

Abstract
Motivation: Functional protein–protein interaction (PPI) networks elucidate molecular pathways underlying complex phenotypes, including those of human diseases. Extrapolation of domain–domain interactions (DDIs) from known PPIs is a major domain-based method for inferring functional PPI networks. However, the protein domain is a functional unit of the protein. Therefore, we should be able to effectively infer functional interactions between proteins based on the co-occurrence of domains.
Results: Here, we present a method for inferring accurate functional PPIs based on the similarity of domain composition between proteins by weighted mutual information (MI) that assigned different weights to the domains based on their genome-wide frequencies. Weighted MI outperforms other domain-based network inference methods and is highly predictive for pathways as well as phenotypes. A genome-scale human functional network determined by our method reveals numerous communities that are significantly associated with known pathways and diseases. Domain-based functional networks may, therefore, have potential applications in mapping domain-to-pathway or domain-to-phenotype associations.
Availability and Implementation: Source code for calculating weighted mutual information based on the domain profile matrix is available from www.netbiolab.org/w/WMI.
Contact: Insuklee@yonsei.ac.kr
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Proteins are major functional molecules that conduct cellular processes. Interactions between proteins are important for understanding the molecular mechanisms of complex traits, including diseases. For example, network-based analysis has recently gained popularity in interpreting data from genome-wide association studies (GWAS) and whole exome sequencing (WES) for human diseases such as cancer (Krogan et al., 2015; Leiserson et al., 2013; Mutation and Pathway Analysis working group of the International Cancer Genome Consortium, 2015; Shim and Lee, 2015). Although a large number of protein–protein interactions (PPIs) have been mapped via experimental detection of physical interactions between proteins, in silico methods are still useful for inferring functional PPIs (Rao et al., 2014) because proteins may work cooperatively without physical interaction.
Interactions between proteins are typically mediated by domain–domain interactions (DDIs); thus, protein domains can be used to infer PPIs (Deng et al., 2002; Reimand et al., 2012; Sprinzak and Margalit, 2001; Wojcik and Schachter, 2001). Indeed, frequently observed domain pairs between interacting proteins have been identified as powerful predictors of PPIs (Deng et al., 2002; Sprinzak and Margalit, 2001). These observations have motivated the systematic extraction of DDIs from known PPIs and extrapolation to new PPIs. Many algorithms have been developed to extract DDIs from PPIs, as well as to infer new PPIs based on these DDIs, which have been collected into meta-databases (Kim et al., 2012; Yellaboina et al., 2011).
Functional PPIs can also be inferred from shared domains between proteins because protein domains are structural, functional and evolutionary units of proteins. In addition, because proteins evolved through gene duplication, recombination, fusion and fission towards specific functions, there exist limited rules for domain combination (Chothia et al., 2003; Moore et al., 2008). However, the presence of shared domains between proteins may not be a sufficient model of functional PPIs. For example, a DNA-binding domain exists in most transcription factors, which regulate many different biological processes. Thus, such a shared domain between two proteins may not be a strong indicator of their functional association with the same pathway. Therefore, we do not expect the prediction of functional PPIs based on domain sharing to be effective. There exists a need for a method to infer functional associations between proteins based on domain co-occurrence.
In this study, we propose a method to infer PPIs based on domain occurrence. Any protein can be represented by a domain profile, which is a vector reflecting the presence or absence of domains. Functional associations between proteins are then measured by the similarity between their domain profiles. Unlike DDI-based inference of PPIs, this approach does not require prior knowledge of the PPIs from which the DDIs are extracted. In addition, to measure domain profile similarity with higher functional relevance, we wanted to account for unequal distribution of functional information content across profiles and domains. Mutual information (MI) differentially compares profile information based on their entropy. We hypothesized that rarer domains tended to be involved in more specific pathways. Therefore, in addition to traditional MI, we tested a weighted MI method that assigned different weights to domains based on their genome-wide frequencies. Here, we demonstrate that weighted MI outperforms traditional MI in the inference of functional PPIs based on domain profile similarity in both yeast and human. We also found that domain profile similarity by weighted MI constructs substantially improved functional networks compared to those based on DDIs. Our domain-based network inference method constructed highly predictive functional networks for complex phenotypes such as human diseases. Finally, we observed that communities of our domain-based network are significantly associated with known pathways or diseases, implicating a potential application of domain-based functional networks in mapping associations between domains and pathways/diseases.
2 Methods
2.1 Pathway and phenotype annotation sets
Inferred functional PPI networks were analyzed using various pathway or phenotype annotations. For pathway analysis, we used Gene Ontology Consortium (2013) biological process (GOBP) annotations for yeast and human. For yeast phenotype analysis, we used a set of literature-based annotations of yeast genes for 100 knockout (KO) phenotypes (McGary et al., 2007). For human phenotype analysis, two disease gene databases were used: Online Mendelian Inheritance in Man (OMIM) (Amberger et al., 2015) and Disease Ontology (DO) (Kibbe et al., 2015).
2.2 Generation of domain profiles for proteins
The InterPro database (Mitchell et al., 2015) (http://www.ebi.ac.uk/interpro), which collects domain annotations from diverse sources, includes over 20 000 entries. We downloaded InterPro entries (v38) for yeast and human proteins via BioMart, and generated domain profiles for 17 013 and 4921 proteins using 8362 and 4261 domains for human and yeast, respectively. A stack of domain profiles, i.e. a protein–domain matrix, was defined as follows:
Definition 1 —
Protein–domain matrix M
Given n proteins and m domains, let a protein–domain matrix M = [cij] be an n m matrix whose elements cij indicate the presence or absence of a domain dj within a protein pi as follows:
2.3 Inference of functional PPIs by MI analysis
Given a protein–domain matrix, the functional association between two proteins can be measured using MI analysis, which measures the mutual dependence between two discrete random variables (e.g. domain profiles) as described in Definition 2.
Definition 2 —
MI I
Given two discrete random variables X and Y,
where H(X), which represents the entropy (i.e. measure of uncertainty) of random variable X, and H(X,Y) represents the joint entropy between discrete random variables X and Y with joint distribution p(X,Y). H(X), H(Y) and H(X,Y) defined as:
where kX and kY are the cardinality of the outcomes of X and Y, respectively.
Higher MI results when the entropy of each random variable is high and the relative entropy between two random variables is low. In addition to traditional MI, we tested weighted MI, which adds more weight to the rarer profile features, as described in the following definitions:
Definition 3 —
Domain-specific weight
Given the n-by-m protein–domain matrix M, the domain-specific weight ωj for each domain j is defined as:
where cij denotes the value of the indicated cell in matrix M.
Definition 4 —
Weighted MI Iω
Given two proteins X and Y,
where and represent the weighted entropy of protein X and protein Y, respectively, and can be calculated as follows:
In addition, represents the weighted joint entropy between X and Y, and can be calculated as follows:
2.4 Functional PPI inference based on DDI
Based on the rationale that domains mediate interactions between proteins, PPIs can be inferred by DDIs. Various algorithms have been applied to extract DDIs from known PPIs. These DDIs have been deposited into databases such as DOMINE (Yellaboina et al., 2011), which collects DDIs from 15 different sources of evidence. Assuming that DDIs from different sources are independent, we assigned a weight to each DDI based on the number of sources; thus, this weight ranged from 0 to 15. The score of a functional interactionbetween protein i and protein j, which is often mediated by multiple DDIs, was calculated using the following definition:
Definition 5 —
The score of each PPI mediated by multiple DDIs was defined as
where is the weight of a DDI between domain u and domain v and N is the number of DDIs involved in the interaction between proteins i and j with weight greater than zero.
2.5 Likelihood of inferred functional PPIs
The functional significance of each inferred PPI was measured by the log likelihood score (LLS), which is based on a Bayesian statistics framework as described previously (Lee et al., 2004). LLS is defined as follows:
where P(L|E) and P(⌐L|E) represent the frequencies of positive (L) and negative (⌐L) gold standard pathway links observed in the given source of evidence (E), and P(L) and P(⌐L) represent the prior expectations (i.e. the total frequencies of all positive and negative gold standard pathway gene pairs, respectively). To avoid over-fitting, we used ‘0.632 bootstrapping’ for all LLS calculations due to its credibility in estimating classifier error rates. For this study, gold standard pathway links were generated by pairing two proteins annotated for the same GOBP terms.
3 Results
3.1 Overview of weighted MI to infer functional PPIs from domain profiles
Given that domains are functional units of proteins, the functions of a protein can be represented by its domain composition. We summarized protein functions as profiles of binary scores that indicate the presence or absence of a given domain. These domain profiles can then be used to infer functional associations between proteins based on their similarity. In this study, we generated domain profiles for proteins using domains registered to a comprehensive domain database, InterPro, as of May 2014 (Mitchell et al., 2015). Figure 1A illustrates an example of a protein–domain matrix composed of multiple domain profiles. Domain profiles are generally sparse because most proteins contain only a few of the several thousand domains annotated in the InterPro database. For example, ∼22% and ∼40% of proteins annotated by InterPro contain a single domain in human and yeast. Meanwhile, only 2% and 0.25% of proteins contain ten or more domains in human and yeast, respectively (Fig. 1B and Supplementary Fig. S1A). To measure profile similarity with high functional relevance, we took into account the unequal distribution of information content across different domains and domain profiles. As shown in Figure 1A, two questions about non-uniformity in information content need to be addressed when measuring profile similarity: (i) Which domain profile is more informative? (ii) Which domain is more informative?
Fig. 1.

Summary of weighted MI analysis to infer domain-based functional associations between genes. (A) An illustration of a protein–domain matrix composed of six proteins (p1–p6) and five domains (d1–d6). Each cell of the matrix has a value of 1 if the given domain exists in the protein, and a value of 0 in all other cases. The distribution of domain occurrence (i.e. domain profiles) are different across proteins. Some domain profiles are more informative than others, and entropy can be used to measure the information content, in which high entropy reflects more informative profiles. Information content across domains is not distributed uniformly. Some domains are associated with more specific functions than others. In general, the more rare the domain, the more informative it is. (B) The distribution of the number of human proteins harbouring k domains. The distribution indicates that most human proteins have only a few domains, and that only a few proteins contain many domains. (C) The distribution of the number of domains that occurred in k proteins, which reveals a power–law distribution (Color version of this figure is available at Bioinformatics online.)
To address the first question of non-uniform information content across domain profiles, we employed MI, which accounts for the entropy within a profile via the similarity measure; profiles with higher entropy are more informative. Using information theory, MI describes how much information is shared between two variables (e.g. two domain profiles). Whereas most commonly used correlation coefficient measures, such as Pearson’s (r), Spearman's (ρ) and Kendall's (τ), can only measure linear relationships (monotonic relationship) between two variables, MI can measure nonlinear dependencies. Therefore, there is no need to specify a theoretical probability distribution or use a mean-variance model to account for non-linear dependencies. In the case of the domain profile, the rate of occurrence change between two proteins is not constant for the entire range of proteins or their component domains. Moreover, MI accounts for the individual entropy of each input, as well as the joint entropy between two variables, which is advantageous in the case of very sparse profiles.
Unequal distribution of information content among domains is illustrated by the power–law distribution of domain occurrence among proteins. From this, we hypothesized that rare domains are associated with relatively specific biological processes while commonly occurring domains contribute to diverse functions (Fig. 1C and Supplementary Fig. S1B). For example, the zinc finger C2H2-like (IPR015880) domain occurs in 793 proteins and plays a role in binding DNA, RNA, protein and/or lipid substrates, such as zinc ion binding and nucleic acid binding. Even if a protein containing the zinc finger domain acts as a core transcription factor, it is difficult to assert that zinc finger domains are associated with a specific pathway. Therefore, we assigned higher weights to the rarer domains during MI calculation based on the assumption that rarer domains have higher information content or pathway specificity (see Definition 4). A similar approach for image registration, in which spatially weighted MI analysis accounted for differential medical importance (Park et al., 2010; Patel et al., 2011; Suh et al., 2010) or pixel contribution (Rivaz and Collins, 2012; Zhuang et al., 2011), was shown to be superior to traditional MI analysis.
3.2 Domain profile similarity by weighted MI outperforms other domain-based PPI inference methods
We compared four different domain-based functional PPI inference methods for yeast and human: (i) PPIs inferred from shared domains, (ii) PPIs inferred from DDIs (see Definition 5), (iii) PPIs inferred from domain profile similarity by traditional MI and (iv) PPIs inferred from domain profile similarity by weighted MI. Homologous proteins generate similar domain profiles. To infer functional PPIs based on domain profile similarity rather than sequence similarity, we excluded paralogous protein pairs (defined by a blastp E-value threshold of 10 − 3) from all domain-based PPI networks. The likelihood that two proteins participate in the same GOBP pathway was measured for every bin of 1000 PPIs ordered from the highest edge score (i.e. from the MI, weighted MI or DDI-based score). If the LLS was greater than zero, the two proteins were more likely to be associated functionally than expected at random. For example, networks inferred from domain profile similarity by weighted MI have 12 000 and 94 000 PPIs before the first bin of 1000 PPIs with negative LLS for yeast and human, respectively.
To test the inference power of each domain-based score for functional PPIs, we performed regression analysis between domain-based scores and LLS of protein pairs that share GOBP pathway annotations. Functional PPIs by shared domains were excluded from this analysis because they were based on binary edge scores, which were not sortable for regression analysis. We tested regressions for the top 15 000 and 200 000 PPIs for all yeast and human networks, respectively. PPI networks were fitted by a sigmoid or linear function. Based on the best-fit relationships for each network, we found that weighted MI was the best regression model for functional PPIs in both yeast and human (Fig. 2).
Fig. 2.
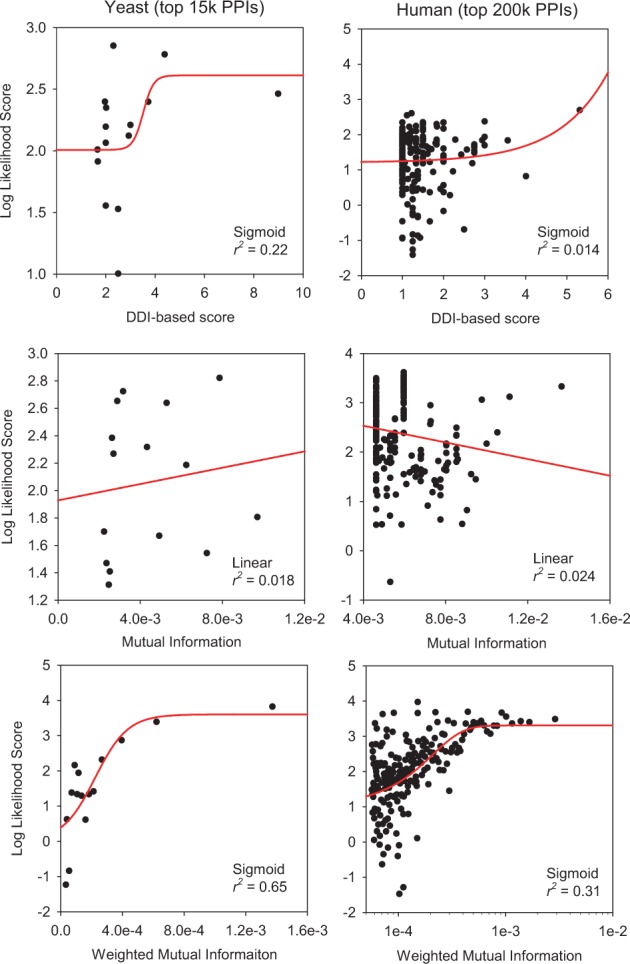
Regression analysis between various domain-based scores and the log likelihood of functional PPIs that share GOBP annotations. To compare the predictive power of three different domain-based network inference scores (DDI-based score, MI and weighted MI) based on regression analysis, we used the same number of links inferred from different methods in yeast (15 000 links) and human (200 000 links). Log likelihood scores were measured for each bin of 1000 protein pairs of networks. The best-fit regression model was identified by the largest R-squared score. The results suggest that weighted MI has the highest predictive power for functional PPIs in both yeast and human
Next, we assessed functional PPI networks by precision-recall analysis, in which we defined recall as the coverage of the coding genome and precision was defined as the probability that the inferred PPIs shared pathway annotations in GOBP. We found that functional PPI networks inferred from domain profile similarity by weighted MI outperformed all other domain-based networks for both yeast and human (Fig. 3). Functional PPIs based on the presence of shared domains between proteins generated the largest PPI networks, which also covered the largest number of coding genes, but with low precision. Functional PPIs based on DDIs or domain profile similarity by traditional MI covered fewer coding genomes. In addition, precision was higher than functional PPIs based on the presence of shared domains, but less than that of weighted MI. Notably, confident networks of 12 000 and 94 000 PPIs by weighted MI covered 45% and 65% of coding genomes in yeast and human, respectively. Therefore, we concluded that domain profile similarity by weighted MI is the best domain-based PPI inference method and can be used to construct genome-scale functional networks in humans.
Fig. 3.
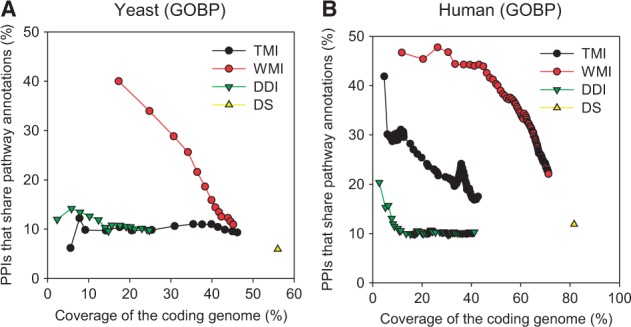
Assessment of functional PPIs by domain-based methods for pathway associations. Precision-recall curves were generated using the percentage of protein pairs that share GOBP annotations and coverage of the coding genome by the given size of the networks for (A) yeast and (B) human. TMI, functional PPIs by traditional MI analysis; WMI, functional PPIs by weighted MI analysis; DDI, functional PPIs by domain-domain interactions; DS, functional PPIs by domain sharing
3.3 Functional PPIs based on domain profile similarity are highly predictive for complex phenotypes
Recently, network-based analysis of candidate genes has become more popular in the genetic dissection of complex phenotypes such as human diseases. For example, PPI networks predictive for human diseases have been used to identify novel disease genes by network association with known disease genes, network-based prioritization candidates within susceptible diseases in the same chromosomal region or GWAS loci. Moreover, PPI networks have been applied to disease modules by searching for subnetworks enriched for patient-specific mutations or disease-associated single nucleotide polymorphisms (SNPs) (Barabasi et al., 2011; Jia and Zhao, 2014; Leiserson et al., 2013; Mutation and Pathway Analysis working group of the International Cancer Genome Consortium, 2015; Shim and Lee, 2015). To test the capability of domain-based functional PPI networks for studying complex phenotypes, we assessed the precision of the networks for connecting proteins that share phenotypes in yeast and humans. Using similar precision-recall analyses as those used to test the predictive power for GOBP pathways, we found that using functional PPIs inferred from domain profile similarity by weighted MI is highly predictive of both yeast KO phenotypes and human diseases in the OMIM database (Fig. 4). These results demonstrate that domain-based functional PPI networks are highly predictive for pathways as well as phenotypes, suggesting that they can be applied towards network-based analysis of complex phenotypes, including human diseases.
Fig. 4.
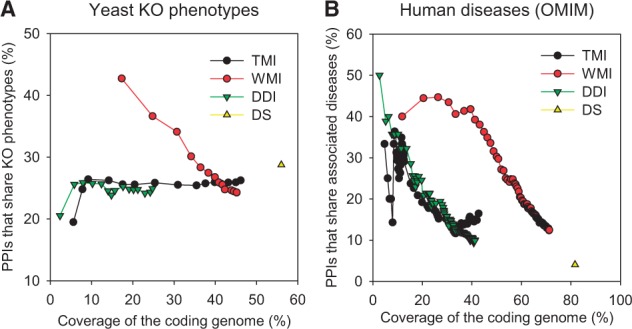
Assessment of functional PPIs by domain-based methods for phenotype associations. Precision-recall curves were generated using the percentage of protein pairs that share knockout phenotypes (KO) for (A) yeast or diseases (OMIM) for (B) human, and coverage of the coding genome by the given size of the networks. Abbreviations for network inference methods are the same as for Figure 3
3.4 Domain-based human functional PPIs elucidate network communities associated with pathways and diseases
The high likelihood of an association between proteins involved in the same pathways may allow genome-scale reconstruction of functional modules represented as network communities. Using CFinder (Derenyi et al., 2005), a software that identifies network communities with overlaps, we searched for all 10 clique-connected subgraphs in the human functional network of 94 000 confident PPIs based on domain profile similarity by weighted MI. A total of 430 communities were identified. We tested these communities for their association with GOBP and DO-Lite gene sets by Fisher’s exact test. Using a criterion of P < 0.01, at least three member genes for the test pathway or disease set and at least two overlapped genes between communities and gene sets, we found that 282 (65%) and 198 (46%) of 430 communities are significantly associated with at least one GOBP pathway and DO-Lite disease, respectively. These results suggest that domain-based network communities tend to be significantly associated with pathways or diseases, implicating the potential application of domain-based functional networks in mapping domain-to-pathway and domain-to-disease associations.
Figure 5 shows 16 of the 20 communities with the strongest association with pathways or diseases in the largest component of the domain-based functional network, which contained 61 133 links among 5676 coding genes. Among the 16 communities, 15 and 10 were significantly associated with GOBP terms (Fig. 5A and Supplementary Table S1) and DO-Lite terms (Fig. 5B and Supplementary Table S2), respectively. Notably, some communities were associated with diseases and their relevant biological processes. For example, two enriched diseases for community C4 (i.e. 4th largest community), epidermolysis bullosa (P = 1.38e−07) and ectodermal dysplasia (P = 1.91e−06), are likely to be influenced by epidermis development (P = 1.84e−27) and ectoderm development (P = 1.91e−06), which are also significantly enriched for C4. As another example, the pathway set linked to the perception of sound (P = 1.08e−03) and the disease set for deafness (P = 2.76e−04) were identified as associated significantly with C20. Furthermore, a pathway term for the antimicrobial humoral response (P = 5.21e−04), as well as a disease term for infection by Cryptococcus neoformans (P = 7.71e−07), were enriched for C18. Many other communities connected diseases and pathways whose relationships are less obvious, but implicated by previous studies. For example, C1 was associated with blood coagulation (P = 3.49e−08) and macular degeneration (P = 2.07e−05); a relationship between anticoagulant medication and massive intraocular haemorrhage in age-related macular degeneration has previously been reported (Tilanus et al., 2000). As another example, community C8 was associated with the lipid metabolic process (P = 1.09e−03) and Cushing syndrome (P = 3.28e−06); elevated triglyceride levels have been observed in patients with Cushing syndrome (Chanson and Salenave, 2010). Furthermore, C13 was associated with the integrin-mediated signalling pathway (P = 5.15e−07) and polyarthritis (P = 1.74e−03); a significant contribution of integrin to inflammatory cartilage destruction has previously been reported (Peters et al., 2012). Finally, C18 was associated with cell adhesion (P = 4.98e−05) and Pseudoxanthoma elasticum infection (P = 6.63e−09); elevated levels of the cell adhesion molecule P-selectin have been observed in patients infected with P.elasticum (Gotting et al., 2008). These results demonstrate the usefulness of domain-based functional networks for studying the underlying molecular mechanisms of diseases.
Fig. 5.
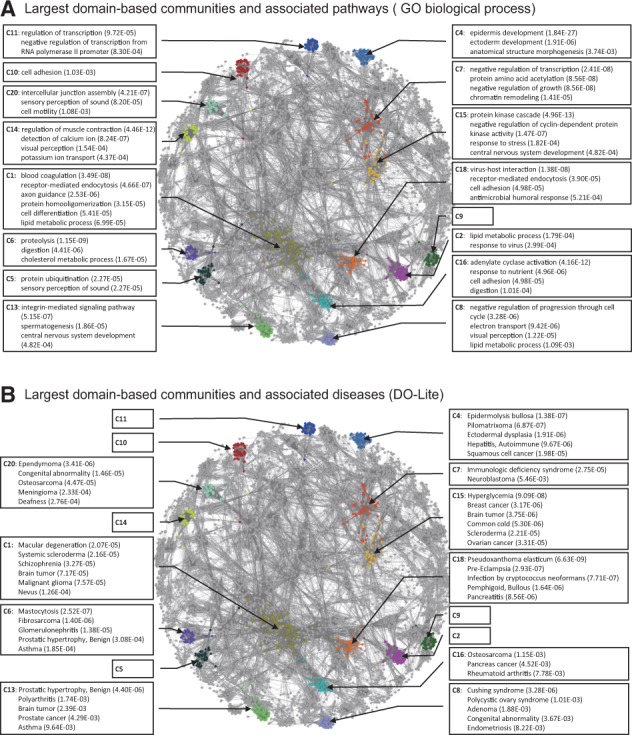
Major communities of a human domain-based functional network. The largest component of the human domain-based network by weighted MI (90 000 links) contains 61 133 links among 5676 coding genes. Among the 20 largest communities identified by CFinder analysis, 16 were included in the largest component network. A few significantly associated GOBP terms (A) and DO-Lite terms (B) for each community are listed along with their significance scores (Color version of this figure is available at Bioinformatics online.)
We also compared communities of PPI networks, one by domain profile similarity based on WMI and the other by DDI-based inferences. We found that the WMI-based network gives substantially more communities with much smaller range of size distribution, and more communities that are associated with diseases or pathways among top 20 communities (Supplementary Fig. S2), indicating superiority of WMI-based network over DDI-based network in reconstructing functional and disease modules.
4 Discussion
The protein domain is a widely accepted functional unit of proteins. Thus, domains can be expected to convey information about the pathways and phenotypes proteins are involved in. However, the use of domains to predict these pathways and phenotypes based on domain occurrence remains limited. Many domains are associated with multiple pathways that are involved in distinct physiological processes. Therefore, domain occurrence may not be a sufficient indicator of domain involvement in specific pathways or phenotypes. Furthermore, some domains appear to be basic components of a large number of proteins. Therefore, domain-based pathway modelling has been challenging. In this study, we demonstrated the feasibility of constructing highly predictive functional PPI networks for pathways and diseases using an information theory approach with differential weights across domains. However, despite superior accuracy, PPI networks by weighted MI exhibit smaller genome coverage than those derived by simple domain sharing, which indicates that many domains are general-purpose components of proteins that do not indicate any specific pathways. These observations suggest that we may be able to use domain-based networks to distinguish domains involved in specific pathways from those that participate in general processes. This enables more reliable pathway prediction based on domain information.
Although we demonstrated the utility of weighted MI to construct domain-based functional PPI for yeast and human in this study, the same method can be easily applied to other organisms and may potentially benefit relatively lesser understood organisms for which only protein sequences with electronic annotation of domains are available.
We also demonstrated that human domain-based network communities tend to be associated with pathways and diseases, suggesting a potential application for domain-based functional networks in mapping domain-to-pathway and domain-to-disease associations. Although we have made vast progress in in silico models to identify associated pathways and diseases for genes, models using domains as indicators remain are less developed. Given that the domain is a more fundamental functional unit of proteins, pathway or disease annotation for each domain would be more relevant to the genome. Therefore, significant efforts have been invested into the manual curation of relationships between domains and pathways, such as InterPro2GO (Burge et al., 2012). We anticipate that the development of an automatic mapper of domains to GOBP terms will accelerate this manual curation project. In addition, the prediction of domain-to-disease associations will provide new insights into the analysis of disease-specific genomics data, such as disease-associated SNPs and mutations, which potentially opens new routes to understanding the molecular mechanisms of human diseases in the future.
Supplementary Material
Funding
This study was supported by the National Research Foundation of Korea (2015R1A2A1A15055859, 2012M3A9B4028641, 2012M3A9C7050151) to I.L. Funding for the open access charge has been provided by the National Research Foundation of Korea (2015R1A2A1A15055859).
Conflict of Interest: none declared.
References
- Amberger J.S. et al. (2015) OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res., 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi A.L. et al. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet., 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge S. et al. (2012) Manual GO annotation of predictive protein signatures: the InterPro approach to GO curation. Database, 2012, bar068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanson P., Salenave S. (2010) Metabolic syndrome in Cushing's syndrome. Neuroendocrinology, 92, 96–101. [DOI] [PubMed] [Google Scholar]
- Chothia C. et al. (2003) Evolution of the protein repertoire. Science, 300, 1701–1703. [DOI] [PubMed] [Google Scholar]
- Deng M. et al. (2002) Inferring domain-domain interactions from protein-protein interactions. Genome Res., 12, 1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derenyi I. et al. (2005) Clique percolation in random networks. Phys. Rev. Lett., 94, 160202. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium. (2013) Gene Ontology annotations and resources. Nucleic Acids Res., 41, D530–D535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotting C. et al. (2008) Circulating P-, L- and E-selectins in Pseudoxanthoma elasticum patients. Clin. Biochem., 41, 368–374. [DOI] [PubMed] [Google Scholar]
- Jia P., Zhao Z. (2014) Network.assisted analysis to prioritize GWAS results: principles, methods and perspectives. Hum. Genet., 133, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe W.A. et al. (2015) Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res., 43, D1071–D1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. et al. (2012) IDDI: integrated domain-domain interaction and protein interaction analysis system. Proteome Sci., 10, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan N.J. et al. (2015) The cancer cell map initiative: defining the hallmark networks of cancer. Mol. Cell, 58, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. et al. (2004) A probabilistic functional network of yeast genes. Science, 306, 1555–1558. [DOI] [PubMed] [Google Scholar]
- Leiserson M.D. et al. (2013) Network analysis of GWAS data. Curr. Opin. Genet. Dev., 23, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary K.L. et al. (2007) Broad network-based predictability of Saccharomyces cerevisiae gene loss-of-function phenotypes. Genome Biol., 8, R258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A. et al. (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res., 43, D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A.D. et al. (2008) Arrangements in the modular evolution of proteins. Trends Biochem. Sci., 33, 444–451. [DOI] [PubMed] [Google Scholar]
- The Mutation Consequences and Pathway Analysis working group of the International Cancer Genome Consortium. (2015) Pathway and network analysis of cancer genomes. Nat. Methods, 12, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.B. et al. (2010) Spatially weighted mutual information image registration for image guided radiation therapy. Med. Phys., 37, 4590–4601. [DOI] [PubMed] [Google Scholar]
- Patel P. et al. (2011) Spatially weighted mutual information (SWMI) for registration of digitally reconstructed ex vivo whole mount histology and in vivo prostate MRI. Conf. Proc. IEEE Eng. Med. Biol. Soc., 2011, 6269–6272. [DOI] [PubMed] [Google Scholar]
- Peters M.A. et al. (2012) The loss of alpha2beta1 integrin suppresses joint inflammation and cartilage destruction in mouse models of rheumatoid arthritis. Arthritis Rheum., 64, 1359–1368. [DOI] [PubMed] [Google Scholar]
- Rao V.S. et al. (2014) Protein-protein interaction detection: methods and analysis. Int. J. Proteomics, 2014, 147648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J. et al. (2012) Domain-mediated protein interaction prediction: From genome to network. FEBS Lett., 586, 2751–2763. [DOI] [PubMed] [Google Scholar]
- Rivaz H., Collins D.L. (2012) Self-similarity weighted mutual information: a new nonrigid image registration metric. Med. Image Comput. Comput. Assist. Interv., 15, 91–98. [DOI] [PubMed] [Google Scholar]
- Shim J.E., Lee I. (2015) Network-assisted approaches for human disease research. Anim. Cells Syst., 19, 231–235. [Google Scholar]
- Sprinzak E., Margalit H. (2001) Correlated sequence-signatures as markers of protein–protein interaction. J. Mol. Biol., 311, 681–692. [DOI] [PubMed] [Google Scholar]
- Suh J.W. et al. (2010) Serial nonrigid vascular registration using weighted normalized mutual information. Proc. IEEE Int. Symp. Biomed. Imaging, 2010, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilanus M.A. et al. (2000) Relationship between anticoagulant medication and massive intraocular hemorrhage in age-related macular degeneration. Graefes. Arch. Clin. Exp. Ophthalmol., 238, 482–485. [DOI] [PubMed] [Google Scholar]
- Wojcik J., Schachter V. (2001) Protein-protein interaction map inference using interacting domain profile pairs. Bioinformatics, 17, S296–S305. [DOI] [PubMed] [Google Scholar]
- Yellaboina S. et al. (2011) DOMINE: a comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res., 39, D730–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X.H. et al. (2011) A nonrigid registration framework using spatially encoded mutual information and free-form deformations. IEEE Trans. Med. Imaging, 30, 1819–1828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.