

Abstract
Accumulating evidence implies that both AKT1 and GABAA receptor (GABAAR) subunit genes are involved in schizophrenia pathogenesis. Activated Akt promotes GABAergic neuron differentiation and increases GABAAR expression on the plasma membrane. To elucidate the role of Akt1 in modulating GABAergic functions and schizophrenia-related cognitive deficits, a set of 6 in vitro and in vivo experiments was conducted. First, an Akt1/2 inhibitor was applied to evaluate its effect on GABAergic neuron-like cell formation from P19 cells. Inhibiting Akt resulted in a reduction in parvalbumin-positive neuron-like cells. In Akt1−/− and wild-type mice, seizures induced using pentylenetetrazol (a GABAAR antagonist) were measured, and GABAAR expression and GABAergic interneuron abundance in the brain were examined. Female Akt1−/− mice, but not male Akt1−/− mice, exhibited less pentylenetetrazol-induced convulsive activity than their corresponding wild-type controls. Reduced parvalbumin-positive interneuron abundance and GABAAR subunit expression, especially in the hippocampus, were also observed in female Akt1−/− mice compared to female wild-type mice. Neuromorphometric analyses revealed significantly reduced neurite complexity in hippocampal pyramidal neurons. Additionally, female Akt1−/− mice displayed increased hippocampal oscillation power and impaired spatial memory compared to female wild-type mice. Our findings suggest that Akt1 deficiency modulates GABAergic interneurons and GABAAR expression, contributing to hippocampus-dependent cognitive functional impairment.
The Akt (also known as protein kinase B) family consists of three serine/threonine kinases (Akt1, Akt2, and Akt3) that are involved in multiple biological functions and cellular processes. The Akt family is highly conserved in mammals, and the amino acid sequences of mouse, rat and human Akts share approximately 95% identity1. Recent advances in genome-wide analyses indicated that there might be a common pathophysiology among several psychiatric disorders2 and that deregulation of Akt signaling pathways is directly associated with some of the most prevalent and incurable human disorders, including schizophrenia3. Accumulating evidence also suggests that multiple susceptibility genes, including Akt1 (PKBα)4 and GABAA receptor (GABAAR) subunit genes5,6, might contribute to the pathogenesis of schizophrenia. Evidence supporting Akt1 as a susceptibility gene for schizophrenia was originally reported in Caucasian families of European descent and was subsequently confirmed in several other ethnic groups4,7,8. Studies of the postmortem brains of schizophrenia patients4,9 and Akt1-deficient mice10,11,12,13,14 as well as functional neuroimaging analyses in humans15,16 support the concept that variations in the Akt1 gene or its encoded protein have epistatic effects on dopamine-related functions. The biological functions of Akt1 and the mechanism by which Akt1 contributes to susceptibility for schizophrenia are currently under intensive investigation.
Schizophrenia is a multifactorial disorder for which there is a strong genetic predisposition, but the exact cause of schizophrenia remains unclear. Complementary to the long-standing dopamine and glutamate hypotheses of schizophrenia, GABA, the primary inhibitory neurotransmitter in the brain, has attracted increasing attention in the search for the neural mechanisms underlying the cognitive deficits observed in schizophrenia17,18. Evidence supporting the role of GABA in schizophrenia development includes the deficiency of GABA synthesis resulting from reduced transcription of the 67-kDa isoform of glutamic acid decarboxylase (GAD67) within parvalbumin-immunoreactive cortical neurons and the reduction in the subpopulation of GABAergic interneurons positive for parvalbumin and the β2 subunit of the GABAA receptor (GABAAR) in animal studies and in the post-mortem brains of schizophrenia patients18,19,20,21,22. More specifically, cognitive dysfunction is considered a core feature of schizophrenia that not only strongly influences the quality of life and functionality of people with this illness23,24 but also predicts the long-term functional outcome of these patients25. The GABA hypothesis further predicts that abnormalities in GABAergic neurons lead to impairments in cortical gamma synchrony and corresponding cognitive control in schizophrenia26. Indeed, schizophrenic patients exhibited deficits in gamma oscillations27, and this finding suggests that altered GABA signalling may associate dysfunction of gamma activity with the pathology of schizophrenia.
Intriguingly, emerging in vitro studies indicated the involvement of Akt signalling in the regulation of GABAergic synapses. It was reported that the activation of Akt signalling facilitated GABAAR trafficking to the membrane via the phosphorylation of β2 GABAAR subunits28. Active PDK1-Akt signalling can promote the differentiation of telencephalic neural precursor cells into GABAergic, but not glutamatergic, neurons29. It was also reported that the regulation of hippocampal neurogenesis by the protein Disrupted-in-Schizophrenia 1 requires GABA-induced depolarization through a convergence onto Akt- mammalian target of rapamycin (mTOR) signalling pathway30. Similarly, our recent findings further revealed reductions in the morphological complexity and alterations in the electrophysiological properties of striatal GABAergic medium spiny neurons in homozygous Akt1-knockout (Akt1−/−) mice12. It is of great interest to investigate how Akt1 regulates GABA signalling and GABA-related cognitive functions that might contribute to the pathogenesis of schizophrenia and its associated cognitive deficits.
As a complement to human studies, animal models provide an indispensable and practical approach to elucidate causal relationships between genetic deficits and functions, especially in genetically modified mice. Distinct sex-specific phenotypes were identified in Akt1-deficient mice11,12, and the Akt1 isoform was reported to be involved in the regulation of hippocampal neuroplasticity and cognition31; this regulatory activity of Akt1 might contribute to the pathogenesis of schizophrenia. Given the importance of Akt1 signalling and GABAergic function in the pathogenesis of schizophrenia and the involvement of Akt1 in the regulation of hippocampal functions, the primary goal of this study was to evaluate whether Akt1 deficiency causes any alteration in GABAergic function or hippocampus-related function at multiple levels. A set of 6 experiments was designed to identify the role of Akt1 in GABAergic signalling and related cognitive functions in vitro and in vivo. Our findings indicated that Akt1 deficiency modulates the number of GABAergic interneurons, GABAAR expression, and neuronal morphology in the hippocampus. These effects of Akt1 deficiency might contribute to the impairment of neuronal oscillations and of hippocampus-dependent cognitive functions in female Akt1−/− mice.
Results
Experiment 1: Akt1/2 inhibitor application disrupted the differentiation of P19 cells into GABAergic neuron-like cells
An Akt1/2 inhibitor was used to evaluate the effect of Akt deficiency on the differentiation of P19 mouse embryonal carcinoma cells (P19 cells) into GABAergic neuron-like cells which were reported to express glutamic acid decarboxylase and multiple GABAAR subunits. As shown in Fig. 1a, Akt1/2 inhibitor application did not affect the Ascl1-induced differentiation of P19 cells into neuron-like cells; a representative image is shown in Figure S1. In contrast, Akt1/2 inhibitor application resulted in a 40% reduction in the proportion of GABAergic neuron-like cells (t(4) = 4.004, p < 0.05; Fig. 1b and Figure S2) and a 60% reduction in the proportion of parvalbumin-positive neuron-like cells (t(4) = 6.990, p < 0.05; Fig. 1c and Figure S3) compared to vehicle treatment. However, Akt inhibitor treatment did not affect the expression of functional GABAARs, as measured by the expression of the GABAAR β2 subunit at plasma membrane (Fig. 1d). These results revealed that inhibition of Akt had no effect on overall neuronal differentiation but resulted in a significant reduction in the proportion of GABAergic neuron-like cells in a P19 cell culture model.
Figure 1. Experiment 1: Inhibiting Akt1/2 had no effect on overall neuronal differentiation but resulted in the proportion of significantly fewer GABAergic neuron-like cells from P19 mouse embryonal carcinoma cells.

(a) Akt1/2 inhibitor application had no effect on the proportion of differentiated neuron-like cells (Tuj1/GFP, mean ± SEM) derived from P19 cells (Tuj1, neuronal marker; GFP, transfection marker; US2, control plasmid for transfection; Ascl1 (Mash1), inducer of neuronal differentiation; Vehicle, vehicle control; Akt inhibitor, Akt1/2 inhibitor). (b) Akt1/2 inhibitor application resulted in a significant reduction of the proportion of GABAergic neuron-like cells (GAD67/GFP, mean ± SEM) derived from P19 cells (GAD67, GABA marker; GFP, transfection marker). (c) Akt1/2 inhibitor application resulted in a significant reduction in the proportion of parvalbumin-positive neuron-like cells (parvalbumin/GFP, mean ± SEM) derived from P19 cells (GFP, transfection marker). (d) Treatment with the Akt1/2 inhibitor had no significant effect on the proportion of cells with GABAAR expression on the plasma membrane (GABAARs/Na+/K+ ATPase, mean ± SEM) (Na+/K+ ATPase, loading control for membrane proteins). *p < 0.05.
Experiment 2: Pentylenetetrazol-induced convulsive activity is reduced in female Akt1 −/− mice compared with female wild-type (WT) mice
As reduced Akt1/2 activity disrupted the differentiation of GABAergic neurons but did not alter overall neuronal differentiation in vitro, we sought to determine whether reducing Akt1 activity changed GABAergic function on a global scale in vivo. For this purpose, we induced seizures using pentylenetetrazol (PTZ), a GABAA receptor antagonist and a CNS convulsant, in both male and female Akt1−/− and WT mice. In female Akt1−/− mice, the onset latency was significantly increased (t(14) = 2.177, p < 0.05; Fig. 2a) and seizure severity was decreased (t(14) = 3.12, p < 0.05; Fig. 2b) compared to female WT littermates. In contrast, no difference in either onset latency (p = 0.253) or seizure severity (p = 0.0856) was found between Akt1−/− and WT males. Thus, Akt1 deficiency modulated the convulsive activity induced by a general GABAAR antagonist, especially in female mice.
Figure 2. Experiment 2: Seizures induced by PTZ (a GABAAR antagonist and a CNS convulsant) in male and female Akt1−/− mice and their WT littermate controls.

Measurement of PTZ-induced seizures in WT control mice (white bar) and Akt1−/− mice (black bar). (a) The latency to initial seizure onset (mean ± SEM, sec) for both males (n = 6 each; Left) and females (n = 8 each; Right). The female Akt1−/− mice exhibited delayed seizure onset compared to the female WT mice. (b) The average seizure severity score (0–4) (mean ± SEM, sec) for both male (n = 6; Left) and female (n = 8; Right) mice. Seizure severity scores: 0, no signs of motor seizures; 1, isolated twitches; 2, tonic-clonic convulsions; 3, tonic extension; and 4, death. Female Akt1−/− mice showed lower seizure severity scores than female WT mice. *p < 0.05.
Experiment 3: Reduced density of parvalbumin-positive interneurons and GABAAR expression in the hippocampus of female Akt1 −/− mice
GABA plays an important role in mediating pre- and post-synaptic inhibition of neuronal activity. Because PTZ-induced seizures were suppressed in female Akt1−/− mice, we examined whether the number of GABAergic interneurons (Experiment 3a) or the GABAAR expression level (Experiment 3b) in the target brain regions was altered in naïve female Akt1−/− mice compared to female WT mice. Among all brain areas examined (Fig. 3a), immunohistochemistry revealed no genotypic difference in the abundance of calretinin-positive interneurons (Fig. 3b). In contrast, female Akt1−/− mice showed significant decreases in the expression of parvalbumin-positive interneurons in the CA1 region (t(14) = 3.074, p < 0.05) and the CA3 region (t(14) = 3.557, p < 0.05) of the dorsal hippocampus (Fig. 3c,d). Western blot analysis revealed a significant reduction in the expression of the GABAAR β2 subunit in the hippocampus (t(8) = 2.652, p < 0.05), but not in the striatum or the cortex (Fig. 3e,f), of Akt1−/− mice compared to WT mice. These results indicated that Akt1 deficiency contributed to a reduction in the density of parvalbumin-positive interneurons and in the expression of functional GABAARs in female mice, especially in the hippocampus.
Figure 3. Experiment 3: Reduced numbers (cell density; cells/mm2; mean ± SEM) of parvalbumin-positive interneurons and reduced expression of GABAA receptors were observed in the hippocampus of female Akt1−/− mice (black bar; n = 6) compared to female WT littermate controls (white bar; n = 8).

(a) Mouse brain atlases highlighting the regions of interest: hippocampal CA1 region (CA1), hippocampal CA3 region (CA3), primary motor cortex (M1), primary auditory cortex (Au1), anterior cingulate cortex, area 1 (aCg1), prelimbic cortex (PrL), and infralimbic cortex (IL). (b) Calretinin-positive interneurons in the different brain regions of female mice. (c) Parvalbumin-positive interneurons in the different brain regions of female mice. The cell density in the CA1 and CA3 areas was lower in female Akt1−/− mice than in female WT mice. (d) Representative immunohistochemical images of parvalbumin-positive interneurons in the dorsal hippocampus (CA1 and CA3 areas; scale bar: 200 μm). (e) The expression of functional GABAARs in the hippocampus, cortex, and striatum of female mice. A significant difference in functional GABAAR expression in the hippocampus was found between Akt1−/− and WT females. (f) Representative Western blot images of functional GABAAR expression (GABAAR: GABAAR β2 subunit; Na+/K+ ATPase: loading control for membrane proteins). *p < 0.05.
Experiment 4: Reduced neurite complexity of hippocampal CA1 pyramidal neurons in female Akt1 −/− mice
The generation of seizures is controlled by the balance of network excitation and inhibition. Given that genetic deletion of Akt1 in mice resulted in a reduction in neuronal morphological complexity in the brain10,11,12 and that GABAergic interneurons primarily innervate the dendrites of hippocampal CA1 pyramidal cells and actively contribute to the input/output signalling of these neurons32,33, we determined the impact of Akt1 deficiency on the morphological characteristics of pyramidal neurons in the dorsal hippocampal CA1 region of Thy1-C57BL6-Tg (GFPm) transgenic mice. Representative tracings of GFP-labelled CA1 pyramidal neurons in the hippocampus of female WT and Akt1−/− mice are shown in Fig. 4a. A quantitative evaluation of the GFP-labelled CA1 pyramidal neurons revealed significant morphological changes in the apical and basal dendritic architecture and its complexity. In apical dendrites, the numbers of dendritic branches (32%, t(22) = 2.887, p < 0.05; left panel of Fig. 4b) and tips (30%, t(22) = 2.887, p < 0.05; middle panel of Fig. 4b) were decreased in the Akt1−/− females compared with the WT females. In basal dendrites, the soma size was significantly increased (20%, t(22) = 2.371, p < 0.05; left panel of Fig. 4c), the number of branches (28%, t(22) = 4.269, p < 0.05), the number of tips (22%, t(22) = 4.224, p < 0.05), and the total dendrite length were reduced (18%, t(22) = 2.688, p < 0.05) in the Akt1−/− females compared to the WT females, as depicted in Fig. 4d. As shown in Fig. 4e, the effect of Akt deficiency on morphological complexity was confirmed via Sholl analysis (F(1, 22) = 7.003, p < 0.05). No significant differences in other morphological variables were found. These results revealed significant reductions in the dendritic complexity of hippocampal CA1 pyramidal neurons in female Akt1−/− mice that might affect neural activity and hippocampal output.
Figure 4. Experiment 4: Examination of the morphological features (mean ± SEM) of pyramidal neurons in the CA1 region of the dorsal hippocampus in female WT mice (n = 13; white bars) and female Akt1−/− mice (n = 11; black bars).

(a) Representative tracings of dorsal hippocampal pyramidal neurons from WT and Akt1−/− mice (scale bar: 50 μm). (b) Apical neuronal properties (left to right: number of branches, number of tips, and total length of the apical tuft (μm)). (c) Basal neuronal properties (left to right: soma size (μm2), number of primary dendrites, and total length of primary dendrites (μm)). (d) Additional basal neuronal properties (left to right: number of branches, number of tips, and total length (μm)). (e) Sholl analysis of basal dendritic complexity within 10-μm concentric spheres around the soma. *p < 0.05.
Experiment 5: Alteration of hippocampal oscillations was detected in anaesthetized female Akt1 −/− mice
The balance between excitation and inhibition plays a critical role in hippocampal neural oscillation34. Based on the observed alterations in the hippocampus of female Akt1−/− mice, local field potentials were recorded in the hippocampal CA1 region. The power spectrum density of the hippocampal local field potentials is shown in Fig. 5a. Compared to female WT mice, female Akt1−/− mice displayed higher oscillation power in the theta (t(8) = 2.629, p < 0.05), alpha (t(8) = 2.497, p < 0.05), beta (t(8) = 2.286, p = 0.052), and gamma (t(8) = 2.585, p < 0.05) frequency ranges but not in the delta frequency range (t(8) = 1.293, p = 0.232), as depicted in Fig. 5b. Alteration of neural oscillations in the CA1 region of the dorsal hippocampus in anaesthetized female Akt1−/− mice was confirmed in this experiment.
Figure 5. Experiment 5: Altered neural oscillations from the hippocampal CA1 region were identified in anaesthetized female Akt1−/− mice compared to female WT littermate controls.
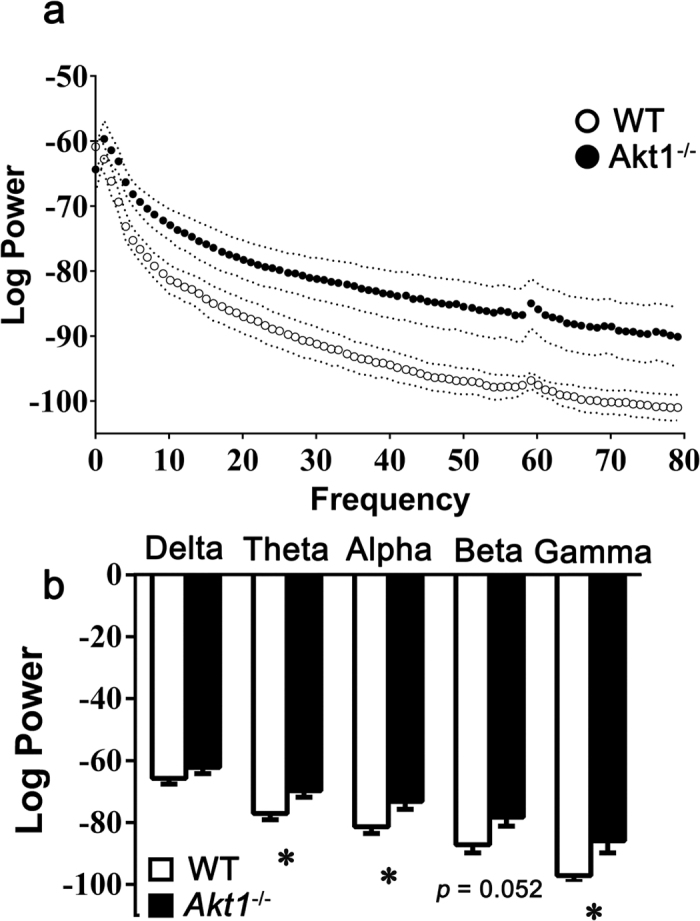
(a) The power spectrum density (mean ± SEM) of hippocampal LFPs in the high frequency ranges was greater in anaesthetized female Akt1−/− mice (white dot; n = 5) than in female WT controls (black dot; n = 5). Original data showing the log oscillation power from low frequency to high frequency (from left to right: 0–80 Hz; delta: 0–4 Hz; theta: 5–8 Hz; alpha: 9–12 Hz; beta: 12–30 Hz; and gamma: 30–80 Hz). (b) The average log oscillation power (mean ± SEM) in female Akt1−/− mice (black bar) was greater than that in WT controls (white bar) in the high frequency ranges. *p < 0.05.
Experiment 6: Female Akt1 −/− mice displayed impairments in hippocampus-related cognitive function
We further investigated whether the observed alterations in hippocampal neural oscillations led to changes in hippocampus-related cognitive functions (Y-maze and Morris water maze task performance) in female Akt1−/− mice. In the Y-maze task (Experiment 6a), female Akt1−/− mice exhibited impairments in the relative time spent (t(14) = 2.075, p = 0.057) and distance travelled in the novel arm (t(14) = 3.082, p < 0.05) compared to their WT controls (Fig. 6a). In the Morris water maze test (Experiment 6b), female Akt1−/− mice learned less rapidly than their WT controls. Female Akt1−/− mice took a significantly longer time to find the platform during the acquisition (F(1,14) = 14.711, p < 0.05) and reversal trials (F(1,14) = 7.141, p < 0.05), as shown in Fig. 6b. On the probe trial (Day 9), female Akt1−/− mice swam a shorter distance in the target quadrant than their WT controls (t(14) = 2.870, p < 0.05), but no difference in swimming activity in the other quadrants was found between the groups (Fig. 6c). Thus, it is evident that female Akt1−/− mice displayed impairments in hippocampus-related cognitive functions on the Y-maze and Morris water maze tasks.
Figure 6. Female Akt1−/− mice exhibited impaired hippocampus-related cognitive performance on the Y-maze (Experiment 6a) and Morris water maze tasks (Experiment 6b).

(a) The percentages (%, mean ± SEM) of time spent and distance travelled in the novel arm of the Y-maze. Female Akt1−/− mice (black bars; n = 8) exhibited reduced relative duration and distance in the novel arm of the Y-maze compared to female WT littermate controls (white bars; n = 8). (b) The escape latency (mean ± SEM sec) to reach the hidden platform in the Morris water maze task. Female Akt1−/− mice (black circles; n = 8) showed impairments in acquisition and reversal learning in the Morris water maze test. (c) Female Akt1−/− mice swam a shorter distance in the target quadrant than female WT controls on the probe test (Day 9). *p < 0.05.
Discussion
Guided by the convergent results of human genetic studies, several assays were performed in this study to evaluate the effect of Akt1 deficiency on the ratio of GABA interneurons, GABAAR expression, and hippocampus-related cognitive functions in vitro and in vivo. Inhibiting Akt had no effect on overall neuronal differentiation but resulted in a significant reduction in the proportion of GABAergic neuron-like cells. Compared to female WT controls, female Akt1−/− mice (1) exhibited less GABAAR antagonist-induced convulsive activity, (2) had fewer parvalbumin-positive interneurons and lower expression of the GABAAR β2 subunit, especially in the hippocampus, (3) displayed morphological alterations in the CA1 pyramidal neurons of the dorsal hippocampus, (4) had increased hippocampal oscillation power, and (5) showed impaired cognitive performance on two hippocampus-related tasks.
Accumulating evidence from human studies and animal models indicates that Akt1 is involved in the regulation of dopaminergic signalling and dopamine-associated functions3,4,10,11,15,35,36, and this evidence suggests the importance of Akt1 in the dopamine hypothesis of schizophrenia. In contrast, the GABA hypothesis of schizophrenia proposes that reduced neuronal GABA concentrations and GABAergic neurotransmission result in cognitive impairments in schizophrenia. The GABA hypothesis allows for the integration of the GABAergic and oscillatory abnormalities into the glutamate hypothesis of schizophrenia, referred to as the “GABAergic interneuron origin hypothesis of schizophrenia”17,18,37. Intriguingly, in Experiments 1–3 of this study, Akt1 deficiency modulated the numbers of parvalbumin-positive interneurons, the convulsive activity induced by a general GABAAR antagonist, and the expression of the GABAAR β2 subunit in the hippocampus. These findings are consistent with the current evidence supporting the GABA hypothesis, in which reductions in the subpopulation of GABAergic interneurons expressing parvalbumin and the GABAAR β2 subunit have been reported in animal models of schizophrenia and in the post-mortem brains of schizophrenia patients18,19,20,21,22. Our findings also support the involvement of Akt1 in the modulation of parvalbumin-positive interneurons and GABAergic signalling, thereby implicating Akt1 in the GABA hypothesis of schizophrenia.
Importantly, the inhibitory activity of parvalbumin-positive interneurons plays a crucial role in generating synchronous gamma oscillations34,37, and accumulating evidence implicates disturbed neuronal synchrony in the gamma frequency range as an important physiological feature of schizophrenia38. It was interesting to observe a specific reduction in the abundance of hippocampal parvalbumin-positive (but not calretinin-positive) interneurons in Experiment 3 of our study, and the difference in the consequences of Akt1 deficiency between parvalbumin-positive and calretinin-positive interneurons may be because these cell types differentiate from different origins of hippocampal neurogenesis during development39. In addition to our observed reductions in the number of hippocampal parvalbumin-positive interneurons and in GABAAR expression in association with Akt1 deficiency, we demonstrated in Experiment 5 that anaesthetized Akt1-deficient mice had increased hippocampal oscillation power in the theta, alpha, beta, and gamma frequency ranges. This alteration in hippocampal neural oscillations might contribute, at least in part, to the impairments in hippocampus-associated cognitive function observed in our final experiment. Brain oscillations in the gamma range (30–80 Hz), which are generated by fast-spiking parvalbumin-positive interneurons, have been proposed to play a crucial role in many cognitive functions40,41. Consequently, disruption of parvalbumin-expressing interneurons is associated with many mental illnesses, including schizophrenia37,42. Supporting findings from studies of schizophrenia patients indicated that these individuals also have deficits in gamma oscillations27. These findings suggest that dysregulation of GABAergic signalling could result in dysfunction of gamma oscillation activity and could consequently lead to the cognitive deficits observed in schizophrenia. Intriguingly, previous preclinical and human studies have commonly shown that deficits in parvalbumin-positive GABAergic neurons result in reduced activity in the gamma frequency range; such findings are somewhat inconsistent with our current finding in Akt1−/− mice and certain findings regarding the baseline oscillation power of schizophrenia patients43,44,45. In the current study, local field potentials were recorded in mice under anaesthesia, somewhat similar to the resting state in humans. A recent study also demonstrated increased resting-state connectivity in the gamma frequency range among individuals with first-episode schizophrenia compared to healthy controls46. Given that the precise roles of gamma oscillations under different states (e.g., resting state vs. conducting cognitive task) remain unclear, in vivo recording of the brains of behaving Akt1−/− mice during a cognitive task would be warranted in a future study.
In addition, it is well characterized that synaptic inhibition in the brain is largely mediated by GABA and that the fast inhibitory effects of GABA are mediated by GABAAR activation. Intriguingly, Chen and colleagues from our group recently reported that Akt1 deficiency significantly affected the intrinsic electrophysiological properties (including enhanced input resistance, decreased rheobase, increased firing frequency, and reduced GABAAR-mediated miniature inhibitory postsynaptic currents) of striatal medium spiny neurons (the principal neurons forming the predominantly GABAergic microcircuit in the striatum) in Akt1−/− mutant mice12; their findings suggest downregulation of functional GABAAR expression or activity in the striatum of Akt1-deficient mice. Complementary to our findings in P19 cells (Experiment 1), it was reported that active Akt can promote the differentiation of neuronal precursor cells into GABAergic neuron-like cells29 and that phosphorylation of GABAAR via insulin-induced Akt signalling can lead to an increase in the number of GABA receptors on the plasma membrane28, thereby increasing inhibitory fast synaptic transmission in neurons. Accordingly, as demonstrated in this study, Akt1 deficiency might contribute to the reductions in functional GABAAR expression and GABAergic interneuron number in the hippocampus, thereby decreasing inhibitory transmission and altering hippocampal oscillations.
Further, as shown in Experiment 4 of this study, Akt1 deficiency not only affected neural activity in the hippocampus but also altered the morphological features of hippocampal CA1 pyramidal neurons. Similar to pyramidal neurons in other cortical areas, CA1 hippocampal pyramidal neurons primarily receive glutamatergic and GABAergic synaptic inputs. These GABAergic inputs, primarily originating from local interneurons, control the firing rate of the pyramidal neurons, modulate their spike timing, and synchronize their activity33. GABAergic interneurons (especially parvalbumin-expressing interneurons) predominantly innervate the dendrites of hippocampal CA1 pyramidal neurons and actively contribute to input/output signalling, signal integration, and synaptic plasticity of these neurons32,33. It is expected that dendritic GABAergic inputs regulate these dendritic signals in pyramidal neurons. As reported in Experiment 4, significant reductions in the morphological features and complexity of hippocampal CA1 pyramidal neurons, especially in the basal dendrites, were observed in Akt1-deficient mice. Moreover, it was reported that Akt1 might be an important regulator of neurite outgrowth47. Further evidence indicates that genetic deletion of Akt1 in mice resulted in a reduction in the morphological complexity of striatal medium spiny neurons12 and of pyramidal neurons in the medial prefrontal cortex10 and the auditory cortex11. Thus, our current finding in the hippocampus is consistent with these previous findings and validates the effect of Akt1 on neuronal morphology. Importantly, the observed alterations in the number of hippocampal parvalbumin-positive interneurons (Experiment 3) and neuromorphological features of pyramidal neurons (Experiment 4) in Akt1-deficent mice could contribute to the shift in the balance between excitation and inhibition in the hippocampus. A proper excitation/inhibition balance is critical for the control of hippocampal oscillations and output32,33. Although we did not concentrate on characterizing electrophysiological properties or excitatory glutamatergic transmission in this study, it was very interesting to find that female Akt1-deficient mice exhibited less general GABAAR antagonist-induced convulsive activity (Experiment 2) and had increased hippocampal oscillation power (Experiment 5). In future studies, further examination of the electrophysiological properties of neurons in hippocampal slices and of hippocampal neural activity in behaving Akt1 mutant mice would be warranted to elucidate the exact mechanism underlying the imbalance between excitation and inhibition in the hippocampus of these mice.
Furthermore, it was reported that mixed-sex Akt1−/− mice exhibited reduced hippocampal neurogenesis and impairments in hippocampus-dependent contextual fear conditioning and recall of spatial memory31. Our current behavioural data are highly consistent with those previous behavioural results regarding hippocampus-dependent functions. We further demonstrated that female Akt1−/− mice had a spatial memory deficit in the Y-maze task and impaired spatial learning, recall, and reversal in the Morris water maze task. One possible explanation for the observed hippocampus-dependent deficits is that Akt1 deficiency affects the morphological features of hippocampal CA1 pyramidal neurons and the abundance of local GABAergic interneurons, resulting in alterations in the firing rate of pyramidal neurons and in the synchronicity of neural activity, as described above. Somewhat surprisingly, we found that female Akt1−/− mice, but not male Akt1−/− mice, are less sensitive to PTZ-induced seizures than their WT controls. Accordingly, we simply focused on female mice in the remainder of our experiments due to time constraints and manpower limitations. However, because we did not conduct all experiments on both males and females, the potential involvement of Akt1 in the regulation of GABAergic interneurons and hippocampal oscillations in males cannot be completely ruled out. Given that the sample size of male mice is somewhat small and that there was a non-significant trend toward reduced severity of PTZ-induced seizures among male Akt1−/− mice compared to male WT mice, caution should be taken when interpreting the sex differences in PTZ-induced convulsions and GABAergic functions observed in the current study. Nonetheless, sex-specific effects of Akt1 deficiency on behavioural phenotypes have been well characterized in Akt1-deficient mice in previous studies11,12. Numerous sex-based differences in the risk of schizophrenia have also been reported in humans48,49, and a sex-based difference in the association of the Akt1 gene with the risk of schizophrenia has been reported, as well50. The mechanisms underlying the sex-specific effect of Akt1 and the effects of hormones (e.g., oestrogen) on GABAergic transmission and the pathogenesis of schizophrenia merit further investigation in future studies.
It is also of great interest to link some of current results to the findings in humans or patients with schizophrenia. Indeed, several studies have reported on the effect of Akt1 on hippocampal function, structure, or postmortem expression in humans, and the reported results are somewhat consistent with our current findings. First, lower Akt1 protein levels were reported in the hippocampus of patients with schizophrenia4. Second, reduced levels of Akt phosphorylated at serine 473 were reported in hilar neurons of the dentate gyrus in postmortem brains of patients with schizophrenia compared with healthy controls31. Third, first-episode schizophrenia patients who did not receive psychotropic medications displayed a decreased Akt phosphorylation ratio in B lymphocytes and exhibited reduced left hippocampal volume compared with healthy controls51. Fourth, the Akt1 rs1130233 variant was linked to differential Akt1 protein expression levels in human lymphoblasts15 and influenced gray matter volume in the medial temporal lobe as well as memory-dependent hippocampal activity52. Taken together, the findings of this study indicate the involvement of Akt1 in the regulation of hippocampal GABAergic interneuron abundance, functional GABAAR expression, neuronal morphology, neuronal activity oscillations, and hippocampal-dependent cognitive functions in a mouse model of schizophrenia. These results indicate the contribution of Akt1 to the pathogenesis of schizophrenia and support the GABA hypothesis of schizophrenia.
Materials and Methods
P19 cell culture
P19 mouse embryonal carcinoma cells (P19 cells) were selected because neuron-like cells derived from P19 cells express glutamic acid decarboxylase53 and multiple GABAAR subunits54. P19 cells were maintained in αMEM with supplements55.
Animals
All adult (2–4-month-old) Akt1−/− and WT mice used in this study were generated from Akt1+/− breeding pairs in the C57BL/6 genetic background (n > 10). All animals were 2–3 months old at the beginning of the experiments. The details of the animal experiments have been described elsewhere11,12 and in the supplementary materials. All animal procedures were performed according to protocols approved by the appropriate Animal Care and Use Committees established by National Taiwan University. The minimum number of mice was used in accordance with the 3R principle of animal use. Adequate measures were utilized to minimize potential pain or discomfort experienced by the mice used in this study.
Experiment 1: The effect of Akt1/2 inhibitor application on neuronal differentiation and the production of GABAergic neuron-like cells from P19 cells
For neuronal differentiation, P19 cells were transfected with the US2-Ascl1 vector to induce neural differentiation, as described elsewhere56. An Akt1/2 inhibitor (1 μM, Calbiochem, San Diego, CA) was mixed into the medium during DIV 0–5. To measure the differentiation of GABAergic neuron-like cells derived from P19 cells, immunocytochemistry was performed at DIV 5 using antibodies against GFP (for transfected cells), Tuj1 (for differentiated neurons), GAD67 (for GABAergic interneurons), and parvalbumin (for parvalbumin-positive neurons). The proportion of neuron-like cells, GABAergic neuron-like cells, and parvalbumin-positive neuron-like cells derived from P19 cells are shown as the proportions of cells expressing Tuj1, GAD67, or parvalbumin among all GFP-positive cells.
To examine functional GABAAR expression, P19 cells were transfected with the US2-puro and US2-Ascl1 vectors. After selecting and extracting the transfected cells, Western blotting was performed using an antibody against the GABAAR β2 subunit and an antibody against the Na+/K+ ATPase as a loading control. Densitometric analysis was performed using NIH ImageJ software. Details are presented in the supplementary methods.
Experiment 2: General evaluation of PTZ-induced seizures in male and female Akt1-deficient mice
PTZ (Sigma-Aldrich, St. Louis, MO) was administered to induce seizures for examination of potential abnormalities in GABA-related phenotypes in both male and female Akt1−/− mice compared to their WT littermates (n = 6 for the male groups; n = 8 for the female groups). PTZ dissolved in saline was administered subcutaneously at a dose of 50 mg/kg (5 mg/ml). After injection, animal behaviours were videotaped immediately for up to 30 min in a clean cage with bedding. The following two behavioural indexes adapted from a previous study57 were used. (A) Latency: the latency (sec) to the initial onset of seizures was recorded. (B) Severity: The severity of seizure activity was scored blindly using the following scale: 0, no signs of motor seizures; 1, isolated twitches; 2, tonic-clonic convulsions; 3, tonic extension; and 4, death.
Experiment 3: Examination of the GABAergic interneuron density and the GABAAR expression level in the brains of female Akt1 −/− mice
Because GABA is mediates pre- and post-synaptic inhibition of neuronal activity, two sub-experiments were designed to examine the number of GABAergic interneurons and the level of functional GABAAR expression in the target brain regions of female mice based on the results of Experiments 1 and 2. Experimental details are provided in the supplementary methods.
Experiment 3a: Examination of the density of GABAergic interneurons in the brains of female mice
Immunohistochemistry was conducted on brain sections from female Akt1−/− mice (n = 6) and their WT littermates (n = 8) to label two major subtypes of GABAergic interneurons using antibodies against parvalbumin and calretinin. Neuronal density in the CA1 and CA3 subregions of the hippocampus and in the cortices (anterior cingulate cortex area 1 (aCg1), prelimbic cortex (PrL), infralimbic cortex (IL), primary motor cortex (M1), and primary auditory cortex (Au1)) was measured using NIH ImageJ software.
Experiment 3b: Examination of the expression of functional GABAA receptors in the target brain areas of female mice
Based on previous findings and our results from Experiments 1 and 2, Western blotting was utilized to examine the functional expression of GABAARs in the hippocampus, cortex, and striatum of adult female Akt1−/− mice and their WT littermates (n = 5 each). The membrane fraction of brain lysates was extracted using a commercial kit (Fermentas), and the expression levels of GABAAR and Na+/K+ ATPase were examined as described for Experiment 1.
Experiment 4: Examination of the morphological features of hippocampal pyramidal neurons in female Akt1 −/− mice
Based on the findings from Experiments 2 and 3, additional female subjects generated from Akt1+/− breeding pairs in a C57BL/6-Tg (GFPm) background were used to investigate neuronal morphology. Morphometric analysis of GFP-labelled pyramidal neurons in the CA1 region of the dorsal hippocampus of adult female Akt1−/− (n = 13) and WT (n = 11) mice was conducted to detect genotypic differences in neuronal morphology. For each complete and available neuron in the CA1 area, 10 morphological variables that were selected based on previous studies11,58 were examined. Details are provided in the supplementary methods.
Experiment 5: Recording of neuronal oscillations in the hippocampus of anaesthetized female mice
Hippocampal oscillatory activity was examined to further assess whether Akt1 deficiency alters neuronal activity in the hippocampus. Adult female Akt1−/− mice and WT littermate controls (n = 5 each) were used, and electrodes were implanted into the CA1 region of the dorsal hippocampus. Details of electrode implantation and histology are provided in the supplementary methods. After recovery, the mice were anaesthetized with isoflurane (1%), and local field potentials (LFPs) were recorded from the hippocampus for 30 min using a Plexon system (Plexon Inc., Dallas, TX). Spectral analysis of LFP power was performed using NeuroExplorer (Plexon Inc.). The power spectra were calculated using Welch’s method (512 frequencies between 1 and 100 Hz, smoothed using a Gaussian Kernel with a bin width of 3). The mean oscillation power was averaged for different frequency ranges (delta: 0–4 Hz; theta: 5–8 Hz; alpha: 9–12 Hz; beta: 12–30 Hz; and gamma: 30–80 Hz).
Experiment 6: Examination of hippocampus-related cognitive function in female Akt1 −/− mice
Based on the results from the above experiments and previous studies of Akt1−/− mice11,12,31, two sub-experiments were conducted to examine the potential genotypic differences in hippocampus-related cognitive function in female Akt1−/− mice compared to female WT mice. Details are provided in the supplementary methods.
Experiment 6a: Examination of spatial memory using the Y-maze task
The retention of spatial memory by naïve female Akt1−/− mice and WT littermates (n = 8 each) was examined using a Y-maze task. The Y-maze test consisted of a 10-min training trial and a 5-min retention trial separated by a one-hour inter-trial interval. Details of the experimental procedure are provided in the supplementary methods. The following two indices of spatial memory retention were calculated: (1) the proportion of time spent in the novel arm and (2) the relative distance travelled in the novel arm.
Experiment 6b: Examination of spatial learning and memory using the Morris water maze task
Spatial learning and memory were further examined using a standard spatial version (hidden platform) of the Morris water maze task (n = 8 per group). Details are provided in the supplementary methods. Briefly, to assess the acquisition and retention of spatial memory, 6 swimming trials per day (with inter-trial intervals of 11–15 min) were performed for 8 consecutive acquisition days. The escape latency (sec) to reach the hidden platform and the path length (cm) were recorded. After 8 days of training, each subject was returned to the pool without a platform for a 1-min probe test on Day 9. The time spent swimming and the swimming distance in each quadrant were recorded. One day after the probe test, each mouse was retrained in the reversal version of the water maze for 5 consecutive days to test reversal learning.
Statistics and Data Analyses
All data are presented as the means ± standard error of the mean (SEM). The data that met the assumptions for normality and homogeneity of variance based on Kolmogorov–Smirnov tests were analysed using parametric tests. All of the data were normally distributed (data not shown). As appropriate, statistical evaluations were performed using Student’s t-test or ANOVA to detect genotypic differences using SPSS 20.0 (SPSS Inc., Chicago, IL, U.S.A.). Post hoc analysis was performed using Fisher’s LSD test when the F values indicated significant differences between groups, and p values of < 0.05 were considered statistically significant.
Additional Information
How to cite this article: Chang, C.-Y. et al. Akting up in the GABA hypothesis of schizophrenia: Akt1 deficiency modulates GABAergic functions and hippocampus-dependent functions. Sci. Rep. 6, 33095; doi: 10.1038/srep33095 (2016).
Supplementary Material
Acknowledgments
This research was supported by grant numbers 102-2420-H-002-008-MY2, 102-2628-H-002-003-MY3, 103-2325-B-002-047, and 104-2325-B-400-014 to WS Lai from the Ministry of Science and Technology in Taiwan, grant numbers 101-042 and 102-053 from the National Taiwan University Hospital, and grant support from the Drunken Moon Lake Integrated Scientific Research Platform (103R104955) as well as the Aim for the Top University Project at National Taiwan University. We thank all members of the Laboratory of Integrated Neuroscience and Ethology (LINE) in the Department of Psychology, National Taiwan University, for their assistance and contributions.
Footnotes
Author Contributions Experiments 1–3 and 5 were conducted by C.-Y.C. Experiments 4 and 6 were performed by Y.-W.C., C.-Y.C. and W.-S.L. wrote and revised the main manuscript. C.-Y.C. prepared Figures 1–3 and 5. Y.-W.C. prepared Figures 4 and 6. T.-W.W. provided technical support and data discussion concerning Experiment 1. All authors discussed and interpreted the results.
References
- Sale E. M. & Sale G. J. Protein kinase B: signalling roles and therapeutic targeting. Cell. Mol. Life Sci. 65, 113–127, 10.1007/s00018-007-7274-9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379, 10.1016/S0140-6736(12)62129-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian E. S. AKT/GSK3 signaling pathway and schizophrenia. Front. Mol. Neurosci. 5, 33, 10.3389/fnmol.2012.00033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian E. S., Hall D., Birnbaum M. J., Karayiorgou M. & Gogos J. A. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nature genetics 36, 131–137, 10.1038/ng1296 (2004). [DOI] [PubMed] [Google Scholar]
- Lo W. S. et al. Association of SNPs and haplotypes in GABAA receptor beta2 gene with schizophrenia. Molecular psychiatry 9, 603–608, 10.1038/sj.mp.4001461 (2004). [DOI] [PubMed] [Google Scholar]
- Petryshen T. L. et al. Genetic investigation of chromosome 5q GABAA receptor subunit genes in schizophrenia. Molecular psychiatry 10, 1074–1088, 1057, 10.1038/sj.mp.4001739 (2005). [DOI] [PubMed] [Google Scholar]
- Norton N., Williams H. J. & Owen M. J. An update on the genetics of schizophrenia. Curr. Opin. Psychiatry 19, 158–164, 10.1097/01.yco.0000214341.52249.59 (2006). [DOI] [PubMed] [Google Scholar]
- Schwab S. G. & Wildenauer D. B. Update on key previously proposed candidate genes for schizophrenia. Curr. Opin. Psychiatry 22, 147–153, 10.1097/YCO.0b013e328325a598 (2009). [DOI] [PubMed] [Google Scholar]
- Zhao Z., Ksiezak-Reding H., Riggio S., Haroutunian V. & Pasinetti G. M. Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr. Res. 84, 1–14, 10.1016/j.schres.2006.02.009 (2006). [DOI] [PubMed] [Google Scholar]
- Lai W. S. et al. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc. Natl. Acad. Sci. USA 103, 16906–16911, 10.1073/pnas.0604994103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. W. & Lai W. S. Behavioral phenotyping of v-akt murine thymoma viral oncogene homolog 1-deficient mice reveals a sex-specific prepulse inhibition deficit in females that can be partially alleviated by glycogen synthase kinase-3 inhibitors but not by antipsychotics. Neuroscience 174, 178–189, 10.1016/j.neuroscience.2010.09.056 (2011). [DOI] [PubMed] [Google Scholar]
- Chen Y. W., Kao H. Y., Min M. Y. & Lai W. S. A sex- and region-specific role of Akt1 in the modulation of methamphetamine-induced hyperlocomotion and striatal neuronal activity: implications in schizophrenia and methamphetamine-induced psychosis. Schizophrenia bulletin 40, 388–398, 10.1093/schbul/sbt031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. C. et al. Akt1 deficiency modulates reward learning and reward prediction error in mice. Genes Brain Behav. 11, p-169, 10.1111/j.1601-183X.2011.00759.x (2012). [DOI] [PubMed] [Google Scholar]
- Huang C. H. et al. Investigation of gene effects and epistatic interactions between Akt1 and neuregulin 1 in the regulation of behavioral phenotypes and social functions in genetic mouse models of schizophrenia. Front. Behav. Neurosci. 8, 455, 10.3389/fnbeh.2014.00455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. Y. et al. Genetic variation in AKT1 is linked to dopamine-associated prefrontal cortical structure and function in humans. J. Clin. Invest. 118, 2200–2208, 10.1172/JCI34725 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. Y. et al. Effective connectivity of AKT1-mediated dopaminergic working memory networks and pharmacogenetics of anti-dopaminergic treatment. Brain 135, 1436–1445, 10.1093/brain/aws068 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D. A. & Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 63, 1372–1376, 10.1001/archneur.63.10.1372 (2006). [DOI] [PubMed] [Google Scholar]
- Yoon J. H. et al. GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. J. Neurosci. 30, 3777–3781, 10.1523/JNEUROSCI.6158-09.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B. et al. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron 54, 889–903, 10.1016/j.neuron.2007.05.015 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T. et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T. et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Molecular psychiatry 13, 147–161, 10.1038/sj.mp.4002011 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneyto M., Abbott A., Hashimoto T. & Lewis D. A. Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb Cortex 21, 999–1011, 10.1093/cercor/bhq169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvevag B. & Goldberg T. E. Cognitive impairment in schizophrenia is the core of the disorder. Crit. Rev. Neurobiol. 14, 1–21 (2000). [PubMed] [Google Scholar]
- Green M. F. Cognitive impairment and functional outcome in schizophrenia and bipolar disorder. J. Clin. Psychiatry 67, e12 (2006). [PubMed] [Google Scholar]
- Green M. F. What are the functional consequences of neurocognitive deficits in schizophrenia? Am. J. Psychiatry 153, 321–330, 10.1176/ajp.153.3.321 (1996). [DOI] [PubMed] [Google Scholar]
- Cho R. Y., Konecky R. O. & Carter C. S. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc. Natl. Acad. Sci. USA 103, 19878–19883, 10.1073/pnas.0609440103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. et al. gamma oscillations in schizophrenia: mechanisms and clinical significance. Brain Res. 1413, 98–114, 10.1016/j.brainres.2011.06.065 (2011). [DOI] [PubMed] [Google Scholar]
- Wang Q. et al. Control of synaptic strength, a novel function of Akt. Neuron 38, 915–928 (2003). [DOI] [PubMed] [Google Scholar]
- Oishi K. et al. Selective induction of neocortical GABAergic neurons by the PDK1-Akt pathway through activation of Mash1. Proc. Natl. Acad. Sci. USA 106, 13064–13069, 10.1073/pnas.0808400106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y. et al. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 148, 1051–1064, 10.1016/j.cell.2011.12.037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu D. T. et al. Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus 22, 230–240, 10.1002/hipo.20887 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megias M., Emri Z., Freund T. F. & Gulyas A. I. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102, 527–540 (2001). [DOI] [PubMed] [Google Scholar]
- Klausberger T. GABAergic interneurons targeting dendrites of pyramidal cells in the CA1 area of the hippocampus. Eur. J. Neurosci. 30, 947–957, 10.1111/j.1460-9568.2009.06913.x (2009). [DOI] [PubMed] [Google Scholar]
- Mann E. O. & Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 30, 343–349, 10.1016/j.tins.2007.05.003 (2007). [DOI] [PubMed] [Google Scholar]
- Beaulieu J. M., Gainetdinov R. R. & Caron M. G. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28, 166–172, 10.1016/j.tips.2007.02.006 (2007). [DOI] [PubMed] [Google Scholar]
- Beaulieu J. M., Gainetdinov R. R. & Caron M. G. Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347, 10.1146/annurev.pharmtox.011008.145634 (2009). [DOI] [PubMed] [Google Scholar]
- Nakazawa K. et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62, 1574–1583, 10.1016/j.neuropharm.2011.01.022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas P. J. & Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 11, 100–113, 10.1038/nrn2774 (2010). [DOI] [PubMed] [Google Scholar]
- Tricoire L. et al. A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J. Neurosci. 31, 10948–10970, 10.1523/JNEUROSCI.0323-11.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basar E., Basar-Eroglu C., Karakas S. & Schurmann M. Gamma, alpha, delta, and theta oscillations govern cognitive processes. Int. J. Psychophysiol. 39, 241–248 (2001). [DOI] [PubMed] [Google Scholar]
- Tallon-Baudry C., Bertrand O., Peronnet F. & Pernier J. Induced gamma-band activity during the delay of a visual short-term memory task in humans. J. Neurosci. 18, 4244–4254 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge D. J., Behrens M. M. & Grace A. A. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. 29, 2344–2354, 10.1523/JNEUROSCI.5419-08.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer K. M. Baseline gamma power during auditory steady-state stimulation in schizophrenia. Front. Hum. Neurosci. 5, 190, 10.3389/fnhum.2011.00190 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L. E. et al. Sensory gating endophenotype based on its neural oscillatory pattern and heritability estimate. Arch. Gen. Psychiatry 65, 1008–1016, 10.1001/archpsyc.65.9.1008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterer G. et al. Prefrontal broadband noise, working memory, and genetic risk for schizophrenia. Am. J. Psychiatry 161, 490–500, 10.1176/appi.ajp.161.3.490 (2004). [DOI] [PubMed] [Google Scholar]
- Andreou C. et al. Increased resting-state gamma-band connectivity in first-episode schizophrenia. Schizophrenia bulletin 41, 930–939, 10.1093/schbul/sbu121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read D. E. & Gorman A. M. Involvement of Akt in neurite outgrowth. Cell. Mol. Life Sci. 66, 2975–2984, 10.1007/s00018-009-0057-8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman A., Kahn R. S. & Selten J. P. Sex differences in the risk of schizophrenia: evidence from meta-analysis. Arch. Gen. Psychiatry 60, 565–571, 10.1001/archpsyc.60.6.565 (2003). [DOI] [PubMed] [Google Scholar]
- McGrath J. et al. A systematic review of the incidence of schizophrenia: the distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med. 2, 13, 10.1186/1741-7015-2-13 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M. Q. et al. Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. J. Clin. Psychiatry 68, 1358–1367 (2007). [DOI] [PubMed] [Google Scholar]
- Szamosi A., Kelemen O. & Keri S. Hippocampal volume and the AKT signaling system in first-episode schizophrenia. J. Psychiatr. Res. 46, 279–284, 10.1016/j.jpsychires.2011.12.005 (2012). [DOI] [PubMed] [Google Scholar]
- Tan H. Y. et al. Epistatic interactions of AKT1 on human medial temporal lobe biology and pharmacogenetic implications. Molecular psychiatry 17, 1007–1016, 10.1038/mp.2011.91 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staines W. A., Morassutti D. J., Reuhl K. R., Ally A. I. & McBurney M. W. Neurons derived from P19 embryonal carcinoma cells have varied morphologies and neurotransmitters. Neuroscience 58, 735–751 (1994). [DOI] [PubMed] [Google Scholar]
- Reynolds J. N., Ryan P. J., Prasad A. & Paterno G. D. Neurons derived from embryonal carcinoma (P19) cells express multiple GABAA receptor subunits and fully functional GABAA receptors. Neurosci. Lett. 165, 129–132 (1994). [DOI] [PubMed] [Google Scholar]
- Farah M. H. et al. Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development 127, 693–702 (2000). [DOI] [PubMed] [Google Scholar]
- Lin Y. T. et al. YAP regulates neuronal differentiation through Sonic hedgehog signaling pathway. Exp. Cell Res. 318, 1877–1888, 10.1016/j.yexcr.2012.05.005 (2012). [DOI] [PubMed] [Google Scholar]
- Erickson J. C., Clegg K. E. & Palmiter R. D. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature 381, 415–421, 10.1038/381415a0 (1996). [DOI] [PubMed] [Google Scholar]
- Pei J. C., Liu C. M. & Lai W. S. Distinct phenotypes of new transmembrane-domain neuregulin 1 mutant mice and the rescue effects of valproate on the observed schizophrenia-related cognitive deficits. Front. Behav. Neurosci. 8, 126, 10.3389/fnbeh.2014.00126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.