

Abstract
Neuronal activity is a high‐energy demanding process recruiting all neural cells that adapt their metabolism to sustain the energy and redox balance of neurons. During neurotransmission, synaptic cleft glutamate activates its receptors in neurons and in astrocytes, before being taken up by astrocytes through energy costly transporters. In astrocytes, the energy requirement for glutamate influx is likely to be met by glycolysis. To enable this, astrocytes are constitutively glycolytic, robustly expressing 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase‐3 (PFKFB3), an enzyme that is negligibly present in neurons by continuous degradation because of the ubiquitin‐proteasome pathway via anaphase‐promoting complex/cyclosome (APC)‐Cdh1. Additional factors contributing to the glycolytic frame of astrocytes may include 5′‐AMP‐activated protein kinase (AMPK), hypoxia‐inducible factor‐1 (HIF‐1), pyruvate kinase muscle isoform‐2 (PKM2), pyruvate dehydrogenase kinase‐4 (PDK4), lactate dehydrogenase‐B, or monocarboxylate transporter‐4 (MCT4). Neurotransmission‐associated messengers, such as nitric oxide or ammonium, stimulate lactate release from astrocytes. Astrocyte‐derived glycolytic lactate thus sustains the energy needs of neurons, which in contrast to astrocytes mainly rely on oxidative phosphorylation. Neuronal activity unavoidably triggers reactive oxygen species, but the antioxidant defense of neurons is weak; hence, they use glucose for oxidation through the pentose‐phosphate pathway to preserve the redox status. Furthermore, neural activity is coupled with erythroid‐derived erythroid‐derived 2‐like 2 (Nrf2) mediated transcriptional activation of antioxidant genes in astrocytes, which boost the de novo glutathione biosynthesis in neighbor neurons. Thus, the bioenergetics and redox programs of astrocytes are adapted to sustain neuronal activity and survival. Developing therapeutic strategies to interfere with these pathways may be useful to combat neurological diseases.
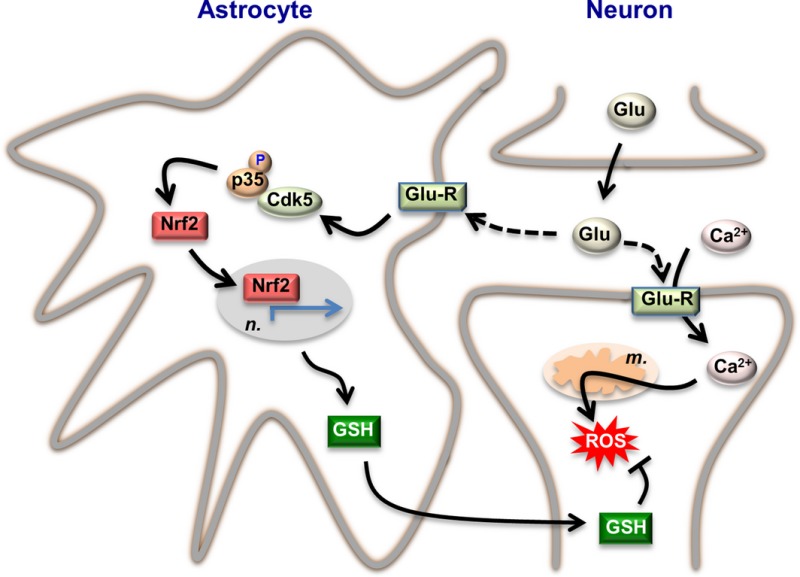
Our current knowledge on brain's management of bioenergetics and redox requirements associated with neural activity is herein revisited. The astrocyte‐neuronal lactate shuttle (ANLS) explains the energy needs of neurotransmission. Furthermore, neurotransmission unavoidably triggers increased mitochondrial reactive oxygen species in neurons. By coupling glutamatergic activity with transcriptional activation of antioxidant genes, astrocytes provide neurons with neuroprotective glutathione through an astrocyte‐neuronal glutathione shuttle (ANGS).
This article is part of the 60th Anniversary special issue.
Keywords: AMPK, Cdh1, Glycolysis, GSH, Nrf2, PFKFB3
Abbreviations used
- 6PG
6‐phosphogluconate
- 6PGD
6‐phosphogluconate dehydrogenase
- AMPK
AMP‐activated protein kinase
- ANGS
astrocyte‐neuronal glutathione shuttle
- ANLS
astrocyte‐neuronal lactate shuttle
- APC/C
anaphase‐promoting complex/cyclosome
- ARE
antioxidant response element
- Cdk5
cyclin‐dependent kinase‐5
- Cul3
cullin‐3
- F2,6P2
fructose‐2,6‐bisphosphate
- G6PD
glucose‐6‐phosphate dehydrogenase
- G6P
glucose‐6‐phosphate
- GCL
glutamate‐cysteine ligase
- GSH
glutathione
- GSSG
oxidized glutathione
- Keap1
Kelch‐like ECH‐associated protein 1
- LDH
lactate dehydrogenase
- MCT
monocarboxylate transporter
- MRC
mitochondrial respiratory chain
- mTOR
mammalian target of rapamycin
- NMDA
N‐methyl‐d‐aspartate
- NMR
nuclear magnetic resonance
- NOS
nitric oxide synthase
- Nrf2
nuclear factor (erythroid‐derived 2)‐like 2
- OXPHOS
oxidative phosphorylation
- PDK
pyruvate dehydrogenase kinase
- PFK1
6‐phosphofructo‐1‐kinase
- PFKFB3
6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase
- PKM2
pyruvate kinase muscle‐2 isoform
- PK
pyruvate kinase
- PPP
pentose‐phosphate pathway
- R5P
ribulose‐5‐phosphate
- ROS
reactive oxygen (and nitrogen) species
- TCA
tricarboxylic acid
Neural activity is energy costly
The brain consumes about 20% of inhaled O2 suggesting a high‐energy‐demanding tissue. Most of the neural energy expenditure is accounted for by the electrical impulses, which require continuous re‐setting of ion (Na+, K+, and Ca2+) gradients across the plasma membrane of dendrites and axons through primary active transporters, including Na+/K+‐ and Ca2+‐ATPase pumps (Cai and Sheng 2009). Accordingly, it is not unexpected that stimulation of glutamate receptors in cortical neurons causes rapid ATP consumption (Almeida and Bolaños 2001), which is immediately followed by a rise in mitochondrial O2 consumption (Thompson et al. 2003; Jekabsons and Nicholls 2004; Garcia et al. 2005). This suggests that the energy requirements of neurons during activity are met by mitochondrial oxidative phosphorylation (OXPHOS) (Nicholls 2008). Interestingly, prior to the rise in O2 consumption, there is an increase in intracellular Ca2+ concentrations (Gleichmann et al. 2009), suggesting an early regulatory role for Ca2+ in the bioenergetics of neurotransmission (Nicholls et al. 2007). Furthermore, mitochondrial influx of Ca2+ alters mitochondrial OXPHOS efficiency; hence, the production of reactive oxygen species (ROS) by this organelle is an inherent phenomenon of neurotransmission (Mattson and Liu 2002).
An issue that has remained controversial over decades is the unequivocal identification of the main energetic precursor for neuronal activity. According to in vivo studies using functional magnetic resonance imaging and positron emission tomography, glucose is the preferred energy substrate for the brain (Hertz et al. 2007). However, the occurrence of a lag between activation of cerebral blood flow and O2 consumption following neural activity (Fox and Raichle 1986) challenges this notion and lays the foundation for a long‐lasting debate (Hertz et al. 2007; Pellerin et al. 2007; Ivanov et al. 2014). The aim of this review was to revisit our current knowledge on the brain's management of the bioenergetics and redox needs that result from neural activity, as well as to identify potential gaps requiring future research.
Energy management of astrocytes during neural activity
Astrocytes are crucial to the metabolic and structural support of the brain (Parpura et al. 1994; Allen and Barres 2009), as they are essential partners in neurotransmission and behavior (Perea et al. 2014; Oliveira et al. 2015). During glutamatergic neurotransmission, astrocytes efficiently take up neuronal‐derived glutamate from the synaptic space through secondary, Na+‐dependent active transporters. Thus, glutamate uptake is energy‐costly for astrocytes, as re‐setting the Na+ gradient across the plasma membrane to the resting levels requires Na+ pumping by the Na+/K+‐ATPase. The mechanism used by astrocytes to couple glutamate uptake with its energy requirement is currently controversial. Intracellular glutamate may follow at least two metabolic fates, namely conversion into α‐ketoglutarate for oxidation within mitochondria via the tricarboxylic acid (TCA) cycle, or conversion into glutamine to be released and taken up by neighbor neurons, which convert it back into glutamate (Fig. 1). Estimations, made on the bases of the energetic efficiencies of substrates, initially concluded that the energy required for glutamate uptake exceeds that provided by the mitochondrial oxidation of glutamate alone (Hertz et al. 2007). Considering this, the increase in the rate of glucose uptake (Loaiza et al. 2003; Porras et al. 2008) coupled to glutamate influx by astrocytes, suggests that invoked glycolysis likely supplements the energy needs of the process, which is part of the so‐called astrocyte‐neuronal lactate shuttle (Pellerin and Magistretti 1994; Magistretti and Allaman 2015) that will be discussed below. Nonetheless, this issue has been the matter of debate. Comparing the use of glucose or lactate in cortical neurons and astrocytes under resting conditions by 13C‐nuclear magnetic resonance (13C‐NMR), it was concluded that lactate was a preferential TCA cycle substrate over glucose in neurons, whereas astrocytes oxidized less lactate indicating a less active oxidative metabolism than neurons (Bouzier‐Sore et al. 2006). This notion has been confirmed in resting cerebellar neurons (Bak et al. 2006), albeit glucose – not lactate – utilization via TCA cycle oxidation increases upon glutamatergic activity in these cells (Bak et al. 2006, 2009, 2012). Therefore, it seems likely that cerebellar and cortical neurons have different intrinsic responses against depolarization. Interestingly, extracellular lactate inhibits glucose usage by astrocytes, an effect that is stronger in resting than in K+‐stimulated conditions (Sotelo‐Hitschfeld et al. 2012). This suggests the occurrence of a negative feedback regulatory mechanism of glucose consumption in astrocytes by lactate that diverts glucose utilization from resting to active zones. Moreover, astrocytes store glycogen particularly in areas of high synaptic activity (Pellerin and Magistretti 2012), and glycogen‐derived lactate (Pellerin et al. 2007) can sustain neuronal activity specially during hypoglycemia (Brown and Ransom 2007; Suh et al. 2007) (Fig. 1). Neurons also synthesize glycogen that is continuously degraded (Vilchez et al. 2007), the physiological function of which is a matter of investigation (Duran and Guinovart 2015).
Figure 1.
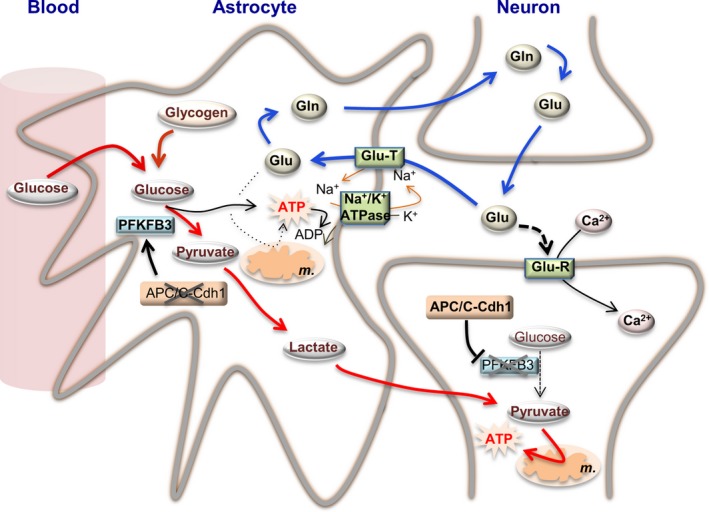
Bioenergetics adaptations of astrocytes to neurotransmission. Synaptic cleft glutamate (Glu) released by the pre‐synaptic neuron acts on glutamate receptors (Glu‐R) placed in the post‐synaptic neurons, triggering the influx of Ca2+ (and Na+, not indicated) and causing plasma membrane depolarization, which is needed to propagate the nervous impulse (neuronal activity). To reset basal levels of glutamate in the synaptic cleft, it is taken up by astrocytes through glutamate transporters (Glu‐T), which require Na+ uptake. Astrocytes convert glutamate into glutamine (Gln), which is released and then taken up by neurons, which convert it back into glutamate (blue arrowed lines). The Na+/K+‐ATPase hydrolyses ATP to conserve its energy to reset Na+ (and K+) homeostasis. Such ATP can be obtained by glutamate oxidation in the tricarboxylic acid cycle in the mitochondria (m.). However, strong evidence indicates that glutamate uptake is coupled, via a yet unknown mechanism, with glucose conversion into lactate through the glycolytic pathway (red arrowed lines). Neuronal activity is also coupled with glucose uptake from the blood, and with glycogen conversion into lactate. As neuronal activity‐associated glycolytic activation in astrocytes is coupled with stoichiometric lactate release, it is likely that glycolytic‐derived ATP would be in charge of sustaining Na+/K+ homeostasis during glutamate uptake. Astrocytes are genetically adapted to support a constitutive glycolytic phenotype in view of their low activity of APC/C‐Cdh1 (anaphase‐promoting complex/cyclosome/Cdh1), the E3 ubiquitin ligase responsible for the degradation of 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase‐3 (PFKFB3), a pro‐glycolytic enzyme. In contrast to astrocytes, neurons express high APC/C‐Cdh1 activity hence continuously degrading PFKFB3 causing their low glycolytic phenotype. Thus, neurons need an energy source different from glucose. Evidence obtained from cultured cells and in vivo strongly suggest that astrocyte‐released lactate support neuronal functions, both serving as a fuel and, possibly, as a signaling molecule. Thus, astrocytes are metabolically adapted to sustain the bioenergetics status of neurons during neural activity. The stoichiometry of the reactions has been omitted for clarity. Likewise, additional factors involved in these adaptations could not be depicted herein and can be found in the main text.
Therefore, it seems likely that cerebellar and cortical neurons have different intrinsic responses against depolarization. (Bittner et al. 2011). However, the nature of the functional coupling between glycolysis and Na+/K+‐ATPase may not be energetic (Fernandez‐Moncada and Barros 2014). The effect of glutamate on glycolysis is delayed and persistent, in contrast to the effect that is observed using K+ at concentrations compatible with those during glutamatergic neurotransmission, which stimulates glycolysis rapidly and reversibly (Bittner et al. 2011). Ammonium (NH4 +), which is produced stoichiometrically with glutamate, can rapidly activate lactate release from astrocytes, as can glutamate (Lerchundi et al. 2015); however, this effect is not coupled with glycolysis but with inhibition of mitochondrial uptake of pyruvate. Thus, the long‐lasting effect of glutamate (Bittner et al. 2011) suggests the action of adaptive metabolic mechanisms of astrocytes to neurotransmission, which will be discussed below. The mechanisms responsible for the short‐term effects on glycolysis and lactate release triggered by K+ and NH4 + deserve further investigation.
Is OXPHOS dispensable for energy generation in astrocytes?
Nitric oxide (•NO) is a neural messenger (Garthwaite et al. 1988) that is formed by neurons following glutamatergic neurotransmission. Experiments performed in astrocytes revealed the inhibition of the mitochondrial respiratory chain (MRC) by a •NO synthase‐mediated mechanism (Bolaños et al. 1994). The most susceptible component of the MRC was found to be cytochrome c oxidase (complex IV) (Bolaños et al. 1994). Subsequent studies confirmed this effect and revealed such inhibition to be reversible (Brown and Cooper 1994; Cleeter et al. 1994; Schweizer and Richter 1994), occurring by competition with O2 (Brown and Cooper 1994). During endogenous •NO production inhibiting the MRC at cytochrome c oxidase in astrocytes, ~ 90–100% of the glucose they consumed was released as lactate, and cells remained alive (Bolaños et al. 1994). These results suggest that OXPHOS inhibition invokes glycolysis as a survival pathway in astrocytes. In fact, endothelial cells‐derived •NO activates glycolytic lactate from neighbor astrocytes through hypoxia‐inducible factor‐1 (HIF‐1)‐mediated induction of glycolytic enzymes and monocarboxylate transporter‐4 expression (Brix et al. 2012). Glycolytic ATP drives the reverse ATP synthase reaction, which pumps H+ into the intermembrane space, thus sustaining the mitochondrial membrane potential (∆Ψm) (Beltran et al. 2000; Almeida et al. 2001). As long as glycolytic ATP sustains ∆Ψm astrocytes avoid apoptosis (Almeida et al. 2001), confirming that glycolysis is a survival pathway (Bolaños et al. 2010). Neurotransmission‐associated NH4 + formation acidifies the mitochondrial matrix in astrocytes leading to inhibition of pyruvate uptake (Lerchundi et al. 2015), which likely leads to TCA cycle impairment and mitochondrial energy stress, thus, contributing to glycolytic activation. Therefore, it is tempting to speculate that glycolysis is essential in astrocytes whereas OXPHOS is dispensable for the generation of energy associated with neurotransmission.
In contrast to astrocytes, neurons are highly vulnerable to the inhibition of the MRC. Incubation of neurons and astrocytes with •NO or its derivative peroxynitrite anion causing identical degree permanent inhibition of cytochrome c oxidase and mitochondrial respiration in both cell types dramatically triggered neuronal death, whereas astrocytes remained intact (Bolaños et al. 1995; Almeida et al. 2001). Interestingly, under these conditions, neurons were unable to up‐regulate glycolysis, and neuronal apoptosis was preceded by ∆Ψm collapse and ATP depletion (Almeida et al. 2001). Furthermore, these features were mimicked by stimulation of glutamate receptors, which immediately caused ATP decline (Almeida and Bolaños 2001; Gleichmann et al. 2009) that did not result in a concomitant up‐regulation of glucose uptake (Porras et al. 2004) or glycolytic activation (Delgado‐Esteban et al. 2000). Thus, during neuronal activity, neurons – in contrast to astrocytes – are tightly dependent on OXPHOS for energy generation and survival.
Astrocytes are constitutively glycolytic
Glycolysis is mainly regulated by the enzymatic activities of hexokinase, 6‐phosphofructo‐1‐kinase (PFK1), and pyruvate kinase (PK). In resting astrocytes, PFK1 specific activity is ~fourfold that found in resting neurons, and the levels of PFK1 positive effector, fructose‐2,6‐bisphosphate (F2,6P2) is twofold (Almeida et al. 2004). There are four isoforms of the enzyme responsible for the synthesis of F2,6P2, 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase (PFKFB). However, isoform 3 (PFKFB3) is, by far, the most abundant one found in astrocytes (Almeida et al. 2004). Interestingly, PFKFB3 has a high, ~ 700‐fold kinase versus bisphosphatase ratio (Pilkis et al. 1995). Hence, PFKFB3 levels are directly proportional to F2,6‐P2 synthesizing activity. PFKFB3 knockdown abolishes the ability of astrocytes to up‐regulate glycolysis upon mitochondrial inhibition, suggesting that PFKFB3 is necessary and sufficient to maintain the glycolytic phenotype of astrocytes (Almeida et al. 2004) (Fig. 1).
The transcriptome of the brain has been analyzed at single‐cell level by RNA‐sequencing, and it was found that PFKFB3, PK muscle‐2 isoform, lactate dehydrogenase B‐isoform (LDHB), and pyruvate dehydrogenase kinase 4‐isoform (PDK4) were highly expressed in astrocytes when compared with neurons (Zhang et al. 2014). This confirms previous findings on PFKFB3 levels and activity (Almeida et al. 2004), strengthening the notion that astrocytes are constitutively glycolytic (at the light of PFKFB3, PK muscle‐2 isoform, and LDHB mRNA levels) and independent on the OXPHOS (as judged by the high mRNA levels of pyruvate dehydrogenase kinase 4‐isoform, which phosphorylates and inhibits pyruvate dehydrogenase). In another study, the isoforms of LDH were shown to be differentially expressed in neurons and astrocytes, with neurons preferentially expressing LDH1 (mainly pyruvate‐producing tissues) and astrocytes expressing LDH5 (associated with high lactate‐producing tissues) (Pellerin et al. 1998). Furthermore, astrocytes were shown to express the monocarboxylate transporters‐1 and ‐4 (MCT1 and MCT4), which are responsible for lactate efflux, whereas neurons express MCT2, which is specialized for lactate influx (Pierre and Pellerin 2005). These data, taken together, support the notion that astrocytes continuously produce lactate through glycolysis and explain the presence of an intracellular lactate reservoir in these cells (Sotelo‐Hitschfeld et al. 2015), which is released through a channel upon extracellular rise in K+ or depolarization (Sotelo‐Hitschfeld et al. 2015).
Upon cellular energy stress, 5′‐AMP concentrations increase and facilitate activation 5′‐AMP‐activated protein kinase (AMPK) by phosphorylation of a Thr172 residue on its catalytic α1 subunit by the liver kinase‐1 (Sanders et al. 2007), which is a protein kinase with tumor suppression activity (Partanen et al. 2009). Active AMPK catalyzes the phosphorylation of several key metabolic regulatory enzymes, including PFKFB (Lage et al. 2008). In astrocytes, AMPK becomes phosphorylated, F2,6P2 levels elevated, and glycolysis activated within minutes of the inhibition of mitochondrial respiration by •NO or cyanide (Almeida et al. 2004). Knockdown of the α1 subunit of AMPK, or PFKFB3, abolishes F2,6P2 elevation and glycolysis activation (Almeida et al. 2004). Thus, energy stress up‐regulates glycolysis through AMPK‐mediated activation of PFKFB3 in astrocytes. There is a likely possibility that the glycolytic activation caused by glutamate uptake (Pellerin and Magistretti 1994) occurs through this AMPK‐PFKFB3 pathway, as suggested before (Ronnett et al. 2009), but this needs to be directly elucidated. Interestingly, the ATP/AMP ratio, as well as O2 levels and nutrients, such as glucose and amino acids, regulate the mammalian target of rapamycin (mTOR) (Wullschleger et al. 2006). Beside controlling key cellular functions, including energy metabolism or autophagy, mTOR regulates synaptic plasticity, memory storage, and cognition, this being the subject of a recent review (Bockaert and Marin 2015). Besides its critical importance in the tissue energy homeostasis, no evidence for the cellular localization and specific functions of mTOR on the bioenergetics and redox adaptations of brain cells to neural activity has been reported so far. The possible involvement of mTOR pathways in the control of brain energy metabolism during neural activity is therefore interesting and worthy of exploration.
Neurons are less glycolytic than astrocytes
While the widely spread notion is that glucose utilization through glycolysis by neurons is low and they depend on OXPHOS for energy generation (Knott et al. 2008), results obtained from different biologic preparations and brain regions are apparently controversial. Studies performed in rat cerebellar neurons in culture (Budd and Nicholls 1996), cortex synaptosomes (Kauppinen and Nicholls 1986; Choi et al. 2009), and retinal neurons (Xu et al. 2007; Bringmann et al. 2009) proposed glucose as the main energetic substrate. Noticeably, fast axonal transport of vesicles has a need for locally supplied ATP, which is met by glycolysis not mitochondria (Zala et al. 2013), and thus is a process that specifically requires glyceraldehyde‐3‐phosphate dehydrogenase. In addition, neurons have recently been reported to require glucose for synaptic activity (Lundgaard et al. 2015), although it remains unclear whether the metabolic fate of neuronally used glucose is glycolysis or pentose‐phosphate pathway (PPP) – known to be essential for antioxidant protection (Herrero‐Mendez et al. 2009) (see below). Moreover, there is question as to the reliability of the fluorescent probe, used in Lundgaard et al. (2015), to specifically assess glucose transport—in contrast to carrier endocytosis (Tadi et al. 2015). Furthermore, recently, using the genetically encoded lactate probe Laconic, it has been provided the first in vivo evidence for a lactate gradient from astrocytes to neurons (Maechler et al. 2016), a prerequisite for the occurrence of the ANLS. Accordingly, glucose does not seem to be used for the energy generation needed during neural activity, at least by cortical and hippocampal neurons, except for in fast vesicle transport through the axons (Zala et al. 2013).
Studies aimed at understanding why neurons do not rely on glycolysis for energy generation found PFKFB3 protein to be virtually absent in neurons (Almeida et al. 2004), both in culture and in vivo, because of the continuous destabilization by the ubiquitin‐proteasome pathway (Herrero‐Mendez et al. 2009). Thus, PFKFB3 – not PFKFB1, ‐2 or ‐4 – contains a 142Lys‐Glu‐Asn (KEN) box that targets it for ubiquitination by the anaphase‐promoting complex/cyclosome (APC/C)‐Cdh1 (Herrero‐Mendez et al. 2009), which is an E3 ubiquitine ligase known for its roles in the regulation of mitosis, meiosis (Pesin and Orr‐Weaver 2008), tumor suppression, and genome stability (Garcia‐Higuera et al. 2008). Besides these roles, APC/C‐Cdh1 regulates important functions in neurons, such as axonal growth (Konishi et al. 2004; Harmey et al. 2009; Huynh et al. 2009), cortical neurogenesis and size (Delgado‐Esteban et al. 2013), and survival (Almeida et al. 2005; Stegmuller and Bonni 2005; Maestre et al. 2008). In cortical neurons, Cdh1 knockdown leads to PFKFB3 accumulation, which is sufficient to increase glycolysis (Herrero‐Mendez et al. 2009). This was the first observation to describe a role for a cell cycle‐related protein (APC/C‐Cdh1) in metabolism, which was mimicked by PFKFB3 full‐length cDNA over‐expression (Herrero‐Mendez et al. 2009). In contrast, Cdh1 protein levels and APC/C‐Cdh1 ubiquitylating activity are very low in astrocytes, which explains their high levels of PFKFB3 and glycolytic activity (Herrero‐Mendez et al. 2009) (Fig. 1).
Metabolic fate of neuronal glucose
The rate of [6‐14C]glucose incorporated into 14CO2 (i.e., an index of glucose that is oxidized in the TCA cycle after having been converted into pyruvate) is negligible in cortical neurons when compared with astrocytes (Garcia‐Nogales et al. 2003; Herrero‐Mendez et al. 2009). Glucose is not actively transformed into lactate either, as judged by the low rate of [U‐14C]glucose incorporation into 14C‐lactate in neurons (Herrero‐Mendez et al. 2009). Lastly, glycolytic rate, assessed as the rate of 3H2O formation from [3‐3H]glucose and thus accurately reflecting the flux of glucose through glycolysis (Bouzier‐Sore and Bolanos 2015), is ~ 4‐5‐fold lower in neurons than in astrocytes (Herrero‐Mendez et al. 2009). It seems, therefore, likely that the neuronal ability to perform glycolysis is limited. Interestingly, over‐expression of PFKFB3 leading to up‐regulation of glycolysis causes oxidative stress and neuronal death (Herrero‐Mendez et al. 2009), whereas over‐activation of glutamate receptors, which inhibits APC/C‐Cdh1 (Maestre et al. 2008), stabilizes endogenous PFKFB3 protein causing neuronal death that can be rescued by knocking down PFKFB3 (Rodriguez‐Rodriguez et al. 2012). Together, these results strongly suggest that high glycolysis is not safe for neurons.
PPP converts glucose‐6‐phosphate (G6P) into ribulose‐5‐phosphate (R5P) in three consecutive steps, the first one catalyzed by G6P dehydrogenase (G6PD), which forms 6‐phosphogluconolactone; 6‐phosphogluconolactone is then converted into 6‐phosphogluconate (6PG) by a lactonase, followed by its decarboxylation into R5P by 6PG dehydrogenase (Wamelink et al. 2008; Bouzier‐Sore and Bolanos 2015). Two of these reactions (G6PD and 6PG dehydrogenase) occur, each, at the expense of one mole of NADPH(H+) regenerated per mole of substrate, and constitute the oxidative branch of the PPP. Thus, this branch of the PPP can regenerate 2 moles of NADPH(H+) per mole of G6P entering the pathway with the loss of one carbon atom as CO2. This branch (the non‐oxidative branch) is followed by a series of reactions that produce, from three moles of R5P, one mole of the glycolytic intermediates fructose‐6‐phosphate (F6P) and glyceraldehyde‐3‐phosphate (Wamelink et al. 2008; Bouzier‐Sore and Bolanos 2015). The oxidative PPP branch is therefore a process that conserves the glucose redox energy in reducing NADP+ to NADPH(H+), a necessary cofactor for antioxidant (GSH) regeneration (Wamelink et al. 2008).
G6PD catalyzes the PPP rate‐limiting reaction (Eggleston and Krebs 1974). A large body of evidence now supports the notion that the PPP has antioxidant and cytoprotective roles. In cortical neurons, exogenous H2O2 stimulates PPP activity and regenerates NADPH(H+) and GSH, thus contributing to neuroprotection (Ben‐Yoseph et al. 1996). By stimulating glutamate receptors in neurons, GSH becomes oxidized to oxidized glutathione, which triggers an increase in the rate of glucose oxidation through the PPP to regenerate neuroprotective NADPH(H+) and GSH (Delgado‐Esteban et al. 2000). At low doses, peroxynitrite activates G6PD, causing an enhancement of neuroprotective PPP activity in neurons (Garcia‐Nogales et al. 2003). Moreover, inhibition of APC/C‐Cdh1 activity leading to PFKFB3 stabilization or PFKFB3 over‐expression shifts glucose‐6‐phosphate utilization from PPP to glycolysis causing neuronal death (Herrero‐Mendez et al. 2009). Taken together, these results strongly suggest that neurons use glucose through the PPP as an essential survival pathway (Vaughn and Deshmukh 2008; Herrero‐Mendez et al. 2009). Thus, by using glucose preferentially through the PPP for antioxidant purposes (Herrero‐Mendez et al.,2009), neurons can mainly rely on ANLS‐derived lactate as metabolic fuel (Kasparov 2016; Maechler et al. 2016). Furthermore, APC/C‐Cdh1 is likely to coordinate the metabolic adaptation of neurons and astrocytes to the astrocyte‐neuronal lactate shuttle (Pellerin and Magistretti 1994, 2012; Bouzier‐Sore et al. 2003; Magistretti 2006; Allaman et al. 2011) (Fig. 1).
An astrocyte‐neuronal glutathione shuttle couples neural activity with redox balance
Neurotransmission unavoidably increases mitochondrial ROS in neurons, possibly because of mitochondrial Ca2+ influx (Mattson and Liu 2002). However, neuronal antioxidant machinery is generally weak (Makar et al. 1994; Bolaños et al. 1995, 1996), albeit provided with some intrinsic defense (Papadia et al. 2008; Deighton et al. 2014; Baxter et al. 2015). The antioxidant defense of neurons is repressed because of continuous protein destabilization of the master antioxidant transcriptional activator, nuclear factor‐erythroid 2‐related factor‐2 (Nrf2), by Cullin 3/Kelch‐like ECH‐associated protein 1 (Bell et al. 2015; Jimenez‐Blasco et al. 2015). In contrast, Nrf2 is highly stable in neighbor astrocytes, which explains their robust antioxidant defense and resistance against oxidative stress (Habas et al. 2013; Jimenez‐Blasco et al. 2015), although this notion has been disputed (Haskew‐Layton et al. 2010). Moreover, astrocytes release GSH precursors, which neurons can use for the de novo GSH biosynthesis (Dringen et al. 1999), a system that contributes to neuroprotective ischemic preconditioning (Bell et al. 2011). However, a definitive answer as to whether astrocytes sense neurotransmission to activate the release of GSH precursors upon neuronal activity has long remained elusive. Like post‐synaptic neurons, astrocytes express glutamate receptors (Conti et al. 1996; Schipke et al. 2001; Seifert and Steinhäuser 2001; Verkhratsky and Kirchhoff 2007; Lee et al. 2010; Palygin et al. 2011), though their function in these glial cells has been enigmatic.
Recently, it was shown that subtle and persistent stimulation of glutamate receptors in astrocytes, through a mechanism not requiring extracellular Ca2+ influx, up‐regulates a signal transduction pathway involving phospholipase C‐mediated endoplasmic reticulum release of Ca2+ and protein kinase Cδ activation. Through phosphorylation, active protein kinase Cδ promotes the stabilization of p35, a cyclin‐dependent kinase‐5 (Cdk5) cofactor. Active p35/Cdk5 complex in the cytosol phosphorylates Nrf2 at Thr395, Ser433, and Thr439 that is sufficient to promote Nrf2 translocation to the nucleus and induce the expression of antioxidant genes. Furthermore, this Cdk5‐Nrf2 transduction pathway boosts GSH metabolism in astrocytes efficiently protecting closely spaced neurons against oxidative damage. These results demonstrate that neural activity is coupled with astrocyte release of GSH for neuronal de novo GSH biosynthesis (astrocyte‐neuronal glutathione shuttle or ANGS) as a strategy for balancing the neuronal redox status (Jimenez‐Blasco et al. 2015) (Fig. 2).
Figure 2.
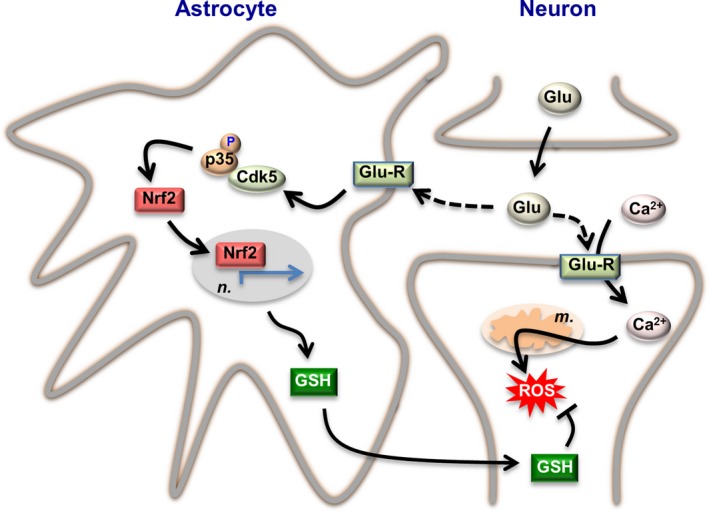
Redox adaptation of astrocytes to neurotransmission. Synaptic cleft glutamate (Glu) released by the pre‐synaptic neuron acts on glutamate receptors (Glu‐R) placed both in the post‐synaptic neuron and astrocytes. Part of intracellular Ca2+ at the post‐synaptic neurons is removed from the cytosol by entering mitochondria, and this causes mitochondrial production of reactive oxygen species (ROS). Synaptic cleft glutamate interacts with its receptors placed in astrocytes, triggering a cascade of events via cyclin‐dependent kinase‐5 (Cdk5)‐mediated phosphorylation of Nrf2 (nuclear factor‐erythroid 2‐related factor‐2), which enters the nucleus (n.) and binds to the antioxidant responsive elements (ARE) to promote the expression of antioxidant genes (green arrowed lines). This pathway leads to the biosynthesis and release of glutathione (GSH), whose precursors are taken up by neurons for the de novo GSH biosynthesis necessary to detoxify neuronal activity‐mediated mitochondrial ROS. Thus, through this astrocyte‐neuronal glutathione shuttle, astrocytes sustain the redox status of neurons during neural activity. The stoichiometry of the reactions has been omitted for clarity. Likewise, additional factors involved in these adaptations could not be depicted herein and can be found in the main text.
Pathophysiological implications of disruption in energy and redox adaptations of astrocytes
Disruption of the metabolic and redox adaptations of astrocytes and neurons to neural activity cause neuronal death and is likely to play important roles in neurological diseases. A condition underlying several of these diseases is the excitotoxic phenomenon, in which there is a Ca2+‐dependent component following glutamate receptor over‐activation‐mediated neuronal death (Choi 1987). Besides the rapid increase in cytosolic Ca2+ by N‐methyl‐d‐aspartate (NMDA) receptor over‐stimulation, there is a delayed Ca2+ deregulation process, which persists even after glutamate removal from the synaptic cleft, which is responsible for the activation of secondary cascades, notably those involving calpain (Brustovetsky et al. 2010). Calpain triggers the proteolytic cleavage of the Na+/Ca2+ exchanger, a major plasma membrane system for Ca2+ extruding, thus impairing Ca2+ homeostasis and leading to neuronal death (Bano et al. 2005; Brustovetsky et al. 2010). Furthermore, the increase in intracellular Ca2+ causes a mitochondrial Ca2+ overload responsible for enhanced ROS formation and cytochrome c release, both playing a crucial role in glutamate‐induced excitotoxicity (Luetjens et al. 2000). Oxidative stress associated with excitotoxicity also leads to mitochondrial fragmentation, an observation that concurs in several neurodegenerative diseases (Knott et al. 2008; Nguyen et al. 2011). Moreover, mitochondrial dynamics imbalance can trigger NMDA receptor up‐regulation, further contributing to propagate the excitotoxic process (Nguyen et al. 2011).
A bioenergetics‐redox component in this excitotoxicity cascade has also been shown. Upon a glutamatergic excitotoxic stimulus in cortical neurons, APC/C‐Cdh1 is inhibited by Cdk5‐mediated phosphorylation of Cdh1 (Maestre et al. 2008), a process that is triggered by Ca2+‐dependent calpain activation (Maestre et al. 2008). This leads to PFKFB3 stabilization triggering neuronal death as a consequence of the PPP inhibition that leads to oxidative stress (Rodriguez‐Rodriguez et al. 2012). Interestingly, PFKFB3 knockdown in cultured human astrocytes leads to extracellular ß‐amyloid accumulation (Fu et al. 2015). Moreover, PFKFB3 is increased in the astrocytes of an Alzheimer's disease mouse model that over‐expresses ß‐amyloid (Fu et al. 2015). Specific inhibition of GSH released from astrocyte triggers neuronal death in co‐culture systems (Jimenez‐Blasco et al. 2015), and disruption of astrocyte function in adult mice causes oxygen and nitrogen redox species imbalance of neurons in vivo (Schreiner et al. 2015). Glucose metabolism is altered in the brains of Alzheimer's disease patients (Silverman et al. 2001), and ß‐amyloid causes an enhancement in the flux of glucose utilization via the PPP (Soucek et al. 2003). Interestingly, increased G6PD levels are found in the surviving pyramidal neurons of hippocampal slices from the post‐mortem brains of Alzheimer's disease patients (Palmer 1999; Russell et al. 1999). In a cell model of Huntington's disease there is evidence for inhibition of the PPP and decreased mitochondrial NADH(H+)/NAD+ ratio (Ferreira et al. 2011). Together, these findings strongly suggest that during the oxidative stress associated with human brain pathologies, the consumption of glucose through the PPP is critical for maintaining the neuronal antioxidant redox status and survival. Thus, besides their critical role in controlling energy metabolism, astrocyte redox metabolism is also adapted to protect neurons against oxidative stress and neurodegeneration, likely playing an important role in neurological disorders.
Concluding remarks and perspectives
Astrocytes and neurons are metabolically programmed to spatiotemporally deal with the energy and redox requirements of neural activity. Astrocytes show a prominent constitutive glycolytic metabolism whereas neurons do not, and this is coordinated by the E3 ubiquitin ligase APC/C‐Cdh1 (Herrero‐Mendez et al. 2009). High Cdh1 levels keep APC/C active, thus destabilizing the pro‐glycolytic enzyme PFKFB3; whereas low Cdh1 levels in astrocytes allows PFKFB3 stabilization responsible for the high glycolytic phenotype (Herrero‐Mendez et al. 2009; Bolaños et al. 2010) (Fig. 1). In addition, messengers such as •NO or NH4 + also contribute to the glycolytic phenotype of astrocytes (Almeida et al. 2004; Brix et al. 2012; Lerchundi et al. 2015), and an appropriate redox balance is critically important to sustain antioxidant protection during neural activity. Neurons spare glucose for its oxidation through the PPP, which serves to regenerate GSH and exert protection against oxidative stress (Vaughn and Deshmukh 2008; Herrero‐Mendez et al. 2009). In addition, neurons constitutively destabilize Nrf2, whereas astrocytes accumulate it (Jimenez‐Blasco et al. 2015). Thus, neurons rely on astrocyte‐derived precursors for de novo GSH biosynthesis (Dringen et al. 1999), and in astrocytes neural activity triggers the signaling pathway needed to activate this redox regulatory system (Jimenez‐Blasco et al. 2015) (Fig. 2). Such mechanisms do not exclude the occurrence of other, still unknown systems helping neural cells adapt their metabolism to the necessary rapid changes occurring during neural activity. These may include mTOR (Bockaert and Marin 2015) and/or hypoxia‐inducible factor‐1 (Brix et al. 2012) signaling pathways, the importance of which is still elusive. In any case, the metabolic status of neural cells would favor the necessary spatiotemporal changes in energy homeostasis following neural activity to satisfy neuronal needs. Recent studies in Drosophila (Volkenhoff et al. 2015) and in mice (Sada et al. 2015; Tadi et al. 2015) confirm the occurrence of the metabolic adaptations of astrocytes in vivo. The use of novel fluorescent probes has confirmed the key role of glycolytic‐derived lactate from astrocytes during neural activity at a real‐time resolution (Sotelo‐Hitschfeld et al. 2015), suggesting dual roles for lactate as metabolic fuel (Pellerin and Magistretti 2012) and intercellular messenger (Barros 2013). Further advances in similar tools would be desirable to investigate roles for neuronal use of glucose through the PPP as an antioxidant strategy during neural activity (Bouzier‐Sore and Bolanos 2015). Thus prominent neurochemical advances have been made over the past few decades in our understanding of the physiological mechanisms that coordinate the metabolic adaptations of neural cells to neurotransmission. Key conserved pathways have been identified that are regulated by specific molecules, the disruption of which causes neural problems. Therefore, it seems reasonable to move forward and develop therapeutic interventions aimed to interfere with these targets for the treatment of neurological disorders.
Acknowledgments and conflict of interest disclosure
J.P.B. is funded by the MINECO (SAF2013‐41177‐R; RTC‐2015‐3237‐1), the Instituto de Salud Carlos III (RD12/0043/0021), the E.U. SP3‐People‐MC‐ITN programme (608381), the E.U. BATCure grant (666918), the European Regional Development Fund, and the NIH/NIDA (1R21DA037678‐01). The author has no conflict of interest to declare.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Allaman I., Belanger M. and Magistretti P. J. (2011) Astrocyte‐neuron metabolic relationships: for better and for worse. Trends Neurosci. 34, 76–87. [DOI] [PubMed] [Google Scholar]
- Allen N. J. and Barres B. A. (2009) Neuroscience: Glia ‐ more than just brain glue. Nature 457, 675–677. [DOI] [PubMed] [Google Scholar]
- Almeida A. and Bolaños J. P. (2001) A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J. Neurochem. 77, 676–690. [DOI] [PubMed] [Google Scholar]
- Almeida A., Almeida J., Bolaños J. P. and Moncada S. (2001) Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically‐generated ATP in astrocyte protection. Proc. Natl Acad. Sci. USA 98, 15294–15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A., Moncada S. and Bolaños J. P. (2004) Nitric oxide switches on glycolysis through the AMP protein kinase and 6‐phosphofructo‐2‐kinase pathway. Nat. Cell Biol. 6, 45–51. [DOI] [PubMed] [Google Scholar]
- Almeida A., Bolaños J. P. and Moreno S. (2005) Cdh1/Hct1‐APC is essential for the survival of postmitotic neurons. J. Neurosci. 25, 8115–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak L. K., Schousboe A., Sonnewald U. and Waagepetersen H. S. (2006) Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J. Cereb. Blood Flow Metab. 26, 1285–1297. [DOI] [PubMed] [Google Scholar]
- Bak L. K., Walls A. B., Schousboe A., Ring A., Sonnewald U. and Waagepetersen H. S. (2009) Neuronal glucose but not lactate utilization is positively correlated with NMDA‐induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 109(Suppl 1), 87–93. [DOI] [PubMed] [Google Scholar]
- Bak L. K., Obel L. F., Walls A. B., Schousboe A., Faek S. A., Jajo F. S. and Waagepetersen H. S. (2012) Novel model of neuronal bioenergetics: postsynaptic utilization of glucose but not lactate correlates positively with Ca2+ signalling in cultured mouse glutamatergic neurons. ASN Neuro. 4, e00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano D., Young K. W., Guerin C. J., Lefeuvre R., Rothwell N. J., Naldini L., Rizzuto R., Carafoli E. and Nicotera P. (2005) Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120, 275–285. [DOI] [PubMed] [Google Scholar]
- Barros L. F. (2013) Metabolic signaling by lactate in the brain. Trends Neurosci. 36, 396–404. [DOI] [PubMed] [Google Scholar]
- Baxter P. S., Bell K. F., Hasel P., Kaindl A. M., Fricker M., Thomson D., Cregan S. P., Gillingwater T. H. and Hardingham G. E. (2015) Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 6, 6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell K. F., Al‐Mubarak B., Fowler J. H. et al (2011) Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proc. Natl Acad. Sci. USA 108, E1–E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell K. F., Al‐Mubarak B., Martel M. A. et al (2015) Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 6, 7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran B., Mathur A., Duchen M. R., Erusalimsky J. D. and Moncada S. (2000) The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc. Natl Acad. Sci. USA 97, 14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Yoseph O., Boxer P. A. and Ross B. D. (1996) Assessment of the role of the glutathione and pentose phosphate pathways in the protection of primary cerebrocortical cultures from oxidative stress. J. Neurochem. 66, 2329–2337. [DOI] [PubMed] [Google Scholar]
- Bittner C. X., Valdebenito R., Ruminot I. et al (2011) Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. J. Neurosci. 31, 4709–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J. and Marin P. (2015) mTOR in Brain Physiology and Pathologies. Physiol. Rev. 95, 1157–1187. [DOI] [PubMed] [Google Scholar]
- Bolaños J. P., Peuchen S., Heales S. J. R., Land J. M. and Clark J. B. (1994) Nitric oxide‐mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J. Neurochem. 63, 910–916. [DOI] [PubMed] [Google Scholar]
- Bolaños J. P., Heales S. J. R., Land J. M. and Clark J. B. (1995) Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary cultures. J. Neurochem. 64, 1965–1972. [DOI] [PubMed] [Google Scholar]
- Bolaños J. P., Heales S. J. R., Peuchen S., Barker J. E., Land J. M. and Clark J. B. (1996) Nitric oxide‐mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic. Biol. Med. 21, 995–1001. [DOI] [PubMed] [Google Scholar]
- Bolaños J. P., Almeida A. and Moncada S. (2010) Glycolysis: a bioenergetic or a survival pathway?. Trends Biochem. Sci. 35, 145–149. [DOI] [PubMed] [Google Scholar]
- Bouzier‐Sore A. K. and Bolanos J. P. (2015) Uncertainties in pentose‐phosphate pathway flux assessment underestimate its contribution to neuronal glucose consumption: relevance for neurodegeneration and aging. Front. Aging Neurosci. 7, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier‐Sore A. K., Voisin P., Canioni P., Magistretti P. J. and Pellerin L. (2003) Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J. Cereb. Blood Flow Metab. 23, 1298–1306. [DOI] [PubMed] [Google Scholar]
- Bouzier‐Sore A. K., Voisin P., Bouchaud V., Bezancon E., Franconi J. M. and Pellerin L. (2006) Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur. J. Neurosci. 24, 1687–1694. [DOI] [PubMed] [Google Scholar]
- Bringmann A., Pannicke T., Biedermann B., Francke M., Iandiev I., Grosche J., Wiedemann P., Albrecht J. and Reichenbach A. (2009) Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem. Int. 54, 143–160. [DOI] [PubMed] [Google Scholar]
- Brix B., Mesters J. R., Pellerin L. and Johren O. (2012) Endothelial cell‐derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF‐1alpha‐mediated target gene activation. J. Neurosci. 32, 9727–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. C. and Cooper C. E. (1994) Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 356, 295–298. [DOI] [PubMed] [Google Scholar]
- Brown A. M. and Ransom B. R. (2007) Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Brustovetsky T., Bolshakov A. and Brustovetsky N. (2010) Calpain activation and Na+/Ca2+ exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 88, 1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd S. L. and Nicholls D. G. (1996) A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J. Neurochem. 66, 403–411. [DOI] [PubMed] [Google Scholar]
- Cai Q. and Sheng Z. H. (2009) Molecular motors and synaptic assembly. Neuroscientist 15, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. (1987) Ionic dependence of glutamate neurotoxicity in cortical cell culture. J. Neurosci. 7, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. W., Gerencser A. A. and Nicholls D. G. (2009) Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem. 109, 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleeter M. W. J., Cooper J. M., Darley‐Usmar V. M., Moncada S. and Schapira A. H. (1994) Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 345, 50–54. [DOI] [PubMed] [Google Scholar]
- Conti F., DeBiasi S., Minelli A. and Melone M. (1996) Expression of NR1 and NR2A/B subunits of the NMDA receptor in cortical astrocytes. Glia 17, 254–258. [DOI] [PubMed] [Google Scholar]
- Deighton R. F., Markus N. M., Al‐Mubarak B., Bell K. F., Papadia S., Meakin P. J., Chowdhry S., Hayes J. D. and Hardingham G. E. (2014) Nrf2 target genes can be controlled by neuronal activity in the absence of Nrf2 and astrocytes. Proc. Natl Acad. Sci. USA 111, E1818–E1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado‐Esteban M., Almeida A. and Bolaños J. P. (2000) D‐Glucose prevents glutathione oxidation and mitochondrial damage after glutamate receptor stimulation in rat cortical primary neurons. J. Neurochem. 75, 1618–1624. [DOI] [PubMed] [Google Scholar]
- Delgado‐Esteban M., Garcia‐Higuera I., Maestre C., Moreno S. and Almeida A. (2013) APC/C‐Cdh1 coordinates neurogenesis and cortical size during development. Nat. Commun. 4, 2879. [DOI] [PubMed] [Google Scholar]
- Dringen R., Pfeiffer B. and Hamprecht B. (1999) Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 19, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J. and Guinovart J. J. (2015) Brain glycogen in health and disease. Mol. Aspects Med. 46, 70–77. doi: 10.1016/j.mam.2015.08.007. [DOI] [PubMed] [Google Scholar]
- Eggleston L. V. and Krebs H. A. (1974) Regulation of the pentose phosphate cycle. Biochem. J. 138, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Moncada I. and Barros L. F. (2014) Non‐preferential fuelling of the Na(+)/K(+)‐ATPase pump. Biochem. J. 460, 353–361. [DOI] [PubMed] [Google Scholar]
- Ferreira I. L., Cunha‐Oliveira T., Nascimento M. V., Ribeiro M., Proenca M. T., Januario C., Oliveira C. R. and Rego A. C. (2011) Bioenergetic dysfunction in Huntington's disease human cybrids. Exp. Neurol. 231, 127–134. [DOI] [PubMed] [Google Scholar]
- Fox P. T. and Raichle M. E. (1986) Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc. Natl Acad. Sci. USA 83, 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W., Shi D., Westaway D. and Jhamandas J. H. (2015) Bioenergetic mechanisms in astrocytes may contribute to amyloid plaque deposition and toxicity. J. Biol. Chem. 290, 12504–12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia O., Almeida A., Massieu L. and Bolaños J. P. (2005) Increased mitochondrial respiration maintains the mitochondrial membrane potential and promotes survival of cerebellar neurons in an endogenous model of glutamate receptor activation. J. Neurochem. 92, 183–190. [DOI] [PubMed] [Google Scholar]
- Garcia‐Higuera I., Manchado E., Dubus P., Canamero M., Mendez J., Moreno S. and Malumbres M. (2008) Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 10, 802–811. [DOI] [PubMed] [Google Scholar]
- Garcia‐Nogales P., Almeida A. and Bolaños J. P. (2003) Peroxynitrite protects neurons against nitric oxide‐mediated apoptosis. A key role for glucose‐6‐phosphate dehydrogenase activity in neuroprotection. J. Biol. Chem. 278, 864–874. [DOI] [PubMed] [Google Scholar]
- Garthwaite J., Charles S. L. and Chess‐Williams R. (1988) Endothelium‐derived relaxing factor release on activation of NMDA receptors suggests a role as intercellular messenger in the brain. Nature 336, 385–387. [DOI] [PubMed] [Google Scholar]
- Gleichmann M., Collis L. P., Smith P. J. and Mattson M. P. (2009) Simultaneous single neuron recording of O2 consumption, [Ca2+]i and mitochondrial membrane potential in glutamate toxicity. J. Neurochem. 109, 644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habas A., Hahn J., Wang X. and Margeta M. (2013) Neuronal activity regulates astrocytic Nrf2 signaling. Proc. Natl Acad. Sci. USA 110, 18291–18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmey D., Smith A., Simanski S., Moussa C. Z. and Ayad N. G. (2009) The anaphase promoting complex induces substrate degradation during neuronal differentiation. J. Biol. Chem. 284, 4317–4323. [DOI] [PubMed] [Google Scholar]
- Haskew‐Layton R. E., Payappilly J. B., Smirnova N. A. et al (2010) Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2‐independent pathway. Proc. Natl Acad. Sci. USA 107, 17385–17390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero‐Mendez A., Almeida A., Fernandez E., Maestre C., Moncada S. and Bolaños J. P. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C‐Cdh1. Nat. Cell Biol. 11, 747–752. [DOI] [PubMed] [Google Scholar]
- Hertz L., Peng L. and Dienel G. A. (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 27, 219–249. [DOI] [PubMed] [Google Scholar]
- Huynh M. A., Stegmuller J., Litterman N. and Bonni A. (2009) Regulation of Cdh1‐APC function in axon growth by Cdh1 phosphorylation. J. Neurosci. 29, 4322–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. I., Malkov A. E., Waseem T., Mukhtarov M., Buldakova S., Gubkina O., Zilberter M. and Zilberter Y. (2014) Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow Metab. 34, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekabsons M. B. and Nicholls D. G. (2004) In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J. Biol. Chem. 279, 32989–33000. [DOI] [PubMed] [Google Scholar]
- Jimenez‐Blasco D., Santofimia‐Castano P., Gonzalez A., Almeida A. and Bolaños J. P. (2015) Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5‐Nrf2 pathway. Cell Death Differ. 22, 1877–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasparov S. (2016) Are Astrocytes the Pressure‐Reservoirs of Lactate in the Brain? (2016) Cell Metabolism, http://dx.doi.org/10.1016/j.cmet.2015.11.001 [DOI] [PubMed] [Google Scholar]
- Kauppinen R. A. and Nicholls D. G. (1986) Failure to maintain glycolysis in anoxic nerve terminals. J. Neurochem. 47, 1864–1869. [DOI] [PubMed] [Google Scholar]
- Knott A. B., Perkins G., Schwarzenbacher R. and Bossy‐Wetzel E. (2008) Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y., Stegmuller J., Matsuda T., Bonni S. and Bonni A. (2004) Cdh1‐APC controls axonal growth and patterning in the mammalian brain. Science 303, 1026–1030. [DOI] [PubMed] [Google Scholar]
- Lage R., Dieguez C., Vidal‐Puig A. and Lopez M. (2008) AMPK: a metabolic gauge regulating whole‐body energy homeostasis. Trends Mol. Med. 14, 539–549. [DOI] [PubMed] [Google Scholar]
- Lee M., Ting K., Adams S., Brew B., Chung R. and Guillemin G. (2010) Characterisation of the expression of NMDA receptors in human astrocytes. PLoS ONE 30, e14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerchundi R., Fernandez‐Moncada I., Contreras‐Baeza Y. et al (2015) NH4 + triggers the release of astrocytic lactate via mitochondrial pyruvate shunting. Proc. Natl Acad. Sci. USA 112, 11090–11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loaiza A., Porras O. H. and Barros L. F. (2003) Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real‐time confocal microscopy. J. Neurosci. 23, 7337–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetjens C. M., Bui N. T., Sengpiel B., Munstermann G., Poppe M., Krohn A. J., Bauerbach E., Krieglstein J. and Prehn J. H. (2000) Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J. Neurosci. 20, 5715–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I., Li B., Xie L. et al (2015) Direct neuronal glucose uptake Heralds activitydependent increases incerebral metabolism. Nat. Commun. 6, 6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P., Wyss M. T., Elsayed M., Stobart J., Gutierrez R., von Faber‐Castell A., Kaelin V., Zuend M., San Martin A., Romero‐Gomez I., Baeza‐Lehnert F., Lengacher S., Schneider B. L., Aebischer P., Magistretti P. J., Barros L. F. and Weber B. (2016) In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metabolism, http://dx.doi.org/10.1016/j.cmet.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Maestre C., Delgado‐Esteban M., Gomez‐Sanchez J. C., Bolaños J. P. and Almeida A. (2008) Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 27, 2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti P. J. (2006) Neuron‐glia metabolic coupling and plasticity. J. Exp. Biol. 209, 2304–2311. [DOI] [PubMed] [Google Scholar]
- Magistretti P. J. and Allaman I. (2015) A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. [DOI] [PubMed] [Google Scholar]
- Makar T. K., Nedergaard M., Preuss A., Gelbard A. S., Perumal A. S. and Cooper A. J. L. (1994) Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurones: evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 62, 45–53. [DOI] [PubMed] [Google Scholar]
- Mattson M. P. and Liu D. (2002) Energetics and oxidative stress in synaptic plasticity and neurodegenerative disorders. Neuromolecular Med. 2, 215–231. [DOI] [PubMed] [Google Scholar]
- Nguyen D., Alavi M. V., Kim K. Y. et al (2011) A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2, e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. G. (2008) Oxidative stress and energy crises in neuronal dysfunction. Ann. N. Y. Acad. Sci. 1147, 53–60. [DOI] [PubMed] [Google Scholar]
- Nicholls D. G., Johnson‐Cadwell L., Vesce S., Jekabsons M. and Yadava N. (2007) Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J. Neurosci. Res. 85, 3206–3212. [DOI] [PubMed] [Google Scholar]
- Oliveira J. F., Sardinha V. M., Guerra‐Gomes S., Araque A. and Sousa N. (2015) Do stars govern our actions? Astrocyte involvement in rodent behavior. Trends Neurosci. 38, 535–549. [DOI] [PubMed] [Google Scholar]
- Palmer A. M. (1999) The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer's disease. J. Neural. Transm. 106, 317–328. [DOI] [PubMed] [Google Scholar]
- Palygin O., Lalo U. and Pankratov Y. (2011) Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br. J. Pharmacol. 163, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S., Soriano F. X., Leveille F. et al (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 11, 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V., Basarsky T. A., Liu F., Jeftinija K., Jeftinija S. and Haydon P. G. (1994) Glutamate‐mediated astrocyte‐neuron signalling. Nature 369, 744–747. [DOI] [PubMed] [Google Scholar]
- Partanen J. I., Nieminen A. I. and Klefstrom J. (2009) 3D view to tumor suppression: Lkb1, polarity and the arrest of oncogenic c‐Myc. Cell Cycle 8, 716–724. [DOI] [PubMed] [Google Scholar]
- Pellerin L. and Magistretti P. J. (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl Acad. Sci. USA 91, 10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L. and Magistretti P. J. (2012) Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 32, 1152–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L., Pellegri G., Bittar P. G., Charnay Y., Bouras C., Martin J. L., Stella N. and Magistretti P. J. (1998) Evidence supporting the existence of an activity‐dependent astrocyte‐neuron lactate shuttle. Dev. Neurosci. 20, 291–299. [DOI] [PubMed] [Google Scholar]
- Pellerin L., Bouzier‐Sore A. K., Aubert A., Serres S., Merle M., Costalat R. and Magistretti P. J. (2007) Activity‐dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262. [DOI] [PubMed] [Google Scholar]
- Perea G., Sur M. and Araque A. (2014) Neuron‐glia networks: integral gear of brain function. Front. Cell. Neurosci. 8, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesin J. A. and Orr‐Weaver T. L. (2008) Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 24, 475–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre K. and Pellerin L. (2005) Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem. 94, 1–14. [DOI] [PubMed] [Google Scholar]
- Pilkis S. J., Claus T. H., Kurland I. J. and Lange A. J. (1995) 6‐Phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase: a metabolic signaling enzyme. Annu. Rev. Biochem. 64, 799–835. [DOI] [PubMed] [Google Scholar]
- Porras O. H., Loaiza A. and Barros L. F. (2004) Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J. Neurosci. 24, 9669–9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras O. H., Ruminot I., Loaiza A. and Barros L. F. (2008) Na+‐Ca2+ cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia 56, 59–68. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Rodriguez P., Fernandez E., Almeida A. and Bolaños J. P. (2012) Excitotoxic stimulus stabilizes PFKFB3 causing pentose‐phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ. 19, 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnett G. V., Ramamurthy S., Kleman A. M., Landree L. E. and Aja S. (2009) AMPK in the brain: its roles in energy balance and neuroprotection. J. Neurochem. 109(Suppl 1), 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. L., Siedlak S. L., Raina A. K., Bautista J. M., Smith M. A. and Perry G. (1999) Increased neuronal glucose‐6‐phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 370, 236–239. [DOI] [PubMed] [Google Scholar]
- Sada N., Lee S., Katsu T., Otsuki T. and Inoue T. (2015) Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367. [DOI] [PubMed] [Google Scholar]
- Sanders M. J., Grondin P. O., Hegarty B. D., Snowden M. A. and Carling D. (2007) Investigating the mechanism for AMP activation of the AMP‐activated protein kinase cascade. Biochem. J. 403, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipke C., Ohlemeyer C., Matyash M., Nolte C., Kettenmann H. and Kirchhoff F. (2001) Astrocytes of the mouse neocortex express functional N‐methyl‐d‐aspartate receptors. FASEB J. 15, 1270–1272. [DOI] [PubMed] [Google Scholar]
- Schreiner B., Romanelli E., Liberski P. et al (2015) Astrocyte depletion impairs redox homeostasis and triggers neuronal loss in the adult CNS. Cell Rep. 12, 1377–1384. [DOI] [PubMed] [Google Scholar]
- Schweizer M. and Richter C. (1994) Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem. Biophys. Res. Commun. 204, 169–175. [DOI] [PubMed] [Google Scholar]
- Seifert G. and Steinhäuser C. (2001) Ionotropic glutamate receptors in astrocytes. Prog. Brain Res. 132, 287–299. [DOI] [PubMed] [Google Scholar]
- Silverman D. H., Small G. W., Chang C. Y. et al (2001) Positron emission tomography in evaluation of dementia: regional brain metabolism and long‐term outcome. JAMA 286, 2120–2127. [DOI] [PubMed] [Google Scholar]
- Sotelo‐Hitschfeld T., Fernandez‐Moncada I. and Barros L. F. (2012) Acute feedback control of astrocytic glycolysis by lactate. Glia 60, 674–680. [DOI] [PubMed] [Google Scholar]
- Sotelo‐Hitschfeld T., Niemeyer M. I., Machler P. et al (2015) Channel‐mediated lactate release by K(+)‐stimulated astrocytes. J. Neurosci. 35, 4168–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek T., Cumming R., Dargusch R., Maher P. and Schubert D. (2003) The regulation of glucose metabolism by HIF‐1 mediates a neuroprotective response to amyloid beta peptide. Neuron 39, 43–56. [DOI] [PubMed] [Google Scholar]
- Stegmuller J. and Bonni A. (2005) Moving past proliferation: new roles for Cdh1‐APC in postmitotic neurons. Trends Neurosci. 28, 596–601. [DOI] [PubMed] [Google Scholar]
- Suh S. W., Bergher J. P., Anderson C. M., Treadway J. L., Fosgerau K. and Swanson R. A. (2007) Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP‐316,819 ([R‐R*, S*]‐5‐chloro‐N‐[2‐hydroxy‐3‐(methoxymethylamino)‐3‐oxo‐1‐(phenylmethyl)propyl]‐1H‐indole‐2 carboxamide). J. Pharmacol. Exp. Ther. 321, 45–50. [DOI] [PubMed] [Google Scholar]
- Tadi M., Allaman I., Lengacher S., Grenningloh G. and Magistretti P. J. (2015) Learning‐induced gene expression in the hippocampus reveals a role of neuron ‐astrocyte metabolic coupling in long term memory. PLoS ONE 10, e0141568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. K., Peterson M. R. and Freeman R. D. (2003) Single‐neuron activity and tissue oxygenation in the cerebral cortex. Science 299, 1070–1072. [DOI] [PubMed] [Google Scholar]
- Vaughn A. E. and Deshmukh M. (2008) Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c . Nat. Cell Biol. 10, 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. and Kirchhoff F. (2007) NMDA Receptors in glia. Neuroscientist 13, 28–37. [DOI] [PubMed] [Google Scholar]
- Vilchez D., Ros S., Cifuentes D. et al (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413. [DOI] [PubMed] [Google Scholar]
- Volkenhoff A., Weiler A., Letzel M., Stehling M., Klambt C. and Schirmeier S. (2015) Glial glycolysis is essential for neuronal survival in drosophila. Cell Metab. 22, 437–447. [DOI] [PubMed] [Google Scholar]
- Wamelink M. M., Struys E. A. and Jakobs C. (2008) The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J. Inherit. Metab. Dis. 31, 703–717. [DOI] [PubMed] [Google Scholar]
- Wullschleger S., Loewith R. and Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484. [DOI] [PubMed] [Google Scholar]
- Xu Y., Ola M. S., Berkich D. A., Gardner T. W., Barber A. J., Palmieri F., Hutson S. M. and LaNoue K. F. (2007) Energy sources for glutamate neurotransmission in the retina: absence of the aspartate/glutamate carrier produces reliance on glycolysis in glia. J. Neurochem. 101, 120–131. [DOI] [PubMed] [Google Scholar]
- Zala D., Hinckelmann M. V., Yu H., Lyra da Cunha M. M., Liot G., Cordelieres F. P., Marco S. and Saudou F. (2013) Vesicular glycolysis provides on‐board energy for fast axonal transport. Cell 152, 479–491. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Chen K., Sloan S. A. et al (2014) An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]