

Abstract
Gene therapy is a promising strategy for the management of various neurological disorders that do not respond adequately to conventional therapeutics. The development of gene vectors with favorable safety profiles that can achieve uniform distribution and high-level transgene expression in the brain remains challenging. The rod-shaped, non-viral gene delivery platform based on poly-L-lysine (PLL) conjugated to a single segment of polyethylene glycol (PEG) has shown safe transfection in human nares and mouse brains in vivo. However, we have previously demonstrated that a denser PEG coating is required for rapid diffusion of nanoparticles in the brain extracellular space. Here, we engineered a densely PEGylated version of this platform based on PLL polymers conjugated to branched PEG via alkyne-azide cycloaddition. We found that the newly developed gene vectors rapidly diffused in the brain parenchyma, providing significantly improved vector distribution and overall transgene expression in vivo compared to the previously developed platform. These brain-penetrating DNA nanoparticles exhibited enhanced cellular uptake presumably due to their ellipsoidal morphology. By simultaneously improving delivery to target cells and subsequent transfection, our densely PEGylated PLL DNA nanoparticles can provide widespread, high levels of transgene expression, essential for effective targeting of highly disseminated brain diseases.
Keywords: Gene therapy, non-viral gene vectors, poly-L-lysine, polyethylene glycol, brain extracellular matrix, convection-enhanced delivery
Graphical abstract
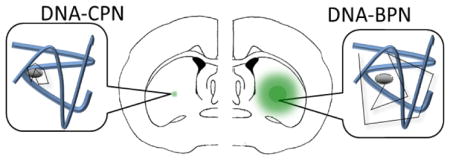
Densely PEGylated poly-L-lysine gene vectors (DNA-BPN) provide more widely distributed transgene expression in the brain, compared to conventionally PEGylated counterparts (DNA-CPN).
1. Introduction
Gene therapy is a widely applicable treatment that targets underlying genetic abnormalities of numerous neurological disorders, including brain tumors and neurodegenerative diseases.1–3 The majority of gene therapy clinical trials currently utilize viral gene vectors due to their relatively high transfection efficiency.1, 4 However, viruses suffer from one or more important limitations, including immunogenicity, insertional mutagenesis, low packaging capacity and difficulty in large-scale production of pharmaceutical-grade products.5, 6 Polymer-based non-viral gene vectors are attractive alternatives that can be tailored to achieve high-level, long-term transfection in the brain while avoiding the aforementioned limitations.7
Cationic polymers, including poly-L-lysine (PLL), have been widely used to stably compact nucleic acids and provide protection from enzymatic degradation.8 Shielding their positive surface charge with neutral, hydrophilic polymers such as polyethylene glycol (PEG) has proven pivotal to improving their safety profile.9–11 A successful example of this approach is the rod-shaped DNA nanoparticle developed by Copernicus Therapeutics, Inc., formulated using di-block copolymers of 30-mer PLL conjugated to a single segment of PEG. This system has demonstrated a favorable safety profile upon administration to the nares of cystic fibrosis patients 12 and mediated long-term transgene expression of glial cell line-derived neurotrophic factor in rodent brains.13, 14 Nevertheless, insufficient surface coating on this conventionally PEGylated system results in adhesive interactions between the nanoparticle and brain extracellular matrix (ECM).11, 15
Once beyond the blood-brain barrier, the brain ECM acts as a hurdle that limits the widespread distribution of gene vectors and their ability to reach and transfect disseminated target cells.16 Even with the pressure-driven flow provided by convection-enhanced delivery (CED), therapeutics remain confined to the point of administration or travel within the perivascular spaces, but fail to efficiently penetrate the brain parenchyma to reach target cells.17–19 We have previously demonstrated that effective surface shielding with a dense layer of hydrophilic and neutrally charged PEG improves diffusion of nanoparticles as large as 100 nm in the brain.15 Based on this finding, we have achieved markedly improved distribution and transfection efficiency of densely PEGylated polyethylenimine-based gene vectors in vivo.11
Here, we sought to optimize the clinically tested, conventionally PEGylated PLL-based nanoparticle system to improve distribution and transfection efficiency in the brain by enhancing gene vector stability and by minimizing the adhesive interactions with the ECM. In order to produce compact and densely PEGylated brain-penetrating gene vectors, we first synthesized a PLL polymer with a high PEG:PLL w/w ratio by conjugating a 30-mer PLL to a house-synthesized branched PEG (BrPEG) via the alkyne-azide cycloaddition. The reaction is a well-established click chemistry notable for selectivity, reliability, high yield and minimal by-product formation.20, 21 We then formulated densely PEG coated DNA nanoparticles with a core of un-PEGylated PLL that has been previously shown to improve nanoparticle compaction and stability.11, 22 This strategy of increasing PEG density was tested for the ability to achieve enhanced DNA nanoparticle distribution and transgene expression in vivo following CED.
2. Experimental
2.1. Polymer Synthesis
BrPEG was synthesized by a two-step reaction. Diethylenetriaminepentaacetic acid (DTPA) anhydride was first reacted with 11-azido-3,6,9-trioxaundecan-1-amine at a 1:1 molar ratio in the presence of 2 molar equivalents of N, N-diisopropylethylamine at 37 °C for 24 h. The azido-DTPA resulting from this reaction was then reacted with 4 molar equivalents of 5 kDa methoxy-PEG-amine (Creative PEGWorks, Winston Salem, NC) in the presence of 40, 5 and 3 molar equivalents of 1-Ethyl-3-(3-methylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS) and 4-dimethylaminopyridine (DMAP), respectively, in dimethylformamide (DMF). The reaction was carried out for 48 h at 37 °C with constant stirring. Subsequently, products were purified by dialyzing (6–8 kDa MWCO, Spectrum Laboratories, Inc., Rancho Dominguez, CA) against ultrapure water for 24 h in 4 °C. Thirty-mer PLL polymers functionalized with terminal alkyne groups (bromide counter-ions) were purchased from Alamanda Polymers Inc. (Huntsville, AL). For the formation of PEGylated PLL polymers, PLL peptides were reacted with PEG (~5 kDa) (Creative PEGWorks, Winston Salem, NC) or BrPEG (~15 kDa) with functionalized azide groups at a molar ratio of 1:1. The click chemistry reaction was carried out at 37 °C for 48 h in the presence of 0.1 molar equivalents of copper acetate, 5 molar equivalents of sodium ascorbate and tris(benzyltriazolylmethyl)amine (TBTA) in 100 mM Tris buffer (pH 7.5). Reaction products, PLL-PEG (di-block polymer of PLL and linear PEG) and PLL-BrPEG (di-block polymer of PLL and branched PEG), were dialyzed against ultrapure water for 24 h using a MWCO of 6–8kDa. To exchange the counter ions, PLL-PEG and PLL-BrPEG were subjected to size exclusion chromatography with Sephadex G15 (MWCO 1500, GE healthcare, Pittsburg, PA) as the stationary phase and 50 mM ammonium acetate buffer (pH 7.4) as the mobile phase. During this process, bromide counter-ions were displaced and acetate counter-ions were associated with PLL side chains as confirmed subsequently by nuclear magnetic resonance (NMR). Peptide concentrations in different fractions were monitored by measuring the absorbance at 220 nm (NanoDrop ND-1000 spectrophotometer, NanoDrop Technologies, Wilmington, DE). High-molecular weight fractions containing peptide were pooled and dialyzed. Dialysis was carried out using dialysis tubes with a MWCO of 6–8 kDa, against ultrapure water over the course of 24 h. Purified products were lyophilized and stored at −80 °C. Final products were assessed for their degrees of PEGylation by quantitative analysis of NMR spectrograms where relative molar ratios of PEG to PLL were calculated. Approximately 1 mg of polymer was dissolved in deuterium oxide, D2O, (Sigma Aldrich, St. Louis, MO) and processed for 1H-NMR (500 MHz). Analysis of the NMR profiles was carried out using ACD/NMR Processor Academic Edition software. The PEG to PLL ratio was calculated by comparing the integrated intensity of proton peaks contributed by PEG and PLL.
2.2. Particle Formulation
Plasmids, pBAL (β-actin driven luciferase; provided by Dr. Mark Cooper; Copernicus Therapeutic, Inc., Cleveland, OH) and pEGFP (cytomegalovirus driven enhanced green fluorescent protein, purchased from Clontech Laboratories Inc., Mountainview, CA) were used for nanoparticle formulation. The plasmids were propagated and purified as previously described.22 To fluorescently label plasmid with Cy3 and Cy5 fluorophores, the Mirus Label IT® Tracker™ Intracellular Nucleic Acid Localization Kit (Mirus Bio, Madison, WI) was used. DNA nanoparticles that mimicked the conventionally PEGylated formulation (DNA-CPN) developed by Copernicus Therapeutics, Inc. were formulated at a nitrogen to phosphorus (N/P) ratio of 2, based on previous literature.22–24 In order to formulate densely PEGylated DNA nanoparticles, or brain penetrating DNA nanoparticles (DNA-BPN), that efficiently compact DNA, despite the steric interference resulting from a high PEG density, we used a blend of 90% PLL-BrPEG and 10% PLL.22 As an additional control for our ex vivo diffusion study we synthesized un-PEGylated DNA nanoparticles (DNA-UPN) at N/P ratio 2 using PLL for DNA compaction. DNA nanoparticles were formed by the drop-wise addition of 10 volumes of plasmid DNA (0.2 mg/ml) to 1 volume of swirling polymer solution. The plasmid/polymer solutions were incubated for 30 min at room temperature. Syringe filtration (0.2 μm) was used for removal of aggregates followed by removal of free polymer and collection of DNA nanoparticles at desired concentration by Amicon® Ultra Centrifugal Filters (100,000 MWCO, Millipore Corp., Billerica, MA). The DNA concentration was determined via absorbance at 260 nm using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE).
2.3. Physicochemical characterization of DNA nanoparticles
The hydrodynamic diameter (Z-average), polydispersity index (PDI) and ζ-potential were measured in 10 mM NaCl at pH 7.0 by dynamic light scattering (DLS) and laser Doppler anemometry using a Nanosizer ZS90 (Malvern Instruments, Southborough, MA). Measurements were performed at 25°C at a scattering angle of 90°. Transmission electron microscopy (TEM) grids for DNA nanoparticles were prepared to visualize morphologies and measure major and minor diameters (n = 3) by TEM (Hitachi H7600, Japan).
Stability of DNA-CPN and DNA-BPN was assessed by recording the Z-average and PDI before and 0, 1 and 24 h after adding DNA nanoparticles in artificial cerebrospinal fluid (aCSF) (Harvard Apparatus, MA) at 37 °C. TEM was conducted concomitantly. The stability of DNA-CPN and DNA-BPN in the infusion solution used for CED was also monitored by TEM.
The heparin displacement assay for DNA-CPN and DNA-BPN was carried out by incubating DNA nanoparticles at different concentrations of heparin (Sigma Aldrich, St. Louis, MO); 0, 0.01, 0.05, 0.1, 0.5 and 1 IU/μg DNA, for 15 min at 37 °C. Gel electrophoresis was carried out for 30 min at 100 V on a 0.8% agarose gel containing 50 μg/ml ethidium bromide.
2.4. Cell Culture
9L rat gliosarcoma cells provided by Dr. Henry Brem (Department of Neurosurgery at Johns Hopkins University) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen Corp., Carlsbad, CA) supplemented with 1% penicillin/streptomycin (Invitrogen Corp., Carlsbad, CA) and 10% heat inactivated fetal bovine serum (FBS, Invitrogen Corp., Carlsbad, CA). Rat brain primary astrocytes were provided by Dr. Arun Venkatesan (Department of Neurology at Johns Hopkins University), isolated as previously published.25 Cells were cultured in DMEM/F12 (Invitrogen Corp., Carlsbad, CA) supplemented with 10% FBS and 1% penicillin-streptomycin. As per assay requirements, cells were trypsinized by incubating with 0.25% Trypsin EDTA (Corning Inc., Tewksbury, MA) for 5 min at 37 °C followed by neutralization with serum containing media and were seeded in 96-well or 24-well plates, and allowed to adhere overnight.
2.5. In vitro Cellular Uptake
For cellular uptake studies, cells were seeded at a density of 50,000 cells per well in 24-well plates, and were treated for 3 h at 37 °C with 1 μg Cy3-labeled DNA/well for different DNA nanoparticle formulations. The use of labeled DNA did not affect the formation of DNA nanoparticles as confirmed by TEM and ζ-potential. For flow cytometry, the cells were treated briefly with 0.04% trypan blue to quench extracellular florescence, washed in phosphate buffered saline (PBS) three times and then trypsinized. The trypsin was neutralized and the cells were collected by centrifugation at 1000 rpm for 10 min. The cell pellet obtained was re-suspended in 100 μl of 10% FBS in PBS and kept on ice until the samples were processed. Nanoparticle cellular uptake was measured using the Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) with a 488 nm laser and a FL2 band-pass filter. Data were analyzed using the BD Accuri C6 software. Thresholds were determined using untreated samples and cells treated with free plasmid served as a control.
2.6. In vitro Transfection
Cells were seeded at a density of 50,000 cells per well in a 24-well plate and treated with 1 μg/well compacted pBAL plasmid for 3 h at 37 °C. The medium was then replaced and the cells were cultured for 3 days before being assayed for luciferase expression. To extract luciferase from the treated cells, the media was removed and the cells were washed twice with 1X PBS. Five hundred microliters of 1X Reporter Lysis Buffer (Promega, Madison, WI) was added to each well and incubated for 10 min at room temperature. The cells from each well were transferred to separate microcentrifuge tubes and subjected to three freeze-thaw cycles to ensure cell lysis, and then centrifuged at 1,000 rpm for 5 min. The supernatant was transferred into a new microcentrifuge tube and assayed immediately for luciferase activity. One hundred microliters of the luciferase substrate-assay buffer mixture (Promega, Madison, WI) was added to polystyrene tubes followed by 20 μl of the supernatant and the luminescence was measured immediately using a 20/20n luminometer (Turner Biosystems, Sunnyvale, CA). The relative light unit (RLU) was normalized by the total protein concentration, measured by Micro-BCA protein assay (Pierce Protein Biology Products, Rockford, IL).
2.7. In vitro Toxicity
In vitro toxicity of DNA nanoparticles was measured using the Cell Counting kit-8 (Dojindo Molecular Technologies, Rockville, MD) as per the manufacturer’s instructions. Cells were seeded at 10,000 cells/well in 96-well plates and treated with different concentrations of compacted DNA for 24 h in DMEM before assessing cell viability.
2.8. Ex vivo DNA nanoparticle diffusion
Ex vivo brain slices were prepared as previously described.11 Briefly, adult female Fischer rats (120–140 g) were euthanized with an overdose of isoflurane, and their brains were rapidly removed and immersed in cold aCSF (Harvard Apparatus, MA) supplemented with 10% glucose for 10 min. The brains were then sliced to 1.5 mm thick slices using a Zivic brain matrix slicer (Zivic Instruments, Pittsburgh, PA) and placed on custom-made slides. Half a microliter of fluorescently labeled DNA nanoparticles was injected in the cerebral cortex at a depth of 1 mm using a 50 μl Hamilton Neuro Syringe (Hamilton, Reno, NV) mounted on a stereotaxic frame. Tissues were covered by a 22 mm x 22 mm coverslip to reduce tissue movement and bulk flow. Particle trajectories were recorded over 20 s at an exposure time of 66.7 ms by an Evolve 512 EMCCD camera (Photometrics, Tucson, AZ) mounted on an inverted epifluorescence microscope (Axio Observer D1, Zeiss; Thornwood, NY) equipped with a 100x/1.46 NA oil-immersion objective. Movies were analyzed by multiple particle tracking (MPT); extracting x, y-coordinates of DNA nanoparticle centroids over time and calculating the mean square displacement (MSD) of each particle as a function of time. At least n = 3 rat brains were used per DNA nanoparticle type and at least 500 DNA nanoparticles were tracked per sample. The geometric mean of the MSD for all nanoparticles was calculated per sample and the average of different rodent brains was calculated as a function of time. Histograms were generated from the MSD of every nanoparticle at a time scale of τ = 1 s. For the analysis of individual MSD histograms, we defined a diffusing fraction as the population of DNA nanoparticles capable of moving at least two times the average of the original hydrodynamic diameter of the three DNA nanoparticles tested.
2.9. In vivo CED injections and fluorescence quantification
To study DNA nanoparticle distribution in vivo following CED, a 20 μl solution of Cy3-labeled DNA-CPN and Cy5-labeled DNA-BPN at individual plasmid concentrations of 0.25 mg/ml in 3% saline was administered in the striatum of female Fischer 344 rats (n=3). Each rodent was anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (7.5 mg/kg). A mid-sagittal incision was made to expose the bregma. A 50 μl Hamilton Neuros syringe (Hamilton, Reon, NV) was positioned 0.5 mm posterior to the bregma, 3 mm lateral to the midline and lowered 3.5 mm below the dura matter to target the striatum. The solution was administered at a rate of 0.33 μl/min, using a Chemyx Inc. Nanojet Stereotaxic syringe pump (Chemyx, Stafford, TX), followed by withdrawal of the syringe at 1 mm per minute. The animal was sutured and placed on a heating pad before returning it to its cage. The rats were sacrificed 2 h after the procedure, and the brains were removed and fixed in formalin overnight. The suspension solution was then changed to 15% sucrose and subsequently to 30% sucrose after 24 h. The brain was sliced using a Leica CM 1905 cryostat (Leica Microsystems, Buffalo Grove, IL) to obtain 100 μm slices. We also assessed transgene expression following administration of EGFP expressing DNA-CPN and DNA-BPN. Infusions were carried out as described for in vivo distribution studies using a plasmid concentration of 1 mg/ml. Animals were sacrificed 48 h following CED administration, harvested brains were fixed as described above and sliced to 50 μm thickness.
Brain slices obtained following in vivo injections were stained with DAPI (Molecular Probes, Eugene, OR) and imaged using the Zeiss LSM 710 Meta Confocal Microscope at 5X and 20X magnification (Carl Zeiss, Hertfordshire, UK). Microscope settings were selected to minimize background fluorescence and were kept constant for all slices. A custom MATLAB code was used to quantify the area of DNA nanoparticle distribution for each slice with a threshold set at 10% of the maximum intensity. Care was taken to avoid quantification in the ventricles or white matter tracts. To determine the total volume of distribution achieved the areas calculated for each slice were multiplied by the slice thickness and summated across all images. For quantitative analysis of transgene expression, the fluorescence intensity was measured as the integrated density. The number of cells transfected per brain slice at different distances from the injection site were counted from 20X microscopic images using the ImageJ Image-based Tool for Counting Nuclei (ITCN) automated plugin.
All animal experiments were carried out at Johns Hopkins University School of Medicine following National Institutes of Health guidelines and local Institutional Animal Care and Use Committee regulations.
2.10. Western Blot Analysis of in vivo transfection
In vivo transfection efficiency was further quantified by western blot analysis following individual CED administrations of EGFP expressing DNA-CPN and DPN-BPN (n = 3). Brain tissues were lysed using brief sonication in ice cold PBS buffer (1 mM PMSF, and 1 μg/ml each of aprotinin, leupeptin, and pepstatin A). Sampling buffer (10% glycerol, 2% SDS, 62.5 mM Tris–HCl, 2% β-mercaptoethanol, pH 6.8) was added to the lysate and samples were boiled at 100 °C for 10 min. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was used to resolve protein bands followed by transfer to a nitrocellulose membrane (Bio-Rad, Hercules, CA) using a semidry blotter (Bio-Rad, Hercules, CA). The membrane was blocked with 3% bovine serum albumin in TBST (10 mM Tris-Cl, pH 8.0, 150 mM NaCl, 0.5% Tween-20) and incubated overnight at 4 °C with primary antibodies. The antibodies, anti-GFP (B-2): sc-9996 and anti-GAPDH (6C5):sc-32233 (Santa Cruz Biotechnology, Santa Cruz, CA), were used for the detection of transgene expression (EGFP) and housekeeping protein (GAPDH), respectively. Immunoblots were visualized by enhanced chemiluminescence method.26 Quantification of western blot results was performed using the gel analysis plugin on ImageJ.
2.11. Statistical Analysis
Statistical analysis of data for particle characterization, in vitro and ex vivo studies was performed by one-way analysis of variance (ANOVA) using R Commander. For in vivo distribution studies statistical analysis of data was performed by paired t-tests between particles injected into the same hemisphere, using R Commander. In vivo transfection data were analyzed by a two-tailed t-test assuming unequal variance. Differences were considered to be statistically significant at a level of p < 0.05.
3. Results and Discussion
3.1. Synthesis and optimization of densely PEGylated PLL-based DNA nanoparticles
The diffuse nature of most central nervous system disorders mandates that nanoparticle therapeutics widely distribute throughout the brain parenchyma in order to be effective. While conventional PLL-based gene vectors have demonstrated promise in pre-clinical and clinical studies12–14, their success in brain gene therapy may be limited by their inability to distribute away from the point of administration most likely due to the insufficient PEG surface coverage.11, 22 To increase the PEG surface density and formulate bio-inert gene vectors, we first synthesized di-block polymers of PLL and BrPEG using a highly efficient alkyne-azide cycloaddition reaction (Fig. S1).27 NMR analysis of purified products revealed that the PEG to PLL ratio for PLL-BrPEG was approximately three fold higher than that of the conventional PLL-PEG di-block polymer (Fig. S2). Densely PEGylated and potentially brain-penetrating DNA nanoparticles, DNA-BPN, were produced by compacting plasmid with a blend of PLL-BrPEG and PLL where 10% of the amine groups were contributed by un-PEGylated PLL polymers (i.e. 90% amine groups from PLL-BrPEG). We also synthesized DNA-UPN from PLL polymers28, 29, and DNA-CPN from PLL-PEG polymers12, 29, 30 as control nanoparticle systems. The hydrodynamic diameters of DNA-UPN, DNA-CPN and DNA-BPN were 108 ± 13 nm, 171 ± 5 nm and 127 ± 3 nm, respectively. While the ζ-potential of uncoated DNA-UPN was positive (10.0 ± 1.2 mV), PEGylated DNA-CPN and DNA-BPN exhibited a near neutral surface charge. In good agreement with previous findings, TEM analysis revealed that DNA-CPN possessed rod-shaped morphology with an average major diameter and aspect ratio of 177 ± 8 nm and 12.0 ± 0.5, respectively23, 24, 30 (Fig. 1A and Table 1). In contrast, both DNA-UPN and DNA-BPN were ellipsoid with significantly smaller major diameters (~85 nm) and aspect ratios (≤ 4) compared to DNA-CPN.
Figure 1. Stability of PLL-based DNA nanoparticles in aCSF.

(A) Representative TEM images of DNA nanoparticles prior to aCSF treatment and at different time points post aCSF treatment; Scale bar: 500 nm. (B) PDI of DNA nanoparticles as measured by DLS prior to incubation with aCSF and at different time points post treatment with aCSF. * Denotes a statistically significant difference from a PDI of 0.5 (p < 0.05), measurements were stopped after PDI significantly exceeded 0.5 (#). (C) Hydrodynamic diameter (nm) of DNA nanoparticles as measured by DLS prior to treatment with aCSF and at different time points post treatment with aCSF; data represents the mean ± standard error of the mean (SEM), * denotes statistical significance (p<0.05). (D) Heparin displacement assay for DNA nanoparticles treated with 0, 0.01, 0.05, 0.1, 0.5 and 1 IU/μg DNA, lanes 1–6 respectively, of heparin for 15 min at 37 °C.
Table 1.
Physicochemical properties of DNA nanoparticles
| PEG to PLL wt. ratioa | Hydrodynamic diameterb (nm) ± SEM | Major diameterc (nm) ± SEM | Minor diameterc (nm) ± SEM | Aspect Ratiod ± SEM | Zeta Potentialb (mV) ± SEM | PDIb | |
|---|---|---|---|---|---|---|---|
| DNA-UPN | - | 108 ± 13 | 82 ± 6 | 46 ± 3 | 2.4 ± 0.4 | 10.0 ± 1.2 | 0.21 |
| DNA-CPN | 1.3:1 | 171 ± 5 | 177 ± 8 | 16 ± 1 | 12.0 ± 0.5 | 1.5 ± 0.6 | 0.31 |
| DNA-BPN | 4.2:1 | 127 ± 3 | 87 ± 6 | 34 ± 2 | 4.0 ± 0.5 | 2.2 ± 1.1 | 0.20 |
PEG to PLL weight ratio of PEGylated polymers used in DNA nanoparticle formulation
Hydrodynamic diameter, ζ-potential and PDI were measured by laser Doppler anemometry and DLS in 10 mM NaCl at pH 7.0 and are represented as the average of three measurements ± SEM
Major and minor diameters were measured from TEM images for a total of at least 500 DNA nanoparticles from three batches using ImageJ and are presented as the average ± SEM
Aspects ratios were calculated for individual particles from their major and minor diameters as measured by TEM and are presented as the average ± SEM
For effective gene delivery to target cells and protection of the DNA cargo in the extracellular space (ECS), it is critical that gene vectors retain their stability in physiological environments. We therefore investigated the physicochemical characteristics of DNA-CPN and DNA-BPN over time in aCSF and the saline solution to be used for subsequent CED studies. We and others have previously shown that the DNA-CPN is stable in saline24, 31 however, the presence of divalent ions in aCSF led to rapid aggregation of a large fraction of the population as evidenced by representative TEM images, a high PDI (0.55 ± 0.06) and markedly elevated hydrodynamic diameter (888 ± 19 nm) (Fig. 1A–C). Aggregation of DNA nanoparticles in the CSF-filled ECS is undesirable as an increase in their size above the ECM mesh-spacing results in steric hindrance and thus limited distribution of nanoparticles.11, 15, 32 In contrast, the dense PEG coatings enhanced the colloidal stability of DNA-BPN in aCSF; PDI (0.19 ± 0.01) and hydrodynamic diameter (168 ± 1 nm) were well retained in aCSF up to 24 h (Fig. 1B, 1C). As expected, both TEM images revealed that both DNA-CPN and DNA-BPN remained colloidally stable following 1 h incubation in the saline infusate solution (Fig. S3).
In addition, a heparin displacement assay revealed improved compaction stability of DNA-BPN. In line with our previously published data the DNA-CPN released plasmid DNA at a heparin concentration of 0.05 IU/μg DNA.31 In contrast, the DNA-BPN remained entirely complexed up to 0.1 IU/μg DNA (Fig. 1D). It should be noted that DNA released in the ECS will be degraded by endogenous nucleases 33 and will not be taken up by cells, due to electrostatic repulsion.34
3.2. In vitro characterization of densely PEGylated PLL DNA nanoparticles
Although toxicity has been raised as a concern for cationic gene vector formulations, a conventionally PEGylated PLL-based gene vector, virtually identical to the DNA-CPN in this study, has been thoroughly characterized and proven to be safe in human lungs12 and in the rodent brain.13, 30 To ensure that our approach to increase surface PEGylation did not introduce particle toxicity, we characterized the in vitro toxicity of DNA-BPN in 9L gliosarcoma cells and rat primary astrocytes and compared it to that of DNA-CPN. As shown in Fig. 2A, for both DNA-CPN and DNA-BPN, DNA concentrations up to 10 μg/ml were non-toxic in 9L gliosarcoma cells, with no statistically significant difference between the two formulations. Similarly, DNA-CPN and DNA-BPN were non-toxic in rat primary astrocytes (Fig. 2B). The favorable safety profiles of these DNA nanoparticles and the previous preclinically and clinically confirmed safety of DNA-CPN12–14 underline the translational potential of DNA-BPN.
Figure 2. In vitro characterization of densely PEGylated PLL-based DNA nanoparticles.

Cell viability of (A) 9L gliosarcoma cells and (B) primary rat astrocytes, treated with different concentrations of DNA nanoparticles for a period of 24 h. Flow cytometric analysis of DNA nanoparticle cellular uptake in (C) 9L gliosarcoma cells and (D) primary rat astrocytes. Luciferase gene expression reported as RLU/mg of protein of (E) 9L cells and (F) primary rat astrocytes. Data represents mean ± SEM. *Denotes statistical significance p < 0.05.
The cellular uptake and transfection efficiency of DNA nanoparticles is largely dependent on physicochemical properties, including size, shape and surface chemistry, as well as cell type.29, 35–37 Previous studies have demonstrated that coating nanoparticles with PEG may reduce cellular uptake and transfection efficiency in vitro.38–40 These observations are most likely attributed to the effective shielding of positive charges which have been shown to be beneficial for the interaction of gene vectors with negatively charged cell membranes.41–43 We investigated the cellular uptake and transfection efficiency of DNA-CPN and DNA-BPN in 9L gliosarcoma cells and primary rat astrocytes. We found that the percentage cellular uptake was significantly higher for DNA-BPN compared to DNA-CPN (Fig. 2C, 2D). In accordance with previous reports, DNA-CPN demonstrated low uptake (1–3%) in both cell lines.31 In contrast, DNA-BPN exhibited 34 ± 2% and 10 ± 1% cellular uptake in 9L cells and primary astrocytes, respectively, equivalent to a 26-fold and 3-fold increases compared to DNA-CPN for corresponding cell lines. The increased DNA-BPN internalization also translated to increased transgene expression in vitro (Fig. 2E, 2F). DNA-BPN exhibited two orders of magnitude greater luciferase transgene expression compared to DNA-CPN in both cell lines.
This phenomenon may be attributed to the differences in particle shape, an important factor that may influence nanoparticle internalization, intracellular trafficking and transgene expression.44–46 Recent studies have suggested that nanoparticles with sub-100 nm minor diameters achieve greater cell internalization at lower aspect ratios.47, 48 For example, polymer-based DNA nanoparticles with an aspect ratio of 2.9 as opposed to 5.6, have demonstrated increased cellular uptake.49 This is in good agreement with our finding where DNA-BPN with lower aspect ratio (~4) exhibited significantly higher cellular uptake and subsequent transfection compared to DNA-CPN with an aspect ratio of ~12. Theoretical models based on energy consideration suggest that rod-shaped particles lying parallel to the cell may be only partially wrapped if excessively elongated50 and particle lengths exceeding the optimal sizes for vesicle formation may hamper receptor mediated endocytosis.51, 52
3.3. Improved brain penetration ex vivo leads to widespread distribution and efficient transfection in vivo
To reach the disseminated target cells and achieve effective therapeutic intervention, gene vectors must efficiently diffuse within the brain ECM. Conventional cationic gene vectors become entrapped in the ECM and diffuse poorly in the brain parenchyma.11, 17, 18 We have previously shown that diffusion behaviors of nanoparticles in brain tissue ex vivo predict their ability to penetrate the brain parenchyma in vivo.53 We first tracked DNA-UPN, DNA-CPN and DNA-BPN in rat brains ex vivo, determined their MSD, and calculated the ensemble averaged geometric MSD (<MSD>) of more than 500 DNA nanoparticles per sample at a time scale (τ) of 1 s. As indicated by the spatially constrained trajectories and low <MSD> (Fig. 3A, 3B), diffusion of uncoated, positively charged, DNA-UPN was largely hindered in brain tissue, likely due to their adhesive interactions with the negatively charged ECM components. The <MSD> of conventionally PEGylated DNA-CPN was only 3-fold higher than that of DNA-UPN and trajectories indicated hindered motion, albeit not being completely immobilized (Fig. 3A,B). DNA-BPN exhibited relatively unhindered diffusion with ~18-fold greater <MSD> compared to DNA-UPN and 5-fold greater <MSD> compared to DNA-CPN (Fig. 3A). Their trajectories were characterized by random walks that spanned much greater distances (Fig. 3B). Histogram analysis revealed that less than 10% of the DNA-UPN population was able to diffuse within the brain parenchyma. In contrast, ~30% of DNA-CPN and ~60% of DNA-BPN were capable of efficiently penetrating brain tissue ex vivo (Fig. 3C). We postulate that the dense PEG coating improved colloidal stability and minimized steric and adhesive interactions with the ECM, thereby leading to improved diffusivity of DNA-BPN.
Figure 3. DNA nanoparticles diffuse in ex vivo rodent brain tissue.

(A) Ensemble-averaged geometric mean of MSD of PLL-based DNA nanoparticles as a function of time. Data represent the ensemble average of at least three independent experiments with >500 particles tracked for each experiment, error bars depict SEM. (B) Representative trajectories over 20 s of respective DNA nanoparticles; Scale bar: 0.25 μm. (C) Histograms of individual MSD for DNA nanoparticles from at least three independent experiments at a timescale of τ = 1 s.
We next proceeded to test whether the favorable ex vivo behavior translated to improved DNA nanoparticle distribution and subsequent transfection in vivo following CED, a widely explored and clinically tested method for local administration to the CNS.54, 55 To this end, we co-infused Cy3-labeled DNA-CPN and Cy5-labeled DNA-BPN in the rat striatum in order to directly compare the conventionally and densely PEGylated DNA nanoparticles. In agreement with our previous studies11, DNA-CPN were confined to the point of administration and less than 1 mm3 volume of distribution was achieved (Fig. 4); this may be attributed to DNA nanoparticle aggregation and nanoparticle-ECM interactions. DNA-BPN provided ~7-fold higher (6.1 ± 0.1 mm3) volume of distribution compared to DNA-CPN (Fig. 4C).
Figure 4. DNA nanoparticle distribution following CED.

(A) Representative distribution of DNA nanoparticles in rodent striatum; blue – nuclear stain (DAPI), green – Cy3 labeled DNA-CPN, red – Cy5 labeled DNA-BPN, yellow – co-localization of DNA-CPN and DNA-BPN; Scale bar: 0.5 mm. (B) Area of distribution of DNA nanoparticles in the coronal plane as a function of distance from injection site and (C) volume of distribution of DNA nanoparticles quantified from confocal images of consecutive 100 mm slices. Data represents the mean ± SEM. * Denotes statistical significance p < 0.05.
The concurrent improvement in brain penetration and in vitro transfection efficiency achieved with DNA-BPN in comparison to DNA-CPN led, as anticipated, to enhanced and more widespread transgene expression in vivo. Following CED, the majority of DNA-CPN were restricted to the point of injection as indicated by the high intensity of the DAPI nucleic acid stain, within the injection site (Fig. 5A). Interestingly, a fraction of the DNA-CPN were convected farther from the injection site where they mediated low-level gene transfer (Fig. 5A). In accordance with our ex vivo results (Fig. 4C), a small percentage of the DNA-CPN population may be diffusive in the brain parenchyma. This may be explained by the size and shape heterogeneity of the population. In addition, current knowledge on the effects of particle shape on transport through porous media suggests that a suitable orientation may have allowed the minor diameter to dictate the movement of a fraction of the DNA-CPN that retained their stability, thus enabling them to be driven further into the tissue.16, 56 In contrast, in vivo transfection images for DNA-BPN revealed widespread and uniform transgene expression over a large area (Fig. 5B). Quantitative analysis of the integrated density and number of cells transfected per brain slice at distances up to 500 μm away from the injection plane, revealed a significant (2–3 fold) increase for DNA-BPN in comparison to DNA-CPN (Fig. 5C, 5D). Western blot analysis for EGFP expression following in vivo CED injections confirmed a 3-fold higher overall level of transgene expression mediated by DNA-BPN compared to DNA-CPN (Fig. 5E, 5F). These results confirm our hypothesis that densely PEGylated PLL-based DNA nanoparticles provide widespread, high-level transgene expression, which may have significant implications for gene therapy of neurological diseases.
Figure 5. DNA nanoparticle transfection following CED injection.

(A, B) Representative EGFP expression in rodent striatum following CED injection of (A) DNA-CPN and (B) DNA-BPN; blue – nuclear stain (DAPI), green – EGFP expression; Scale bar: 1 mm. (C) Fluorescence intensity per brain slice at different distances from injection plane (D) Number of cells transfected per brain slice at different distances from injection plane. (E, F) Western blot analysis for transgene expression following CED injection of DNA-CPN and DNA-BNP, EGFP expression was normalized against GAPDH for quantification. * Denotes statistical significance p < 0.05.
4. Conclusion
We modified a widely used and clinically tested gene delivery platform in order to achieve both rapid brain penetration and high-level transgene expression in vivo following CED. DNA-BPN demonstrated favorable colloidal stability and rapid diffusion in the brain parenchyma while achieving improved cellular uptake and transgene expression compared to conventional DNA-CPN. The combination of rapid diffusion and efficient transgene delivery of DNA-BPN resulted in uniformly widespread distribution and enhanced overall level of transgene expression. Further, the clinically confirmed safety profile of conventional DNA-CPN sets an excellent precedence for the translational potential of this similarly safe DNA-BPN.
Supplementary Material
Acknowledgments
The funding was provided by the National Institutes of Health (R01CA164789, R01EB020147, R01CA197111, R01CA204968 and P30EY001765), Focused Ultrasound Foundation and W.W. Smith Charitable Trust (J.S.S). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Abhijit Date for insightful discussion involving polymer synthesis.
References
- 1.Pulkkanen KJ, Yla-Herttuala S. Mol Ther. 2005;12:585–598. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- 2.Allen PJ, Feigin A. Neurotherapeutics. 2014;11:60–67. doi: 10.1007/s13311-013-0233-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowers WJ, Breakefield XO, Sena-Esteves M. Hum Mol Genet. 2011;20:R28–41. doi: 10.1093/hmg/ddr110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotterman MA, Schaffer DV. Nat Rev Genet. 2014 doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verma IM, Somia N. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 6.Thomas CE, Ehrhardt A, Kay MA. Nat Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 7.Tiera MJ, Shi Q, Winnik FM, Fernandes JC. Current Gene Therapy. 2011;11:288–306. doi: 10.2174/156652311796150408. [DOI] [PubMed] [Google Scholar]
- 8.Thomas M, Klibanov AM. Applied microbiology and biotechnology. 2003;62:27–34. doi: 10.1007/s00253-003-1321-8. [DOI] [PubMed] [Google Scholar]
- 9.Lv H, Zhang S, Wang B, Cui S, Yan J. J Control Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Frohlich E. International journal of nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mastorakos P, Zhang C, Berry S, Oh Y, Lee S, Eberhart CG, Woodworth GF, Suk JS, Hanes J. Adv Healthc Mater. 2015;4:1023–1033. doi: 10.1002/adhm.201400800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konstan MW, Davis PB, Wagener JS, Hilliard KA, Stern RC, Milgram LJ, Kowalczyk TH, Hyatt SL, Fink TL, Gedeon CR, Oette SM, Payne JM, Muhammad O, Ziady AG, Moen RC, Cooper MJ. Human gene therapy. 2004;15:1255–1269. doi: 10.1089/hum.2004.15.1255. [DOI] [PubMed] [Google Scholar]
- 13.Yurek DM, Fletcher AM, Smith GM, Seroogy KB, Ziady AG, Molter J, Kowalczyk TH, Padegimas L, Cooper MJ. Mol Ther. 2009;17:641–650. doi: 10.1038/mt.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher AM, Kowalczyk TH, Padegimas L, Cooper MJ, Yurek DM. Neuroscience. 2011;194:220–226. doi: 10.1016/j.neuroscience.2011.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. Science Translational Medicine. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sykova E, Nicholson C. Physiological reviews. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacKay JA, Deen DF, Szoka FC., Jr Brain research. 2005;1035:139–153. doi: 10.1016/j.brainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Kenny GD, Bienemann AS, Tagalakis AD, Pugh JA, Welser K, Campbell F, Tabor AB, Hailes HC, Gill SS, Lythgoe MF, McLeod CW, White EA, Hart SL. Biomaterials. 2013;34:9190–9200. doi: 10.1016/j.biomaterials.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 19.Mamot C, Nguyen JB, Pourdehnad M, Hadaczek P, Saito R, Bringas JR, Drummond DC, Hong K, Kirpotin DB, McKnight T, Berger MS, Park JW, Bankiewicz KS. J Neurooncol. 2004;68:1–9. doi: 10.1023/b:neon.0000024743.56415.4b. [DOI] [PubMed] [Google Scholar]
- 20.Franc G, Kakkar AK. Chem Soc Rev. 2010;39:1536–1544. doi: 10.1039/b913281n. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Aneja R, Chaiken I. Molecules. 2013;18:9797–9817. doi: 10.3390/molecules18089797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suk JS, Kim AJ, Trehan K, Schneider CS, Cebotaru L, Woodward OM, Boylan NJ, Boyle MP, Lai SK, Guggino WB, Hanes J. J Control Release. 2014;178:8–17. doi: 10.1016/j.jconrel.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suk JS, Boylan NJ, Trehan K, Tang BC, Schneider CS, Lin JM, Boyle MP, Zeitlin PL, Lai SK, Cooper MJ, Hanes J. Mol Ther. 2011;19:1981–1989. doi: 10.1038/mt.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziady AG, Gedeon CR, Miller T, Quan W, Payne JM, Hyatt SL, Fink TL, Muhammad O, Oette S, Kowalczyk T, Pasumarthy MK, Moen RC, Cooper MJ, Davis PB. Mol Ther. 2003;8:936–947. doi: 10.1016/j.ymthe.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Hosmane S, Tegenge MA, Rajbhandari L, Uapinyoying P, Kumar NG, Thakor N, Venkatesan A. J Neurosci. 2012;32:7745–7757. doi: 10.1523/JNEUROSCI.0203-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alegria-Schaffer A, Lodge A, Vattem K. Methods Enzymol. 2009;463:573–599. doi: 10.1016/S0076-6879(09)63033-0. [DOI] [PubMed] [Google Scholar]
- 27.Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 28.Dhanoya A, Chain BM, Keshavarz-Moore E. J Biotechnol. 2011;155:377–386. doi: 10.1016/j.jbiotec.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 29.Mannisto M, Vanderkerken S, Toncheva V, Elomaa M, Ruponen M, Schacht E, Urtti A. J Control Release. 2002;83:169–182. doi: 10.1016/s0168-3659(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 30.Walsh M, Tangney M, O’Neill MJ, Larkin JO, Soden DM, McKenna SL, Darcy R, O’Sullivan GC, O’Driscoll CM. Molecular Pharmaceutics. 2006;3:644–653. doi: 10.1021/mp0600034. [DOI] [PubMed] [Google Scholar]
- 31.Boylan NJ, Kim AJ, Suk JS, Adstamongkonkul P, Simons BW, Lai SK, Cooper MJ, Hanes J. Biomaterials. 2012;33:2361–2371. doi: 10.1016/j.biomaterials.2011.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goula D, Remy JS, Erbacher P, Wasowicz M, Levi G, Abdallah B, Demeneix BA. Gene Therapy. 1998;5:712–717. doi: 10.1038/sj.gt.3300635. [DOI] [PubMed] [Google Scholar]
- 33.Grigsby CL, Leong KW. J R Soc Interface. 2010;7(Suppl 1):S67–82. doi: 10.1098/rsif.2009.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amiji MM. Polymeric gene delivery : principles and applications. CRC Press; Boca Raton: 2005. [Google Scholar]
- 35.Rimann M, Luhmann T, Textor M, Guerino B, Ogier J, Hall H. Bioconjugate chemistry. 2008;19:548–557. doi: 10.1021/bc7003439. [DOI] [PubMed] [Google Scholar]
- 36.Fischer D, Bieber T, Li Y, Elsasser HP, Kissel T. Pharmaceutical research. 1999;16:1273–1279. doi: 10.1023/a:1014861900478. [DOI] [PubMed] [Google Scholar]
- 37.Choi YH, Liu F, Kim JS, Choi YK, Park JS, Kim SW. J Control Release. 1998;54:39–48. doi: 10.1016/s0168-3659(97)00174-0. [DOI] [PubMed] [Google Scholar]
- 38.Mishra S, Webster P, Davis ME. European journal of cell biology. 2004;83:97–111. doi: 10.1078/0171-9335-00363. [DOI] [PubMed] [Google Scholar]
- 39.Ogris M, Steinlein P, Carotta S, Brunner S, Wagner E. AAPS PharmSci. 2001;3:E21. doi: 10.1208/ps030321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verma A, Stellacci F. Small. 2010;6:12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 41.Luo X, Feng M, Pan S, Wen Y, Zhang W, Wu C. J Mater Sci Mater Med. 2012;23:1685–1695. doi: 10.1007/s10856-012-4632-4. [DOI] [PubMed] [Google Scholar]
- 42.Chen L, McCrate JM, Lee JC, Li H. Nanotechnology. 2011;22:105708. doi: 10.1088/0957-4484/22/10/105708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue ZG, Wei W, Lv PP, Yue H, Wang LY, Su ZG, Ma GH. Biomacromolecules. 2011;12:2440–2446. doi: 10.1021/bm101482r. [DOI] [PubMed] [Google Scholar]
- 44.Champion JA, Katare YK, Mitragotri S. J Control Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albanese A, Tang PS, Chan WC. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Kroger M, Liu WK. Nanoscale. 2015;7:16631–16646. doi: 10.1039/c5nr02970h. [DOI] [PubMed] [Google Scholar]
- 47.Qiu Y, Liu Y, Wang L, Xu L, Bai R, Ji Y, Wu X, Zhao Y, Li Y, Chen C. Biomaterials. 2010;31:7606–7619. doi: 10.1016/j.biomaterials.2010.06.051. [DOI] [PubMed] [Google Scholar]
- 48.Chithrani BD, Ghazani AA, Chan WC. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 49.Shi J, Choi JL, Chou B, Johnson RN, Schellinger JG, Pun SH. ACS Nano. 2013;7:10612–10620. doi: 10.1021/nn403069n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Decuzzi P, Ferrari M. Biophysical Journal. 2008;94:3790–3797. doi: 10.1529/biophysj.107.120238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao H, Shi W, Freund LB. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9469–9474. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. Science translational medicine. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, Shaffrey M, Ram Z, Piepmeier J, Prados M, Croteau D, Pedain C, Leland P, Husain SR, Joshi BH, Puri RK. Neuro Oncol. 2010;12:871–881. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eberling JL, Jagust WJ, Christine CW, Starr P, Larson P, Bankiewicz KS, Aminoff MJ. Neurology. 2008;70:1980–1983. doi: 10.1212/01.wnl.0000312381.29287.ff. [DOI] [PubMed] [Google Scholar]
- 56.Chauhan VP, Popovic Z, Chen O, Cui J, Fukumura D, Bawendi MG, Jain RK. Angew Chem Int Ed Engl. 2011;50:11417–11420. doi: 10.1002/anie.201104449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.