

Abstract
Chronic pain is a highly prevalent and poorly managed human health problem. We used microarray-based expression genomics in 25 inbred mouse strains to identify dorsal root ganglion (DRG)-expressed genetic contributors to mechanical allodynia, a prominent symptom of chronic pain. We identified expression levels of Chrna6, which encodes the α6 subunit of the nicotinic acetylcholine receptor (nAChR), as highly associated with allodynia. We confirmed the importance of α6* (i.e., α6-containing) nAChRs by analyzing both gain- and loss-of-function mutants. We find that mechanical allodynia associated with neuropathic and inflammatory injuries is significantly altered in α6* mutants, and that α6* but not α4* nicotinic receptors are absolutely required for peripheral and/or spinal nicotine analgesia. Furthermore, we show that Chrna6’s role in analgesia is at least partially due to direct interaction and cross-inhibition of α6* nAChRs with P2X2/3 receptors in DRG nociceptors. Finally, we establish relevance of our results to humans by the observation of genetic association in patients suffering from chronic postsurgical pain and temporomandibular pain.
INTRODUCTION
Chronic pain in the clinic manifests itself mainly in the form of spontaneous pain and mechanical allodynia, a sensitized response to an innocuous stimulus. Patients suffering from the latter symptom sometimes find clothing touching their skin or a light breeze to be very painful. Current medications are mostly inadequate to treat such symptoms, and an in-depth understanding of molecular mechanisms of mechanical allodynia is still lacking.
Here we used an unbiased approach to identify genes involved in mechanical allodynia. Specifically, we correlated mechanical allodynia phenotypes of 25 inbred mouse strains to genome-wide gene expression levels in dorsal root ganglia of these strains. This expression genomics strategy has been adopted previously in pain research (1–4), but using a much smaller number of strains. We provide evidence for the expression of the Chrna6 gene encoding the nicotinic α6 subunit as a major determinant of variable mechanical allodynia after nerve injury.
Neuronal nAChRs are hetero- or homopentameric ligand-gated ion channels composed of α (α2–α7, −9 and −10) and β (β2–β4) subunits. They have been the target of analgesic drug discovery for many years, with progress being hindered by a narrow therapeutic window and side effects. Attention has been focused largely on α4β2* (i.e., α4- and β2-containing) nAChRs (5), the most highly expressed subtype in the CNS but effects on pain of α3* (6), α7 (7) and α9* (8, 9) nAChRs have also been demonstrated. The α6* nAChRs have been mysterious until the recent elucidation of their involvement in the mesolimbic dopaminergic system, in which they activate dopamine neurons causing locomotor hyperactivity (10), and visual system, in which they modulate glutamate and γ-amino-butyric acid release in the superior colliculus (11). The α6 subunit is known to be localized in sensory ganglia (12–15). There are no reported agonists that discriminate well between α6* and α4* nAChRs, raising the possibility that the α6 subunit plays an unappreciated role in nicotinic analgesia in the spinal cord or periphery.
RESULTS
Association between DRG Chrna6 expression and neuropathic mechanical allodynia in mice
Mechanical allodynia induced by spared nerve injury (SNI) was quantified in 25 inbred mouse strains using von Frey filaments, and compared to basal DRG expression of 45,101 mRNA transcripts using microarray gene-expression profiling (Affymetrix MOE430v2 chip) in these same 25 strains. All strains displayed ipsilateral allodynia (Fig. 1A,B, fig. S1), but highly significant effects of strain (F24,89=9.1, P<0.001) and strain x repeated measures interaction (F144,534=2.2, P<0.001) were observed. That is, strains displayed different extents and time courses of allodynia (see fig. S1). A significant strain x sex x repeated measures interaction (F144,534=1.4, P<0.05, Greenhouse-Geisser corrected) was also evinced. This interaction appeared to be largely due to robust sex differences in the SM/J (see below) and C3H/HeJ strains (16).
Fig. 1. Correlation of DRG expression of Chrna6 and mechanical allodynia after SNI in inbred mice.
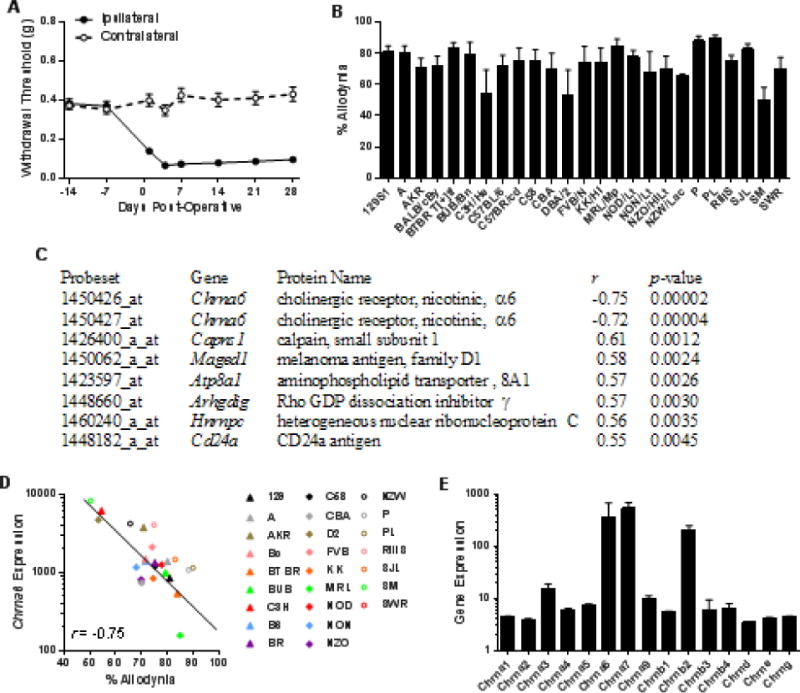
SNI surgery was performed on 25 inbred mouse strains, and withdrawal thresholds of the ipsilateral hind paw to von Frey fiber stimulation were measured. Symbols in A (n=139) represent mean ± SEM paw withdrawal threshold on each testing day; bars in B (n=4–6 mice/strain) represent mean ± SEM percentage of maximum possible allodynia (see Materials and Methods online). (C) The top 7 correlations (Pearson’s r; all P<0.005) between the strain means shown in B and basal DRG expression levels of ≈45,000 probesets in these same strains. (D) The correlation between allodynia and basal DRG expression (in arbitrary units) of probeset 1450426_at (Chrna6); symbols represent individual strain values. Symbol abbreviations are self-explanatory except for: Bc, BALB/cBy; B6, C57BL/6; BR, C57BR/cd; D2, DBA/2. (E) Average expression across all strains of all Chrn* genes. Bars represent basal DRG expression (in arbitrary units) ± SD.
Correlating the overall allodynia data (Fig. 1B) with the Affymetrix chip data revealed 10 correlations at the P<0.005 level (uncorrected) (Fig. 1C). The top two highest correlations (in either direction), genome-wide, were with two different probes for Chrna6 (r=−0.75 and r=−0.72) (Fig. 1C,D) such that higher expression of Chrna6 was associated with lower levels of allodynia development. These associations are both significant at 0.05 using false discovery rate correction for multiple comparisons; no other associations were significant, and thus no attempts were made to evaluate the candidacy of the other genes in Fig. 1C. The Chrna6 correlations were sex-dependent, being considerably higher in males (r=−0.67, −0.63) than in females (r=−0.37, −0.33). Table S1 shows strain-dependent expression levels of all Chrn* genes coding for nicotinic subunits; none other than Chrna6 featured suggestive correlations with allodynia or baseline nociception. Average DRG expression of Chrn* genes across-strain is shown in Fig. 1E. As can be appreciated by the error bar, Chrna6 displayed more genotypic variation than any other subunit gene; with a coefficient of variation ≈50% higher than the next most variable subunit (Chrnb3) and more than 10-fold higher than Chrna6 (see table S1).
Conventional haplotype mapping was also performed, correlating mechanical allodynia strain means with approximately 156,000 genomic haplotypes (17). Of the top 10 correlated haplotypes genome-wide (see table S2), two of them were located just upstream of the Chrna6 gene on mouse chromosome 8. Other potentially associated genes include Kcnv1, Ubc, Aldh7a1, Gfra2, and Chrna3 (located very near Chrna6 on chromosome 14).
Chrna6 is expressed in a subset of DRG neurons
qPCR experiments revealed detectable expression of Chrna6 mRNA in whole brain, DRG, and eye, but not lung (Table 1). However, relative expression levels varied in these tissues, with DRG expression >10-fold higher than expression in whole brain and >2-fold higher than in the eye (both P<0.001). In situ hybridizations performed at the Allen Institute for Brain Science showed the presence of Chrna6 expression in small- to medium-diameter DRG neurons (not shown), and this was confirmed using α6*-GFP BAC transgenic mice (Fig. 2). DRGs were double-stained with GFP and neuronal sensory marker antibodies, including NF200 that marks mostly myelinated Aβ neurons, as well as IB4 and CGRP that mark distinct populations of nociceptive neurons. Sixty-six percent of the GFP-positive neurons also expressed NF200 (203/306 out of 840 total neurons counted), whereas 37% of NF200 positive neurons expressed GFP (123/336). Twenty-six percent of the GFP population also stained for IB4 (135/512 out of 1286), while 58% of IB4 population expressed GFP (169/293). Finally, 8% of the GFP neurons also expressed CGRP (39/496 from a total of 1213), while 35% of CGRP positive neurons expressed GFP (91/258). These results suggest that Chrna6 is expressed in various functionally distinct DRG subtypes. A previous study observed a similar range of co-labelling of Chrna6 mRNA and peptidergic nociceptor-related (i.e., CGRP or transient receptor potential, V1) immunoreactivity in rat trigeminal ganglion (18).
Table 1. Expression of Chrna6 in multiple tissues.
Values represent mean ± SEM expression normalized to Actb (β-actin).
| Tissue | Expression |
|---|---|
| DRG | 0.42 ± 0.06 |
| Eye | 0.16 ± 0.01 |
| Whole Brain | 0.04 ± 0.02 |
| Lung | n.d. |
n.d., not detectable
Fig. 2. Chrna6 mRNA expression in a subset of DRG neurons.
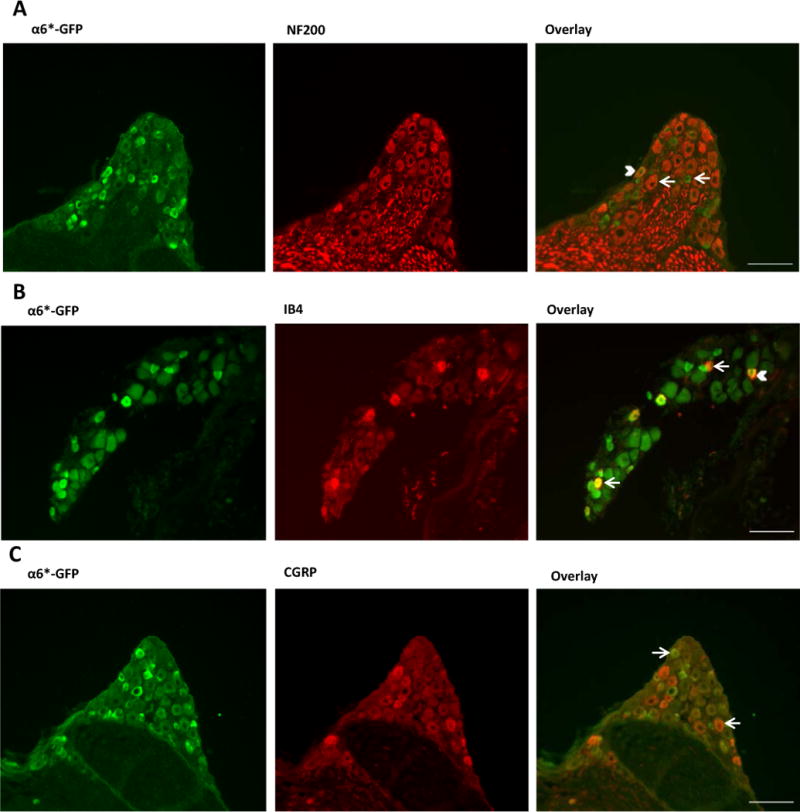
DRG neurons from adult α6*-GFP mice were stained with antibodies against GFP (green) and sensory neuron markers NF200 (A), IB4 (B) and CGRP (C) (all red). Arrows indicate neurons expressing either GFP or the sensory marker. Filled arrow heads indicate neurons co-expressing both markers. Scale bars= 100 μm.
We also confirmed genotype- and sex-dependent Chrna6 expression in the DRG in three mouse strains using qPCR. As was observed in the gene chip experiment (fig. S2A), Chrna6 expression appeared to be robustly strain-dependent, and in one strain, strongly sex-dependent, with male SM/J mice displaying >30-fold higher expression than female mice of the same strain (fig. S2B). This was an intriguing observation, since we also observed a marked sex difference in SM/J mice in the development of mechanical allodynia after spared nerve injury (female > male; sex x repeated measures: F6,78=3.7, P<0.005) (fig. S2C).
Nerve injury-induced down-regulation of Chrna6 and mechanical allodynia
In an independently performed experiment to identify genes associated with chronic pain (2), microarray gene expression profiling (also using the Affymetrix MOE430v2 chip) was performed in the DRGs of five inbred mouse strains after sham surgery or spinal nerve ligation (SNL), another common preclinical assay of neuropathic pain associated with mechanical allodynia. The two Chrna6 probe sets appeared in the top 10 highest fold-regulations by SNL compared to sham surgery (fig. S3A), with down-regulations of 67.3-fold and 41.6-fold, respectively. The downregulation was highly correlated (r=0.90, P<0.05) with basal DRG expression (fig. S3B), but even after downregulation by SNL a strongly negative correlation (r=−0.93) between Chrna6 expression and allodynia was observed in the three strains tested behaviorally (fig. S3C), suggesting that Chrna6 expression protects against allodynia after nerve injury as well.
Confirmation of Chrna6 involvement in mutant mice
To provide causal evidence of the involvement of the α6 subunit in neuropathic pain, we tested transgenic Chrna6 null mutant mice (19) and Chrna6 gain-of-function L9’S mutant mice (10) for mechanical allodynia after SNI. To investigate whether α6* nAChRs play a similar role in chronic inflammatory pain, we also tested these mutants for mechanical allodynia after intraplantar CFA injection (Fig. 3). All genotypes displayed expected time courses of allodynia. For both SNI and CFA, Chrna6 KO mice displayed higher overall levels of allodynia compared to WT mice (t21=3.2, P<0.005, t10=2.5, P<0.05, respectively). For both SNI and CFA, Chrna6 L9’S mutant mice displayed lower overall levels of allodynia compared to their WT controls (t9=3.0, P=0.01, t16=2.2, P<0.05, respectively). There were no significant genotype x sex interactions observed in any data set, although strong trends were noted for genotype differences being larger in male versus female mice (not shown). In an experiment performed independently, in a different laboratory, using Chrna6 KO mice and another neuropathic assay (CCI), the increased mechanical allodynia of KO mice was confirmed (fig. S4A). A separate head-to-head experiment using CCI and CFA in Chrna6 and Chrna4 KO mice confirmed the significantly increased allodynia in Chrna6 KOs, but revealed no differences between Chrna4 KOs and their wildtype controls (fig. S4B,C). The α6 subunit appears to play a highly specific role in the modulation of mechanical allodynia, as Chrna6 KO mice displayed statistically equivalent responses to wildtype mice on a battery of acute and tonic nociceptive assays (fig. S5).
Fig. 3. Differential mechanical allodynia after nerve injury and chronic inflammation in Chrna6 mutant mice.
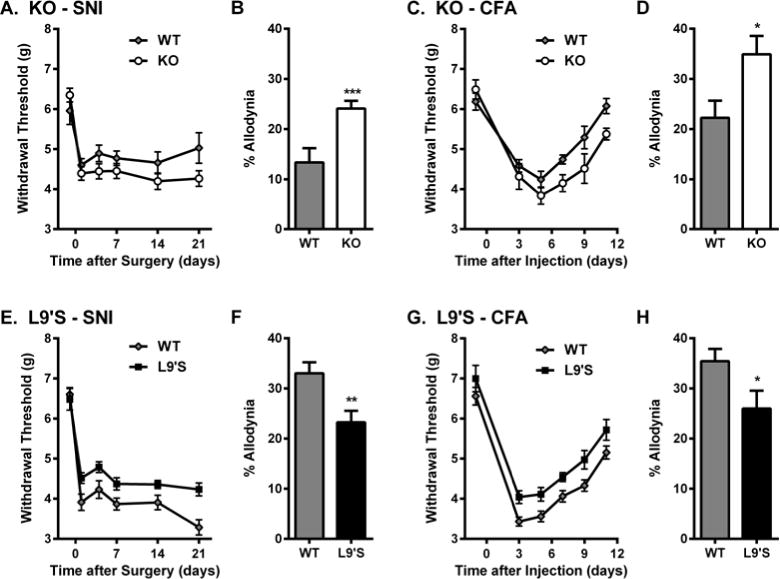
Increased mechanical allodynia after SNI surgery (A,B) and CFA injection (C,D) in Chrna6 KO mice, and decreased mechanical allodynia after SNI (E,F) and CFA (G,H) in Chrna6 L9’S gain-of-function mutants. In all graphs, symbols (n=5–12 mice/genotype) represent mean ± SEM paw withdrawal threshold (g) on each testing day; bars represent mean ± SEM percentage of maximum possible allodynia (see Materials and Methods). *P<0.05, **P<0.01, ***P<0.001 compared to other genotype. A replication of the KO data, using a different neuropathic assay, can be found in fig. S4.
Relevance of α6 to anti-allodynic effects of nicotine
Nicotine itself exerts anti-allodynic effects after both inflammatory and neuropathic injuries (5). We tested the ability of systemic, intracerebroventricular (i.c.v.), intrathecal (i.t.) and peripheral (intraplantar; i.pl.) (−)-nicotine to reverse mechanical allodynia produced by both SNI and CFA in WT, KO and L9’S mice. Although potency and efficacy varied by route of administration, nicotine was significantly and dose-dependently effective against both types of allodynia in wildtype mice by all injection routes (Fig. 4; table S3). Gain-of-function L9’S mutants showed similar or significantly increased efficacy, but Chrna6 KO mice displayed no significant nicotine-induced anti-allodynia in either assay by any route. We then performed a head-to-head comparison of supraspinal, spinal and peripheral nicotine-induced anti-allodynia (25 μg, i.c.v.; 17 μg, i.t.; 50 μg, i.pl.) in Chrna6 and Chrna4 null mutants after neuropathic (CCI) or inflammatory (CFA) injury. All routes of administration produced robust reversal of both types of mechanical allodynia in both wildtype lines at these doses (Fig. 5). Supraspinal nicotine anti-allodynia was significantly reduced in Chrna6 mutants (CCI: t9 = 4.9, p<0.001; CFA: t10 = 2.3, p<0.05), and completely abolished in Chrna4 mutants (CCI: t8 = 4.1, p<0.01; CFA: t10 = 3.5, p<0.01) (Fig. 5A). By contrast, spinal nicotine anti-allodynia was abolished in Chrna6 mutants (CCI: t20 = 5.7, p<0.001; CFA: t9 = 3.2, p=0.01), and preserved in Chrna4 mutants (CCI: t8 = 1.9, n.s.; CFA: t10 = 2.0, n.s.) (Fig. 5B). Similarly, anti-allodynia resulting from injection of nicotine directly into the hind paw was abolished in Chrna6 mutants (CCI: t10 = 6.4, p<0.001; CFA: t10 = 3.7, p<0.01), and preserved in Chrna4 mutants (CCI: t8 = 0.6, n.s.; CFA: t10 = 1.7, n.s.) (Fig. 5C). These data suggest that nicotine blocks mechanical allodynia in the periphery and/or spinal cord in a wholly α6-specific manner, except supraspinally, where both α6* and α4* nicotinic receptors appear to contribute.
Fig. 4. Altered anti-allodynic potency and efficacy of nicotine in Chrna6 mutant mice.
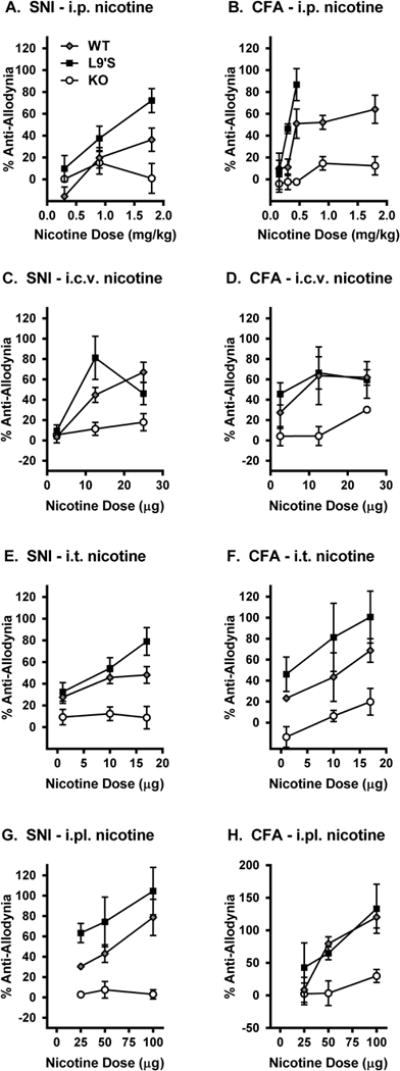
Shown are dose-response relationships for the ability of systemic (i.p.; A,B), intracerebroventricular (i.c.v.; C,D), intrathecal (i.t.; E,F), and peripheral (intraplantar; i.pl.; G,H) nicotine to reverse already-developed (and maximal) mechanical allodynia produced by SNI (day 7 post-surgery; A,C,E,G) and CFA (day 3 post-injection; B,D,F,H). Symbols (n=4–8 mice/dose/genotype) represent mean ± SEM percentage of maximum possible anti-allodynia, based on the pre-SNI/CFA and post-SNI/CFA withdrawal thresholds of each mouse (see Materials and Methods). Statistical analyses are shown in table S3.
Fig. 5. Dependence of spinal and/or peripheral nicotine anti-allodynia on α6.
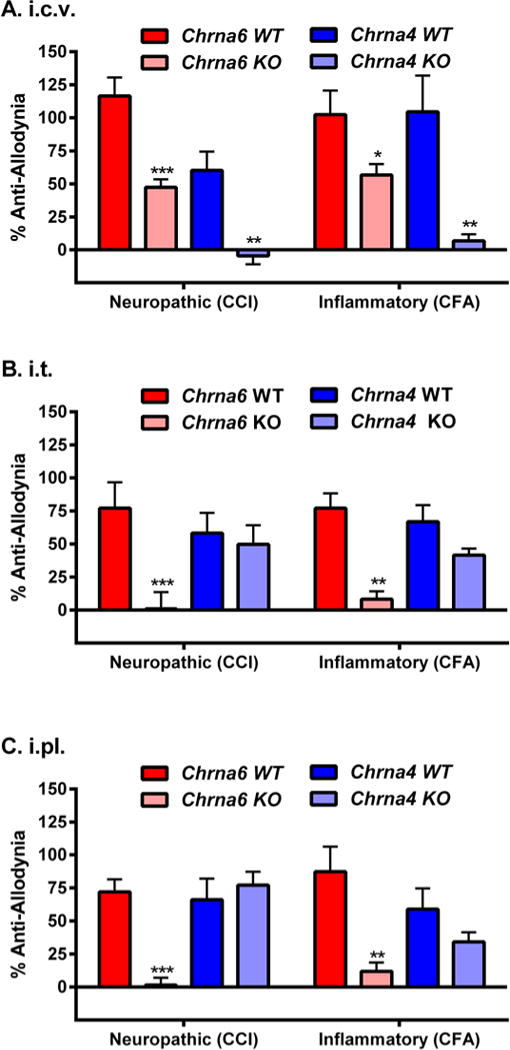
A head-to-head comparison of supraspinal (25 μg, i.c.v.; graph A), spinal (17 μg, i.t.; graph B) and peripheral (50 μg, i.pl.; graph C) nicotine anti-allodynia against neuropathic (CCI) and inflammatory (CFA) pain in Chrna6 (α6*) and Chrna4 (α4*) WT and KO mice tested using identical parameters at the peak of allodynia (14 days post-CCI, 3 days post-CFA). Bars (n=5–6 mice/genotype/injury) represent mean ± SEM percentage of maximum possible anti-allodynia (see Materials and Methods). **P<0.01, **P<0.01, ***P<0.001 compared to analogous WT.
Electrophysiological measurement of α6* and P2X2/3 receptor interactions
An anti-allodynic effect of α6* activation (after DRG gene expression) suggests a functional interaction between α6* nAChRs and another pain-relevant molecular target in the spinal cord or periphery. Previous data show that several subtypes of nAChRs interact, both functionally and physically, with several subtypes of P2X receptors (20–22). We therefore considered the hypothesis that α6* nAChRs interact with P2X2 and P2X3 receptors, known to be involved in pain (23, 24), and, like α6* nAChRs (see Fig. 2), to be expressed in the IB4-positive subpopulation of nociceptors (24).
We tested three combinations of nAChR subunits (α6β4, α6β4β3 and α6β2), co-expressed with most of the possible combinations of P2X2 and P2X3 subunits (P2X2, P2X3, and P2X2/3 receptors). Most α6* nAChRs yield very small agonist-induced current in heterologous expression experiments, vitiating accurate measurements; we overcame these problems by using gain-of-function α6 subunits (α6(L9’S) for α6β4 (10), or gain-of-function β3 subunits (β3-(V13’S) for α6β4β3 (25), or the combination α6(L9’S)β2(L9’S)LFM/AAQA (26). P2X2 receptors and P2X2/3 receptors express robustly in oocytes; the latter are activated selectively by α,βme-ATP (27).
With seven of the eight combinations of α6* receptors and P2X receptors studied we found functional interactions, in the form of cross-inhibition between these two classes of ligand-gated receptors. In the first type of interaction, when ACh and ATP are co-applied, the agonist-induced currents are less than the sum of individual currents. This type of interaction was previously observed between P2X receptors and several other pentameric receptors. When α6* nAChRs were expressed alone, they showed no direct responses to ATP; the addition of ATP (320 μM) produced < 15% change in the ACh-evoked currents at any concentration. We found as well that P2X2, P2X3, or P2X2/3 currents were not affected by ACh (100 μM). In four of the six cases where we could study dose-response relations, we found only minor (<2-fold) changes in the EC50 values for each agonist when we co-expressed these receptors (table S4); an exception is described below. Despite these minimal interactions at the dose-response levels, when ACh and ATP are co-applied, the agonist-induced currents are less than the sum of individual currents (Fig. 6A–C). This pattern was observed with all types of α6* nAChR expressed with P2X2 (table S4), or with P2X2/3 receptors (Fig. 6A–C). Cross-inhibition was also observed between α6(L9’S)β4 and P2X3(K65A) receptors (28); the P2X3(K65A) mutation was employed because it decreases the rate of desensitization (29).
Fig. 6. Electrophysiological detection of crosstalk between P2X and α6* receptors in Xenopus oocytes.
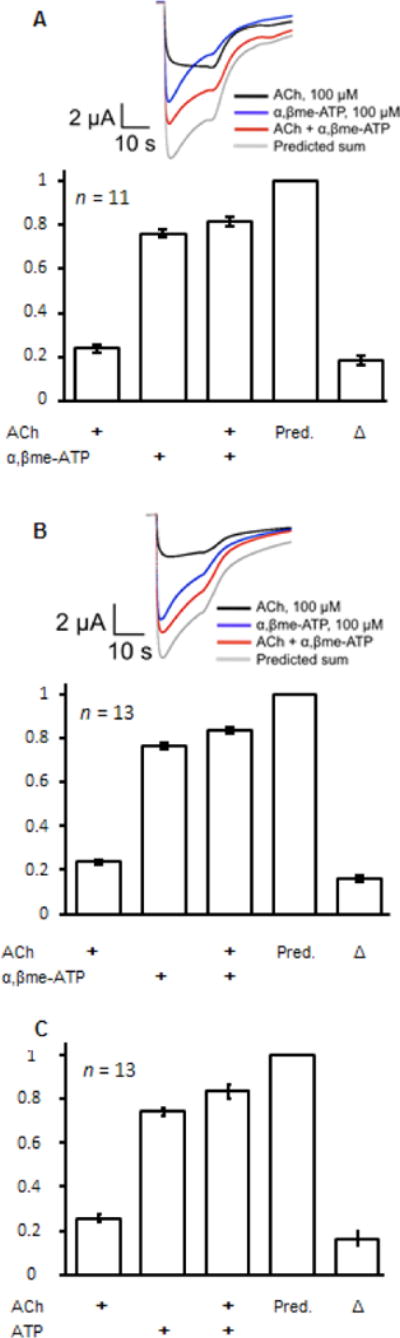
(A) Co-expressed P2X2/3 and α6β4 receptors; (B) co-expressed P2X2/3 and α6β4β3 receptors. Exemplar inward currents are shown, evoked with 100 μM α,βme-ATP, 100 μM ACh, or a mixture of 100 μM α,βme-ATP+ACh. (C) Co-expressed P2X2 and α6β2 receptors were tested with 100 μM ATP, 10 μM ACh, or a mixture of the two agonists. All graphs summarize experiments from n = 11–13 cells. The ‘predicted’ (Pred.) current for each cell is the arithmetic sum of the Iα,βme-ATP and IACh currents. The ‘Δ’ current for each cell is the predicted current minus the observed Iα,βme-ATP+ACh current. Error bars represent SEM. To provide measureable responses, several subunits were mutated as described in Methods.
A second type of cross-talk occurs between α6β2 or α6β4 nAChRs and P2X receptors. The presence of co-expressed α6* nAChRs changes the dose-response relation of the P2X3 receptor (28). This type of interaction has been previously reported only for the interaction between α3β4 nAChR and P2X2 (22). The EC50 of the P2X3 receptor is 2–3-fold higher, and the response has decreased apparent cooperativity, revealed by a reduced Hill coefficient. As a result, responses to ATP in the concentration range 10–100 μM are reduced by approximately half to two-thirds, when normalized to maximal responses. These data are summarized in table S4 [see also (28)].
FRET measurement of interaction between α6* and P2X3 receptors
We tested for physical interactions between α6β4* nAChRs and P2X receptors in cultured mouse cortical neurons, using FRET as previously performed for P2X2 and α4β2 nAChRs (20). FRET typically reveals interactions between fluorophores that are less than 80 Å apart, implying a macromolecular complex. We tested for interactions between eYFP- and mCherry-labeled receptors, using fluorescence life time imaging microscopy. Results show that P2X3 and α6β4 receptors physically interact, with a FRET efficiency of ≈50% (fig. S6) and a binding fraction of ≈40%.
We tested FRET with and without incorporation of non-fluorescent β3 subunit into the α6β4 receptor (fig. S6B,C). The incorporation of β3 did not alter the binding fraction or the FRET efficiency. To test whether the β3 simply did not become incorporated into the α6β4 receptor, we measured FRET in cells transfected with α6, β3-eYFP, and β4 subunits and with P2X3-mCherry receptors where the fluorophores were located on the β3 and P2X3. This resulted in FRET efficiency of ≈50%, indicating that the incorporation of the β3 subunit does not change the FRET efficiency between P2X3 and α6-containing nAChRs. Because some of the electrophysiological data were obtained using the gain-of-function α6 mutant, FRET imaging was also performed in the mutant, expressed with β4-eYFP subunit and P2X3-mCherry. The FRET obtained with α6(L9’S)β4 receptor with P2X3 did not differ statistically from the data obtained with the wild type α6 subunit. A range of control experiments (including FRET determination of soluble eYFP and P2X3-mCherry, P2X3-eYFP and α6-mCherry, P2X3-eYFP and β4-mCherry, and P2X3-eYFP and plasma membrane anchored mCherry) were negative.
Behavioral measurement of α6* and P2X2/3 receptor interactions
As P2X3 receptors mediate both neuropathic and inflammatory pain (23, 24), and there is precedent for the ability of a protein (P2X7) to affect pain indirectly via downregulation of P2X3 receptors (30), we assessed whether P2X3-dependent pain could be affected by activation of α6* receptors, as suggested by the observed cross-inhibitions in a heterologous expression system. The P2X3 agonist, α,βme-ATP, injected into the hind paw produced frank nocifensive (licking) behavior of equivalent intensity in all three genotypes (Fig. 7A). The pain behavior was dose-dependently reversed by systemic nicotine in WT (F3,18=9.1, P<0.001) and L9’S mice (F3,11=6.1, P=0.01), as well as the P2X3 receptor antagonist A-317491 (t10=4.6, P<0.001). In Chrna6 KO mice statistically significant reversal (F3,28=3.1, P=0.04) was only achieved at the highest dose, and to a lesser degree than in the other two genotypes (P<0.01) (Fig. 7A). α,βme-ATP also produced A-317491-reversible mechanical allodynia of equal magnitude in the three genotypes, which was completely reversed by nicotine (0.9 mg/kg) in L9’S mice, partially reversed in WT mice, and unaffected by nicotine in KO mice (P<0.05 compared to L9’S) (Fig. 7B).
Fig. 7. Modulation of P2X2/3 agonist-induced pain and hypersensitivity by nicotine in Chrna6 mutant mice.
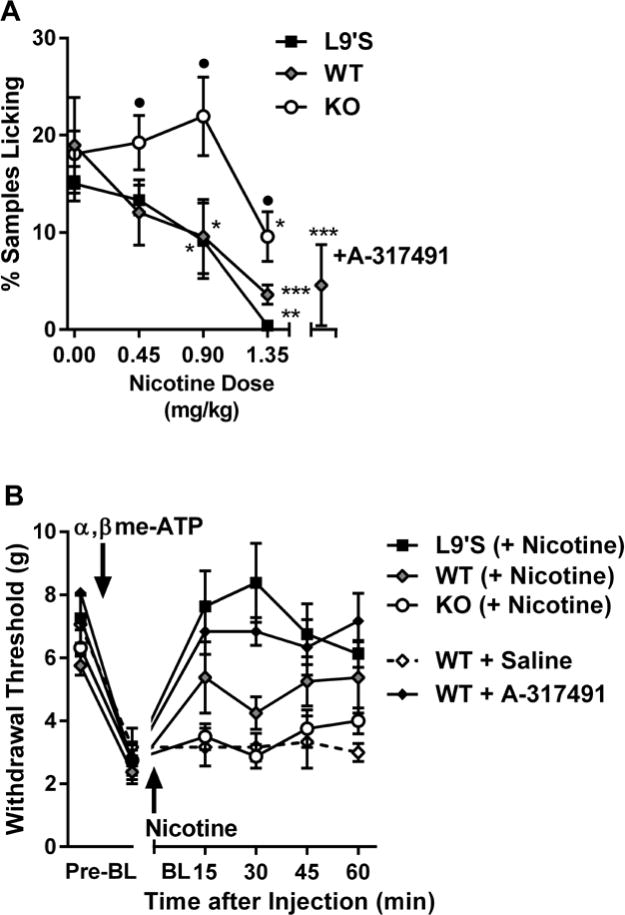
(A) Intrathecal administration of α,βme-ATP produces nocifensive (licking) behavior inhibited by the P2X3 antagonist, A-317491 (300 nmol, intraplantar), and systemic nicotine in WT and L9’S but not KO mice. Symbols (n=4–11 mice/dose/genotype) represent mean ± SEM samples featuring licking behavior (see Materials and Methods). *P<0.05, **P<0.01, ***P<0.001 compared to 0 dose within genotype. •P<0.05 compared to other genotypes within dose. (B) Intrathecal administration of α,βme-ATP produces mechanical allodynia reversed by A-317491 (300 nmol, intraplantar) and nicotine (0.9 mg/kg) in WT and L9’S but not KO mice. Symbols (n=4 mice/genotype or drug) represent mean ± SEM paw withdrawal threshold at each time point.
CHRNA6 and variable chronic pain in humans
A human cohort of 429 adults who underwent herniotomy (31) was genotyped at three CHRNA6 polymorphisms that cover haplotypic diversity in the gene locus to test association of the gene with clinical pain. One promoter region SNP (rs7828365) was found to be associated with changes in pain susceptibility under a recessive inheritance model, in which the minor allele homozygote (TT) showed an increased risk of persistent pain at 6 months post-surgery (odds ratio=12.0, standard error=1.1, P=0.03; Fig. 8A). Only eight TT homozygotes were present in the cohort, and thus the association P-value was computed by a permutation t-test, which is robust in the presence of small expected counts. To replicate this finding, we genotyped rs7828365 in another cohort where the clinical pain phenotype was thoroughly characterized, a cohort a cohort of 159 Caucasian females with TMD (32). Although only two individuals were TT homozygotes, in agreement with findings from the post-surgical pain cohort these TT-carrying TMD patients experienced substantially higher intensity and greater duration of clinical head and orofacial pain symptoms on a normalized composite score incorporating multiple domains of the Chronic Pain Symptom Questionnaire (33). The pain increase was significant as tested by a permutation t-test (P=0.03; Fig. 8B). This cohort was also tested for association with SNPs in the CHRNA4 (3 SNPs), CHRNA5 (8 SNPs) and CHRNB2 (2 SNPs) genes; no P-value was lower than 0.40.
Fig. 8. Human clinical pain is affected by a promoter SNP (rs7828365) in CHRNA6.
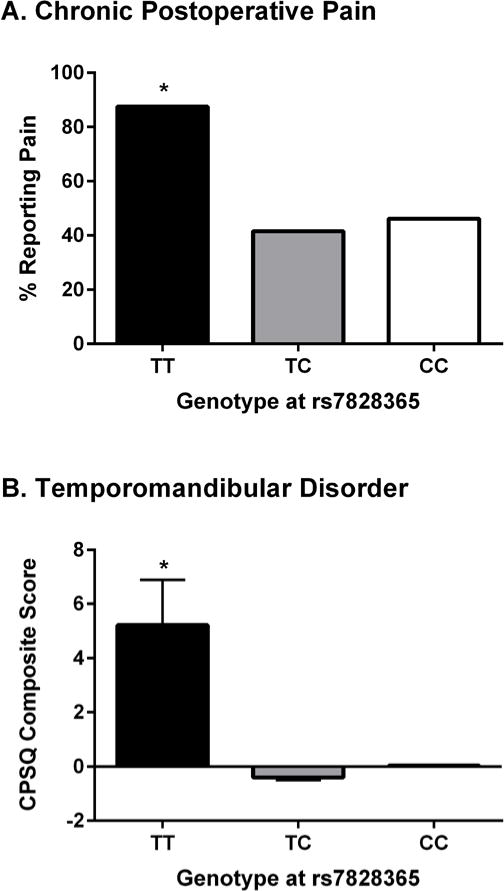
(A) Percentage of herniotomy patients reporting persistent pain 6 months after surgery, stratified by rs7828365 genotype (TT: n=8; TC: n=82; CC: n=325). (B) CPSQ composite pain scores (see Materials and Methods) of temporomandibular disorder patients stratified by rs7828365 genotype (TT: n=2; TC: n=38; CC: n=117). *P<0.05 compared to other genotypes.
To estimate the overall effect of the genotype TT on chronic pain in human subjects, results of the two human studies were combined, yielding P=0.002, which remains significant (P=0.02) after adjusting for three SNPs and three inheritance models examined.
DISCUSSION
The current studies reveal, via expression genomics performed in the mouse, an unexpected role of the Chrna6 gene and α6* receptors in mediating mechanical allodynia after nerve injury or inflammation, and in the reversal of such mechanical allodynia by nicotine. We show that: a) α6* receptors are expressed in a subset of nociceptors within the DRG; b) Chrna6 expression in the DRG correlates with mechanical allodynia across mouse strains; c) mutant mice showing null- or over-expression of Chrna6 display more and less mechanical allodynia, respectively; and, d) nicotine’s spinal and peripheral anti-allodynic effects are mediated by α6* rather than α4* receptors, although both subunits participate in supraspinal effects of nicotine. Further, we have defined a plausible mechanism (although not necessarily the only one) whereby α6* receptors can ameliorate chronic pain, via cross-inhibition with P2X2/3 receptors facilitated by direct contact between the proteins, demonstrated in vitro and behaviorally. Nicotine may be employing this α6*- and P2X2/3-dependent mechanism to produce pain relief, although the statistically significant (but incomplete) analgesia at 1.35 mg/kg in Chrna6 null mutants suggests that alternative mechanisms may also be recruited.
In addition, we have demonstrated the relevance of the CHRNA6 gene in humans; the fact that TT homozygotes report substantially higher clinical pain in two very different chronic pain disorders reinforces the notion that α6* receptors are playing a similar role, qualitatively, in pain biology in mice and humans. The rare frequency of the TT genotype suggests that CHRNA6 is not a primary explanation of chronic pain variability in our species as it appears to be in the mouse. This is very likely simply due to species differences in frequencies of the trait-relevant alleles. The utility of rare variants in the validation of molecular targets for pain is well accepted (34, 35). Nonetheless, the highly limited sample size of TT homozygotes in the present study represents a limitation of the human genetic findings. The true role of CHRNA6 in pain awaits investigation in more highly powered cohorts.
After the discovery of the remarkably high analgesic potency of the frog alkaloid, epibatidine (36), pioneering investigations on epibatidine analogs were interpreted in terms of agonism of α4β2* nAChRs (37, 38). Preclinical and limited clinical evidence do suggest that α4β2* subtypes play an important role in nociception. α4β2* nAChRs are expressed in many CNS regions that modulate pain transmission and α4β2* agonists induce increases in inhibitory tone in the spinal cord. Several high affinity α4β2* nAChR agonists were reported to have potent analgesic activity in rodent models of acute and chronic pain. Furthermore, evidence from studies using KO mice for the α4* and β2* nicotinic subunits showed the dependency of nicotinic analgesia on this subtype (39), although we note that this conclusion was largely based on nicotine inhibition of acute, thermal pain. Much less is known about the composition of other nAChRs mediating analgesia. Various additional nAChR subunits, including α5, α6, α7, β3, and β4 subunits participate in subtypes (for example α4α6β2*, α6β2*, and α6β4*) which have been identified in the spinal cord and DRG tissues. Previous pharmacological and genetic evidence has challenged the assumption that the α4β2 subtype is the main analgesic target (40–43), as does the recently reported clinical trial failure of ABT-894 (44).
The present observations suggest that α6* nicotinic receptors produce their analgesic effects by cross-inhibition of P2X2/3 receptors involving direct contact between the proteins. Other nAChR–P2X receptor interactions increase with the densities of the receptors (21). The details of the contacts, the receptor states involved, and the possible roles of ion flux are not fully known; but modifications to desensitization could play a role (28, 45). Purinergic receptors are important pain processing molecules known to be expressed on nociceptive small diameter neurons in the DRG (46), with important roles having been demonstrated for P2X2/3 (23, 24, 47), P2X4 (48), and P2X7 (49). P2X2/3 receptors have been shown to contribute to multiple pain modalities, including inflammatory pain (23, 24, 47, 50), neuropathic pain (51), visceral pain (52), musculoskeletal pain (53), cancer pain (54), and migraine (55). Presumably the α6* nicotinic receptors interacting in the periphery with P2X2/3 receptors are activated endogenously by acetylcholine, which exists abundantly in mammals both neuronally and non-neuronally, for example in keratinocytes (56). Expression levels of choline acetyltransferase in mouse epidermis exceed that of any other tissue (biogps.org). Furthermore, experiments examining the pharmacokinetic profile of A-317491, and using the rat skin-nerve preparation, have suggested a peripheral site of action of P2X2/3 receptors on pain processing (57).
Our current data demonstrate that, in both chronic inflammatory and neuropathic pain models, nicotine blocks mechanical allodynia—a more important clinical symptom (58) than the acute thermal pain that has been often studied in this area—in an α6*-specific manner, and that the role of α4* in this phenomenon is limited or absent, except in the supraspinal compartment. It is therefore possible that the modest efficacy of some α4β2* agonists reported in animal models of chronic inflammatory pain (43) and initial clinical studies (38) may be related to their insufficient binding and/or functional activity at α6* subtypes, including both α6β2* and α6β4* subtypes studied here; Hone et al. (12) found that the latter have larger responses to ACh. In fact, a very recent paper shows equal binding affinity of ABT-894 to α6β2* and α6β4* nAChRs in monkey striatum (59), but functional selectivity and efficacy were not reported. We believe that the refocusing of nAChR analgesic development on α6*-containing receptors could lead to much more efficacious compounds, which due to the comparatively limited distribution of Chrna6 expression should display a favorable side-effect profile compared to current drugs. Peripheral targeting of such compounds would make them even more attractive, as our findings suggest that efficacy would be preserved while side-effects—for example related to enhanced dopamine release (60) and engagement of brain reward pathways (61)—would be reduced yet further. Development of α6*-acting drugs for the treatment of Parkinson’s disease and nicotine addiction have been hampered by the inability to achieve heterologous expression of α6*-containing receptors and designing ligands that discriminate α6* from α3*, but mutation-based improvements have recently been reported (62, 63). Our findings do reveal a potential side effect of α6* antagonists being contemplated for the treatment of tobacco addiction (64).
MATERIALS AND METHODS
Study Design
This study represents a series of experiments using multiple techniques—including genetics, pharmacology, and electrophysiology—in Xenopus oocytes, mice and humans. All in vivo studies and tissue harvests were performed in accordance with national and institutional guidelines, and were approved by animal care and use committees at McGill University, Virginia Commonwealth University and the California Institute of Technology. In pharmacological studies, mice were assigned to experimental groups using within-cage randomization. Blinding to genotype was in general not possible because of coat color variation; experimenters were however blinded to drug and dose. Power analyses were in general not possible because the effect size of genotype and drug effects were not predictable a priori; sample sizes in this study are consistent with norms in the field (65). Data from three mice in the strain survey were omitted from further analysis because they were identified as statistical outliers (Studentized residual > 3).
Mice
Mice in the 25-strain survey were naïve, adult (6–14 week old) mice of both sexes obtained from The Jackson Laboratory. Strains included: 129S1, A, AKR, BALB/cBy, BTBR T+ tf, BUB/Bn, C3H/He, C57BL/6, C57BR/cd, C58, CBA, DBA/2, FVB/N, KK/Hl, MRL/Mp, NOD/Lt, NON/Lt, NZO/HlLt, NZW/Lac, P, PL, RIIIS, SJL, SM and SWR; all “J” substrains. Mouse strains used in the Persson et al. (2) experiment, some of whose results are reanalyzed here, included AKR/J, C3H/HeJ, C57BL/7J, C58/J, and CBA/J. Subjects of all subsequent experiments were naïve, adult (6–14 week old) C57BL/6J mice bred in our vivarium from breeders obtained from The Jackson Laboratory, mice with L9’S gain-of-function of α6* nAChRs and their wildtype (WT) controls (10), transgenic Chrna6 null mutant (KO) mice and their WT controls (19), α6*-GFP BAC transgenic mice (11), or Chrna4 (α4*) KO mice and their WT controls (66). All mutants have been bred fully congenic (>10 generations) to C57BL/6. All mice were housed in standard polycarbonate cages in groups of 2–5 same-sex littermates in a temperature-controlled (20 ± 1 °C) environment (14:10 h light/dark cycle; lights on at 07:00 h); tap water and food (Harlan Teklad 8604) were available ad lib.
Microarray gene-expression profiling
DRGs were dissected from naïve 2-month-old mice of both sexes (n=3 mice/sex/strain) between 09:00–12:00 h. Total RNA was isolated from tissues using Trizol (Invitrogen) followed by RNeasy (Qiagen, Valencia, CA). RNA quality was examined on an Experion (BioRad) instrument. cDNA and amplified antisense RNA was made from 1.4 μg of pooled total RNA, using the Affymetrix Single Amplification protocol. Affymetrix MOE430v2 arrays were hybridized, washed, stained, and scanned using standard Affymetrix protocols.
Haplotype mapping
Haplotype association mapping was done as described (17, 67). Briefly, local haplotypes were computed for all inbred mouse strains by analyzing a sliding window of SNP genotypes. Strains were grouped based on haplotype group assignment, and the F-statistic was used to quantify the association between that local haplotype and the phenotype of interest. The significance of that F-statistic was computed non-parametrically based on a weighted bootstrap method that accounted for the inherent population structure in the panel of inbred mouse strains (17).
Quality control was achieved as follows. Total RNA samples were only used for pools if the 28S rRNA bands were at least twice the intensity of 18S rRNA bands. Arrays were only included in the final dataset if the following metrics from the Affymetrix MAS5 algorithm were met: 1) percent present calls ≥40; and 2) scaling factors, percent present calls, and background were all within two standard deviations of the mean. Signal intensity histograms, displayed in the MAS5 software, contained no outliers when observed visually. Hierarchical clustering of samples was also examined for outliers which might indicate arrays and RNA with good quality but from poorly dissected tissue; there were no such outliers.
Immunofluorescence
Adult male α6*-GFP transgenic mice were perfused with 4% PFA and DRGs from all levels were quickly dissected. The DRG were post-fixed overnight in 30% sucrose and embedded in OCT. DRGs were sectioned with a cryostat at 10-μm thickness and mounted on super frost plus slides and stored at −80 °C.
Frozen slides were air dried at room temperature for 1 h. Slides were blocked for 1 h at room temperature in PBS plus Triton X-100-containing 3% BSA and 10% goat serum or donkey serum, and overnight at 4 °C with primary antibodies diluted in the blocking solution. The primary antibodies used were: 1:500 rabbit anti-GFP (Life Technologies, Cat. A11122), 1:150 goat anti-GFP (LifeSpan Biosciences; Cat. LS-C48996), 1:1000 rabbit anti-CGRP (Abcam; Cat. ab-47027); 1:500 rabbit anti-NF200 (Sigma; Cat. N4142). The sections were then washed three times in PBS with Triton X-100 and incubated at room temperature for 1 h with secondary antibodies conjugated to Alexa-488 or 568 fluorochromes (Life Technologies) diluted 1:200 in blocking solution. To detect IB4 staining, GS-IB4-Alexa 568 (Life Technologies, Cat. I21412) was diluted 1:200 and incubated during secondary antibody incubations. Sections were then washed three times in PBS with Triton X-100 and mounted in SlowFade gold anti-fade medium with DAPI (Life Technologies).
Image Acquisition and Quantification
Fluorescence images were acquired using an AX70 microscope (Olympus). Images were taken using identical acquisition parameters and raw images were analyzed with Metamorph software. Neurons were considered GFP-positive if the mean fluorescence intensity, measured as arbitrary units, was higher than the mean background fluorescence. This was set as the threshold to include all the GFP-positive cells. Regions were drawn around the GFP-positive cells and these regions were transferred over to the other sensory marker to image co-expressing neurons. Cells were considered positive for NF200, IB4 or CGRP if the mean fluorescence intensity was higher than the mean background fluorescence.
Real-time qPCR
For tissue comparison experiments, DRGs were freshly isolated from adult male C57BL/6J mice and snap frozen on dry ice, and total RNA was isolated using Trizol treatment. Total RNA from all other tissues was purchased from Zyagen. For strain comparison experiments, DRGs from different inbred strains were isolated (n=3 mice/sex/strain) and treated similarly. Two hundred ng of total RNA was used to generate the first-strand cDNA using the Quantitect Reverse transcript kit (Qiagen). A real time Taqman PCR assay for Chrna6 (Assay ID: Mm00517529_m1) was purchased from Life Technologies, with a FAM reporter dye and a non-fluorescent quencher. FastStart Universal probe master mix (Rox) from Roche Diagnostics was used. The reaction was run, in triplicate, in the ABI 7900HT fast real time system using 0.5 μl of the cDNA in a 10-μl reaction as per the manufacturer’s instructions.
Calibrations and normalizations were done using the 2−∆∆CT method. The target gene was Chrna6, while the reference gene was Actb (β-actin). The calibrator for the tissue comparisons was the DRG; the calibrator for the strain comparisons was the DBA/2 strain.
Oocyte Expression and Analysis
Rat α6, rat β2, and mouse β3 nAChR subunits were in the pGEM vector, and rat β4 nAChR was in the pAMV vector. All P2X cDNAs were in the pcDNA3 vector. Site-directed mutagenesis was performed using the Stratagene QuikChange protocol and verified through sequencing. Circular cDNA was linearized, then used as a template for in vitro transcription. Stage V–VI Xenopus laevis oocytes were injected with 50 nl of mRNA solution. To express the α6β4 combination, we used a hypersensitive α6 subunit containing a serine mutation at the leucine9’ in the M2 domain (residue 279). To express the α6β4β3 combination, we used the wild-type α6 and β4 in combination with the hypersensitive β3 containing a serine mutation at the valine13’ in M2 (residue 283). When α6β4* nAChR and P2X receptors were co-expressed, equal volumes of corresponding mRNA solutions were mixed prior to the oocyte injection. To express the α6β2 combination, we used the hypersensitive α6 subunit, as well as a hypersensitive β2 subunit containing a serine mutation at the leucine9’ in M2 and two endoplasmic reticulum export-enhancing mutations (26). To study P2X3, we used the K65A mutation, which accelerated the rate of recovery from desensitization. The α6β2P2X2:α6Lβ2P2X3 mRNA injection ratios were 10:10:1 and 1:1:1 respectively, at 5 ng/oocyte total mRNA. P2X2/3 was expressed by co-injection of 1:10 ratio of P2X2:P2X3 mRNA. After mRNA injection, oocytes were incubated for 12–72 h at 18 °C in culture medium (ND96+ with 5% horse serum).
Two-electrode voltage-clamp recordings used the OpusXpress 6000A (Axon Instruments). For cross-inhibition experiments on P2X3(K65A), the concentration of ATP was 100 μM for cells expressing P2X3(K65A) and α6β4β3(V13’S) or 320 μM for P2X3(K65A) and α6(L9’S)β4. To investigate cross-interaction between P2X2/3 receptor and α6β4* nAChRs, the P2X2/3 receptor was activated by 100 μM α,βme-ATP, and the α6* nAChR by 100 μM ACh. Peak currents from at least three traces were averaged from the same cell for data analysis.
All dose-response data were normalized to the maximal current (Imax = 1) of the same cell and then averaged. EC50 and Hill coefficient (nH) were determined by fitting averaged, normalized dose-response relations to the Hill equation. Dose-response relationships of individual oocytes were also examined and used to determine outliers.
For all cross-interaction data involving P2X2 or P2X2/3, the predicted current from agonist co-application was calculated from the arithmetic sum of IACh and IATP (or Iα,βme-ATP) from the same cell. The actual, observed current upon co-application of the agonists was subtracted from the prediction value of the same cell, and this difference was designated as the Δ. All current data and Δ were normalized to the prediction value of the same cell, and then the normalized data were averaged across ≥ 7 cells from ≥ 2 batches of oocytes.
For all cross interaction data on the P2X3(K65A) receptor, co-application of the agonists used the “prolonged plus brief pulse” protocol (28). Averaged ATP-evoked peak current during ACh application (IATP*) was subtracted from averaged ATP-evoked current in the absence of ACh (IATP) from the same cell to obtain a Δ*. All current data and Δ* were normalized to (IATP) and averaged across ≥ 8 cells from ≥ 2 batches of oocytes.
Neuronal Cultures
Cortical neurons were extracted from day 17 mouse embryos and plated on 35-mm Mattek polylysine-coated glass bottom culture dishes in a neuronal medium containing Neurobasal, B27 (Invitrogen), and Glutamax supplemented with 3% equine serum. Neurons were plated at a density of 60,000 cells per dish. On day 4 of culture, neurons were treated with 1 μM cytosine arabinoside. Neurons were maintained via 50% exchange with feeding medium (Neurobasal, B27, and Glutamax) twice per week. On day 7 in culture, plasmids were mixed in 100 μl of OptiMEM, although 4 μl of Lipofectamine-2000 was mixed with a separate 100 μl aliquot of OptiMEM. After 5 min at 22 °C, the separate solutions were mixed together and kept at room temperature for an additional 25 min. Neurons were transfected with 500 ng of each nAChR plasmids (α6, β3 and β4) and 1000 ng of P2X3 plasmid wild type or labeled with fluorescent protein. After 3 h at 37 °C, transfection medium was replaced with neuronal feeding medium.
FRET Analysis
Mouse E17 cortical neurons were transiently transfected with the indicated constructs on day 5 in culture, and measurements were made 1–3 days later. Before an imaging session, cell culture medium was replaced with phenol red to n2-independent Leibovitz (L-15) medium (Invitrogen). FRET was analyzed by fluorescence lifetime imaging microscopy (FLIM) using a 60x oil immersion objective on a C1si laser-scanning confocal microscope (Eclipse; Nikon) equipped with a 60r-scanning confocal microscope. Samples were scanned at a rate of 6 μs per pixel for a 256 × 256 pixel image. A 480 nm picosecond pulsed diode laser (PDL 800-D, PicoQuant GmbH) provided the excitation light (40 MHz), and emitted light was directed to a single-photon photomultiplier (SPCM-AQR SPAD; Perkin Elmer). A time-correlated single photon counting module and event timer (PicoHarp 300, PicoQuant GmbH) was used to record photon arrival times. Histograms of the time delay between the laser excitation pulse and photon arrival events were fit to exponential decays to extract fluorescence lifetimes for each pixel using PicoHarp 2.0, SymPhoTime software. The extracted lifetimes were used to determine the FRET efficiency (E) where E= 1- τda/τd (τd = donor lifetime in the absence of the acceptor and τda = donor lifetime in the presence of the acceptor). Binding fractions were determined from the coefficients of each exponential component in the fit.
Nociceptive Assays
von Frey Test
In the strain survey, mice were tested on the von Frey test using the up-down staircase method of Dixon (68). Mice were placed on a metal mesh floor within small Plexiglas cubicles (9 × 5 × 5 cm high), and a set of eight calibrated von Frey fibers (Stoelting Touch Test Sensory Evaluator Kit #2 to #9; ranging from 0.007 g to 1.40 g of force) were applied to the plantar surface of the hind paw until they bowed. The presence or absence of a withdrawal response in the next 3 s was scored, and determined the next fiber to be applied. In all subsequent experiments, an automated von Frey test was used (Ugo Basile Dynamic Plantar Aesthesiometer). In this assay, pressure is gradually increased by the device until the mouse withdraws its hind paw; the maximal pressure at that point is displayed. We have found this method to feature less variability than the up-down technique. Relative strain sensitivities are preserved using both methods (J.S. Mogil, unpublished data). In all experiments, measurements were taken in both ipsilateral and contralateral hind paws. Except for in Fig. 1A, only ipsilateral hind paw responses are presented. There were no significant main effects of surgery, genotype or drug on contralateral hind paw withdrawal thresholds in any experiment.
Neuropathic Surgeries
After testing on two separate occasions (averaged) for baseline mechanical sensitivity as described above, some mice received experimental surgeries featuring damage to peripheral nerves serving the hind paw. In different studies either the spared nerve injury (SNI) (69, 70), spinal nerve ligation (SNL) (71), or chronic constriction injury (CCI) (72) was used. In the SNI we spared the sural nerve, and thus von Frey testing occurred on the lateral aspect of the hind paw. Mice were retested for mechanical sensitivity on postoperative days 1, 4, 7, 14, 21 and 28 in experiments evaluating allodynic severity, and on day 7 in experiments evaluating drug anti-allodynia. In the latter, (−)-nicotine (Sigma) was injected either systemically (0.15–1.8 mg/kg, i.p.), intracerebroventricularly (2.5–25 μg, i.c.v.; (73), intrathecally (1–17 μg, i.t.; (74), or subcutaneously into the mid-plantar hind paw (25–100 μg; intraplantar; i.pl.) immediately after “baseline” testing on day 7, and retested 15, 30, 45 and 60 min later. In the experiment shown in Fig. 5e, mice were retested 5, 15 and 30 min after i.t. nicotine injection.
Inflammatory Assay
After testing on two separate occasions (averaged) for baseline mechanical sensitivity as described above, mice were injected with complete Freund’s adjuvant (CFA; 50%; Sigma) into one hind paw. Mice were retested 3, 5, 7, 9 and 11 days post-injection in experiments evaluating allodynic severity, and on day 3 in experiments evaluating drug anti-allodynia. In the latter, (−)-nicotine (Sigma) was injected either systemically (0.30–1.8 mg/kg, i.p.), intracerebroventricularly (2.5–25 μg, i.c.v.; (73), intrathecally (1–17 μg, i.t.; (74), or subcutaneously into the mid-plantar hind paw (25–100 μg; intraplantar; i.pl.) immediately after “baseline” testing on day 3, and retested 15, 30, 45 and 60 min later. In the experiment shown in Fig. 5e, mice were retested 5, 15 and 30 min after i.t. nicotine injection.
α,βme-ATP-induced Pain Behaviors
In some experiments, mice pretreated 20 min earlier with nicotine (0–1.35 mg/kg, i.p.) or A-317491 (300 nmol, i.pl.; Tocris Bioscience) were injected with 40 nmol of α,βme-ATP (Tocris) into one hind paw, and nocifensive licking/biting behaviors were measured over the next 60 min by sampling the first 10 s of every 1-min time period. In other experiments, mice were tested for mechanical sensitivity as described above immediately prior to and 15 min after 40 nmol α,βme-ATP (to confirm the presence of mechanical allodynia), followed immediately by systemic injection of nicotine (0.9 mg/kg) or A-317491 (300 nmol, intraplantar). Mechanical sensitivity was then measured at 15, 30, 45, 60, 90 and 120 min post-drug.
Pain Test Battery
Details of the battery of acute and tonic assays are provided in Mogil et al. (65).
Quantification of Allodynia and Anti-allodynia
Allodynia over the multiple testing days was calculated as area over the withdrawal threshold × time curve using the trapezoidal rule; percentage of maximum possible allodynia (% allodynia) was calculated for each mouse as compared to a hypothetical subject with the same baseline threshold and maximal allodynia (i.e., a threshold of 0 g) at all post-surgery or post-CFA time points.
Drug anti-allodynia over 60 min was calculated as area under the curve using the trapezoidal rule, with respect to the pre-injury (pre-surgery or pre-CFA) baseline and the pre-drug (post-surgery or post-CFA) baseline. Percentage of maximum possible anti-allodynia (% anti-allodynia) was calculated for each mouse as compared to a hypothetical subject with the same pre-injury and pre-drug baseline thresholds and complete resolution of allodynia at all post-drug time points.
Human Clinical Cohorts
Persistent Post-herniotomy Pain Cohort
This prospective cohort was comprised of 429 Danish (n=242) and German (n=187) adult male patients of Caucasian origin (mean age: 55.1 years; SD=13.3) who underwent open or laparoscopic transabdominal pre-peritoneal elective groin hernia repair (31). The main outcome for association analysis was the presence of moderate/severe postoperative 6-month pain (yes=46.6%/no=53.4%). There was no difference in preoperative nociceptive function assessed by quantitative sensory testing between the Danish and the German cohort (31). Genotype-phenotype analysis was done using a pre-specified regression equation, incorporating our assumption that one or two copies of the rare allele would affect the pain score in different genetic models, and adjusted by the following covariates: patients’ age, surgery type and Activity Assessment Scale (AAS) score (“0%” if no pain-related activity impairment was reported, and “100%” for maximum impairment) at baseline. All subjects donated a blood sample for DNA extraction; 14 samples could not be confidently assigned to a genotype. The study was approved by local ethics committees (Hørsholm Hospital, Denmark and Centre for Minimal Invasive Surgery, Germany).
Temporomandibular Disorder (TMD) Cohort
Subjects were non-Hispanic white females (n=159), aged 18 to 60 (mean: 36.8 years), recruited for a case-control study at the UNC Orofacial Pain Clinic between 2005 and 2009. As described previously (32), TMD cases had to report facial pain for at least 5 days during the previous 2 weeks and be diagnosed with TMD arthralgia or myalgia during a standardized clinical examination that used the Research Diagnostic Criteria for TMD (75). Study participants who completed the Chronic Pain Symptom Questionnaire and provided blood for DNA extraction were included in this analysis. The CPSQ is a self-report questionnaire designed to ascertain the presence and characteristics of multiple pain symptoms, and the lifetime presence of multiple pain conditions (33). To derive a single composite value representing pain of the head and neck, seven individual responses (duration of facial pain, intensity of current facial pain, intensity of greatest pain in the last 6 months, intensity of average pain over the last 6 months, primary headache characteristics, percentage of lifetime suffering from primary headache, and count of comorbid pain conditions) were normalized by conversion to z-scores, and then summed. All subjects provided signed informed consent for study procedures including blood draw and genotypic assessment, and this study was approved by the UNC Biomedical Institutional Review Board.
Human Genotyping
Genomic DNA was extracted from each blood sample using QIAamp DNA Bloodkit (Qiagen, CA). Three tagging SNPs were identified within CHRNA6 gene locus using the Haploview Tagger program: rs892413 (MAF=0.21); rs1072003 (MAF= 0.18), and rs7828365 (MAF=0.12). Tagging SNPs were genotyped using the 5’ nuclease method (76) and predesigned ABI SNP assays. Allele-specific signals were distinguished by measuring endpoint 6-FAM or VIC fluorescence intensities at 508 nm and 560 nm, respectively; genotypes were generated using StepOnePlus System Software (Applied Biosystems). The genotyping error rate was directly determined by re-genotyping 25% of the samples, randomly chosen, for each locus. Data cleaning and analysis were implemented using PLINK software v1.07 (77). Standard genotyping quality filters were imposed (call rate >95%, Hardy-Weinberg equilibrium P>5×10−5).
Statistical Analyses
Statistical analyses for mouse studies were conducted using an α level of 0.05. ANOVAs or t-tests were performed as appropriate after determining the normality of the experimental data (Shapiro-Wilk test), followed by Tukey’s or Dunnett’s posthoc tests, as appropriate. One-tailed testing was used where a priori expectations of direction of effect (e.g., analgesia from a known analgesic compound like nicotine) existed. Analgesic ED50s and associated 95% confidence intervals were calculated using the method of Tallarida and Murray (78) as implemented by the FlashCalc 40.1® macro (M.H. Ossipov, University of Arizona). In expression and haplotype genomic mapping studies, multiple testing was controlled using false discovery rate.
Due to the small expected counts for TT homozygotes observed in the two human samples, permutation t-tests were used to assess significance of genetic associations (79). In the case of the herniotomy sample, where the response is binary, the usage of the t statistic in permutations is equivalent to the permutation test based on the χ2 statistic (80). Association P-values for two human studies were combined by a modification of the inverse normal method (81), where study-specific directional P-values are combined and the result is converted to a two-sided P-value.
Supplementary Material
Fig. S1. Hind paw withdrawal thresholds to von Frey fiber stimulation measured before (−14, −7 days) and after (1, 4, 7, 14, 21 and 28 days) SNI surgery.
Fig. S2. Sex differences in Chrna6 DRG mRNA expression in SM mice, and their correlation with sex differences in mechanical allodynia.
Fig. S3. Downregulation of Chrna6 by nerve injury, and correlation with mechanical allodynia.
Fig. S4. Increased ipsilateral mechanical allodynia in Chrna6 (α6 KO) but not Chrna4 null mutants (α4 KO).
Fig. S5. No altered sensitivity of Chrna6 KO mice in a battery of acute and tonic nociceptive assays.
Fig. S6. Physical contacts between P2X3 and α6β4* nAChRs revealed by fluorescence lifetime imaging microscopy.
Table S1. Affymetrix gene expression data of all Chrn* probes, and correlation with baseline nociceptive sensitivity and mechanical allodynia.
Table S2. Top 10 correlated haplotypes (by p-value), genome-wide, with SNI-induced mechanical allodynia in 25 mouse strains.
Table S3. Nicotine anti-allodynic ED50s in all genotypes.
Table S4. Dose-response characteristics (EC50 and Hill coefficients; mean ± S.E.M.) for various combinations of α6-containing nicotinic receptors and P2X receptors expressed in oocytes.
Acknowledgments
We thank Taryn Earley, Takashi Miyamoto, and Matt Petrus for assistance with DRG dissection. We thank Sanjeev Ranade and Valerie Uzzell for data analysis.
Funding: Supported by the Canadian Institutes for Health Research and the Louise and Alan Edwards Foundation (J.S.M.), and the U.S. National Institutes of Health (D.A.D., H.A.L., A.P.).
Footnotes
Author contributions: The study was conceived by A.P. and J.S.M., and designed by J.S.W., L.D., I.B., M.I.D., H.A.L., A.P., and J.S.M. J.S.W., J.Y., W.L., M.R.P., M.A.-Q., R.E.S., L.J.M., K.F., J.-S.A., J.Z., J.M., P.M.S., S.C., J.J., S.A.S., E.K.A., R.B., C.I.R., H.K., and J.W. collected data. D.V.Z., S.B.S., F.D., R.M.D., S.K.S., W.L., G.D.S., and A.I.S. analyzed data. W.M., J.-P.C. and M.D. provided reagents or data. W.M., L.D., I.B., S.C.R.L., M.I.D., H.A.L., A.P., and J.S.M. supervised the collection of data, contributed to its interpretation, and edited the manuscript. J.S.W. and J.S.M. wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The expression genomics data for this study have been deposited into the GeneNetwork Database (www.genenetwork.org).
References
- 1.Li X, Sahbaie P, Zheng M, Ritchie J, Peltz G, Mogil JS, Clark JD. Expression genetics identifies spinal mechanisms supporting formalin late phase behaviors. Mol Pain. 2010;6:11. doi: 10.1186/1744-8069-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Persson A-K, Gebauer M, Jordan S, Metz-Weidmann C, Schulte AM, Schneider H-C, Ding-Pfennigdorff D, Thun J, Xu X-J, Wiesenfeld-Hallin Z, Darvasi A, Fried K, Devor M. Correlational analysis for identifying genes whose regulation contributes to chronic neuropathic pain. Mol Pain. 2009;5:7. doi: 10.1186/1744-8069-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persson A-K, Xu X-J, Wiesenfeld-Hallin Z, Devor M, Fried K. Expression of DRG candidate pain molecules after nerve injury – a comparative study among five inbred mouse strains with contrasting pain phenotypes. J Peripher Nerv Syst. 2010;15:26–39. doi: 10.1111/j.1529-8027.2010.00249.x. [DOI] [PubMed] [Google Scholar]
- 4.Nissenbaum J, Devor M, Seltzer Z, Gebauer M, Michaelis M, Tal M, Dorfman R, Abitbul-Yarkon M, Lu Y, Elahipanah T, delCanho S, Minert A, Fried K, Persson A-K, Shpigler H, Shabo E, Yakir B, Pisante A, Darvasi A. Susceptibility to chronic pain following nerve injury is genetically affected by CACNG2. Genome Res. 2010;20:1180–1190. doi: 10.1101/gr.104976.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vincler M. Neuronal nicotinic receptors as targets for novel analgesics. Expert Opin Invest Drugs. 2005;14:1191–1198. doi: 10.1517/13543784.14.10.1191. [DOI] [PubMed] [Google Scholar]
- 6.Young T, Wittenauer S, McIntosh JM, Vincler M. Spinal α3β2* nicotinic acetylcholine receptors tonically inhibit the transmission of nociceptive mechanical stimuli. Brain Res. 2008;1229:118–124. doi: 10.1016/j.brainres.2008.06.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.AlSharari SD, Freitas K, Damaj MI. Functional role of alpha7 nicotinic receptor in chronic neuropathic and inflammatory pain: studies in transgenic mice. Biochem Pharmacol. 2013;86:1201–1207. doi: 10.1016/j.bcp.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 8.McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol. 2009;78:693–702. doi: 10.1016/j.bcp.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohammadi S, Christie MJ. α9-Nicotinic acetylcholine receptors contribute to the maintenance of chronic mechanical hyperalgesia, but not thermal or mechanical allodynia. Mol Pain. 2014;10:64. doi: 10.1186/1744-8069-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, Marks MJ, Lester HA. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity α6* nicotinic acetylcholine receptors. Neuron. 2008;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackey EDW, Engle SE, Kim MR, O’Neill HC, Wageman CR, Patzlaff NE, Wang Y, Grady SR, McIntosh JM, Marks MJ, Lester HA, Drenan RM. α6* Nicotinic acetylcholine receptor expression and function in a visual salience circuit. J Neurosci. 2012;32:10226–10237. doi: 10.1523/JNEUROSCI.0007-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hone AJ, Meyer EL, McIntyre M, McIntosh JM. Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the α6β4* subtype. FASEB J. 2011;26:917–926. doi: 10.1096/fj.11-195883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, Chang GQ, Jiao YQ, Simon SA. Neuronal nicotinic acetylcholine receptors in rat trigeminal ganglia. Brain Res. 1998;809:238–245. doi: 10.1016/s0006-8993(98)00862-2. [DOI] [PubMed] [Google Scholar]
- 14.Keiger CJ, Walker JC. Individual variation in the expression profiles of nicotinic receptors in the olfactory bulb and trigeminal ganglion and identification of alpha2, alpha6, alpha9, and beta3 transcripts. Biochem Pharmacol. 2000;59:233–240. doi: 10.1016/s0006-2952(99)00326-3. [DOI] [PubMed] [Google Scholar]
- 15.Genzen JR, Van Cleve W, McGehee DS. Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol. 2001;86:1773–1782. doi: 10.1152/jn.2001.86.4.1773. [DOI] [PubMed] [Google Scholar]
- 16.Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin J-S, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. 2011;31:15450–15454. doi: 10.1523/JNEUROSCI.3859-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClurg P, Janes J, Wu C, Delano DL, Walker JR, Batalov S, Takahashi JS, Shimomura K, Kohsaka A, Bass J, Wiltshire T, Su AI. Genomewide association analysis in diverse inbred mice: power and population structure. Genetics. 2007;176:675–683. doi: 10.1534/genetics.106.066241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dussor GO, Leong AS, Gracia NB, Kilo S, Price TJ, Hargreaves KM, Flores CM. Potentiation of evoked calcitonin gene-related peptide release from oral mucosa: a potential basis for the pro-inflammatory effects of nicotine. Eur J Neurosci. 2003;18:2515–2526. doi: 10.1046/j.1460-9568.2003.02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Champtiaux N, Han Z-Y, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux J-P. Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22:1208–1217. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khakh BS, Fisher JA, Nashmi R, Bowser DN, Lester HA. An angstrom scale interaction between plasma membrane ATP-gated P2X2 and α4β2 nicotinic channels measured with FRET and TIRF microscopy. J Neurosci. 2005;25:6911–6920. doi: 10.1523/JNEUROSCI.0561-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khakh BS, Zhou X, Sydes J, Galligan JJ, Lester HA. State-dependent cross-inhibition between transmitter-gated cation channels. Nature. 2000;406:405–410. doi: 10.1038/35019066. [DOI] [PubMed] [Google Scholar]
- 22.Decker DA, Galligan JJ. Molecular mechanisms of cross-inhibition between nicotinic acetylcholine receptors and P2X receptors in myenteric neurons and HEK-293 cells. Neurogastroenterol Motil. 2010;22:901–908. doi: 10.1111/j.1365-2982.2010.01505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa LC, Suzuki R, Carpenter K, Dickenson AH, Boyce S, Hill R, Nebenius-Oosthuizen D, Smith AJH, Kidd EJ, Wood JN. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017. doi: 10.1038/35039526. [DOI] [PubMed] [Google Scholar]
- 24.Cockayne DA, Hamilton SG, Zhu Q-M, Dunn PM, Zhong Y, Novakovic S, Malmberg AB, Cain G, Berson A, Kassotakis L, Hedley L, Lachnit WG, Burnstock G, McMahon SB, Ford APDW. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. doi: 10.1038/35039519. [DOI] [PubMed] [Google Scholar]
- 25.Dash B, Bhakta M, Chang Y, Lukas RJ. Identification of N-terminal extracellular domain determinants in nicotinic acetylcholine receptor (nAChR) α6 subunits that influence effects of wild-type or mutant β3 subunits on function of α6β2*- or α6β4*-nAChR. J Biol Chem. 2011;286:37976–37989. doi: 10.1074/jbc.M111.263673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao C, Srinivasan R, Drenan RM, Mackey ED, McIntosh JM, Lester HA. Characterizing functional α6β2 nicotinic acetylcholine receptors in vitro: mutant β2 subunits improve membrane expression, and fluorescent proteins reveal responsive cells. Biochem Pharmacol. 2011;82:852–861. doi: 10.1016/j.bcp.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virginio C, North RA, Suprenant A. Calcium permeability and block at homomeric and heteromeric P2X2 and P2X3 receptors, and P2X receptors in rat nodose neurones. J Physiol. 1998;510(Pt 1):27–35. doi: 10.1111/j.1469-7793.1998.027bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Limapichat W, Dougherty DA, Lester HA. Subtype-specific mechanisms for functional interaction between α6β4* nicotinic acetylcholine recpeotrs and P2X receptors. Mol Pharmacol. 2014;86:263–274. doi: 10.1124/mol.114.093179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pratt EB, Brink TS, Bergson P, Voigt MM, Cook SP. Use-dependent inhibition of P2X3 receptors by nanomolar agonist. J Neurosci. 2005;25:7359–7365. doi: 10.1523/JNEUROSCI.5189-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Zhang X, Wang C, Li G, Gu Y, Huang L-YM. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proc Natl Acad Sci U S A. 2008;105:16773–16778. doi: 10.1073/pnas.0801793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aasvang EK, Gmaehle E, Hansen JB, Gmaehle B, Forman JL, Schwarz J, Bittner R, Kehlet H. Predictive risk factors for persistent postherniotomy pain. Anesthesiology. 2010;112:957–969. doi: 10.1097/ALN.0b013e3181d31ff8. [DOI] [PubMed] [Google Scholar]
- 32.Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain. 2011;152:2802–2812. doi: 10.1016/j.pain.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohrbach R, Fillingim RB, Mulkey F, Gonzalez Y, Gordon S, Gremillion H, Lim PF, Riberio-Dasilva M, Greenspan JD, Knott C, Maixner W, Slade G. Clinical findings and pain symptoms as potential risk factors for chronic TMD: descriptive data and empirically identified domains from the OPPERA case-control study. J Pain. 2011;12(11 Suppl):T27–T45. doi: 10.1016/j.jpain.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stodberg T, Hennings JC, Bergmann M, Altmuller J, Thiele H, Wetzel A, Nurnberg P, Timmerman V, de Jonghe P, Blum R, Schaible H-G, Weis J, Heinemann SH, Hubner CA, Kurth I. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet. 2013;45:1399–1404. doi: 10.1038/ng.2767. [DOI] [PubMed] [Google Scholar]
- 36.Daly JW. Nicotinic agonists, antagonists, and modulators from natural sources. Cell Mol Neurobiol. 2005;25:513–552. doi: 10.1007/s10571-005-3968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bannon AW, Decker MW, Holladay MW, Curzon P, Donnelly-Roberts D, Puttfarcken PS, Bitner RS, Diaz A, Dickenson AH, Porsolt RD, Williams M, Arneric SP. Broad-spectrum, non-opioid analgesic activity by selective modulation of neuronal nicotinic acetylcholine receptors. Science. 1998;279:77–81. doi: 10.1126/science.279.5347.77. [DOI] [PubMed] [Google Scholar]
- 38.Arneric SP, Holladay M, Williams M. Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem Pharmacol. 2007;74:1092–1101. doi: 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 39.Marubio LM, del Mar Arroyo-Jiminez M, Cordero-Erausquin M, Lena C, Novere N Le, de Kerchove d’Exaerde A, Huchet M, Damaj MI, Changeux J-P. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 40.Damaj MI, Fei-Yin M, Dukat M, Glassco W, Glennon RA, Martin BR. Antinociceptive responses to nicotinic acetylcholine receptor ligands after systemic and intrathecal administration in mice. J Pharmacol Exp Ther. 1998;283:1058–1065. [PubMed] [Google Scholar]
- 41.Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA, Damaj MI. Role of α5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. J Pharmacol Exp Ther. 2010;334:134–146. doi: 10.1124/jpet.110.165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Damaj MI, Fonck C, Marks MJ, Deshpande P, Labarca C, Lester HA, Collins AC, Martin BR. Genetic approaches identify differential roles for α4β2* nicotinic receptors in acute models of antinociception in mice. J Pharmacol Exp Ther. 2007;321:1161–1169. doi: 10.1124/jpet.106.112649. [DOI] [PubMed] [Google Scholar]
- 43.Gao B, Hierl M, Clarkin K, Juan T, Nguyen H, Valk M, Deng H, Guo W, Lehto SG, Matson D, McDermott JS, Knop J, Gaida K, Cao L, Waldon D, Albrecht BK, Boezio AA, Copeland KW, Harmange JC, Springer SK, Malmbert AB, McDonough SI. Pharmacological effects of nonselective and subtype-selective nicotinic acetylcholine receptor agonists in animal models of persistent pain. Pain. 2010;149:33–49. doi: 10.1016/j.pain.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Rowbotham DJ, Arslanian A, Nothaft W, Duan WR, Best AE, Pritchett Y, Zhou Q, Stacey BR. Efficacy and safety of the α4β2 neuronal nicotinic receptor agonist ABT-894 in patients with diabetic peripheral neuropathic pain. Pain. 2012;153:862–868. doi: 10.1016/j.pain.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Hausmann R, Bahrenberg G, Kuhlmann D, Schumacher M, Braam U, Bieler D, Schlusche I, Schmalzing G. A hydrophobic residue in position 15 of the rP2X3 receptor slows desensitization and reveals properties beneficial for pharmacological analysis and high-throughput screening. Neuropharmacology. 2014;79C:603–615. doi: 10.1016/j.neuropharm.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Vulchanova L, Riedl MS, Shuster SJ, Buell G, Surprenant A, North RA, Elde R. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36:1229–1242. doi: 10.1016/s0028-3908(97)00126-3. [DOI] [PubMed] [Google Scholar]
- 47.Cockayne DA, Dunn PM, Zhong Y, Rong W, Hamilton SG, Knight GE, Ruan H-Z, Ma B, Yip P, Nunn P, McMahon SB, Burnstock G, Ford APDW. P2X2 knockout mice and P2X2/P2X3 double knockout mice reveal a role for the P2X2 receptor subunit in mediating multiple sensory effects of ATP. J Physiol. 2005;567:621–639. doi: 10.1113/jphysiol.2005.088435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nat Neurosci. 2012;15:1068–1073. doi: 10.1038/nn.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorge RE, Trang T, Dorfman R, Smith SB, Beggs S, Ritchie J, Austin J-S, Zaykin DV, Vander Meulen H, Costigan M, Herbert TA, Yarkoni-Abitbul M, Tichauer D, Livneh J, Gershon E, Zheng M, Tan K, John SL, Slade GD, Jordan J, Woolf CJ, Peltz G, Maixner W, Diatchenko L, Seltzer Z, Salter MW, Mogil JS. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat Med. 2012;18:595–599. doi: 10.1038/nm.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honore P, Mikusa J, Bianchi B, McDonald H, Cartmell J, Faltynek C, Jarvis MF. TNP-ATP, a potent P2X3 receptor antagonist, blocks acetic acid-induced abdominal constriction in mice: comparison with reference analgesics. Pain. 2002;96:99–105. doi: 10.1016/s0304-3959(01)00434-1. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Zhang X, Guo QL, Zou WY, Huang CS, Yan JQ. Cyclooxygenase inhibitors suppress the expression of P2X3 receptors in the DRG and attenuate hyperalgesia following chronic constriction injury in rats. Neurosci Lett. 2010;478:77–81. doi: 10.1016/j.neulet.2010.04.069. [DOI] [PubMed] [Google Scholar]
- 52.Kiyatkin ME, Feng B, Schwartz ES, Gebhart GF. Combined genetic and pharmacological inhibition of TRPV1 and P2X3 attenuates colorectal hypersensitivity and afferent sensitization. Am J Physiol Gastrointest Liver Physiol. 2013;305:G638–G648. doi: 10.1152/ajpgi.00180.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noma N, Shinoda M, Honda K, Kiyomoto M, Dezawa K, Nakaya Y, Komiyama O, Imamura Y, Iwata K. Interaction of IL-1β and P2X3 receptor in pathologic masseter muscle pain. J Dent Res. 2013;92:456–460. doi: 10.1177/0022034513483770. [DOI] [PubMed] [Google Scholar]
- 54.Liu M, Yang H, Fang D, Yang JJ, Cai J, Wan Y, Chui DH, Han JS, Xing GG. Upregulation of P2X3 receptors by neuronal calcium sensor protein VILIP-1 in dorsal root ganglions contributes to the bone cancer pain in rats. Pain. 2013;154:1551–1568. doi: 10.1016/j.pain.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 55.Hullugundi SK, Ferrari MD, Van den Maagdenberg AM, Nistri A. The mechanism of functional up-regulation of P2X3 receptors of trigeminal sensory neurons in a genetic mouse model of familial hemiplegic migraine type 1 (FHM-1) PLoS One. 2013;8:e60677. doi: 10.1371/journal.pone.0060677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wessler I, Kilbinger H, Bittinger F, Unger R, Kirkpatrick CJ. The non-neuronal cholinergic system in humans: expression, function and pathophysiology. Life Sci. 2003;72:2055–2061. doi: 10.1016/s0024-3205(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 57.Wu G, Whiteside GT, Lee G, Nolan S, Niosi M, Pearson MS, Ilyin VI. A-317491, a selective P2X3/P2X2/3 receptor antagonist, reverses inflammatory mechanical hyperalgesia through action at peripheral receptors in rats. Eur J Pharmacol. 2004;504:45–53. doi: 10.1016/j.ejphar.2004.09.056. [DOI] [PubMed] [Google Scholar]
- 58.Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci. 2009;10:283–294. doi: 10.1038/nrn2606. [DOI] [PubMed] [Google Scholar]
- 59.Zhang D, Bordia T, McGregor M, McIntosh JM, Decker MW, Quik M. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Mov Disord. 2014;29:508–517. doi: 10.1002/mds.25817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Lee JW, Oh G, Grady SR, McIntosh JM, Brunzell DH, Cannon JR, Drenan RM. Enhanced synthesis and release of dopamine in transgenic mice with gain-of-function α6* nAChRs. J Neurochem. 2014;129:315–327. doi: 10.1111/jnc.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Biasi M, Dani JA. Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci. 2011;34:105–130. doi: 10.1146/annurev-neuro-061010-113734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rasmussen AH, Strobaek D, Dyhring T, Jensen ML, Peters D, Grunnet M, Timmermann DB, Ahring PK. Biophysical and pharmacological characterization of α6-containing nicotinic acetylcholine receptors expressed in HEK293 cells. Brain Res. 2014;1542:1–11. doi: 10.1016/j.brainres.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 63.Hone AJ, Ruiz M, Scadden M, Christensen S, Gajewiak J, Azam L, McIntosh JM. Positional scanning mutagenesis of α-conotoxin PeIA identifies critical residues that confer potency and selectivity for α6/α3β2β3 and α3β2 nicotinic acetylcholine receptors. J Biol Chem. 2013;288:25428–25439. doi: 10.1074/jbc.M113.482059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brunzell DH. Preclinical evidence that activation of mesolimbic alpha 6 subunit containing nicotinic acetylcholine receptors supports nicotine addiction phenotype. Nicotine Tob Res. 2012;14:1258–1269. doi: 10.1093/ntr/nts089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mogil JS, Ritchie J, Sotocinal SG, Smith SB, Croteau S, Levitin DJ, Naumova AK. Screening for pain phenotypes: analysis of three congenic mouse strains on a battery of nine nociceptive assays. Pain. 2006;126:24–34. doi: 10.1016/j.pain.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 66.Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J. Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu C, Delano DL, Mitro N, Su SV, Janes J, McClurg P, Batalov S, Welch GL, Zhang J, Orth AP, Walker JR, Glynne RJ, Cooke MP, Takahashi JS, Shimomura K, Kohsaka A, Bass J, Saez E, Wiltshire T, Su AI. Gene set enrichment in eQTL data identifies novel annotations and pathway regulators. PLoS Genet. 2008;4:e1000070. doi: 10.1371/journal.pgen.1000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia evoked by unilateral ligation of the fifth and sixth lumbar nerves in the rat. J Neurosci Meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 69.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 70.Shields SD, Eckert WA, III, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain. 2003;4:465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- 71.Kim SH, Chung JM. An experimental model for peripheral neuropathic produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 72.Bennett GJ, Xie Y-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 73.Laursen SE, Belknap JK. Intracerebroventricular injections in mice: some methodological refinements. J Pharmacol Meth. 1986;16:355–357. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- 74.Hylden JLK, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 75.Dworkin S, LeResche L. Research diagnostic criteria for temporomandibular disorders: review, criteria, examinations and specifications, critique. J Craniomandib Disord. 1992;6:301–355. [PubMed] [Google Scholar]
- 76.Shi MM, Myrand SP, Bleavins MR, de la Iglesia FA. High throughput genotyping for the detection of a single nucleotide polymorphism in NAD(P)H quinine oxidoreductase (DT diaphoresis) using TaqMan probes. Mol Pathol. 1999;52:295–299. doi: 10.1136/mp.52.5.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tallarida RJ, Murray RB. Manual of Pharmacologic Calculation. Springer-Verlag; New York: 1981. [Google Scholar]
- 79.Good PI. Permutation tests: a practical guide to resampling methods for testing hypotheses. Springer; New York: 2000. [Google Scholar]
- 80.D’Agostino RB. Relation between chi-squared and ANOVA tests for testing equality of k independent dichotomous populations. Am Statistician. 1972;26:30. [Google Scholar]
- 81.Zaykin DV. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J Evol Biol. 2011;24:1836–1841. doi: 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Hind paw withdrawal thresholds to von Frey fiber stimulation measured before (−14, −7 days) and after (1, 4, 7, 14, 21 and 28 days) SNI surgery.
Fig. S2. Sex differences in Chrna6 DRG mRNA expression in SM mice, and their correlation with sex differences in mechanical allodynia.
Fig. S3. Downregulation of Chrna6 by nerve injury, and correlation with mechanical allodynia.
Fig. S4. Increased ipsilateral mechanical allodynia in Chrna6 (α6 KO) but not Chrna4 null mutants (α4 KO).
Fig. S5. No altered sensitivity of Chrna6 KO mice in a battery of acute and tonic nociceptive assays.
Fig. S6. Physical contacts between P2X3 and α6β4* nAChRs revealed by fluorescence lifetime imaging microscopy.
Table S1. Affymetrix gene expression data of all Chrn* probes, and correlation with baseline nociceptive sensitivity and mechanical allodynia.
Table S2. Top 10 correlated haplotypes (by p-value), genome-wide, with SNI-induced mechanical allodynia in 25 mouse strains.
Table S3. Nicotine anti-allodynic ED50s in all genotypes.
Table S4. Dose-response characteristics (EC50 and Hill coefficients; mean ± S.E.M.) for various combinations of α6-containing nicotinic receptors and P2X receptors expressed in oocytes.