

Peptide lattices were designed using computational methods and controlled by tuning self-assembly solution conditions.
Keywords: Peptide, self-assembly, computational design, de novo design, nanomaterials, soft matter
Abstract
Folded peptides present complex exterior surfaces specified by their amino acid sequences, and the control of these surfaces offers high-precision routes to self-assembling materials. The complexity of peptide structure and the subtlety of noncovalent interactions make the design of predetermined nanostructures difficult. Computational methods can facilitate this design and are used here to determine 29-residue peptides that form tetrahelical bundles that, in turn, serve as building blocks for lattice-forming materials. Four distinct assemblies were engineered. Peptide bundle exterior amino acids were designed in the context of three different interbundle lattices in addition to one design to produce bundles isolated in solution. Solution assembly produced three different types of lattice-forming materials that exhibited varying degrees of agreement with the chosen lattices used in the design of each sequence. Transmission electron microscopy revealed the nanostructure of the sheetlike nanomaterials. In contrast, the peptide sequence designed to form isolated, soluble, tetrameric bundles remained dispersed and did not form any higher-order assembled nanostructure. Small-angle neutron scattering confirmed the formation of soluble bundles with the designed size. In the lattice-forming nanostructures, the solution assembly process is robust with respect to variation of solution conditions (pH and temperature) and covalent modification of the computationally designed peptides. Solution conditions can be used to control micrometer-scale morphology of the assemblies. The findings illustrate that, with careful control of molecular structure and solution conditions, a single peptide motif can be versatile enough to yield a wide range of self-assembled lattice morphologies across many length scales (1 to 1000 nm).
INTRODUCTION
Self-assembly of designed molecules in solution provides marked potential for efficiently achieving complex, robust materials with nanometer precision. Traditional nanomaterial assembly strategies have used small molecules (1, 2) or polymeric (3–5) amphiphiles. Recently developed assembly methods can produce complexity in structure and composition through chemical variation of the assembling molecules (6–9) or the use of hierarchical solution assembly protocols (10–13). Biopolymers offer unique capabilities to encode both local molecular building block structure and long-range material morphology via the design of specific sequences; this design has been applied to DNA (14, 15), polypeptides (16–22), and polymer-biomolecule hybrids (14, 23). Solution assembly of peptides can readily produce “one-dimensional” nanostructures such as fibrils (24–26) and tubes (27–29). New peptide nanostructure formation strategies have used nonnatural peptide sequences (19) and biomimetic strategies using modified natural proteins (30, 31). Much of the peptide work involves the synthesis of new systems and subsequent characterization of the structures they form. However, the a priori design of proteins and peptides that form targeted assemblies is subtle because of the complexities and subtleties of folding and protein-protein interactions. Moreover, these assemblies can be highly sensitive to sequence and mutation. These difficulties have motivated the experimental use of interactions that are more easily programmed at protein interfaces, such as metal coordination (32), to drive intermolecular assembly.
Theoretical and computational methods provide a way to approach the design of intermolecular, noncovalent interactions between self-assembling peptides or proteins in solution to produce materials with predetermined morphologies, including desired point and space-group symmetries (33–36). Nearly all these efforts in assembly design have used variants of natural proteins as building blocks, and different tertiary and quaternary structures are often used for different local geometries in the assembly. Here, we present the computational de novo design of peptides that are robust, easily synthesized, and versatile.
Our aim is to explore the extent to which the information required for folding and intermolecular long-range order can be designed de novo into short peptide sequences, as opposed to the redesign of large natural proteins. The effort is focused on (i) the de novo design of a homotetrameric helix bundle motif that is robust with respect to variation of exterior residues; (ii) the design of the exterior residues to guide the solution assembly of variants of this motif into distinct lattices having rectangular, square, or hexagonal local symmetries; (iii) the experimental characterization and determination of the extent to which the nanostructures are robust with respect to solution-phase conditions; and (iv) the exploration of how solution conditions can be used to control micrometer-scale morphology.
RESULTS
The designed building block motif consisted of a helical homotetramer of four 29-residue peptides arranged with D2 symmetry (Fig. 1). Although many similar oligomeric helical proteins have been designed and investigated, we seek a structure that is robust with respect to variation of exterior residues, and we opt to design the structure and the hydrophobic core de novo. Candidate bundles were generated via a multiparameter mathematical model of helical coiled coils, with the final bundle structure specified by a set of five defining geometric parameters (see the Supplementary Materials). For each candidate bundle motif, a probabilistic approach was applied to calculate the site-specific probabilities of the amino acids at variable residues (33). The calculations also yield an average energy, E, over sequence probabilities for a given bundle structure (33). E was used as an objective function in a Monte Carlo search over helix bundle parameters. A helical peptide structure and 11 interior hydrophobic residues were specified (highlighted in gray in Table 1), providing the tetrameric helix bundle motif, or building block, for subsequent design of the material assemblies. The remaining 18 residues were designed in the context of four predetermined material nanostructures: an isolated, water-soluble helix bundle not expected to self-associate (Fig. 1A) and three material assemblies derived from P622, P422, and P222 space-group symmetries (Fig. 1, B to D). Each of these layered space-group symmetries contains D2 symmetric positions on which the individual peptide bundles were positioned (Fig. 1, B to D). Calculations were performed using only a single, isolated layer from the corresponding space group. For a given nanostructure symmetry, the variation of the unit cell parameters produced a set of possible lattice structures consistent with the chosen symmetry; the amino acid probabilities and E were calculated for each assembly structure. From the resulting energy landscape for each type of assembly, energy minima were identified. Within these minima, sequences were identified, where the amino acid with the largest calculated probability was selected at each variable residue position.
Fig. 1. Computationally designed, helical, homotetramer assemblies.
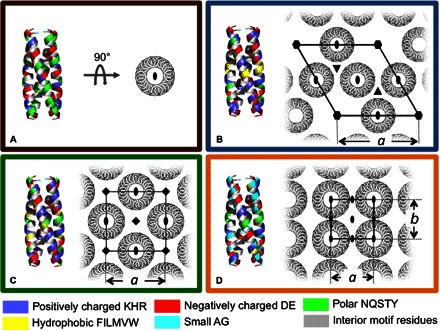
(A to D) Models of peptides forming distinct nanostructures using a de novo designed helical homotetramer motif, which comprises both the backbone coordinates of the D2 symmetric tetramer and interior hydrophobic residues. On the left of each panel, designed exterior residues are colored according to chemical properties: positively charged KHR (blue), negatively charged DE (red), polar NQSTY (green), hydrophobic FILMVW (yellow), and small AG (cyan). Interior hydrophobic residues common to all the sequences are gray. On the right of each panel, the targeted assemblies are rendered along with symmetry axes (C2, oval; C3, triangle; C4, square; C6, hexagon) and the unique dimensions of the unit cell, a and b. (A) D2 symmetric tetramer designed in isolation and targeted to remain not assembled in solution. The exterior residues of the remaining proteins were designed in the context of a single layer from the corresponding space groups (B) P622, (C) P422, and (D) P222.
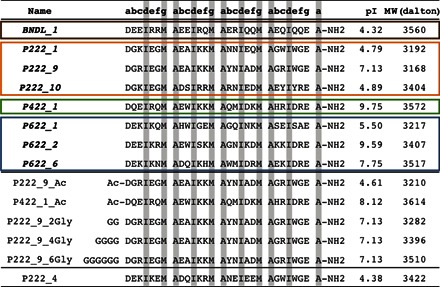
Table 1. Table of computationally determined peptides for solution assembly.
Colored rectangles contain eight candidate sequences that were experimentally characterized. Sequences were theoretically designed to produce tetrahelical bundles. BNDL_1 was designed in the absence of any lattice assembly and is expected to remain soluble (brown). The P222 (orange), P422 (green), and P622 (blue) sequences were designed in the presence of lattices of corresponding symmetry. The remaining P222_9 and P422_1 sequences contain covalently modified termini. P222_4 is the only sequence candidate that did not behave as predicted and could not be assembled into a nanostructure in the solution conditions used for the other peptides. The heptad repeat positions (abcdefg) of all peptides are shown in the table heading. The designed, hydrophobic interior residues of the motif shown are highlighted in gray. MW, molecular weight.
In solution, all but one of the computationally designed peptides exhibited the intended assembly properties: the peptide designed using the isolated tetramer template (Fig. 1A) remained soluble and did not form a higher-order structure, whereas those designed in the context of a lattice (Fig. 1, B to D) formed some sort of regular array. Nine designed sequences were selected for experimental investigation (Table 1). The single sequence designed to form isolated bundles (BNDL_1) formed soluble homotetramers that did not undergo further interbundle assembly. Seven molecules computationally designed to form material assemblies with specific, interbundle packing produced sheetlike nanomaterials. No assembly was observed for the sequence P222_4 (Table 1).
Figure 2 presents representative data from four different peptide molecules theoretically designed to produce the four chosen nanostructures in Fig. 1. Figure 2A shows small-angle neutron scattering (SANS) results for BNDL_1 modeled as a short cylinder with dimensions consistent with the designed, tetrameric coiled coil. Analytical ultracentrifugation (AUC) supports the presence of tetrameric helix bundles coexisting with a minority of monomeric peptide (fig. S14). BNDL_1 solutions were experimentally monitored for 3 months, and no interbundle lattice structure was observed. Figure 2B shows the nanostructure formed from assembled P622_6 molecules, which is consistent with hexagonal local symmetry and the targeted unit cell parameters (Table 2). The structure is stabilized by two unique bundle-bundle interfaces that arise from the design around the threefold symmetry axis (Fig. 1B). Both interfaces, one between two antiparallel helices and the other between two parallel helices, are populated by salt bridges, hydrogen bonds, and hydrophobic interactions in the computational design. Figure 2C shows the materials formed from the self-assembly of P422_1 that have the targeted fourfold-like symmetry. Although the fourfold symmetry is clear in Fig. 2B, the experimentally observed unit cell dimensions are larger than predicted (Table 2). Figure 2D shows the materials formed from the self-assembly of P222_1. The observed morphology consists of regular nanosheets that have the targeted rectangular structure, but differences are observed in symmetry and unit cell spacings from what was theoretically designed (Table 2). Although the targeted lattice parameters are not precisely recovered experimentally in each case, together, the data support the use of computational design to realize a set of homotetrameric helix bundles that have predetermined self-assembly properties (for example, sheet-forming tetrameric peptide bundles versus soluble bundles) and distinct, local ordering that is determined by the exterior surface residues of the helical bundle.
Fig. 2. Structural characterization of assemblies comprising designed peptides.

(A) Small-angle neutron scattering data and nanocylinder fit (black curve) of BNDL_1 assembled from 5 mM peptide solution in borate buffer (pH 10). Fit provides a cylinder length of ~3.5 nm and a radius of ~1 nm, consistent with tetrameric coiled coil soluble bundle design. (B to D) Peptide solutions were heated to 80°C to obviate intermolecular or intramolecular structures and then allowed to cool to room temperature for intermolecular assembly. (B) Left: Low-magnification cast-film transmission electron microscopy (TEM) image of P622_6 assembled from 1 mM peptide solution in phosphate buffer (pH 7). (B) Right: High-magnification image of negatively stained lattice consistent with P622 symmetry. Upper inset is the fast Fourier transform (FFT) calculated from the high-magnification TEM data, whereas the lower inset is the inverse FFT (IFFT) calculated using the FFT maxima. (C) Left: Low-magnification cast-film TEM image of P422_1 assembled from 1 mM peptide solution in borate buffer (pH 10). (C) Right: High-magnification image of negatively stained lattice. Upper and lower insets are the FFT and IFFT, respectively. (D) Left: Low-magnification cast-film image of P222_1 assembled from 1 mM peptide solution in phosphate buffer (pH 7). (D) Right: High-magnification image of positively stained lattice. Upper and lower insets are the FFT and IFFT, respectively.
Table 2. Lattice parameters of the self-assembling peptides from the design in comparison with those determined from analysis of Fourier transforms of the TEM images in Fig. 2.
a and b denote the dimensions of the two-dimensional unit cell, and γ denotes the interior angle defined by sides a and b.
| Design | TEM | |||||
| a (nm) | b (nm) | γ (°) | a (nm) | b (nm) | γ (°) | |
| P622_6 | 4.57 | 4.57 | 120 | 4.5 ± 0.3 | 4.5 ± 0.3 | 112.7 ± 0.4 |
| P422_1 | 3.12 | 3.12 | 90 | 4.2 ± 0.2 | 3.9 ± 0.2 | 88.9 ± 0.9 |
| P222_1 | 2.09 | 2.00 | 90 | 3.3 ± 0.3 | 3.2 ± 0.3 | 100.4 ± 0.9 |
The robustness and versatility of the designed bundle-forming peptides were further explored by changing solvent conditions. Different solution conditions, selected on the basis of the physicochemical properties of the bundles, could be used to manipulate interbundle assembly and alter the resulting superstructural morphology of the nanomaterials. For example, Fig. 3 (A and B) reveals that the micrometer-scale morphology of P622_2 can be manipulated simply by first melting the secondary structure at 80°C and subsequently quenching the solution to two different temperatures. Smaller particles were formed at the higher quenching temperature (50°C) than at the lower quenching temperature (25°C). The data suggest that a higher temperature results in a much slower assembly process. Figure 3 (C and D) reveals the sensitivity of select bundle assemblies to changes in pH. The molecule P222_9 has a theoretical isoelectric point (pI) = 7 and was assembled under two solution conditions that differed only in pH. The assembly at pH 7 resulted in two-dimensional, plate-like growth, whereas the assembly at pH 10 yielded anisotropic growth and the formation of long, needle-like structures. The thickness of the needles prevented clear, high-magnification lattice imaging, but the layer spacings of the underlying lattice (fig. S15) were consistent with the nanostructure observed for the molecule P222_9 (Figs. 1 and 2). The results suggest that, at pH 10, there is a clear preference in growth direction during helix bundle solution assembly.
Fig. 3. Cast-film TEM examples of morphology control with manipulation of solution assembly conditions and peptide primary structure.

All sample solutions heated to above 80°C for 1 hour to obviate any assembled or secondary structure before respective cooling treatment. (A and B) P622_2 peptide (0.5 mM) at pH 7 (phosphate buffer) quenched to (A) 50°C versus (B) 25°C and imaged after 1 day. (C and D) P222_9 (1.0 mM) ambiently cooled to room temperature at (C) pH 7 (phosphate buffer) versus (D) pH 10 (borate buffer) showing a clear difference in superstructure growth. (E and F) Plates grown from 0.1 mM peptide solutions with peptide primary structure altered through acetylation of the N terminus. Ambient cooling to room temperature allowing assembly of (E) P222_9_Ac at a low pH of 4.5 in sodium acetate buffer and of (F) P422_1_Ac quenched to 50°C at pH 8 in phosphate buffer. (G and H) Plates grown from 1.0 mM peptide solutions at pH 7 (phosphate buffer) after ambient cooling to room temperature, with P222_9 peptide primary structure altered through addition of (G) four glycines versus (H) six glycines to the N terminus of the P222_9 peptide molecule.
Covalent alteration of the original designed peptides can also be used to probe the robustness of the assembly and to modulate assembly at particular solution conditions. For example, acetylation of the N terminus of P222_9 (denoted P222_9_Ac; Table 1) reduces its theoretical pI from pI = 7 (P222_9) to pI = 4 (P222_9_Ac), allowing for assembly at low pH. At pH 4.5, P222_9 remains dissolved, whereas P222_9_Ac (Fig. 3E) assembles into the same nanostructure as that observed for P222_9 at pH 7 (fig. S16). Similarly, P422_1_Ac forms plate nanostructures at pH 8 (Fig. 3F), whereas P422_1 assembles at pH 10 (Fig. 2B). Therefore, one can use covalent modifications of the designed peptide sequences to alter solution conditions in which nanostructures can be formed. Adding residues to the termini of the originally predicted sequences did not disrupt solution self-assembly. Specifically, sequences of two, four, and six glycine residues were added to the N terminus of P222_9 (Table 1), resulting in quite uniform assemblies (Fig. 3, G and H) with similar lattice structures to those observed for the unmodified P222_9 (fig. S17). These observations reveal the robust nature of these theoretically designed peptides and their material assemblies. Together, the data from Fig. 3 exemplify that multiple modifications of the theoretically predicted sequences remain competent for forming the selected nanostructures.
DISCUSSION
The data from Figs. 2 and 3 and the Supplementary Materials support the fact that theoretically designed sequences listed in Table 1 self-assemble to form predetermined sheet nanostructures or soluble bundles. Specifically, the sequence BNDL_1, targeted to form soluble, nonassociating bundles, formed homotetramers in solution but did not exhibit further lattice assembly. Similarly, the sequence P622_6 was designed to form two-dimensional sheets with hexagonal local symmetry and formed a nanostructure consistent with that identified in the computational design of the sequences; this consistency is evidenced in the similarity of the unit cell parameters of the theoretical model to those derived from analysis of TEM data. As shown in Fig. 2, the molecules designed to produce two-dimensional sheets with P422 symmetry assemble into a two-dimensional sheet nanomaterial with local fourfold bundle packing symmetry. However, the lattice spacing formed experimentally is larger than that expected from the computational design. Similarly, the molecules predicted to yield the two-dimensional materials with P222 symmetry assembled into sheet nanostructures. However, the lattice symmetry and unit cell parameters were different from those identified in the computational modeling. Additional calculations using the P422_1 molecule sequence within a related fourfold symmetry, P4, reveal a local energy minimum at the experimentally determined unit cell parameters (fig. S18). The distance between the C2 symmetry axes of neighboring bundles within each model structure is essentially indistinguishable: 2.2 nm (P4) versus 2.2 nm (P422). With the P4 structure, the unit cell contains four helical bundles (16 peptides) as opposed to two helical bundles (8 peptides) in the original model with P422 symmetry, which is consistent with the larger unit cell dimensions observed experimentally. Similar calculations involving P222_1 within a related twofold symmetry, P2, reveal a local minimum that is consistent with the experimental observations (fig. S19). More effort is needed to refine design methods, recognize deterministic molecular attributes, and develop assembly pathways that will reproducibly funnel the molecules into predetermined nanostructures during solution assembly. Nonetheless, the designed assemblies are robust over a wide range of pH and temperature values and are resilient with respect to chemical modification at the peptide termini. Thus, the control of solution conditions (pH and temperature), primary sequence, and chemical modification of peptides can be used to control solution-phase assembly and the final material morphology.
We have presented an approach for the de novo design of peptide assemblies that assemble into nanomaterials with predetermined local structures. We computationally designed a versatile, helical, homotetrameric building block structure that was stabilized largely by the formation of a hydrophobic interior. Subsequent computational determination of the bundle exterior residues resulted in solution-assembled materials with predetermined morphologies (two-dimensional sheet versus soluble bundle) and distinct, local nanostructures that varied with the bundle exterior sequence. Integrated theoretical and experimental studies will guide future refinements for predictably achieving specific, desired nanostructures and elucidating the principles underlying their formation. In addition to achieving symmetric assemblies of biopolymers, the display of a wide variety of chemical functional groups within designed assemblies offers abundant opportunities for hierarchical pathways for nanomaterial production, such as biopolymer-templated growth and/or assembly of inorganic phases with nanoscale precision. Computational design combined with the experimental control of assembly pathways has the potential to provide exquisite control over new materials with desired nanostructures.
MATERIALS AND METHODS
Peptide synthesis
Peptides were prepared at a 0.25-mmol scale on a Rink amide resin using an AAPPTec Focus XC synthesizer (AAPPTec). Standard Fmoc-based protocols were used. Peptides were deprotected for 5 min and then for 10 min with 20% piperidine in dimethylformamide (DMF). The coupling reaction was conducted for 40 min with 4 eq of the appropriate amino acid dissolved in N-methyl-2-pyrrolidone (NMP) (5 ml), 3.8 eq of O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU) dissolved in DMF (2.5 ml), and 8 eq of diisopropylethylamine dissolved in NMP (1 ml). Five washes were performed in between steps with 50:50 (v/v) DMF/methylene chloride (12 ml) for the first two washes and with DMF (10 ml) for the last three. Amino acids, resins, and activators were purchased from ChemPep and were used as received. All solvents were of analytical grade (Fisher Scientific). Peptide cleavage was achieved by shaking the peptide solutions for 2 hours in a cleavage cocktail comprising (by volume) 95% trifluoroacetic acid (TFA), 2.5% triisoproylsilane, and 2.5% Milli-Q water. The peptide was then precipitated by adding the cleavage cocktail and cleaved peptide to diethyl ether. The mixture was then centrifuged, and the supernatant was discarded. The process of suspending in diethyl ether, centrifuging, and discarding the supernatant was repeated a total of three times. The resulting peptide was then dissolved in water and lyophilized.
Peptide purification
Purification was performed via reversed-phase high-performance liquid chromatography (HPLC) using a BEH130 Prep C18 10-μm column (XBridge, Waters Corporation). Crude peptides were dissolved in Milli-Q water containing 0.1% (v/v) TFA and were filtered (0.20-μm filter, Corning Inc.) before HPLC injection. The products were subjected to an elution gradient [Quaternary Gradient Module (Waters 2545), Waters Corporation] of 100% solvent A [0.1% (v/v) TFA] to 30% solvent A within 60 min; solvent B consisted of acetonitrile with 0.1% (v/v) TFA. Fractions were detected using ultraviolet-visible detection at 214 nm (Waters 2489, Waters Corporation) and collected (Waters Fraction Collector III, Waters Corporation). The collected fractions were examined by electrospray ionization–mass spectrometry (LCQ Advantage Mass Spectrometer System, Thermo Finnigan) with an autosampler system (Surveyor Autosampler, Thermo Finnigan). Pure fractions were combined and lyophilized.
Analytical HPLC
Purity analysis was performed via reversed-phase analytical-scale HPLC using a BEH C4 3.5-μm column (XBridge, Waters Corporation). Peptides were dissolved in 80:20 (v/v) Milli-Q water/acetonitrile containing 0.1% (v/v) TFA and filtered with a 0.20-μm filter (Corning Inc.) before injection. The products were subjected to a linear elution gradient (Waters 600 Controller, Waters Corporation) of 80% solvent A [Milli-Q water with 0.1% (v/v) TFA] to 5% solvent A in 70 min; solvent B consisted of acetonitrile with 0.1% (v/v) TFA. Fractions were detected using a photodiode array detector (Waters 2996, Waters Corporation) tuned at 214 nm.
Circular dichroic spectroscopy
Secondary structures and the temperature-dependent behavior of the synthetic peptides were analyzed using circular dichroic spectroscopy on a Jasco J-820 spectropolarimeter (JASCO Inc.). Sample solutions were prepared at 0.1 mM concentration in 10 mM buffer appropriate for the desired pH and were transferred into an absorption cuvette with a path length of 1 mm (110-QS, Hellma Inc.). Pure buffer solutions were used for the background correction. Sample spectra were recorded from 190 to 250 nm at desired temperatures. Data points for the wavelength-dependent circular dichroic spectra were recorded at every nanometer with a bandwidth of 1 nm and a response time of 4 s for each data point. The ellipticity at 222 nm was used to monitor the temperature-dependent unfolding and refolding of the peptides. Data points for the scans were recorded at 222 nm at 1-min intervals. The mean residue ellipticity, [θ]MRE (deg cm2 dmol−1), was calculated using the peptide concentration, number of amino acid residues, and cell path length.
Transmission electron microscopy
Carbon-coated 200-mesh copper grids (CF200-Cu, Electron Microscopy Sciences Inc.) were freshly treated by glow discharge using a plasma cleaner (PDC-32G, Harrica Plasma Inc.) before sample preparation. Three microliters of the sample suspension was applied onto the grids. After ~5 min, any remaining liquid was blotted from the edge of the grids using filter paper. Then, 3 μl of Milli-Q water was applied to the grids and blotted immediately to remove excess unassembled peptides and buffer salts. The grids were incubated under ambient conditions for another 30 min before TEM observation (Tecnai 12, FEI) or staining. To negatively stain the grids, 3 μl of an aqueous solution of uranyl acetate (1 weight percent) was applied to the cast-film grids, incubated for 20 to 30 s, and then blotted with filter paper. The stained grids were allowed to sit for at least 10 min under ambient conditions before TEM observation. Positive staining of the samples was achieved by exposing the dried cast-film grids to ruthenium tetroxide aqueous solution vapor [0.5% (w/v)] for 5 to 10 min. The transmission electron microscope was operated at an acceleration voltage of 120 kV, and all images were recorded digitally using a Gatan 791 MultiScan side-mounted charge-coupled device camera. Lattice plane spacings were determined by conducting an FFT of the area of interest of a TEM micrograph using DigitalMicrograph software version 2.3 (Gatan Inc.) and the PASAD plug-in (37). FFT intensity was plotted relative to radial distance from the origin as well as relative to azimuthal angle. Corresponding real space values of interplanar distances and angles were calculated to estimate unit cell parameters. Uncertainties of the unit cell parameters were approximated by the size of the maxima in the FFT. Fourier-filtered images (lower insets in Fig. 2) were obtained by applying masks around Fourier frequencies with distinct intensity maxima within the respective FFT and then performing an inverse FFT.
Small-angle neutron scattering
SANS measurements were conducted at the National Center for Neutron Research [National Institute of Standards and Technology (NIST), Gaithersburg, MD] on the NG-7 30-m SANS beamline. Samples were dissolved in 10 mM borate buffer (pH 10) prepared in D2O and heated to 80°C for 1 hour to melt any organized secondary structure/aggregated structure and were subsequently cooled to room temperature for intermolecular assembly. A neutron beam with a mean wavelength of 6 Å was defined using a mechanical velocity selector. The wavelength spread (Δλ/λ) was 0.15 at full width at half maximum. The 640 mm × 640 mm 3He proportional counter used has a spatial resolution of 5.08 mm × 5.08 mm. Sample-to-detector distances of 1, 4, and 13 m were used to provide a q range of approximately 0.004 to 0.500 Å−1, where q is the scattering wave vector defined by q = (4/λ) sin(θ/2). Data obtained with this instrument were corrected for background noise and radiation, detector inhomogeneity, and empty cell scattering. Intensities were normalized to an absolute scale relative to the empty beam transmission. The uncertainties of individual data points were calculated statistically from the number of averaged detector counts. See the Supplementary Materials for a description of data fitting and analysis.
Analytical ultracentrifugation
AUC experiments were run on a Beckman Coulter ProteomeLab XL-I instrument. Four hundred microliters of 0.5 mM peptide solution in buffer (10 mM borate buffer with extra 50 mM NaCl to screen long-range Coulombic interactions; pH 10) was transferred into a two-channel cell equipped with sapphire windows. The cells were mounted into a four-cell An-60 Ti analytical rotor and equilibrated at 20°C for 2 hours in the rotor chamber. Sedimentation velocity experiments were carried out at 50,000 rpm at 20°C using 800 scans with 1 scan per minute per sample. The changes of the concentration profiles along the cell radius were monitored using Rayleigh interference optics. All data were analyzed using the SEDFIT package version 14.81 (38).
Supplementary Material
Acknowledgments
We thank C. Von Bargen and C. MacDermaid for assistance with computational studies and P. Schuck (NIH) for the fruitful discussions about the AUC data and analysis. We acknowledge the help of Y.-S. Wu of the University of Delaware (UD) Centers of Biomedical Research Excellence (COBRE) with AUC measurements as well as the help of C.-y. Ni, F. Deng, and J. Sloppy of the UD Keck Electron Microscopy Laboratory. Funding: We acknowledge support from NSF DMREF (Designing Materials to Revolutionize and Engineer our Future) program under awards DMR-1234161 and DMR-1235084. J.G.S. thanks the Penn Nano/Bio Interface Center for infrastructural support (NSF DMR-0832802). J.G.S. acknowledges additional support from the Penn Laboratory for Research on the Structure of Matter (NSF DMR-1120901). This work used the Extreme Science and Engineering Discovery Environment, which is supported by NSF grant no. ACI-1053575, under grant no. TG-CHE110041. D.J.P. acknowledges cooperative agreement 70NANB12H239 from NIST, the U.S. Department of Commerce, and the UD-NIST Center for Neutron Science. The statements, findings, conclusions, and recommendations are those of the author(s) and do not necessarily reflect the view of NIST or the U.S. Department of Commerce. The National Center for Neutron Science facilities were also supported in part by the NSF under agreement no. DMR-1508249. F.P. thanks the Deutsche Forschungsgemeinschaft for financial support. The UD COBRE NIH-COBRE 1P30 GM110758 is acknowledged for partial support of the Keck electron microscopy facility. Author contributions: H.V.Z., J.G.S., and D.J.P. designed the peptides and nanostructures for solution assembly. H.V.Z. and J.A.V. performed the computational studies. F.P., M.J.H., and Y.T. performed peptide synthesis, purification, solution assembly, and electron microscopy characterization. M.J.H. carried out SANS characterization of BNDL_1. F.P., M.J.H., Y.T., D.J.P., and K.L.K. analyzed the experimental data. H.V.Z., F.P., M.J.H., Y.T., K.L.K., D.J.P., and J.G.S. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1600307/DC1
Supplementary Materials and Methods
fig. S1. Representative backbone configurations of the helix bundle motif building block illustrating the variation of the geometric parameters associated with the bundle.
fig. S2. Side view and top view of the selected low-energy helix bundle motif with the most probable amino acids at the interior sites shown in space-filling representations.
fig. S3. P422_1 analytical HPLC.
fig. S4. P422_1_ac analytical HPLC.
fig. S5. P222_9 analytical HPLC.
fig. S6. P222_9 sequence (3168 daltons).
fig. S7. P422_1 sequence (3572 daltons).
fig. S8. BNDL_1 (3560 daltons).
fig. S9. P622_6 (3517 daltons).
fig. S10. P222_9 (0.1 mM) in borate buffer (pH 10) on heating.
fig. S11. P622_6 (0.1 mM) in phosphate buffer (pH 7) on heating.
fig. S12. BNDL_1 (0.1 mM) in borate buffer (pH 10) on heating.
fig. S13. P422_1 (0.1 mM) in borate buffer (pH 10).
fig. S14. AUC data and analysis of BNDL_1.
fig. S15. High-magnification TEM of 1.0 mM P222_9 ambiently cooled to room temperature from 80°C at pH 7 (left) and pH 10 (right).
fig. S16. High-magnification TEM of (left) lattice of P222_9_Ac at pH 7 ambiently cooled to room temperature from 80°C and (right) P422_1_Ac at pH 8 quenched to 40°C from 80°C.
fig. S17. High-magnification TEM of lattice of P222_9_6Gly at pH 7 ambiently cooled to room temperature from 80°C.
fig. S18. Putative structure of assembly of P422_1 helix bundles packed with P4 symmetry, which is a local minimum within the structure energy landscape with respect to variation of the unit cell parameter.
fig. S19. Putative assembly structures of P222_1 assemblies with P2 symmetry located at a local minimum of the structure energy landscape with respect to variation of the unit cell parameters.
REFERENCES AND NOTES
- 1.Danino D., Talmon Y., Zana R., Vesicle-to-micelle transformation in systems containing dimeric surfactants. J. Colloid Interface Sci. 185, 84–93 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Vinson P. K., Bellare J. R., Davis H. T., Miller W. G., Scriven L. E., Direct imaging of surfactant micelles, vesicles, discs, and ripple phase structures by cryo-transmission electron microscopy. J. Colloid Interface Sci. 142, 74–91 (1991). [Google Scholar]
- 3.Discher D. E., Ortiz V., Srinivas G., Klein M. L., Kim Y., Christian D., Cai S., Photos P., Ahmed F., Emerging applications of polymersomes in delivery: From molecular dynamics to shrinkage of tumors. Prog. Polym. Sci. 32, 838–857 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LoPresti C., Lomas H., Massignani M., Smart T., Battaglia G., Polymersomes: Nature inspired nanometer sized compartments. J. Mater. Chem. 19, 3576–3590 (2009). [Google Scholar]
- 5.Brinkhuis R. P., Rutjes F. P. J. T., van Hest J. C. M., Polymeric vesicles in biomedical applications. Polym. Chem. 2, 1449–1462 (2011). [Google Scholar]
- 6.Rosales A. M., Segalman R. A., Zuckermann R. N., Polypeptoids: A model system to study the effect of monomer sequence on polymer properties and self-assembly. Soft Matter 9, 8400–8414 (2013). [Google Scholar]
- 7.Hudson Z. M., Boott C. E., Robinson M. E., Rupar P. A., Winnik M. A., Manners I., Tailored hierarchical micelle architectures using living crystallization-driven self-assembly in two dimensions. Nat. Chem. 6, 893–898 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Moughton A. O., Hillmyer M. A., Lodge T. P., Multicompartment block polymer micelles. Macromolecules 45, 2–19 (2012). [Google Scholar]
- 9.Huang M. J., Hsu C.-H., Wang J., Mei S., Dong X. H., Li Y. W., Li M. X., Liu H., Zhang W., Aida T. Z., Zhang W.-B., Yue K., Cheng S. Z. D., Selective assemblies of giant tetrahedra via precisely controlled positional interactions. Science 348, 424–428 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Gröschel A. H., Walther A., Löbling T. I., Schacher F. H., Schmalz H., Müller A. H. E., Guided hierarchical co-assembly of soft patchy nanoparticles. Nature 503, 247–251 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Rupar P. A., Chabanne L., Winnik M. A., Manners I., Non-centrosymmetric cylindrical micelles by unidirectional growth. Science 337, 559–562 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Liu C., Hillmyer M. A., Lodge T. P., Evolution of multicompartment micelles to mixed corona micelles using solvent mixtures. Langmuir 24, 12001–12009 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Pochan D. J., Zhu J., Zhang K., Wooley K. L., Miesch C., Emrick T., Multicompartment and multigeometry nanoparticle assembly. Soft Matter 7, 2500–2506 (2011). [Google Scholar]
- 14.Jones M. R., Seeman N. C., Mirkin C. A., Programmable materials and the nature of the DNA bond. Science 347, 840 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Wang T., Schiffels D., Cuesta S. M., Fygenson D. K., Seeman N. C., Design and characterization of 1D nanotubes and 2D periodic arrays self-assembled from DNA multi-helix bundles. J. Am. Chem. Soc. 134, 1606–1616 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krejchi M. T., Atkins E. D., Waddon A. J., Fournier M. J., Mason T. L., Tirrell D. A., Chemical sequence control of beta-sheet assembly in macromolecular crystals of periodic polypeptides. Science 265, 1427–1432 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Yu S. M., Conticello V. P., Zhang G., Kayser C., Fournier M. J., Mason T. L., Tirrell D. A., Smectic ordering in solutions and films of a rod-like polymer owing to monodispersity of chain length. Nature 389, 167–170 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Padilla J. E., Colovos C., Yeates T. O., Nanohedra: Using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 2217–2221 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fletcher J. M., Harniman R. L., Barnes F. R. H., Boyle A. L., Collins A., Mantell J., Sharp T. H., Antognozzi M., Booth P. J., Linden N., Miles M. J., Sessions R. B., Verkade P., Woolfson D. N., Self-assembling cages from coiled-coil peptide modules. Science 340, 595–599 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vargo K. B., Parthasarathy R., Hammer D. A., Self-assembly of tunable protein suprastructures from recombinant oleosin. Proc. Natl. Acad. Sci. U.S.A. 109, 11657–11662 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holowka E. P., Sun V. Z., Kamei D. T., Deming T. J., Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat. Mater. 6, 52–57 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Brodin J. D., Smith S. J., Carr J. R., Tezcan F. A., Designed, helical protein nanotubes with variable diameters from a single building block. J. Am. Chem. Soc. 137, 10468–10471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shu J. Y., Panganiban B., Xu T., Peptide-polymer conjugates: From fundamental science to application. Annu. Rev. Phys. Chem. 64, 631–657 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Hume J., Sun J., Jacquet R., Renfrew P. D., Martin J. A., Bonneau R., Gilchrist M. L., Montclare J. K., Engineered coiled-coil protein microfibers. Biomacromolecules 15, 3503–3510 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Loo Y., Zhang S., Hauser C. A. E., From short peptides to nanofibers to macromolecular assemblies in biomedicine. Biotechnol. Adv. 30, 593–603 (2012). [DOI] [PubMed] [Google Scholar]
- 26.O’Leary L. E. R., Fallas J. A., Bakota E. L., Kang M. K., Hartgerink J. D., Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat. Chem. 3, 821–828 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Xu C., Liu R., Mehta A. K., Guerrero-Ferreira R. C., Wright E. R., Dunin-Horkawicz S., Morris K., Serpell L. C., Zuo X., Wall J. S., Conticello V. P., Rational design of helical nanotubes from self-assembly of coiled-coil lock washers. J. Am. Chem. Soc. 135, 15565–15578 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Burgess N. C., Sharp T. H., Thomas F., Wood C. W., Thomson A. R., Zaccai N. R., Brady R. L., Serpell L. C., Woolfson D. N., Modular design of self-assembling peptide-based nanotubes. J. Am. Chem. Soc. 137, 10554–10562 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Egelman E. H., Xu C., DiMaio F., Magnotti E., Modlin C., Yu X., Wright E., Baker D., Conticello V. P., Structural plasticity of helical nanotubes based on coiled-coil assemblies. Structure 23, 280–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brodin J. D., Ambroggio X. I., Tang C., Parent K. N., Baker T. S., Tezcan F. A., Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat. Chem. 4, 375–382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai Y.-T., Reading E., Hura G. L., Tsai K.-L., Laganowsky A., Asturias F. J., Tainer J. A., Robinson C. V., Yeates T. O., Structure of a designed protein cage that self-assembles into a highly porous cube. Nat. Chem. 6, 1065–1071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sontz P. A., Song W. J., Tezcan F. A., Interfacial metal coordination in engineered protein and peptide assemblies. Curr. Opin. Chem. Biol. 19, 42–49 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Lanci C. J., MacDermaid C. M., Kang S.-g., Acharya R., North B., Yang X., Qiu X. J., DeGrado W. F., Saven J. G., Computational design of a protein crystal. Proc. Natl. Acad. Sci. U.S.A. 109, 7304–7309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King N. P., Sheffler W., Sawaya M. R., Vollmar B. S., Sumida J. P., André I., Gonen T., Yeates T. O., Baker D., Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 336, 1171–1174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King N. P., Bale J. B., Sheffler W., McNamara D. E., Gonen S., Gonen T., Yeates T. O., Baker D., Accurate design of co-assembling multi-component protein nanomaterials. Nature 510, 103–108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonen S., DiMaio F., Gonen T., Baker D., Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces. Science 348, 1365–1368 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Gammer C., Mangler C., Rentenberger C., Karnthaler H. P., Quantitative local profile analysis of nanomaterials by electron diffraction. Scr. Mater. 63, 312–315 (2010). [Google Scholar]
- 38.Schuck P., Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grigoryan G., Degrado W. F., Probing designability via a generalized model of helical bundle geometry. J. Mol. Biol. 405, 1079–1100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quine J. R., Helix parameters and protein structure using quaternions. J. Mol. Struct. (THEOCHEM) 460, 53–66 (1999). [Google Scholar]
- 41.Deng Y., Liu J., Zheng Q., Eliezer D., Kallenbach N. R., Lu M., Antiparallel four-stranded coiled coil specified by a 3-3-1 hydrophobic heptad repeat. Structure 14, 247–255 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calhoun J. R., Kono H., Lahr S., Wang W., DeGrado W. F., Saven J. G., Computational design and characterization of a monomeric helical dinuclear metalloprotein. J. Mol. Biol. 334, 1101–1115 (2003). [DOI] [PubMed] [Google Scholar]
- 43.MacKerell A. D. Jr, Bashford D., Dunbrack R. L. Jr, Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M., All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998). [DOI] [PubMed] [Google Scholar]
- 44.O’Neil K. T., DeGrado W. F., A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250, 646–651 (1990). [DOI] [PubMed] [Google Scholar]
- 45.Testa O. D., Moutevelis E., Woolfson D. N., CC+: A relational database of coiled-coil structures. Nucleic Acids Res. 37, D315–D322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu X., Kono H., Saven J. G., Probabilistic approach to the design of symmetric protein quaternary structures. Protein Eng. 16, 971–977 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J., CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 50.SasView, www.sasview.org.
- 51.A. Guinier, G. Fournet, Small-angle scattering of X-rays (John Wiley and Sons, New York, 1955). [Google Scholar]
- 52.Kline S. R., Reduction and analysis of SANS and USANS data using IGOR Pro. J. Appl. Crystallogr. 39, 895–900 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1600307/DC1
Supplementary Materials and Methods
fig. S1. Representative backbone configurations of the helix bundle motif building block illustrating the variation of the geometric parameters associated with the bundle.
fig. S2. Side view and top view of the selected low-energy helix bundle motif with the most probable amino acids at the interior sites shown in space-filling representations.
fig. S3. P422_1 analytical HPLC.
fig. S4. P422_1_ac analytical HPLC.
fig. S5. P222_9 analytical HPLC.
fig. S6. P222_9 sequence (3168 daltons).
fig. S7. P422_1 sequence (3572 daltons).
fig. S8. BNDL_1 (3560 daltons).
fig. S9. P622_6 (3517 daltons).
fig. S10. P222_9 (0.1 mM) in borate buffer (pH 10) on heating.
fig. S11. P622_6 (0.1 mM) in phosphate buffer (pH 7) on heating.
fig. S12. BNDL_1 (0.1 mM) in borate buffer (pH 10) on heating.
fig. S13. P422_1 (0.1 mM) in borate buffer (pH 10).
fig. S14. AUC data and analysis of BNDL_1.
fig. S15. High-magnification TEM of 1.0 mM P222_9 ambiently cooled to room temperature from 80°C at pH 7 (left) and pH 10 (right).
fig. S16. High-magnification TEM of (left) lattice of P222_9_Ac at pH 7 ambiently cooled to room temperature from 80°C and (right) P422_1_Ac at pH 8 quenched to 40°C from 80°C.
fig. S17. High-magnification TEM of lattice of P222_9_6Gly at pH 7 ambiently cooled to room temperature from 80°C.
fig. S18. Putative structure of assembly of P422_1 helix bundles packed with P4 symmetry, which is a local minimum within the structure energy landscape with respect to variation of the unit cell parameter.
fig. S19. Putative assembly structures of P222_1 assemblies with P2 symmetry located at a local minimum of the structure energy landscape with respect to variation of the unit cell parameters.