another for growth niches and other resources (20, 21). CDI may contribute to inter-strain competition because every B. pseudomallei isolate carries at least one cdi gene cluster (16). B. pseudomallei CdiA proteins share 59–99.5% pairwise sequence identity over the N-terminal 2,770–2,829 residues but diverge abruptly after a conserved (E/Q)LYN peptide motif that demarcates the CdiA-CT region. At least 10 distinct CdiA-CT/CdiI sequence types are found in the species, but only three toxin activities have been characterized experimentally (16). The type I CdiA-CT is homologous to the nuclease domain from colicin E5 and exhibits anticodon nuclease activity against tRNAHis, tRNATyr, tRNAAsp, and tRNAAsn (16, 22). The type II toxin cleaves all tRNAs between conserved residues T54 and Ψ55 in the T-loop; and the type V toxin preferentially cleaves near the 3′-end of tRNAAla (16, 17). Type VII and VIII CdiA-CT sequences are closely related in sequence and are predicted RNA deaminases (14). There are no predicted activities for the other B. pseudomallei CDI toxins, although the type X toxin contains a conserved RES (Arg-Glu-Ser) domain of unknown function (Pfam: PF08808).
Here, we present the crystal structure of the type II toxin/immunity protein complex from B. pseudomallei isolate E479 (16, 20). The C-terminal tRNase domain of CdiA-CTE479 is built upon an α/β-fold characteristic of PD(D/E)XK nucleases. The PD(D/E)XK superfamily includes most restriction endonucleases and other enzymes involved in DNA recombination and repair (23). A DALI server (24) search reveals that the type V toxin domain from B. pseudomallei 1026b is the closest structural homolog of CdiA-CTE479. Although CdiA-CTE479 and CdiA-CT1026b nuclease domains do not share significant sequence identity, they have very similar active-site architectures with catalytic residues contributed by α1, β2, β3, and α2 elements of the PD(D/E)XK core. By contrast, CdiIE479 and CdiI1026b are unrelated in sequence and structure, but each immunity protein inactivates cognate toxin by binding within the nuclease active-site cleft to prevent access to tRNA. To gain insight into substrate specificity, we used molecular docking approaches to model each toxin domain bound to tRNA. The resulting models fit reasonably well into low resolution electron-density envelopes generated by small-angle x-ray scattering analysis of toxin/tRNA complexes. Although most PD (D/E)XK nucleases are DNA-specific phosphodiesterases, our findings indicate that these enzymes are commonly used as toxic RNases in bacterial competition. Because PD(D/E)XK nucleases can be difficult to identify through sequence analyses (23, 25), it is possible that many other uncharacterized CDI toxins also belong to the superfamily.
Results
Structure of the CdiA-CT/CdiIE479 Toxin/Immunity Protein Complex
We previously reported that overexpression of the CdiA-CT/CdiIE479/His6 complex in E. coli cells leads to tRNA degradation and concomitant growth arrest (16). These observations suggest that the expression construct produces insufficient immunity protein to neutralize tRNase activity. Because CdiA-CTE479 activity precludes protein overproduction, we inactivated the toxin with the D285A mutation to allow high level expression of the toxin/immunity protein complex (16). We purified and crystallized the SeMet-labeled complex and used SAD phasing to produce an initial partial model. The model was subsequently improved using molecular replacement with a native dataset, resulting in a final resolution of 2.0 Å (Table 1). The final model includes CdiA-CTE479 residues Arg-201–Lys-316 (numbered from Glu-1 of the ELYN peptide motif), CdiIE479 residues Ala-2–Gly-105, and 155 water molecules. The final Rwork/Rfree (%) was 19.3/23.7 with 98.8% of dihedral angles in favorable regions and the remaining 1.2% within allowed regions as estimated by Ramachandran plot.
TABLE 1.
X-ray diffraction data and atomic refinement for the CdiA-CT·CdiIE479 complex
NA means not applicable.
| Se-SAD dataset | Native dataset | |
|---|---|---|
| Space group | I4 | P22121 |
| Unit cell dimensions (Å) | 117.2 × 117.2 × 111.6 | 54.5 × 73.3 × 110.0 |
| pH of crystallization condition | 6.5 | 7.0 |
| Protein concentration (mg/ml) | 20 | 20 |
| Dataset | ||
| Wavelength, Å | 0.97591 | 0.97591 |
| Resolution range | 50–3.3 | 50–2.0 |
| Unique reflections (total) | 22,290 (329,892) | 30,121 (322,487) |
| Completeness, %a | 100 (100) | 99.24 (99.28) |
| Redundancya | 14.8 (14.9) | 10.7 (10.9) |
| Rmergea,b | 0.159 (0.48) | 0.071 (0.494) |
| Rmeasa,c | 0.157 (0.467) | 0.075 (0.519) |
| Rp.i.m. a,d | 0.041 (0.121) | 0.023 (0.156) |
| CC1/2a | 0.999 (0.993) | 0.998 (0.966) |
| I/σa | 18.03 (6.95) | 29.99 (5.79) |
| Figure of merit | 0.408 | NA |
| No. of selenium sites | 18 | NA |
| NCS copies | 3 | 2 |
| Model refinement | ||
| Resolution range, Å | 43.7–2.0 | |
| No. of reflections (working/free) | 30,094 | |
| No. of protein atoms | 3,382 | |
| No. of water molecules | 155 | |
| Residues in model | CdiA-CT 201–316; CdiI 2–105 | |
| Rwork/Rfree %e | 19.3/23.7 | |
| Room mean square deviations | ||
| Bond lengths, Å | 0.008 | |
| Bond angles | 1.09 | |
| Ramachandran plot | ||
| Most favorable region, % | 98.8 | |
| Additional allowed region, % | 1.2 | |
| Disallowed region | 0 | |
| PDB code | 5J4A | |
a Statistics for the highest resolution shell are given in parentheses.
b Rmerge = ΣhklΣi|Ii(hkl) − (I(hkl))|/ΣhklΣi Ii(hkl).
c Rmeas = Σhkl {N(hkl)/(N(hkl) − 1)}1/2 Σi|Ii(hkl) − (I(hkl))|/ΣhklΣi Ii(hkl).
d Rp.i.m (precision-indicating Rmerge) = Σhkl {1/(N(hkl) − 1)}½ Σi|Ii(hkl) − (I(hkl))|/ΣhklΣi Ii(hkl).
e Rwork = Σ|Fobs − Fcalc|/ΣFobs. Rfree was computed identically except where all reflections belong to a test set of 5% randomly selected data.
Like other CdiA-CT constructs (17, 26, 27), the N-terminal region of CdiA-CTE479 (residues Glu-1–Phe-200) is not resolved in the final model. This unresolved region corresponds to the “translocation” domain, which is postulated to mediate CdiA-CT transport across the cytoplasmic membrane of target bacteria (28). The resolved C-terminal domain corresponds to the tRNase domain responsible for growth inhibition activity (16). The CdiA-CTE479 nuclease domain consists of a five-stranded mixed β-sheet decorated by four α-helices (Fig. 1A). The sheet forms a half β-barrel-like structure with helix α1 running through its central cavity (Fig. 1A). The C-terminal half of helix α2 (α2b) is bent 90° with respect to the N-terminal portion (α2a). The CdiIE479 immunity protein consists of a slightly curved three-stranded antiparallel β-sheet decorated with four α-helices (Fig. 1A). The CdiA-CT/CdiIE479 interface is largely electrostatic with 19 direct salt bridges and hydrogen bonds mediating the interaction (Fig. 1B and Table 2). Helix α2b of the nuclease domain sits in the curve of the CdiIE479 β-sheet. Additionally, helices α1′, α4′, and the β-sheet of CdiIE479 interact with the end of the nuclease β-sheet and the extended loop connecting α2′ and β3′ (Fig. 1B). This protein-protein interface buries 1015 Å2 of surface area, corresponding to 14% of the nuclease domain and 18% of the immunity protein total surface area. In accord with this extensive interaction surface, biolayer interferometry showed that CdiA-CTE479 and CdiIE479 form a relatively high affinity complex with an apparent equilibrium dissociation constant of 72 ± 23 nm (Fig. 2).
FIGURE 1.

Structure of the CdiA-CT/CdiIE479 toxin/immunity protein complex. A, CdiA-CT/CdiIE479 complex is shown as a schematic with secondary structure elements of CdiI indicated with prime symbols. B, CdiA-CT/CdiIE479 complex formation is mediated by electrostatic interactions. Interacting residues are in stick representation with nitrogen and oxygen atoms colored in blue and red (respectively), and bonds are shown as black dashed lines. The view is rotated 180° about the x axis relative to A.
TABLE 2.
Direct intermolecular hydrogen bonds and salt bridges in the CdiA-CT·CdiIE479 complex
| CdiA-CTE479 | CdiIE479 | Distance |
|---|---|---|
| Å | ||
| Hydrogen bonds | ||
| Glu-204(OE2) | Thr-17 (OG1) | 2.39 |
| Gly-225(O) | Gln-19 (NE2) | 3.79 |
| Ser-226(O) | Ser-16 (NE2) | 3.37 |
| Asp-229(OD1) | Thr-17(N) | 2.79 |
| Gln-253(NE2) | Gly-50(O) | 2.98 |
| Asn-267(ND2) | Glu-57 (OE2) | 2.66 |
| Asn-270(ND2) | Lys-9(O) | 3.70 |
| Thr-271(OG1) | Ser-4(OG) | 2.39 |
| His-275(NE2) | Gly-3(O) | 2.62 |
| Salt bridges | ||
| Asp-229(OD2) | Ala-2(N) | 3.83 |
| Lys-263(NZ) | Glu-48 (OE1) | 3.64 |
| Lys-263(NZ) | Glu-48 (OE2) | 2.47 |
| Asp-274(OD1) | Arg-12(NE) | 3.78 |
| Asp-274(OD2) | Arg-12(NE) | 2.82 |
| Asp-274(OD1) | Arg-12 (NH2) | 2.97 |
| Asp-274(OD2) | Arg-12 (NH2) | 3.47 |
FIGURE 2.
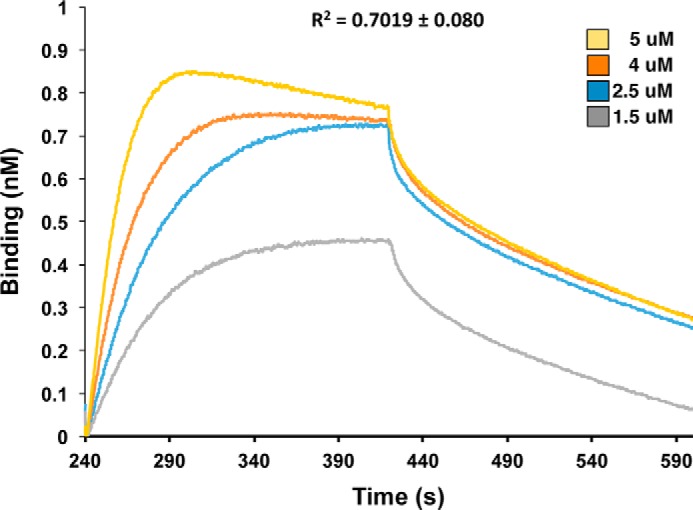
Biolayer interferometry of the CdiA-CT/CdiIE479 binding interaction. Immobilized CdiIE479-His6 was exposed to varying concentrations (1.5–5 μm) of CdiA-CTE479, and the binding interaction and dissociation monitored a wavelength shift (nm). Representative association and dissociation curves are presented with the overall correlation coefficient (R2) shown for the fit.
CdiA-CTE479 Toxin Domain Is a Member of the PD(D/E)XK Superfamily
We used the DALI server (24) to identify proteins with structural similarity to the CdiA-CTE479 nuclease domain. This search revealed that two other CdiA-CT nuclease domains from B. pseudomallei 1026b and Yersinia pseudotuberculosis YPIII exhibit the greatest similarity with CdiA-CTE479 (Table 3). The CdiA-CT1026b and CdiA-CTE479 nuclease domains superimpose with a root-mean-square deviation (r.m.s.d.) of 3.7 Å over 89 of 132 α-carbons, corresponding to a DALI Z-score of 7.0. The CdiA-CTYPIII nuclease domain exhibits comparable structural similarity, although its β4-β5 hairpin element is absent from the CdiA-CTE479 nuclease domain (27). Other proteins identified during this search include two closely related XisH endonucleases from cyanobacteria, another CdiA-CT nuclease domain from Escherichia coli EC869, and the AspBHI restriction endonuclease from Azoarcus sp. BH72 (Table 3). All of these domains share the core structure of the PD(D/E)XK nuclease superfamily, which includes most type II restriction endonucleases and various enzymes involved in DNA recombination and repair. The PD(D/E)XK core domain is a mixed β-sheet flanked by two α-helices with αβββαβ topology. CdiA-CTE479 and CdiA-CT1026b share this core fold, but the CdiA-CTE479 nuclease domain contains an insertion that forms helix α2a (Fig. 3, A and B). We also note that helix α1 from CdiA-CTE479 is significantly shorter than the corresponding helix in the CdiA-CT1026b structure (Fig. 3, A and B).
TABLE 3.
DALI server search results
| Search input | Structural homolog | Organism | PDB code | Z-score | r.m.s.d.a |
|---|---|---|---|---|---|
| Å | |||||
| CdiA-CTE479 | CdiA-CTII1026b | B. pseudomallei 1026b | 4G6V | 7.0 | 3.7 (89/132)b |
| CdiA-CTYPIII | Y. pseudotuberculosis YPIII | 4ZQU | 7.0 | 3.0 (95/124) | |
| XisH | Nostoc punctiforme PCC 73102 | 2INB | 6.4 | 2.7 (87/128) | |
| AVA_3312 | Anabaena variabilis | 2OKF | 6.4 | 2.9 (90/129) | |
| CdiA-CTo11EC869 | E. coli EC869 | 4G6U | 5.8 | 3.3 (96/213) | |
| AspBHI | Azoarcus sp. BH72 | 4OC8 | 5.2 | 3.2 (91/387) | |
| CdiIE479 | NS3 | Rice hoja blanca virus (RHBV) | 3AJF | 5.8 | 2.7 (75/90) |
a r.m.s.d. is root-mean-square deviation.
b Data were aligned over number of α-carbons out of number of residues.
FIGURE 3.

Sequence and structure alignment of CdiA-CTE479 and CdiA-CT1026b nuclease domains. A, sequence alignment of CdiA-CTE479 and CdiA-CT1026b toxins with proposed active-site residues highlighted in red and conserved residues in blue. Secondary structure elements are colored gold and green for CdiA-CTE479 and CdiA-CT1026b, respectively. B, superimposition of CdiA-CTE479 and CdiA-CT1026b nuclease domains. Secondary structure elements that superimpose are color-coded in gold (CdiA-CTE479) and green (CdiA-CT1026b), and those that do not align rendered in white (CdiA-CTE479) and gray (CdiA-CT1026b). C, active site of CdiA-CTE479 and CdiA-CT1026b nuclease domain. Predicted active-site residues are shown in stick representation (nitrogen and oxygen atoms are colored blue and red, respectively).
Identification of the CdiA-CTE479 Active Site
We previously suggested that Asp-280 and Asp-285 of CdiA-CTE479 may function in catalysis, because mutation of these residues abrogated toxicity (16). However, structural alignment with the nuclease domain from CdiA-CT1026b indicates that residues Glu-204, Asp-229, Asp-243, and His-275 of CdiA-CTE479 are more likely to catalyze tRNA cleavage (Fig. 3A). These residues overlay well with active-site residues Glu-187, Asp-214, Asp-223, and Lys-242 of CdiA-CT1026b (Fig. 3C). To test this prediction, we mutated CdiA-CTE479 residues His-275, Asp-243, Asp-229, and Glu-204 to Ala individually and examined the growth inhibition activity of each toxin variant. Induction of wild-type CdiA-CTE479 expression in E. coli cells resulted in immediate growth inhibition (Fig. 4A). By contrast, induction of domains that carry the H275A, D243A, D229A, or E204A mutations had no effect on cell growth (Fig. 4A), consistent with the loss of toxic nuclease activity. We also purified each toxin domain and tested its tRNase activity in vitro. Wild-type CdiA-CTE479 toxin cleaved a large proportion of tRNA molecules as assessed by denaturing PAGE analysis, and this activity was blocked when purified CdiIE479 immunity protein was included in the reaction (Fig. 4B). This tRNase activity was confirmed by Northern blot analysis, which showed cleavage of tRNAGly molecules in the reactions (Fig. 4B). By contrast, none of the other CdiA-CTE479 mutant variants exhibited detectable RNase activity (Fig. 4B). To determine whether the mutations adversely affect toxin structure, we tested whether the refolded CdiA-CTE479 proteins still interact with cognate immunity protein using affinity co-purification. Each mutant toxin co-eluted with CdiIE479-His6 during Ni2+-affinity chromatography (Fig. 4C), indicating that the nuclease domains retain their native fold. Taken together with the crystal structure, these findings suggest that CdiA-CTE479 residues His-275, Asp-243, Asp-229, and Glu-204 participate in catalysis. Because CdiIE479 binds directly over this cluster of residues, the immunity protein presumably neutralizes toxin activity by blocking access to tRNA substrates. We previously showed that CdiA-CTE479 cleaves between residues T54 and Ψ55 in the T-loop of tRNA molecules (16). To test whether post-transcriptional modifications at positions 54 and 55 are required for CdiA-CTE479 activity, we examined toxin activity on unmodified E. coli tRNAAsp and tRNAGln substrates prepared by in vitro transcription. Each substrate was cleaved efficiently by purified nuclease and the activity completely neutralized by CdiIE479 immunity protein (Fig. 4D). Thus, the universal T-loop modifications are not required for CdiA-CTE479-mediated tRNase activity.
FIGURE 4.

CdiA-CTE479 growth inhibition and tRNase activities. A, growth inhibition activity of CdiA-CTE479 variants. The indicated toxins were expressed in E. coli cells from a rhamnose-inducible promoter as described under “Experimental Procedures.” Expression was induced at 0 min, and cell growth was monitored by measuring the optical density at 600 nm (OD600). The curve labeled repressed corresponds to un-induced cells carrying the wild-type CdiA-CTE479 construct. The average ± S.E. from three independent biological replicates is presented. B, in vitro nuclease assays. The indicated CdiA-CTE479 variants were purified and incubated with total E. coli RNA. Reactions were run on denaturing 6% polyacrylamide gels and stained with ethidium bromide. C, mutant CdiA-CTE479 domains bind to CdiIE479 immunity protein. Isolated toxin domains were mixed with purified CdiIE479-His6 and then subjected to Ni2+-affinity chromatography. Lanes labeled input show the protein mixtures loaded onto the column; free lanes show proteins that failed to bind the column, and bound indicates proteins eluted from the column with imidazole. Prior work has shown that CdiA-CTE479 does not bind to Ni2+-NTA-agarose resin (16). D, CdiA-CTE479 cleaves unmodified tRNAs produced by in vitro transcription. E. coli tRNAGln and tRNAAsp transcripts were incubated with purified CdiA-CTE479 and CdiIE479, and reactions were analyzed on denaturing 6% polyacrylamide gels stained with ethidium bromide. Experiments in B–D were repeated twice with essentially identical results. Representative data are shown for each experiment.
Structural Comparison of CdiIE479 and CdiI1026b Immunity Proteins
Although the CdiA-CTE479 and CdiA-CT1026b nuclease domains share a common fold and active site, the corresponding immunity proteins appear to be unrelated. Using iterative PSI-BLAST, we were unable to establish a link between CdiIE479 and CdiI1026b sequences. Moreover, structural superimposition of the two immunity proteins reveals a poor fit between the central β-sheets and misalignments of most α-helical elements (Fig. 5, A and B). CdiIE479 and CdiI1026b align with an r.m.s.d. of 3.42 Å over only 42 of 103 α-carbons (Z-score of 1.8) indicating low structural similarity. We next used the DALI server to search for proteins with structural similarity to CdiIE479. The only hit with a Z-score of >5 was the N-terminal domain of protein NS3 from rice hoja blanca tenuivirus (Table 3). NS3 suppresses RNA interference pathways in host cells by binding to both siRNA and miRNAs (29, 30). Although these proteins superimpose with r.m.s.d. of 2.7 Å over 75 of 90 α-carbons, the NS3 domain is entirely α-helical and lacks the central β-sheet found in CdiIE479.
FIGURE 5.

Sequence and structure comparison of CdiIE479 and CdiI1026b immunity proteins. A, structure-based sequence alignment of CdiIE479 (blue) and CdiI1026b (cyan) with secondary structure elements indicated above and below the sequence alignment. Conserved residues are highlighted in blue. B, superimposition of CdiIE479 and CdiI1026b structures. Secondary elements that partially or fully superimpose are labeled.
CdiA-CT/CdiI Complexes Have Unique Electrostatic Interfaces
Both toxin/immunity complexes interact primarily via electrostatic interactions and shape complementation; and intriguingly, both immunity proteins use the N-terminal α-amino group to form a salt bridge with a catalytic Asp residue in the toxin active site (Table 2) (17). However, the size, charge distribution, shape, and position of these patches differ between the two immunity proteins (Fig. 6). The electrostatic surface map shows that CdiIE479 has a positively charged protrusion (formed from α1′, α2′, and the β-sheet) that is complementary to the negatively charged active-site groove within the CdiA-CTE479 nuclease domain (Fig. 6A). In addition, a small negatively charged patch on CdiIE479 interacts with a positive patch on the cognate toxin. Much like CdiIE479, the CdiI1026b immunity protein inserts a positively charged protrusion (formed from β3′, α3′, and the connecting loop region) into the negatively charged active-site cleft of its cognate toxin. CdiI1026b also contains a negatively charged patch, which complements a positive patch adjacent to the Lys-242 active-site residue within CdiA-CT1026b. Collectively, these electrostatic and shape complementation interactions ensure that each nuclease domain is only neutralized by its cognate immunity protein.
FIGURE 6.
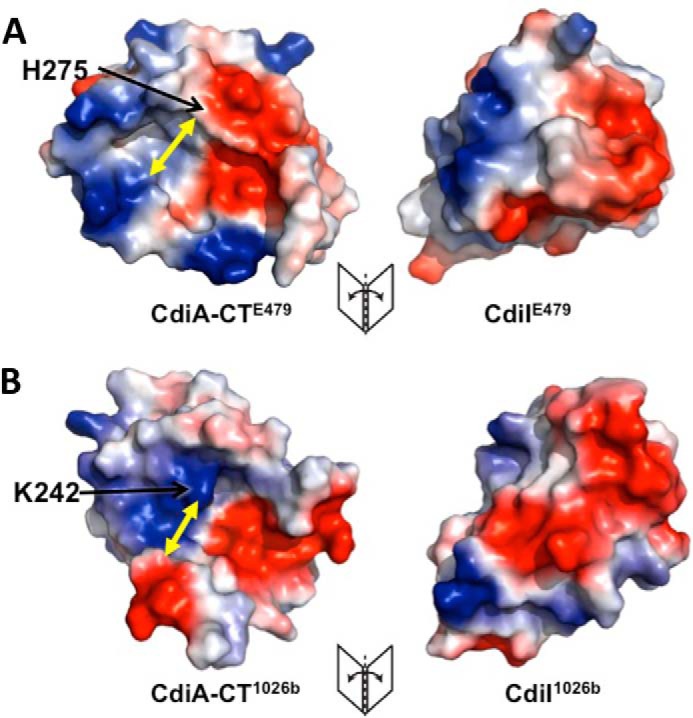
CdiA-CT/CdiIE479 and CdiA-CT/CdiI1026b complexes interact through distinct electrostatic surfaces. A, electrostatic surface map of the CdiA-CT/CdiIE479 complex interface. Negative, positive, and neutral surfaces regions are shown in red, blue, and white, respectively. B, electrostatic surface map of the CdiA-CT/CdiI1026b complex interface. Yellow arrows indicate the minimal width of each active-site pocket.
Nuclease Active-site Architecture and tRNA Specificity
CdiA-CTE479 and CdiA-CT1026b cleave tRNA at distinct positions, and presumably, this specificity is dictated by the shape of the active-site pocket. The CdiA-CTE479 nuclease domain has a slightly larger open pocket with a diameter of 12.6 Å measured from Gln-253 to His-275 (Fig. 6A). By contrast, the active-site cleft of CdiA-CT1026b is narrower with a diameter of 10.4 Å from Glu-187 to Lys-242 (Fig. 6B). To explore how the nuclease domains interact with substrate, we used Hex 8.0 to dock each toxin onto the structure of E. coli tRNACys. We used experimentally determined cleavage sites for each nuclease to guide the docking searches. The models obtained from the simulations show that each nuclease active-site is positioned near the scissile bond in the tRNA substrate (Fig. 7, A and C). The calculated interaction energy for the tRNA/CdiA-CTE479 complex was −802 kcal/mol, and that of the tRNA/CdiA-CT1026b complex was −1,267 kcal/mol. These energies are comparable with those calculated for Hex-assisted docking of each toxin domain with its cognate immunity protein as follows: −953 kcal/mol for CdiA-CT/CdiIE479 complex and −1,199 kcal/mol for the CdiA-CT/CdiI1026b complex. A negative control simulation using CdiIE479 and CysK, which do not interact with one another, yielded a considerably higher energy of −368 kcal/mol. Simulations using non-cognate toxin/immunity protein pairs produced models with lower than anticipated energies (−742 kcal/mol for CdiA-CTE479/CdiI1026b and −626 kcal/mol for CdiA-CT1026b/CdiIE479), but in each instance the solutions did not position the immunity proteins over the nuclease active sites.
FIGURE 7.

Computational modeling and SAXS analysis of tRNA/CdiA-CT complexes. A, CdiA-CT1026b (green schematic) binding with its active-site residues adjacent to the backbone of the tRNACys (PDB code 1B23) amino acceptor stem loop with active-site residues shown as spheres (oxygen and nitrogen atoms colored red and blue, respectively). B, SAXS electron density envelope (white mesh) fitted with the docking solution showing the monomeric CdiA-CT1026b toxin (red schematic) bound to tRNACys (green and blue). C, CdiA-CTE479 (green schematic) binding with its active-site residues adjacent to the backbone of the tRNACys T-loop with active-site residues shown as spheres (oxygen and nitrogen atoms colored red and blue, respectively). D, SAXS electron density envelope (white mesh) fitted with the docking solution showing the tetrameric CdiA-CTE479 toxin bound to four molecules of tRNACys (green and blue).
Small-angle X-ray Scattering (SAXS) Analysis of Toxin/tRNA Complexes
To test the computational docking models, we sought to produce stable tRNA/toxin complexes for structural analysis using SAXS. We overproduced catalytically inactive toxins that carry N-terminal His6 tags and purified the proteins by Ni2+-affinity chromatography. Remarkably, large quantities of endogenous tRNA co-purified with each inactive toxin (Fig. 8A), indicating that the tRNA/toxin complexes are indeed stable. The tRNA/CdiA-CT1026b complex migrated at ∼45 kDa on size-exclusion chromatography, indicative of a 1:1 complex with tRNA in solution (Fig. 8B). By contrast, size-exclusion chromatography showed that the tRNA/CdiA-CTE479 complex is ∼150 kDa (Fig. 8C), suggestive of a higher order complex containing four nuclease domains bound to four tRNA molecules. We used SAXS to generate low resolution electron density envelopes of each nucleoprotein complex. DAMAVER (31) was used to calculate normalized spatial discrepancies (NSD) of 0.979 ± 0.038 for tRNA/CdiA-CT1026b and 0.959 ± 0.081 for the tRNA/CdiA-CTE479 complex, with no restorations rejected. These average NSD values imply reasonable stability of the solutions. The Hex-generated models for each tRNA/toxin complex were then fitted into the respective electron density envelopes using Chimera (Fig. 7, B and D) (32). Supcomb (33) was used to calculate the NSD values between the averaged and filtered shape from SAXS and the structural models of 0.9371 for tRNA/CdiA-CT1026b and 0.9013 for the tRNA/CdiA-CTE479 complex. These NSD values suggest that the average SAXS envelopes and the structural models agree quite well with each other. The four CdiA-CTE479 nuclease domains form a donut-like structure. Each nuclease domain fits helix α3 into the curvature of the β-sheet (strands β1–β3) on the adjacent domain. Interdomain contacts also occur between helix α4 and β5 and the loop connecting strands β2 and β3. Within the tetramer, the four α1 helices are directed toward the center, and the nuclease active sites project outward. The buried surface area of each monomer is consistent with a stable oligomeric state (34). The complex is further stabilized by interactions between tRNA molecules, which pack together with their aminoacyl acceptor stems pointing into the center of the complex. Similar tight packing interactions have been observed in the crystal structure of tRNAAsp (35).
FIGURE 8.

Inactive CdiA-CTE479 and CdiA-CT1026b toxin domains bind to endogenous tRNA. A, agarose gel analysis of catalytically inactive CdiA-CTE479 and CdiA-CT1026b toxins purified under non-denaturing conditions. Control RNA is from yeast (Sigma). B, size-exclusion chromatography of the purified tRNA/CdiA-CT(D214A)1026b complex. C, size-exclusion chromatography of the purified tRNA/CdiA-CT(D243A)E479 complex. Chromatography migration standards are as follows: bovine thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa), and vitamin B12 (1.3 kDa). Each experiment was carried out in triplicate with similar results. Representative data are shown for each experiment.
Discussion
The results presented here demonstrate that the CdiA-CTE479 nuclease domain is a member of the PD(D/E)XK nuclease superfamily. Together with the previously characterized CdiA-CT1026b and CdiA-CTo11EC869 nuclease domains (17, 27), there are at least three CDI toxin classes that share the PD(D/E)XK core fold. The amino acid sequences of these toxins are distinct (15–18% pairwise sequence identity) and show no apparent relationship to one another through iterative PSI-BLAST analyses. However, structural superimposition of the CdiA-CTE479, CdiA-CT1026b, and CdiA-CTo11EC869 toxins reveals significant similarities. The PD(D/E)XK fold consists of a central four-stranded mixed β-sheet flanked by two α-helices with a characteristic α1β1β2β3α2β4 topology. The core structure serves as a scaffold to arrange catalytic residues. The canonical PD(D/E)XK active site found in type II restriction endonucleases is built from a conserved Asp residue at the N terminus of β2 and the (D/E)XK sub-motif within β3 of the core (23, 36). However, there are several variations in the active-site configuration, with catalytic residues migrating to other secondary structure elements during evolution (23, 37, 38). For the CdiA-CTE479 nuclease domain, Asp-229 and Asp-243 occupy canonical positions within β2 and β3, but Glu-204 and His-275 are contributed by α1 and α2, respectively. This arrangement is very similar to the active site of CdiA-CT1026b and the type IIS restriction endonuclease BspD6I (17, 39). The DNase domains of CdiA-CTo11EC869 and CdiA-CTYPIII have yet another configuration that was first described for EcoO109I (17, 27, 40, 41). In these latter enzymes, Glu of the (D/E)XK sub-motif has migrated from β3 to helix α1 to produce an alternative E-PDXXK motif. For the CdiA-CTo11EC869 class of toxins, the active sites use an E(F/Y)DSXK sequence motif (17, 27). Interestingly, these latter DNases contain an additional β-hairpin inserted between α2 and β4 of the PD(D/E)XK core. This β-hairpin constitutes much of the binding interface with the cognate immunity protein, and its sequence varies between family members (27). Analogous insertions into the PD(D/E)XK core have been detected in other superfamily members (23), again underscoring the flexibility of the core fold.
Most PD(D/E)XK enzymes are phosphodiesterases involved in DNA restriction, transposon excision, recombination, and repair. By contrast, there are relatively few family members with RNase activity. EndA/Sen15 tRNA splicing endonucleases were the first PD(D/E)XK enzymes to be implicated in RNA metabolism (23, 42). More recently, Rai1 has been reported to act as a phosphodiesterase to remove 5′-cap structures from eukaryotic mRNAs (43). Our findings show that the PD(D/E)XK fold has been adopted to produce RNases with novel specificities. The CdiA-CTE479 nuclease domain cleaves tRNA between residues T54 and Ψ55 of the conserved TΨC-loop (16). Positions 54 and 55 are modified to thymidine and pseudouracil in eubacterial tRNAs, but these universal post-transcriptional modifications are not required for CdiA-CTE479 activity in vitro. CdiA-CT1026b is a novel RNase that cleaves near the 3′-end of tRNA (16, 17). The computational docking studies reported here represent the first steps toward a detailed understanding of tRNA-binding specificity. Docking of tRNA onto the CdiA-CT1026b domain provides a reasonable model for toxin binding to the aminoacyl-acceptor stem. The interaction between CdiA-CTE479 and substrate appears to be more complicated, and it is unclear why CdiA-CTE479/tRNA complexes oligomerize in solution. It should be noted that CdiA-CTE479 in the absence of tRNA also forms a tetramer (data not shown). Although the models are still vague, it is tempting to speculate that the additional helix α2a within CdiA-CTE479 contributes to T-loop binding specificity. Helix α2a forms a ridge along the lower edge of the putative tRNA-binding surface. Residues Phe-260 and Phe-261, which form a prominent hydrophobic patch adjacent to the nuclease-active site, may participate in substrate binding by stacking onto nucleobases. An elucidation of specific contacts must await high resolution structural studies of toxin/substrate complexes. Given that inactive versions of each nuclease domain bind to tRNA with high affinity, it should be possible to generate specific nucleoprotein complexes for high resolution crystallography.
We have now reported crystal structures for four different CDI toxin classes. As described above, three of these toxins are nucleases of the PD(D/E)XK superfamily. The other toxin, CdiA-CTECL from Enterobacter cloacae ATCC 13047, is an Ntox21 family member and adopts a fold common to barnase, endonuclease poly(U)-specific, colicin E5/D, and RelE (BECR) toxins (14, 26). Sequence analyses by Aravind and co-workers (14, 15) indicate that CDI systems encode several other toxin families with distinct protein folds and activities. However, most CdiA-CT sequences do not have Pfam designations nor predictions for their biochemical activities (3). Given that the CdiA-CTE479, CdiA-CT1026b, and CdiA-CTYPIII toxins were not identified as PD(D/E)XK nucleases by prior computational surveys, it remains possible that other uncharacterized CDI toxins also belong to the superfamily. Because of extreme sequence variability and catalytic residue migration, PD(D/E)XK enzymes are notoriously difficult to identify through computational approaches (23, 25, 38). This problem is compounded by insertions and circular permutations of the core structure (23). Aravind and co-workers (14) recently predicted five new restriction endonuclease-like domains (Tox-REase-2, -3, -5, -7, and -9) that are associated with prokaryotic competition systems. Only the Tox-REase-7 family (Pfam PF15649) is found in CdiA effectors, and these CDI toxins appear to be limited to Pseudomonas and Acinetobacter species. These systems are under considerable positive selection to diversify, presumably due to the competitive advantage obtained with novel toxins. Similar pressures are postulated to drive the impressive diversity of restriction endonucleases, which is the result of the complex interplay between bacteria and their phages (44). Thus, it is not surprising that the versatile PD(D/E)XK core structure has been adopted by CDI and other prokaryotic competition systems.
Experimental Procedures
Plasmid Constructions
Plasmids used in this study are listed in Table 4. Constructs for the overproduction of CdiA-CT/CdiI1026b-His6 (pCH7590), CdiA-CT(D285A)/CdiIE479-His6 (pCH8288), and wild-type CdiA-CT/CdiIE479-His6 (pCH7770) complexes have been described previously (16). Active-site mutations were made in the CdiA-CTE479 nuclease domain using mega-primer PCR. Plasmid pCH7770 was amplified with primer E479-cdiI-Spe-rev (5′-TTT ACT AGT ATT CCC CGA AAC TCC GAG CC) in conjunction with mutagenic forward primers: E479-E204A-for (5′-AAA TTT AGA CCA GGT GCA GCC GGA GCA GCG GC), E479-D229A-for (5′-GGC TCC TCG GTT GCC TTT GTA TTC AGC TCC), E479-D243A-for (5′-AAC GGT AAG ACC GTG GCT TTT ATG CTT ACG CC), and E479-H275A-for (5′-GAA CAC TCT TTC GGA TGC TGC GGC TGC TGC GG). The resulting products were used as mega-primers in subsequent reactions with forward primer E479-Nco-for (5′-CGG CCA TGG CAT CGA ACG TCG AGC TTT AC). The final products were digested with NcoI and SpeI and then ligated to plasmid pET21 to generate mutant CdiA-CT/CdiIE479-His6 expression constructs. These plasmids were used as templates to amplify cdiA-CTE479 coding sequences with primers E479-Nco-for and E479-CT-Xho-rev (5′-GCC ACT CGA GCC TTA CTT GAT CAG AAT AAT C). The products were digested with NcoI and XhoI and then ligated to plasmid pSCRhaB2 (45) to generate l-rhamnose-inducible expression constructs to monitor growth inhibition activities. Plasmid pCH8479 was amplified with oligonucleotides 1026b-Spe-for (5′-ATA ACT AGT GCA TCG AAC GTC GAG C) and 1026b-CT-Xho-rev (5′-AAT CTC GAG TTA ATT CCC CTT TGG), and the resulting fragment was ligated into plasmid pSH21 to generate a construct that overproduces inactive His6-CdiA-CT(D214A)1026b. The cdiA-CT(D285A)E479 coding sequence was amplified from pCH8427 with primers E479-CT-NdeI-H6-for (5′-GAT CAT ATG ATG GGG GCA AGC TCA GGT AGT AAT ATC) and E479-CT-EcoRI-rev (5′-GAT GAA TTC TCA CTT GAT CAG AAT AAT CTT CGC CTG CAG TTT). The product was digested with NdeI/EcoRI and ligated to pET28b. The D243A mutation was introduced via site-directed mutagenesis using primers E479-CT-D243A-for (5′-CGG TAA GAC CGT GGC GTT TAT GCT TAC GCC-3′) and E479-CT-D243A-rev (5′-GGC GTA AGC ATA AAC GCC ACG GTC TTA CCG-3′) to produce an expression construct that overproduces His6-CdiA-CTE479 carrying the D243A and D285A mutations.
TABLE 4.
Bacterial strains and plasmids
| Strains or plasmids | Descriptiona | Ref. or source |
|---|---|---|
| Strain | ||
| BL21 (DE3) | F′ ompT gal dcm lon hsdSB (rB− mB−) | Novagen |
| X90 | F′ lacIq lac′ pro′/ara Δ(lac-pro) nal1 argE(amb) rifr thi-1, RifR | 61 |
| CH2016 | X90 (DE3) Δrna ΔslyD::kan, RifR KanR | 51 |
| Plasmids | ||
| pET21b | Isopropyl 1-thio-β-d-galactopyranoside-inducible T7 RNA polymerase expression vector, AmpR | Novagen |
| pSCRhaB2 | Rhamnose-inducible expression vector, TpR | 45 |
| pTrc99A | Isopropyl 1-thio-β-d-galactopyranoside-inducible expression vector, AmpR | GE Healthcare |
| pCH7590 | pET21::cdiA-CT (G123)-cdiIII1026b, AmpR | 16 |
| pCH7770 | pET21::cdiA-CT-cdiIE479, AmpR | 16 |
| pCH8479 | pET21::cdiA-CT (G123/D214A)-cdiIII1026b, AmpR | 16 |
| pCH7913 | pET21::cdiIE479, AmpR | 16 |
| pCH8288 | pET21::cdiA-CT (D285A)-cdiIE479, AmpR | 16 |
| pCH8427 | pET21::cdiA-CT (G157/D285A)-cdiIE479, AmpR | 16 |
| pPJ100 | pET28::his6-cdiA-CT (G157/D243A/D285A)E479, AmpR | This study |
| pCH10115 | pET21::his6-cdiA-CT (G123/D214A)1026b, AmpR | This study |
| pCH11617 | pET21::cdiA-CT (D229A)-cdiIE479, AmpR | This study |
| pCH11618 | pET21::cdiA-CT (H275A)-cdiIE479, AmpR | This study |
| pCH11619 | pET21::cdiA-CT (D243A)-cdiIE479, AmpR | This study |
| pCH11620 | pET21::cdiA-CT (E204A)-cdiIE479, AmpR | This study |
| pCH11648 | pSCRhaB::cdiA-CT (D229A)E479, TpR | This study |
| pCH11649 | pSCRhaB::cdiA-CT (H275A)E479, TpR | This study |
| pCH11650 | pSCRhaB::cdiA-CT (D243A)E479, TpR | This study |
| pCH11651 | pSCRhaB::cdiA-CT (E204A)E479, TpR | This study |
| pCH11669 | pSCRhaB::cdiA-CTE479, TpR | This study |
a The abbreviations used are as follows: AmpR, ampicillin-resistant; KanR, kanamycin-resistant; RifR, rifampicin-resistant; TpR, trimethoprim-resistant.
Protein Overexpression and Purification
CdiA-CTE479 (from residue Gly-157, numbered from Glu-1 of the ELYN motif) was co-expressed with CdiIE479-His6 and overproduced in E. coli BL21 (DE3) cells grown aerobically at 37 °C in LB medium supplemented with 50 μg/ml ampicillin. Protein expression was induced by addition of isopropyl β-d-thiogalactoside to 1 mm final concentration once the culture reached an absorbance at 600 nm (A600) of ∼0.8. Induced cells were incubated for 4 h and then harvested by centrifugation at 5,100 × g for 20 min. The cell pellet was resuspended in 20 mm sodium phosphate (pH 7.0), 200 mm NaCl supplemented with 10 mg/ml lysozyme, and 1 mm phenylmethylsulfonyl fluoride (PMSF), and the cells were broken by sonication. The lysate was clarified by centrifugation at 14,000 × g for 30 min, and the soluble fraction passed through a 0.22-μm filter before loading onto a Ni2+-charged Hi-trap column (GE Healthcare). The column was washed with 20 mm sodium phosphate (pH 7.0), 200 mm NaCl, 15 mm imidazole, and the CdiA-CT/CdiIE479-His6 complex eluted with a linear gradient of 15–250 mm imidazole. The purified complex was concentrated with a 10-kDa centrifugal concentrator and then run on a Superdex 200 size-exclusion column equilibrated in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl. SeMet-labeled proteins were overproduced in E. coli BL21 (DE3) cells grown in M9 minimal medium supplemented with l-leucine, l-isoleucine, and l-valine at 50 mg/liter; l-phenylalanine, l-lysine, and l-threonine at 100 mg/liter; and SeMet at 75 mg/liter as described (46). The SeMet-labeled CdiA-CT/CdiIE479-His6 complex was purified as described above.
Crystallization and Structure Determination
CdiA-CT/CdiIE479 was crystallized by hanging drop-vapor diffusion against a 1-ml reservoir of crystallization buffer (0.1 m HEPES (pH 7.0), 20 mm MgCl2, 30% (w/v) polyacrylic acid). Polyacrylic acid (Sigma) with average molecular mass of 5,100 Da was prepared as a 50% (w/v) solution, filtered, and used to facilitate crystallization. Hanging drops were prepared from a 1:1 (v/v) mixture of protein solution (20 mg/ml) and crystallization buffer supplemented with 20 μg/ml chymotrypsin. Crystals were soaked in cryo-protectant solution containing 1:1 mixture of 40% (v/v) glycerol and crystallization buffer and then collected by flash freezing. A native dataset was acquired at 70 K at 0.97591 Å on beamline 7-1 at Stanford Synchrotron Radiation Lightsource. Data were processed using HKL2000 (47), resulting in a 99.24% complete dataset to 2.0 Å resolution. The CdiA-CT/CdiIE479 complex crystallized in space group P22121 with two complexes per asymmetric unit and unit cell dimensions of 54.5 × 73.3 × 110.0 Å. Diffraction data were initially indexed and scaled to P2221; however, the best solution obtained by molecular replacement using Phaser in the PHENIX suite was P22121. The SeMet-labeled complex was crystallized using 0.1 m MES (pH 6.5), 0.01 m ZnCl2, 15% (w/v) PEG-6000 as the buffer. SeMet-labeled CdiA-CT/CdiIE479 crystallized in space group I4 with three complexes per asymmetric unit and unit cell dimensions of 117.2 × 117.2 × 111.6 Å. A SAD was collected at 70 K at 0.97591 Å on beamline 7-1 at Stanford Synchrotron Radiation Lightsource. Data were processed using HKL2000, yielding a 99.9% complete dataset to 3.3 Å resolution. We used Autosol in the PHENIX suite (48) to detect 18 selenium atoms with a figure of merit of 0.408 and overall score of 36.2 ± 14.2. Autosol also built a partial model consisting of 416 residues with an Rwork/Rfree (%) of 38.4/44.9. This model showed little secondary structure except for α1 of CdiA-CTE479 and α2′ and α3′ of CdiIE479. Molecular replacement was carried out with Phaser in the PHENIX suite using the partial model together with higher resolution data from native crystals. The Phaser-generated model was then subjected to Autobuild and phenix.refine (48). The final model includes residues Arg-201–Lys-316 of CdiA-CTE479 and residues Ala-2–Gly-105 of CdiIE479 with a final Rwork/Rfree (%) 19.3/23.7. The Ramachandran plot shows 98.8% in the favorable allowed regions and the other 1.2% in the allowed regions. Data collection and refinement statistics are presented in Table 1. Intermolecular hydrogen bonds and salt bridges were determined using PDBePISA (49).
Toxin Immunity Protein Binding Kinetics
The apparent equilibrium dissociation constant for the CdiA-CT/CdiIE479 complex was determined by biolayer interferometry using a BLitz instrument (ForteBio) (50). CdiA-CTE479 was separated from CdiIE479-His6 by Ni2+-affinity chromatography under denaturing conditions. The isolated toxin was refolded by dialysis and run on a Superdex S200 size-exclusion column. CdiIE479-His6 was purified by Ni2+-affinity chromatography as described previously (16). CdiIE479-His6 was loaded onto a Ni2+-NTA-coated biosensor in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl at 25 °C. Sensor-bound immunity protein was then incubated with 1.5–5 μm CdiA-CTE479 toxin for 180 s. The sensor was then washed with buffer, and toxin dissociation was monitored over 180 s. Curve fitting was run following reference subtraction using the BLitz Pro Software to calculate dissociation constants. Local fit analyses were performed for individual association-dissociation curves, followed by averaging to obtain the final apparent Kd value and standard deviation.
Growth Inhibition Assays
E. coli X90 cells harboring rhamnose-inducible CdiA-CTE479 expression plasmids (Table 4) were grown to mid-log phase in LB media supplemented with 100 μg/ml trimethoprim. Cells were then diluted to A600 = 0.05 in fresh LB media supplemented with 100 μg/ml trimethoprim and either 0.4% d-glucose to repress or 0.4% l-rhamnose to induce CdiA-CTE479 expression. Cultures were incubated with shaking at 37 °C, and cell growth was monitored by measuring the A600 every 30 min.
In Vitro tRNase Assays
Purified CdiA-CT/CdiIE479-His6 complexes were denatured in binding buffer supplemented with 6 m guanidine-HCl and CdiA-CTE479 isolated from the void volume during Ni2+-affinity chromatography (1). Toxins were refolded by dialysis against binding buffer, and all purified proteins were quantified by absorbance at 280 nm. Total RNA was isolated from E. coli X90 cells as described (51) and used as a substrate for in vitro nuclease assays. E. coli RNA (5 μg) was incubated with CdiA-CTE479 variants (5 μm) in reaction buffer (20 mm Tris-HCl (pH 7.5), 100 mm NaCl, 10 mm MgCl2, 0.2 mg/ml BSA) for 1 h at 37 °C. Where indicated, CdiIE479 was included at 17.5 μm final concentration. tRNAAsp and tRNAGln substrates were generated by in vitro transcription using RNA polymerase from bacteriophage T7 RNA. The tRNAAsp template was prepared with oligonucleotides 5′-tRNA-Asp (5′-AAT TCC TGC AGT AAT ACG ACT CAC TAT AGG AGC GGT AGT TCA GTC GGT TAG AAT ACC TG) and 3′-tRNA-Asp (5′-TGG CGG AAC GGA CGG GAC TCG AAC CCG CGA CCC CCT GCG TGA CAG GCA GGT ATT CTA AC), and the tRNAGln template with oligonucleotides 5′-tRNA-Gln (5′-AAT TCC TGC AGT AAT ACG ACT CAC TAT AGG GGG TAT AGG GGG TAT CGC CAA GCG GTA AGG CAC CGG) and 3′-tRNA-Gln (5′-TGG CTG GGG TAC GAG GAT TCG AAC CTC GGA ATG CCG GAA TCA GAA TCC GGT GCC TT). Annealed oligonucleotides were end-filled with Klenow fragment of DNA polymerase I. Templates were incubated with T7 RNA polymerase, 2 mm NTP, 10 mm dithiothreitol, and 10 mm MgCl2 for 3 h at 37 °C. Template DNA was removed with RNase-free DNase I, and the transcripts purified with the Direct-zol RNA MiniPrep kit (Genesee Scientific). Nuclease reactions were analyzed by denaturing electrophoresis on 50% urea, 6% polyacrylamide gels in 1× Tris borate-EDTA (TBE) buffer. Gels were stained with ethidium bromide or transferred to nylon membrane for Northern blot hybridization with 5′-radiolabeled oligonucleotide glyV probe (5′-CTT GGC AAG GTC GTG CT) as described (16, 51).
Molecular Docking
Hex 8.0 (52–54) was used to dock CdiA-CT nuclease domains onto tRNA to generate models of enzyme/substrate complexes. CdiA-CT1026b and CdiA-CTE479 nuclease domains were docked onto the structure of E. coli tRNACys (PDB code 1B23) (55). The active site of each nuclease was positioned adjacent to the known scissile bond and the origin set to sample multiple orientations in search of the low energy interactions. Positive control docking simulations were performed using cognate and non-cognate toxin/immunity proteins pairs from B. pseudomallei E479 and 1026b. As a negative control, CdiIE479 and CysK from Salmonella typhimurium LT2 (PDB code 1OAS (56)) were docked onto one another.
SAXS
Inactive CdiA-CT(D243A/D285A)E479 and CdiA-CT(D214A)1026b (17) toxins carrying N-terminal His6 epitope tags were purified by Ni2+-affinity chromatography under non-denaturing conditions. Under these conditions, endogenous tRNA co-purifies with the inactive nuclease domains. Toxin/tRNA complexes were exchanged into 20 mm sodium phosphate (pH 7.4), 150 mm NaCl using a Superdex S200 size-exclusion column and diluted to several concentrations ranging from 0.5 to 5 mg/ml for SAXS analysis. SAXS data were collected on SIBYLS beamline 12.3.1 at the Advanced Light Source using a Pilatus3 2 M detector with exposure times of 0.5, 1, 2, and 4 s. Buffer subtracted data were analyzed using PRIMUS (31), following modification with GNOM (57); P(r) output files with dmax of 91 and 190 for tRNA/CdiA-CT1026b and tRNA/CdiA-CTE479 complexes (respectively) were used to generate electron density envelopes via GASBOR (58). Density envelopes (12 per tRNA/toxin complex) were averaged using DAMAVER (31), and docking solutions were fitted into the final envelopes using Chimera (32) and Crysol (59). SAXS parameters and statistics are provided in Table 5 according to (60). Crysol outputs, together with Guinier, Kratky and P(r) plots, are presented in Fig. 9.
TABLE 5.
Data collection and scattering-derived parameters for tRNA·CdiA-CT complexes
| Data collection parameters | tRNA/CdiA-CT1026b | tRNA/CdiA-CTE479 |
| Beam line | ALS 12.3.1 | ALS 12.3.1 |
| Wavelength (Å) | 1.0 | 1.0 |
| q range (Å−1) | 0.012–0.324 | 0.012–0.324 |
| Exposure time (seconds) | 0.5, 1, 2, 4 | 0.5, 1, 2, 4 |
| Concentration range (mg ml−1) | 0.5–5 | 2–10 |
| Temperature (K) | 283 | 283 |
| Structural parametersa | ||
| I(0) (cm−1) (from P(r)) | 248.1 ± 13.2 | 595.9 ± 13.4 |
| Rg (Å) (from P(r)) | 27.3 ± 1.3 | 46.9 ± 3.4 |
| I(0) (cm−1) (from Guinier) | 246.8 ± 14.9 | 590.6 ± 20.8 |
| Rg (Å) (from Guinier) | 27.1 ± 1.7 | 46.1 ± 4.0 |
| Dmax (Å)b | 91 | 190 |
| Porod volume estimate (Å3) | 43,041 ± 2,353 | 256,350 ± 2,517 |
| Dry volume calculated from Crysol14 (Å3)c | 45,510 | 43,112 (176,000)d |
| Molecular mass determinationa | ||
| Molecular mass (from Primus) (Da) | 41,539 ± 1,675 | 16,5473 ± 6,083 |
| Calculated monomeric mass from sequence (Da)c | 43,215 | 41,827 (167,308) |
a Data are reported for 5 mg ml−1 measurements.
b Dmax is a model parameter in the P(r) calculation.
c Values for tetramer are shown in parentheses.
d The estimated volume of the tRNA·CdiA-CTE479 complex is greater than the calculated volume from sequence due to the void volume assumed by the tetramer.
FIGURE 9.

SAXS analyses of tRNA/CdiA-CT complexes. Plots for tRNA/CdiA-CT1026b (I) and tRNA/CdiA-CTE479 (II) SAXS data. A, log I(q) versus q plot with experimental SAXS profile shown in blue and the corresponding structural model fitted data via Crysol (41) shown in green. B, Guinier plots. C, Kratky plots. D, P(r) plots.
Author Contributions
P. M. J., G. C. G., F. G.-S., T. W., L.-W. H., C. S. H., and C.W.G. conceived and designed experiments and analyzed data; P. M. J., G. C. G., F. G.-S., T. W., and L.-W. H. performed the experiments; P. M. J., G. C. G., F. G.-S., T. W., L.-W. H., C. S. H., and C. W. G. wrote the paper.
Acknowledgments
We thank the Stanford Synchrotron Radiation Lightsource for their invaluable help in data collection. We thank the beam line staff members at the beam lines 12.3.1 (BL12.3.1) at the Advanced Light Source for their help in SAXS data collection. The Advanced Light Source is operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies program, supported by Department of Energy, Office of Biological and Environmental Research.
This work was supported in part by National Institutes of Health Grants AI099687 and GM102318 (to C. W. G. and C. S. H.) and National Institutes of Health Project MINOS Grant GM105404. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5J4A) have been deposited in the Protein Data Bank (http://www.pdb.org/).
- CDI
- contact-dependent growth inhibition
- r.m.s.d.
- root-mean-square deviation
- SAXS
- small-angle x-ray scattering
- SeMet
- selenomethionine
- SAD
- single-wavelength anomalous dataset
- NSD
- normalized spatial discrepancy
- for
- forward
- rev
- reverse
- PDB
- Protein Data Bank.
References
- 1. Aoki S. K., Diner E. J., de Roodenbeke C. T., Burgess B. R., Poole S. J., Braaten B. A., Jones A. M., Webb J. S., Hayes C. S., Cotter P. A., and Low D. A. (2010) A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468, 439–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruhe Z. C., Low D. A., and Hayes C. S. (2013) Bacterial contact-dependent growth inhibition. Trends Microbiol. 21, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Willett J. L., Ruhe Z. C., Goulding C. W., Low D. A., and Hayes C. S. (2015) Contact-dependent growth inhibition (CDI) and CdiB/CdiA two-partner secretion proteins. J. Mol. Biol. 427, 3754–3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Makhov A. M., Hannah J. H., Brennan M. J., Trus B. L., Kocsis E., Conway J. F., Wingfield P. T., Simon M. N., and Steven A. C. (1994) Filamentous hemagglutinin of Bordetella pertussis. A bacterial adhesin formed as a 50-nm monomeric rigid rod based on a 19-residue repeat motif rich in β strands and turns. J. Mol. Biol. 241, 110–124 [DOI] [PubMed] [Google Scholar]
- 5. Kajava A. V., Cheng N., Cleaver R., Kessel M., Simon M. N., Willery E., Jacob-Dubuisson F., Locht C., and Steven A. C. (2001) β-Helix model for the filamentous haemagglutinin adhesin of Bordetella pertussis and related bacterial secretory proteins. Mol. Microbiol. 42, 279–292 [DOI] [PubMed] [Google Scholar]
- 6. Aoki S. K., Malinverni J. C., Jacoby K., Thomas B., Pamma R., Trinh B. N., Remers S., Webb J., Braaten B. A., Silhavy T. J., and Low D. A. (2008) Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol. Microbiol. 70, 323–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruhe Z. C., Townsley L., Wallace A. B., King A., Van der Woude M. W., Low D. A., Yildiz F. H., and Hayes C. S. (2015) CdiA promotes receptor-independent intercellular adhesion. Mol. Microbiol. 98, 175–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruhe Z. C., Wallace A. B., Low D. A., and Hayes C. S. (2013) Receptor polymorphism restricts contact-dependent growth inhibition to members of the same species. mBio 4, e00480–e00413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aoki S. K., Pamma R., Hernday A. D., Bickham J. E., Braaten B. A., and Low D. A. (2005) Contact-dependent inhibition of growth in Escherichia coli. Science 309, 1245–1248 [DOI] [PubMed] [Google Scholar]
- 10. Aoki S. K., Poole S. J., Hayes C. S., and Low D. A. (2011) Toxin on a stick: modular CDI toxin delivery systems play roles in bacterial competition. Virulence 2, 356–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayes C. S., Aoki S. K., and Low D. A. (2010) Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44, 71–90 [DOI] [PubMed] [Google Scholar]
- 12. Poole S. J., Diner E. J., Aoki S. K., Braaten B. A., t'Kint de Roodenbeke C., Low D. A., and Hayes C. S. (2011) Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 7, e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Webb J. S., Nikolakakis K. C., Willett J. L., Aoki S. K., Hayes C. S., and Low D. A. (2013) Delivery of CdiA nuclease toxins into target cells during contact-dependent growth inhibition. PLoS ONE 8, e57609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang D., de Souza R. F., Anantharaman V., Iyer L. M., and Aravind L. (2012) Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Direct. 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang D., Iyer L. M., and Aravind L. (2011) A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 39, 4532–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nikolakakis K., Amber S., Wilbur J. S., Diner E. J., Aoki S. K., Poole S. J., Tuanyok A., Keim P. S., Peacock S., Hayes C. S., and Low D. A. (2012) The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol. Microbiol. 84, 516–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morse R. P., Nikolakakis K. C., Willett J. L., Gerrick E., Low D. A., Hayes C. S., and Goulding C. W. (2012) Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc. Natl. Acad. Sci. U.S.A. 109, 21480–21485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lazar Adler N. R., Govan B., Cullinane M., Harper M., Adler B., and Boyce J. D. (2009) The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS Microbiol. Rev. 33, 1079–1099 [DOI] [PubMed] [Google Scholar]
- 19. Limmathurotsakul D., and Peacock S. J. (2011) Melioidosis: a clinical overview. Br. Med. Bull. 99, 125–139 [DOI] [PubMed] [Google Scholar]
- 20. Chantratita N., Wuthiekanun V., Limmathurotsakul D., Vesaratchavest M., Thanwisai A., Amornchai P., Tumapa S., Feil E. J., Day N. P., and Peacock S. J. (2008) Genetic diversity and microevolution of Burkholderia pseudomallei in the environment. PLoS Negl. Trop. Dis. 2, e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ngamdee W., Tandhavanant S., Wikraiphat C., Reamtong O., Wuthiekanun V., Salje J., Low D. A., Peacock S. J., and Chantratita N. (2015) Competition between Burkholderia pseudomallei and B. thailandensis. BMC Microbiol. 15, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogawa T., Tomita K., Ueda T., Watanabe K., Uozumi T., and Masaki H. (1999) A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 283, 2097–2100 [DOI] [PubMed] [Google Scholar]
- 23. Steczkiewicz K., Muszewska A., Knizewski L., Rychlewski L., and Ginalski K. (2012) Sequence, structure and functional diversity of PD(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 40, 7016–7045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kosinski J., Feder M., and Bujnicki J. M. (2005) The PD(D/E)XK superfamily revisited: identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function. BMC Bioinformatics 6, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beck C. M., Morse R. P., Cunningham D. A., Iniguez A., Low D. A., Goulding C. W., and Hayes C. S. (2014) CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure 22, 707–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morse R. P., Willett J. L., Johnson P. M., Zheng J., Credali A., Iniguez A., Nowick J. S., Hayes C. S., and Goulding C. W. (2015) Diversification of β-augmentation interactions between CDI toxin/immunity proteins. J. Mol. Biol. 427, 3766–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Willett J. L., Gucinski G. C., Fatherree J. P., Low D. A., and Hayes C. S. (2015) Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc. Natl. Acad. Sci. U.S.A. 112, 11341–11346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang X., Tan S. H., Teh Y. J., and Yuan Y. A. (2011) Structural implications into dsRNA binding and RNA silencing suppression by NS3 protein of Rice Hoja Blanca Tenuivirus. RNA 17, 903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hemmes H., Lakatos L., Goldbach R., Burgyán J., and Prins M. (2007) The NS3 protein of Rice hoja blanca tenuivirus suppresses RNA silencing in plant and insect hosts by efficiently binding both siRNAs and miRNAs. RNA 13, 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 34, 33–41 [Google Scholar]
- 34. Nooren I. M., and Thornton J. M. (2003) Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 325, 991–1018 [DOI] [PubMed] [Google Scholar]
- 35. Moras D., Comarmond M. B., Fischer J., Weiss R., Thierry J. C., Ebel J. P., and Giegé R. (1980) Crystal structure of yeast tRNAAsp. Nature 288, 669–674 [DOI] [PubMed] [Google Scholar]
- 36. Pingoud A., and Jeltsch A. (2001) Structure and function of type II restriction endonucleases. Nucleic Acids Res. 29, 3705–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Knizewski L., Kinch L. N., Grishin N. V., Rychlewski L., and Ginalski K. (2007) Realm of PD(D/E)XK nuclease superfamily revisited: detection of novel families with modified transitive meta profile searches. BMC Struct. Biol. 7, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gupta R., Capalash N., and Sharma P. (2012) Restriction endonucleases: natural and directed evolution. Appl. Microbiol. Biotechnol. 94, 583–599 [DOI] [PubMed] [Google Scholar]
- 39. Kachalova G. S., Rogulin E. A., Yunusova A. K., Artyukh R. I., Perevyazova T. A., Matvienko N. I., Zheleznaya L. A., and Bartunik H. D. (2008) Structural analysis of the heterodimeric type IIS restriction endonuclease R. BspD6I acting as a complex between a monomeric site-specific nickase and a catalytic subunit. J. Mol. Biol. 384, 489–502 [DOI] [PubMed] [Google Scholar]
- 40. Hashimoto H., Shimizu T., Imasaki T., Kato M., Shichijo N., Kita K., and Sato M. (2005) Crystal structures of type II restriction endonuclease EcoO109I and its complex with cognate DNA. J. Biol. Chem. 280, 5605–5610 [DOI] [PubMed] [Google Scholar]
- 41. Feder M., and Bujnicki J. M. (2005) Identification of a new family of putative PD(D/E)XK nucleases with unusual phylogenomic distribution and a new type of the active site. BMC Genomics 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bujnicki J. M., and Rychlewski L. (2001) Unusual evolutionary history of the tRNA splicing endonuclease EndA: relationship to the LAGLIDADG and PD(D/E)XK deoxyribonucleases. Protein Sci. 10, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jiao X., Xiang S., Oh C., Martin C. E., Tong L., and Kiledjian M. (2010) Identification of a quality-control mechanism for mRNA 5′-end capping. Nature 467, 608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stern A., and Sorek R. (2011) The phage-host arms race: shaping the evolution of microbes. BioEssays 33, 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cardona S. T., and Valvano M. A. (2005) An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid 54, 219–228 [DOI] [PubMed] [Google Scholar]
- 46. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., and Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 [DOI] [PubMed] [Google Scholar]
- 47. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 48. Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., and Hung L. W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 50. Concepcion J., Witte K., Wartchow C., Choo S., Yao D., Persson H., Wei J., Li P., Heidecker B., Ma W., Varma R., Zhao L. S., Perillat D., Carricato G., Recknor M., et al. (2009) Label-free detection of biomolecular interactions using BioLayer interferometry for kinetic characterization. Comb. Chem. High Throughput Screen. 12, 791–800 [DOI] [PubMed] [Google Scholar]
- 51. Garza-Sánchez F., Janssen B. D., and Hayes C. S. (2006) Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J. Biol. Chem. 281, 34258–34268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ritchie D. W. (2003) Evaluation of protein docking predictions using Hex 3.1 in CAPRI rounds 1 and 2. Proteins 52, 98–106 [DOI] [PubMed] [Google Scholar]
- 53. Ritchie D. W., and Kemp G. J. (2000) Protein docking using spherical polar Fourier correlations. Proteins 39, 178–194 [PubMed] [Google Scholar]
- 54. Ritchie D. W., and Venkatraman V. (2010) Ultra-fast FFT protein docking on graphics processors. Bioinformatics 26, 2398–2405 [DOI] [PubMed] [Google Scholar]
- 55. Nissen P., Thirup S., Kjeldgaard M., and Nyborg J. (1999) The crystal structure of Cys-tRNACys-EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Structure 7, 143–156 [DOI] [PubMed] [Google Scholar]
- 56. Burkhard P., Rao G. S., Hohenester E., Schnackerz K. D., Cook P. F., and Jansonius J. N. (1998) Three-dimensional structure of O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 283, 121–133 [DOI] [PubMed] [Google Scholar]
- 57. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 58. Svergun D. I., Petoukhov M. V., and Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stovgaard K., Andreetta C., Ferkinghoff-Borg J., and Hamelryck T. (2010) Calculation of accurate small angle x-ray scattering curves from coarse-grained protein models. BMC Bioinformatics 11, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jacques D. A., Guss J. M., Svergun D. I., and Trewhella J. (2012) Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr. D Biol. Crystallogr. 68, 620–626 [DOI] [PubMed] [Google Scholar]
- 61. Beckwith J. R., and Signer E. R. (1966) Transposition of the lac region of Escherichia coli. I. Inversion of the lac operon and transduction of lac by phi80. J. Mol. Biol. 19, 254–265 [DOI] [PubMed] [Google Scholar]