

Abstract
A single polypeptide chain may provide an astronomical number of conformers. Nature selected only a trivial number of them through evolution, composing an alphabet of scaffolds, that can afford the complete set of chemical reactions needed to support life. These structural templates are so stable that they allow several mutations without disruption of the global folding, even having the ability to bind several exogenous cofactors. With this perspective, metal cofactors play a crucial role in the regulation and catalysis of several processes. Nature is able to modulate the chemistry of metals, adopting only a few ligands and slightly different geometries. Several scaffolds and metal-binding motifs are representing the focus of intense interest in the literature. This review discusses the widespread four-helix bundle fold, adopted as a scaffold for metal binding sites in the context of de novo protein design to obtain basic biochemical components for biosensing or catalysis. In particular, we describe the rational refinement of structure/function in diiron–oxo protein models from the due ferri (DF) family. The DF proteins were developed by us through an iterative process of design and rigorous characterization, which has allowed a shift from structural to functional models. The examples reported herein demonstrate the importance of the synergic application of de novo design methods as well as spectroscopic and structural characterization to optimize the catalytic performance of artificial enzymes.
Keywords: Enzyme mimics, Metalloenzymes, Protein design, Helical structures, Iron, DF models
1. Introduction
Metal ions are of key importance for numerous proteins to function. In fact, about half of all known proteins contain a metal ion, performing a plethora of essential tasks, spanning from protein structure stabilization to catalysis, electron transfer, dioxygen carrier and activation, signal transduction, and nitrogen fixation.[1,2] It is quite remarkable that, despite the myriad of diverse reactions performed in the living cell, Nature uses only a handful of metal ions and structural motifs. Thus, understanding at a molecular level how metal cofactors and the surrounding protein environment mutually influence their properties is a fundamental issue in bioinorganic chemistry.[3–5] In this respect, the effective power of de novo metalloprotein design is constantly emerging, not only to improve our knowledge of the mechanisms of fundamental biological processes, but also to engineer novel metalloproteins with programmed properties.[6–8]
De novo metalloprotein design, or design “from scratch”, combines the fundamental knowledge of protein design and bioinorganic chemistry, and can be defined as design “from first principles”. In its purest and most challenging form, de novo metalloprotein design involves the construction of a polypeptide that is not directly related to any natural protein, yet folds into a predicted well-defined three-dimensional structure, incorporates a metal co-factor into a precise geometry, and/or is capable of performing desired functions.[6,9] De novo design can be considered a way to isolate and investigate the active site of functional metalloproteins in a smaller, defined model system, thus allowing researchers to accurately evaluate first-and second-shell interactions, which are crucial for structural and functional properties of metal binding sites.
In addition, de novo metalloprotein design allows scientists to explore beyond what has already appeared in Nature, and to develop structures and functions that are possible, but not yet discovered.[10–13] Several structural motifs, such as β sheets, α/β motifs, and especially α helices and helical bundles, have been designed with the highest degree of confidence. In particular, the “rules” that control stability, oligomerization, and helix–helix orientation are now well established and a variety of de novo designed α-helical coiled coils and bundles, with native-like structures, have been reported.[6,14–16] Furthermore, numerous functions have been designed into these structural scaffolds.[17–22]
One significant advantage of de novo design is that the designed systems, although providing a sufficient structural or functional complexity, are simpler than natural systems, thus amenable to systematic study. However, due to the complexity of the design problem, a trial-and-error approach is often necessary for designing de novo a metalloprotein with a native-like structure. Through several cycles of design, synthesis, characterization, and redesign, it is possible to fine-tune the structural properties of the initial model and to tailor the functional metal site into the interior.[23–25]
The ability of de novo designed four-helix bundles to bind heme and related porphyrins has been extensively studied. In the first pioneering work,[26] Choma and De-Grado and co-workers designed a four-helix bundle capable of incorporating a heme group into the interior. Afterwards, Dutton and co-workers reached impressive results in the design of four-helix bundles, named maquettes, which house arrays of cofactors and reproduce native-like functions.[27] Among these, worth mentioning is the construction of an O2 transport protein, made up of a de novo designed four-helix bundle encompassing bis-His-ligated Schemes.[28]
De novo designed proteins provide a unique opportunity to probe the functional potential of sequences that are stably folded, but were neither explicitly designed nor evolutionarily selected to perform any particular type of activity. Hecht and co-workers applied laboratory evolution techniques to de novo designed sequences to develop heme-binding four-helix bundles that exhibited peroxidase activities.[29–32]
By using the binary patterning approach, two libraries of sequences were tested. In the first round of the design, out of the 30 binary code sequences tested, 15 were found to bind heme with a broad range of affinities and with spectroscopic features resembling those of natural cytochromes.[29] Subsequently, several binary code proteins exhibited peroxidase activity at rates rivaling natural peroxidases[30] or carbon monoxide binding.[31] In this last achievement, Patel and Hecht aimed to mimic natural selection by introducing random mutations and screening for variants with improved activity.[32] The screening of a relatively small number (hundreds or thousands) of variants yielded novel sequences with improved peroxidase activity. Biochemical characterization of the purified proteins showed that the evolved variants were nearly three-fold more active than the parental sequence. These results demonstrate that de novo designed proteins can be utilized as a novel feedstock for the evolution of enzyme activity.
Recently, a four-helix bundle protein was designed to bind Fe4S4 in its hydrophobic core. This is particularly noteworthy given that natural Fe4S4-binding proteins are not α-helical and generally bind the ligand in flexible loops.[33] The successful engineering of a blue copper site into a de novo designed four-helical coiled-coil protein was reported by Tanaka and co-workers.[34] A huge amount of data have been reported by Pecoraro and co-workers, demonstrating that three-stranded coiled coils are helpful scaffolds for understanding the biochemistry of different heavy metals, such as CdII, HgII, AsIII, and PbII. In an elegant contribution, more recently, they described the design and structural characterization of a protein (TRIL9CL23H) that approaches the catalytic performance of the natural enzyme carbonic anhydrase (CA).[21] The research activity of Ogawa and co-workers further expanded the repertoire of coiled-coil motifs utilized as protein cages for housing metal cofactors. In an effort to incorporate redox activity into designed metalloproteins, Ogawa and co-workers designed a coiled-coil polypeptide, C16C19-GGY [Ac-K(IEALEGK)2-(CEACEGK)(IEALEGK)GGY-NH2], in which the Cys residues were placed at the a and d positions of the third heptad repeat (positions 16 and 19 in the peptide sequence).[35,36] The resulting peptide underwent a significant conformational change from monomeric random coil to a metal-bridged coiled coil upon binding to a variety of soft metal ions, such as CdII, HgII, AgI, AuI, and CuI.[36] Interestingly, the inorganic cofactors were not only able to induce peptide self-assembly, but also to dictate the nature of the oligomerization state.[37,38]
Beyond these examples, other successful metalloprotein designs have appeared in the literature;[39–44] and readers can refer to other recent reviews for a comprehensive description.[10,12,17,45]
In this review we describe our results of the de novo design of four-helix bundles housing a diiron site. A brief description of the design process we used is reported, together with an analysis of the naturally occurring diiron–oxo proteins. Finally, we outline all the steps that we performed on the way to shift from structural to functional artificial diiron oxo proteins.
2. Nature-Inspired De Novo Design
2.1. Four-Helix Bundles and the Designability Concept
The four-helix bundle motif is very common in nature.[46] It is classically viewed as an α-helical coiled coil.[47] Coiled coils are often characterized by a seven-residue (heptad) repeat; they rotate around the helix axis almost two full turns (700°), lagging by only 20°.[48] This lag implies supercoiling between helices, with the almost ubiquitous left-handed superhelical twist.[6] Residues composing the repeat are classically alphabetically ordered from a to g, with the a and d positions always directed towards the interior of the bundle (Figure 1). Even though the hydrophobic interactions by the a and d positions are essential to direct the association of the helices, they do not recapitulate the full pattern of interactions needed to drive the correct folding of coiled coils.
Figure 1.
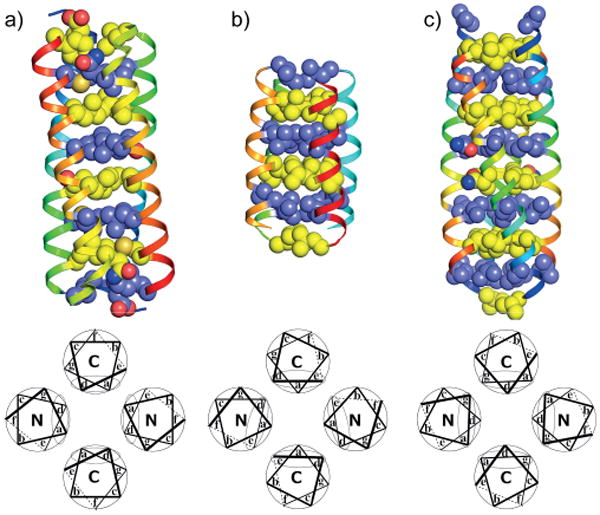
Structures (top) and wheel (bottom) representations of the three main core packing in tetrameric antiparallel coiled coils: a) a-d, b) a-d-e, and c) a-d-g core packing. PDB IDs: 1ROP, 1TLF, and 2B22. The structures and the wheel representations were generated with PyMol[52a] and DrawCoil,[52b] respectively.
In the special case of the four-helix bundle, four complementary-packed amphiphilic helices coil together, segregating the hydrophobic core from the solvent, thus probably constituting the easiest example of a globular protein. The pioneering work of Harbury and co-workers[49] on GCN4 mutants demonstrated how precise tuning of the side-chain packing in the interior of the protein tunes the properties of the bundle. They showed that leucine at position a and isoleucine at position d drive the tetramerization due to the complementarity of these side-chains through the bundle. However, such a recursive pattern may lead to a mixed orientation between parallel or antiparallel states. As evidenced by the work of Yadav et al.[47] and Deng et al.,[50] the e and g positions are crucial in the relative ratio between the two orientations. Polar side-chains selectively direct the orientation and the homo/heterotetramerization, even by destabilizing the alternative orientations.[51] Although polar interactions do not influence the packing of the hydro-phobic interior, hydrophobic substitutions at g positions lead only to antiparallel tetramers with different amino acid mutual dispositions and interdigitated helix–helix interfaces following a knobs-into-holes pattern.[47] Limited to the antiparallel case, three preferential topologies arise from the mutual orientation of the helices composing the four-helix bundle: The classical a-d core as found in repressor of primer (ROP) (Figure 1, a),[53] the a-d-e core as found in the Lac repressor protein (Figure 1, b),[54] and the a-d-g core as found in the hydrophobic g mutants of GCN4 (Figure 1, c).[55]
Such a deep understanding of the factors involved in the folding of the four-helix bundle inspired several authors to design new artificial proteins adopting this scaffold in a practice that is commonly identified as de novo design.[6]
De novo design in its purest form involves the ability to design a protein from scratch, shaping all the secondary and tertiary structural features only from the first principles. This approach has also been referred to as the inverse folding problem,[7] because the designer starts with the idea of a folded polypeptide chain possessing a determined, probably non-natural, function and ends with a sequence encoding the desired information for correct structure/function. Several outstanding papers have appeared in the literature describing the successful design of proteins with several global folds.[55] The first design of a protein with an unprecedented fold not yet found in nature was achieved with Top7.[56] A complete dissertation of the successful examples of de novo design is far from the target of this review; it just outlines the main problems behind the design concept, and which folds should be preferred as targets.
To simplify the complexity of such a problem, it is possible to divide it into three fundamental steps: 1) Selection of the structural folds/motifs to design, generally inspired by nature, 2) parametrization/idealization of the selected tertiary arrangement, and 3) the search for the best sequence encoding it.
De novo protein design would be impossible without hypothesizing that the native states are sufficiently low in energy to avoid becoming trapped in local misfolded energy minima.[7] From a thermodynamic point of view, a unique structure requires a large free-energy gap between the native state and the ensemble of non-native folded states and partially folded states,[57] often denoted as molten globules.[58–60] These non-native folded states exhibit extensive secondary structure but lack well-defined tertiary structure. The population of each state is dictated by the Boltzmann distribution. For a native protein structure, the free-energy gap, here referred to as Δ, must be large enough to significantly populate a distinct native state and fold into a unique three-dimensional structure. Thus, successful protein design requires sufficient thermodynamic stability to favor the folded state over the unfolded states, as well as a large free-energy gap (relative to the thermal energy, kT) to ensure conformational specificity. A hypothetical free-energy diagram for a protein is shown in Figure 2.
Figure 2.
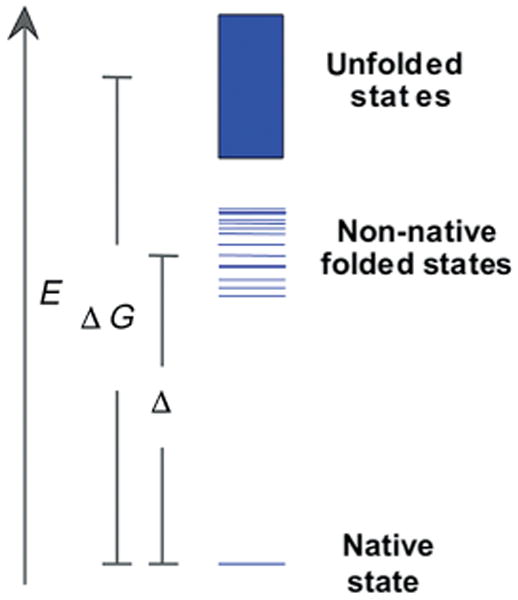
Hypothetical free-energy diagram for a protein. Each line or bar represents a distinct conformational state, with the native state as the lowest energy state and the unfolded states a densely populated, nearly isoenergetic ensemble. Between these extremes are non-native folded states, oftentimes referred to as the molten globule ensemble. (Adapted from ref.[61]).
For natural proteins, Δ has evolved to be much larger than in de novo designed proteins. Thus, Nature explored the sequence space and selected some robust tridimensional organizations for peptide chains, which can be encoded by sequences, sharing even a very low number of residues,[62] generating what is known as the “fold alphabet”.[63]
In the context of folding and inverse folding problems, this robustness of structure in the sequence space has been referred to as designability.[64–66] The final aim of a protein designer is to a priori limit oneself to engineering only reasonably designable structure. In fact, the relationship between structure and designability is a fundamental problem in protein design and the proper selection of structural folds/motifs is the crucial first step in de novo protein design as it dramatically enhances the chances of success. Several automated methods are now available to identify designable structure, and two approaches are delineated in the literature that can be viewed as complementary: A bottom-up and a top-down approach.
The first approach can be described as a statistical evaluation of designability, based on the frequency of finding a target topology in a representative subset of the Protein Data Bank (PDB). Zhang and Grigoryan adopted a cross-distance matrix to compute the similarity of the target backbone against a generated database of distance maps. Mining the ever-growing structural database, the MaDCaT software assesses the convenience of a certain tertiary motif, even suggesting sequence patterns that may encode it.[67] More recently, they developed a new and more efficient search tool, named MASTER. MASTER is a RMSD-based search engine; by enhancing 10-fold the speed, it allows the designer to identify the most common mutual orientation for the desired topology. Moll et al. developed an algorithm for substructure matching, named Label-Hash.[68] This algorithm is implemented in the UCSF Chimera molecular graphics,[69] and allows short-sequence motifs within some structural requirements to be identified. DeepView, or Swiss-PDBViewer,[70] allows the search for more complicated structural motifs based on tridimensional orientation even for noncontiguous residues. By means of such structural search engines, Johansson et al.[71] showed that such noncontiguous recurrent structural motifs (RSMs) can cover up to 60% of the structure of an unknown function protein (PDB ID: 2in5), which has a very low sequence identity in the whole PDB. Almost the full set of contacts for this protein, already categorized as New Fold in the SCOP classification, can be covered by RSMs. Computational alanine scanning of these RSMs further confirmed the role of these structural contexts in stabilizing the global fold. Gonzalez et al. implemented the Suns search engine,[72] which makes an even faster search for structural contexts in the whole PDB, visualizing already superimposed results in the PyMOL molecular graphics software. Suns outperforms the state-of-the-art all-atom search engine Erebus,[73] giving the designer the opportunity to search for specific, chemically meaningful, residue fragments.
The top-down approach in the identification of design-able structures, on the other hand, is related to the definition of “folding rules” by means of physical or knowledge-based chemical force fields. This approach has been extensively adopted and is reviewed elsewhere.[74] Recently, Koga et al. adopted a computational approach to identify several structural rules for the stabilization of α-β folds[75] by means of ab initio folding calculations in the Rosetta suite environment.
The second step of de novo protein design (parametrization/idealization of the selected tertiary arrangement) involves generating backbone coordinates from scratch, taking care of what has been learned from the previous analysis. Generally, this is possible through mathematical parametrization of the identified structural motif,[76–79] or by idealization of the selected topology by fragment-based tertiary structure composition.[80–83]
The concept of designability in the context of four-helix bundles and coiled coils in general is a classic example of a structural motif with strong geometric preferences.[84] This leads to easy mathematical formulation, fully describing the global organization of the peptide chains, which are then crucial for the de novo design of proteins from scratch. It was recently demonstrated which coiled coils are designable by a generalized mathematical model for backbone parametrization, producing deviation between ideal and real structures within 1 Å.[84] This model is the Crick parametrization of coiled coils, which can be easily expressed as the relationship between the superhelical frequency ω0 and the superhelical radius R0 according to the equation R0ω0 = dsinα, in which α is the pitch angle and d is the rise per residue.
Several key factors are remarkable in the choice of the overall set of parameters, directing future work on the design of coiled coils and four-helix bundles and also providing a useful web-server for the automated fit and generation of coiled coils based on Crick parametrization, namely CCCP (www.grigoryanlab.org/cccp/).[84]
The last step of de novo protein design (search for the best sequence encoding it) points to achieving minimal frustration of the selected fold by the careful identification of sequences and their properties.[85] The exponential number of possible sequences determines the complexity of this search because 20N sequences are possible for an N-residue protein, taking into account only the 20 natural amino acids. The designer may count on several computational methods that help to narrow the search. Deterministic methods, such as the dead-end elimination (DEE),[86] self-consistent mean field,[87] or site-specific probabilistic methods (e.g., SCADS[88]), help to select a set of sequences with a higher confidence to give the desired conformation. Recently, new methods for sequence design have emerged that are not strictly related to physically meaningful features. Based on an evolutionary profile search, EvoDesign[89] generates sequences that are suitable for the input backbone, comparing it to PDB structures presenting the same global fold.
Unfortunately, none of these algorithms can identify a single solution, not only because of the intrinsic limits of the algorithms themselves, but mostly because there may not be a single solution to such a combinatorial problem. The inner flexibility of protein backbones, also known as “template flexibility”,[90] has been widely considered either implicitly, by scaling down the van der Waals radii,[91] or explicitly, by generating ensembles of structures for successive rounds of sequence design.[92,93] Even though a general understanding of hydrophobic core packing is well established, there are still several issues relating to the role of the residues exposed to solvent and the influence they exert on the destabilization of alternative folding (negative design). Few examples reported in the literature adopted explicit negative design,[51,94] probably because starting structures are generally biased precluding this possibility, even though in some cases it is possible to demonstrate that it is not necessary.[95,96]
With this perspective, four-helix bundles and more generally coiled coils are particularly amenable for design purposes as they are less affected by template flexibility,[48,90] leading to rationalization of a complete set of designs as the basis for further functional implementations.[97,98]
In the next sections we will focus on our personal efforts in the design of functional non-heme de novo designed metalloproteins, highlighting the three steps of design (motif identification, template structure generation, sequence selection), and concluding with the latest achievements concerning the final efforts towards fully functional de novo metalloprotein catalysts.
2.2. Naturally Occurring Four-Helix Bundle Metalloproteins
As already outlined in the previous section, the four-helix bundle motif is a common structural motif among natural, functionally diverse proteins and metalloproteins. For example, four-helix bundles are involved in the RNA-binding process[53] and are found in several proteins, such as tobacco mosaic virus protein,[99,100] growth hormones,[101] and cytokines.[102] Finally, four-helix bundle motifs are found in different classes of metalloproteins: Heme proteins, such as cytochrome C′,[103] cytochrome b562,[104] and carboxylate-bridged diiron proteins.[105–107]
In this section we focus on the carboxylate-bridged diiron protein family to highlight the fundamental role of the four-helix bundle in fine-tuning protein functions, illustrating how the same metal cofactor can be engaged in a number of different roles.
Carboxylate-bridged diiron proteins belong to a protein family involved in essential and different physiological processes, ranging from dioxygen transport and activation to phosphoryl transfer and iron storage.[106–110] Among them, particularly relevant are the following: Ferritin[111,112] and bacterioferritin, which use iron as a substrate for ferroxidation and iron storage,[113] the growing subclass of bacterial multicomponent monooxygenases (BMMs;[114,115] also referred to as soluble diiron monooxygenases, SDIMOs),[116] including soluble methane monooxygenase (MMO), toluene/o-xylene monooxygenases (ToMO), phenol hydroxylase (PH), and alkene monooxygenase (AMO), which hydroxylate a variety of organic substrates; hemerythrin (Hr) and myohemerythrin, which reversibly bind and transport oxygen;[117] the ribonucleotide reductase R2 subunit (RNR-R2), which generates a tyrosyl radical essential for the reduction of ribonucleotides to deoxyribonucleotides in DNA biosynthesis;[108,118–120] the stearoyl-acyl carrier protein (ACP) Δ9-desaturase, which introduces a double bond into saturated fatty acids.[121,122] More recently, the diiron carboxylate protein family was broadened to include p-amino-benzoate N-oxygenase (AurF), which is involved in the biosynthesis of antibiotic aureothin, catalyzing the formation of p-nitrobenzoic acid from p-aminobenzoic acid,[123–125] and four membrane-associated enzymes, first identified only on the basis of six conserved amino acids, four carboxylate residues, and two histidines, which constitute the iron-binding motif. These proteins are alternative oxidase (AOX), plastid terminal oxidase (PTOX), 5-demethoxyquinone hydroxylase (DMQ hydroxylase), and Mg protoporphyrin IX monomethyl ester hydroxylase (MME hydroxylase).[126] More recently, the crystal structures of the trypanosomal AOX, in the absence and presence of ascofuranone derivatives, were solved.[127]
Diiron proteins are highly divergent in their amino acid sequences, with sequence identities generally too low for conventional phylogenetic analysis. However, when the analysis is restricted to the four helices containing the active-site coordination ligands, the amino acid similarity rises to 16–31%.[128]
Extensive structural studies revealed that their active sites contain a non-heme diiron center located within a very simple D2-symmetric hydrophobic four-helix bundle. It was pointed out that a simple model for the four-helix bundle in terms of a D2 symmetric tetrameric coiled coil could satisfy the structural features of the natural proteins, with a slight RMSD deviation of around 1 Å (Table 1).
Table 1.
Retrostructural analysis of diiron four-helix bundle proteins.[a]
| Protein | RMSD [Å] | Symmetry | Coil radius, R0 [Å] | Coil frequency, ω0 [°/res] | Pitch angle, α [°] | Coil phase offset, Δϕ0 [°] | Rise per residue, d [Å] | Axial offset, ΔZaa′ [Å] |
|---|---|---|---|---|---|---|---|---|
| Bacterioferritin[b] | 1.12 | D2 | 7.2 | −2.5 | −12.0 | 73.8 | 1.51 | 3.11 |
| Rubrerythrin[c] | 1.86 | D2 | 7.4 | −2.5 | −12.9 | 70.9 | 1.48 | 0.45 |
| Ribonucleotide reductase (R2)[d] | 1.54 | D2 | 7.6 | −3.4 | −18.3 | 80.8 | 1.46 | 1.56 |
| Methane monooxygenase[e] | 2.08 | D2 | 7.5 | −4.0 | −21.0 | 75.6 | 1.46 | 1.93 |
| Toluene o-xylene monooxygenase[f] | 2.27 | D2 | 7.7 | −4.0 | −21.2 | 66.9 | 1.49 | 2.25 |
| Toluene 4-monooxygenase[g] | 2.25 | D2 | 7.6 | –3.9 | −20.6 | 69.4 | 1.50 | 2.19 |
| ACP Δ9-desaturase[h] | 1.52 | D2 | 7.8 | −3.1 | −15.6 | 70.9 | 1.56 | 3.00 |
| Hemerythrin[i] | 2.19 | D2 | 7.4 | −2.1 | −10.6 | −86.6 | 1.50 | −7.10 |
| p-Aminobenzoate N-oxygenase[j] | 1.84 | D2 | 7.6 | −3.8 | −19.1 | 86.5 | 1.52 | 1.69 |
| DF1[k] | 0.86 | D2 | 7.4 | −3.2 | −15.8 | 83.4 | 1.52 | 2.55 |
| Repressor of primer (ROP)[l] | 0.34 | D2 | 6.9 | −2.7 | −12.2 | 81.7 | 1.52 | −6.30 |
This table is inspired by the analysis in ref.[77] and updated parameters have been evaluated by coiled Crick parametrization, enforcing D2 symmetry according to ref.[84] for a 15-residue window around binding Glu residues in each of the four helices composing the bundle. PDB IDs are given in the following footnotes.
4AM5.
1J30.
1SYY.
1MTY.
2INC.
3DHG.
1OQ9.
1I4Y.
3CHH.
1EC5 (A chain).
1ROP.
All the proteins share the so-called ExxH motif, in which a glutamate and histidine residue, involved in the metal binding, occupy the a and d positions of the coil, respectively.
Nature selected the four-helix bundle scaffold and the ExxH motif because of their ability to fine-tune the chemistry of the metals by means of 1) the global orientation of the bundle, 2) the choice of residues in the first coordination sphere, 3) the choice of interacting residues in the second coordination sphere (proton shuttling, hydrophobic environment, pKa of the ligands, redox properties of the metal), and 4) the ability to bind exogenous cofactors and substrates.
The majority of these enzymes are involved in dioxygen activation, and their dimetal sites are bridged by a combination of oxo, hydroxo, or carboxylate donors; two histidine and four carboxylate ligands represent a mostly conserved protein-derived ligand set. The only exception is hemerythrin (Hr), which contains a relatively rigid His-rich metal-binding site and functions as an oxygen carrier in invertebrate marine organisms, similarly to hemoglobin in vertebrates.[117]
The active-site structures of carboxylate-bridged diiron proteins and their catalyzed reactions are summarized in Figure 3. Brief descriptions of some examples of oxygen-activating diiron–oxo proteins are reported below.
Figure 3.
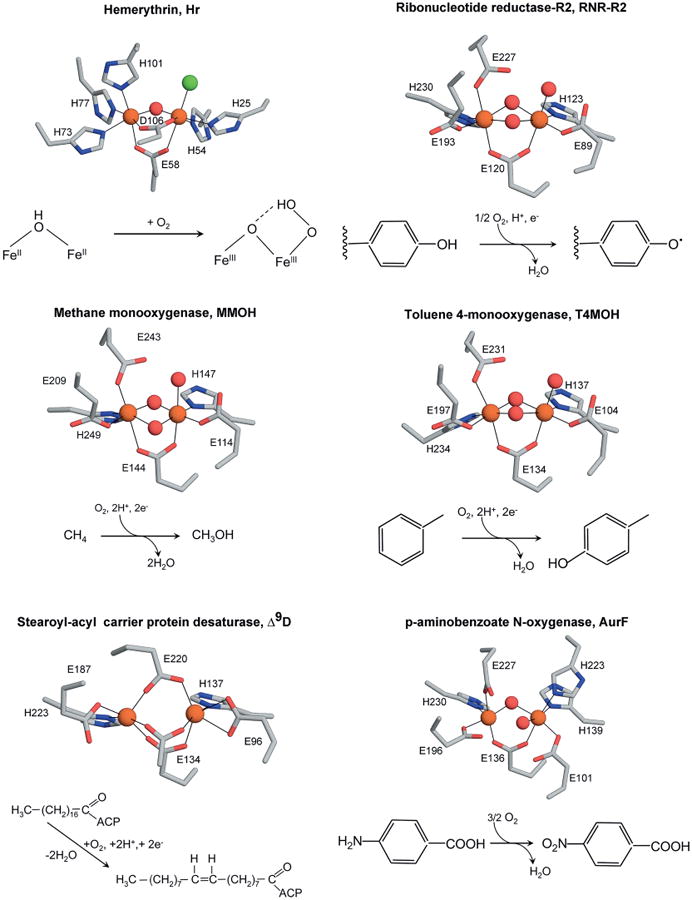
Metal binding sites of selected binuclear non-heme iron enzymes, and the reactions they catalyze. Pictures generated with PyMOL.[52a]
Ribonucleotide reductases (RNRs) are enzymes responsible for the conversion of ribonucleotides into their deoxyribonucleotide counterparts, thereby providing the precursors needed for both the synthesis and the repair of DNA. These proteins perform the reaction with the help of organic free radicals, which are stored by the enzyme until required for catalysis. Four distinct classes of RNRs have been found[129] that differ in their cofactor. Each metal co-factor is proposed to function as a radical chain initiator to generate a transient thiyl radical located on a cysteine residue within the protein, which initiates the nucleotide reduction. Class I RNRs are composed of two different sub-units, homodimeric proteins R1 and R2. The R2 proteins contain the dinuclear iron site that generates, stores, and delivers the radical essential for activity. The iron center is only involved in the activation of the Tyr122 radical. The stable tyrosyl radical (Tyr122·) is generated by oxidation of the binuclear ferrous center of R2, and plays the catalytic role, transferring its radical character to a remote cysteine in the R1 subunit during the catalytic turnover.
Soluble methane monooxygenases (MMOs) are the most notable members of bacterial multicomponent monooxygenases, a diiron-dependent family of enzymes found in many bacteria and Archaea. The catalytic chemistry of these enzymes is extraordinary in that they can catalyze the cleavage of the most stable bond in Nature, the C–H bond in methane, promoting the formation of methanol. They also catalyze the oxidation of a remarkable variety of substrates (saturated, nonsaturated, aromatic, heterocyclic halogenated hydrocarbons, etc.), and promote the formation of numerous chemically different products.[130]
MMOs are made up of three components: The hydroxylase subunit (MMOH), which houses the diiron site, the reductase component (MMOR), which accepts electrons from NADH and transfers them to the hydroxylase (for the reduction of the diiron site), and the regulatory protein (MMOB), which couples electron transfer to substrate oxidation.[106]
Stearoylacyl carrier protein Δ9-desaturase (Δ9-D) is a key enzyme of fatty acid synthetic metabolism in higher plants, performing dehydrogenation reactions by activating molecular oxygen. Its diiron site, which is highly symmetric, is involved in a two-electron oxidation in which double bonds are formed in long-chain fatty acids. Located in plastid stroma, it catalyzes the desaturation of stearoyl-ACP, introducing a double bond into the fatty-acid chain between positions 9 and 10 to form oleoyl-ACP.[131]
p-Aminobenzoate N-oxygenase (AurF) catalyzes the stepwise oxidation of the arylamine via hydroxylamine and nitroso intermediates to yield nitroarenes. The active site of this enzyme represents a dimetal cluster that contains either dimanganese[123] or diiron,[124,132] as shown by the crystal structure analyses of two variants of AurF. In addition, the presence of a heteronuclear Fe/Mn cluster has been discussed.[133] Irrespective of the metal involved, AurF is active with all three possible cofactors, but shows maximum activity in its diiron form. The crystal structure of diiron AurF shows protein ligands reminiscent of other diiron–oxo enzymes. An additional histidine coordinates one iron to afford a 3-histidine/4-carboxylate metal coordination site (see Figure 3).
From the previous analysis, it emerges that, despite the striking structural similarities, the diiron–oxo proteins display a large functional diversity, which can be ascribed to different mechanisms in their interaction with dioxygen.
Most of these enzymes bind dioxygen in the diferrous (bis-Fe2+) state. This dioxygen complex evolves to the diferric–peroxide intermediate, the fate of which depends on the specific environment of the diiron site.[106,109,110] Fo r example, in hemerythrins the rigid and His-rich metal binding site allows cycling between the diferrous and diferric/peroxo states, which constitutes the molecular basis for reversible oxygen binding. In the fully reduced deoxy state of Hr, one ferrous ion (Fe1) is coordinatively saturated (six-coordinate) and one iron (Fe2) is five-coordinate. Thus, the binding and the concomitant two-electron reduction of O2 occur at Fe2, the open coordination site, generating oxyhemerythrin, a species that is best described as a peroxo adduct, with the iron ions in the ferric oxidation state.[134] The rigidity of the Hr diiron site allows only terminal O2 binding.
In MMOH, RNR-R2, and Δ9-D, the flexible and carboxylate-rich metal binding site allows changes in the carboxylate binding mode, giving rise to the so-called carboxylate shift, which occurs upon changes in the oxidation state of the diiron site. In these diiron sites, ligands coordinate to the Fe centers differently, depending on the enzymatic needs. Thus they coordinate as bidentate ligands (bridging or chelating) when saturation of the first coordination sphere of Fe ions is required, and as monodentate (terminal) ligands when one (or more) vacant coordination site(s) is needed in the Fe center (e.g., upon dioxygen or substrate coordination) to enable a certain reaction step to take place.
The consequence of these structural changes is that both five-coordinate iron(II) ions react with O2, thereby allowing O2 to bridge the two irons, and subsequently leading to a diferric intermediate with a symmetrical bridging peroxo group.[135–137] The catalytic relevance of the carboxylate shifts occurring in these enzymes is that they tend to consume oxygen, rather than binding it reversibly.[138] In fact, in these enzymes the diferric–peroxide intermediate evolves towards the formation of a high-valent iron–oxo intermediate. This species then decays to the diferric–oxo or –hydroxo complex and is subsequently reduced back to the original diferrous complex. In ferritins, the diferric–peroxo complex may decay directly to the diferric–oxo species in a process known as the ferroxidase reaction. The catalytic cycle of MMO is presented in Figure 4, as an example.
Figure 4.
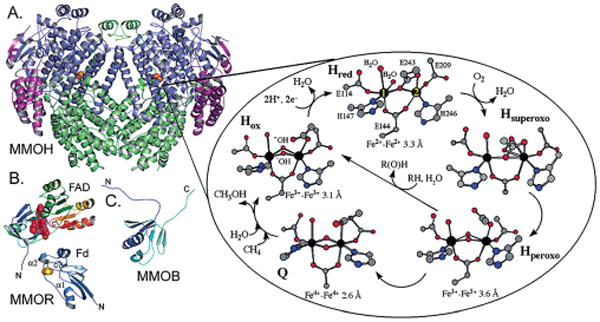
Structures of MMO components and proposed reaction cycle. A) MMOH, B) the MMOR FAD and ferredoxin (Fd) domains, and C) MMOB. The MMOH α, β, and γ subunits are colored blue, green, and purple, respectively. Iron, sulfur, and FAD are colored orange, yellow, and red, respectively, and are depicted as spheres. The MMO reaction cycle is shown on the right with atoms colored according to type [iron (black), carbon (gray), oxygen (red), and nitrogen (blue)]. (Reprinted with permission from ref.[139] Copyright 2006 American Chemical Society.).
The reaction between ferrous iron and oxygen is not simply a reaction, but the stabilization of high-valence intermediates, and the specific oxidation of substrates can only occur in a highly optimized protein frame. Moreover, the coordination sphere and the environment surrounding the diiron site in these proteins have apparently evolved to optimize a variety of reactions with O2.
In summary, the structural/functional analysis carried out on the carboxylate-bridged diiron proteins demonstrates the uniqueness of the four-helix bundle scaffold in hosting a metal cofactor and in providing the tunable structural flexibility needed to exert difficult chemical transformations.
3. Carboxylate-Bridged Dimetal Sites in de novo Designed Four-Helix Bundles: The Due Ferri Family
3.1. The Quest for Structural Robustness
As outlined above, Nature selected the four-helix bundle, as a common structural motif, for chemical processes needed to support life. This scaffold is compact, thermodynamically stable, and able to tolerate residue substitutions, deletions, and insertions without perturbing the global three-dimensional structure. As a consequence, the bundle orientation and the active-site environment (first and second coordination sphere) can be modified to fine-tune the chemistry of the metals and to accomplish specific functions.
As a consequence, the four-helix bundle represents a useful designable structure for structure/function relationship analysis.
We used a de novo design approach for the development of artificial diiron–oxo proteins. Starting from a retrostructural analysis of the natural diiron–oxo proteins (as mentioned in the previous paragraph), we developed the DF (due ferri, two-iron) family of artificial metalloproteins, minimal models for diiron and dimanganese protein.[15,16,140,141]
We divided the problem of designing catalytic metalloproteins into several parts: The first involves the design of stable, uniquely folded proteins that contain metal binding sites. This was followed by the engineering of a substrate recognition site that is capable of binding small molecules proximal to the metal center. The geometry, chemical reactivity, and electronic properties of the metal center were then finely tuned to provide catalytic functions.
The design started by taking advantage of the pseudo-C2 symmetry of this class of proteins. This symmetry has been shown to be particularly advantageous, simplifying the design and their structural characterization, and reducing the size of the synthesized peptides. The first member of this family, DF1, is made up of two 48-residue helix-loop-helix (α2) motifs that specifically self-assemble into an antiparallel four-helix bundle and bind a dimetal cofactor near the protein center.[61,77,142] The amino acid sequence of the α2 motif (entry 1) together with the intended helical secondary structure are presented in Table 2.
Table 2.
Peptide sequence of the DF family. Coordinating residues are in bold, helical regions are underlined, and active-site access residues are in italic.
| Sequence | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| g | abcdefg | abcdefg | abcdefg | abcdefg | abcdefg | abcdefg | abcdefg | abcdef | ||
| 1 | DF1 | DY | LRELLKL | ELQLIKQ | YREALEYV--KL | -----PV | LAKILED | EEKHIEW | LETILG | |
| 2 | DF2 | M | DY | LRELYKL | EQQAMKL | YREASERV--GD | -----PV | LAKILED | EEKHIEW | LETING |
| 3 | DF2t | M | DY | LRELYKL | EQQAMKL | YREASEK--ARN | -PEKKSV | LQKILED | EEKHIEW | LETING |
| 4 | DF3 | DY | LRELLKG | ELQGIKQ | YREALEYT--HN | -----PV | LAKILED | EEKHIEW | LETILG | |
| 5 | DFsc(l-28) | DE | LRELLKA | EQQAIKI | YKEVLKKA--KE | -----GD | ||||
| …(29-59) | EQELARL | IQEIVKA | EKQAVKV | YKEAAE----KA | -----RN | |||||
| …(60-88) | PEKRQV | IDKILED | EEKHIEW | LKAASK----Q | -----GN | |||||
| …(89-114) | AEQFASL | VQQILQD | EQRHVEE | IEKKN | ||||||
| 6 | 3H-DFsc(l-28) | DE | LRELLKG | EQQGIKI | LKEVLKKA--KE | -----GD | ||||
| …(29-59) | EQELARL | NQEIVKG | EKQGVKV | YKEAAE----KA | -----RN | |||||
| …(60-88) | PEKRQV | IDKILED | EEKHIEW | HKAASK-----Q | -----GN | |||||
| …(89-114) | AEQFASL | VQQHLQD | EQRHVEE | IEKKN | ||||||
| 7 | DFtet: | |||||||||
| A | K | LKELKSK | LKELLKL | ELQAIKQ | YKELKAE | LKEL | ||||
| Aa | E | LKELKSE | LKELLKL | ELQAIKQ | FKELKAE | LKEL | ||||
| Ab | K | LKKLKSR | LKKLLKL | ELQAIHQ | YKKLKAR | LKKL | ||||
| B | E | LEELESE | LEKILED | EERHIEW | LEKLEAK | LKEL | ||||
To provide the ligands of the first coordination shell, two helices (helices 2 and 2′) bear the ExxH motif, as found in natural proteins, in which a bridging Glu residue (Glu36/Glu36′) is in the a position and a terminal His (His39/His39′) is in the d position of the coiled coil describing the four-helix bundle. A second Glu residue is placed in the other two helices (helices 1 and 1′), thereby providing a fourth ligand (Glu10/Glu10′) for each metal ion. Other keystone residues of the second coordination shell are intended to stabilize the polar metal binding site in the middle of the protein as in the natural counterparts. Thus, a Lys/Asp/His hydrogen-bonding network was designed similar to the Arg/Asp/His pattern found in BMMs, and a Tyr residue donates a proton to the nonbridging carboxylate ligand (Figure 5). Each metal is five-coordinate in order to host an exogenous ligand.
Figure 5.
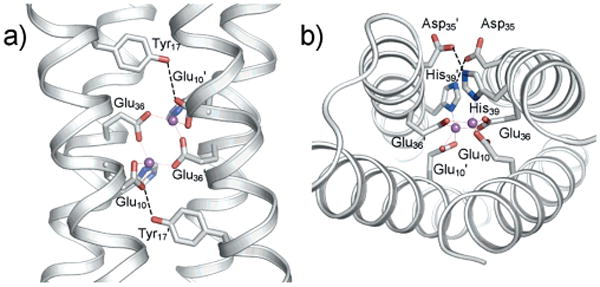
X-ray structure of di-ZnII-DF1 (PDB ID 1EC5). a,b) Second-shell interactions observed in the X-ray structures of DF1. (Reprinted with permission from ref.[144] Copyright 2005 American Chemical Society.).
All of the residues in the core and those in contact with the solvent were placed to achieve good packing and the correct antiparallel topology. Finally, an idealized γ-αL-β interhelical loop was included to stabilize the α2 motif.
DF1 was demonstrated to bind ZnII, CoII, MnII, or FeII. The crystal structures of di-ZnII-DF1,[77] di-MnII-DF1, and di-CoII-DF1,[142] reveal that the metal binding site and surrounding secondary interactions are very similar to the intended design. Furthermore, the solution NMR structure of the apo form[143] demonstrates that the metals bind the protein in a preorganized environment, as in the natural proteins. To the best of our knowledge, DF1 is the first protein, entirely designed from scratch, for which both crystallo-graphic and NMR structures have been obtained.
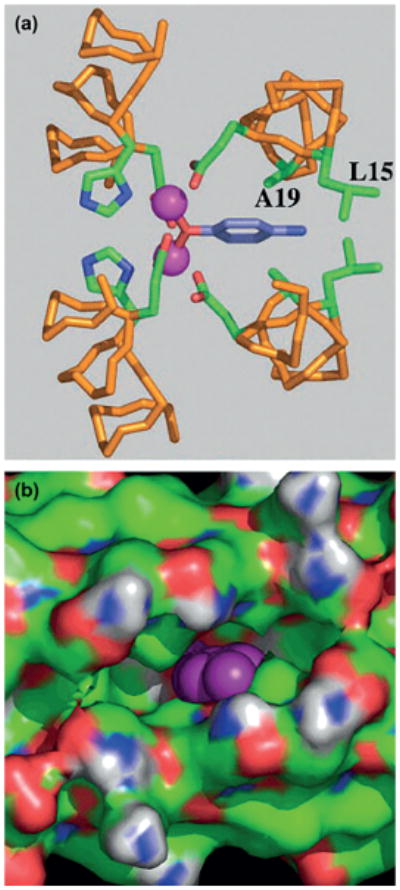
Although DF1 behaves as a native-like protein irrespective of its ligation state, it had low solubility in water, and unfortunately was not able to support any function because the access to its active site was hampered by a pair of Leu residues in the d positions opposite the His-binding residues (Leu13 and Leu13′), thereby forming a compact hydrophobic core around the metal center.[77,144]
In the first round of redesign, the DF1 subset of analogues was mainly devoted to overcoming the limits of DF1 in active-site accessibility. Thus, DF1 was redesigned to carve out a larger access channel to the active site by mutating each of the Leu residues to smaller side-chain amino acids, such as Ala and Gly.[144,145] The crystal structures of di-MnII-L13A-DF1 and di-MnII-L13G-DF1 confirm the presence of a designed cavity, which increases in size on going from the larger Leu to Ala and the smaller Gly. A substrate access channel to the dimetal site is occupied by ordered water molecules, of which there are more in di-MnII-L13G-DF1 (Figure 6). In addition, a ligating dimethyl sulfoxide molecule, coming from the crystallization buffer, forms a monatomic bridge between the metal ions and occupies the cavity in di-MnII-L13A-DF1.
Figure 6.
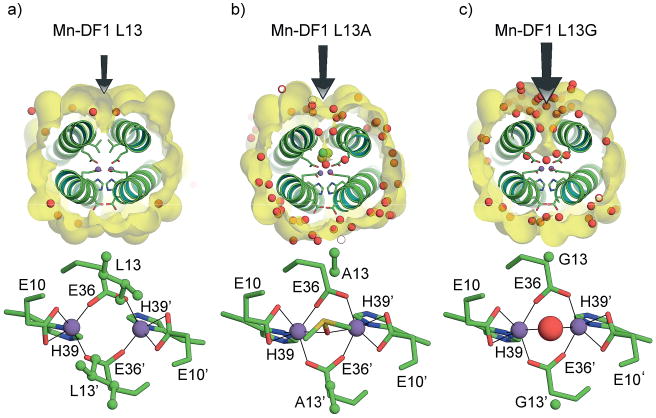
Top view of the active-site cavity and relative coordination geometry of the dimetal center in a) di-MnII-DF1, b) di-MnII-DF1–L13A, and c) di-MnII-DF1–L13G. The access channel, indicated by black arrows, expands upon Leu to Ala to Gly mutation. The red spheres indicate the water molecules and the yellow spheres (in DF1–L13A) indicate the DMSO molecule. The different residues in position 13 are depicted as balls and sticks. Pictures generated with PyMOL.[52a]
The presence of an exogenous bridging organic ligand in the di-MnII-L13A-DF1 active site is well precedented in the active sites of structurally characterized diiron and dimanganese proteins. For example, dimethyl sulfoxide can coordinate directly to iron in the mixed-valence state of methane monooxygenase.[146] An exogenous acetate molecule is observed in the diiron site of the hydroxylase component of MMO, which coordinates the metal ions in a η2-bridging manner. It has been suggested that this ion may occupy the position normally occupied by substrates during a catalytic cycle.[114,147]
Although the crystallographic data described above provide evidence that the L13A and L13′A mutations result in small molecule (DMSO) and solvent access to the dimetal center, experimental solution data were also desirable. Because this helical bundle system has been designed to serve as a minimal model for both natural diiron and dimanganese proteins, it is of interest to know whether the conclusions concerning small molecule and solvent access to the metal center from the crystallographic studies with di-MnII-L13A-DF1 would extend to the diiron derivative. To this end, we examined the binding of azide, a spectroscopic homologue of the anionic dioxygen species,[148,149] and acetate, a molecule known to inhibit manganese catalase,[150] to the diferric form of DF2[144] (see Table 2, entry 2), a variant of L13A-DF1 with increased water solubility (DF1 = 10 μm; DF2 = 0.5 mm).
The UV/Vis spectrum of di-FeIII-DF2 (Figure 7) is similar to those of proteins and inorganic complexes that contain FeIII–O–FeIII systems. These natural proteins and model complexes exhibit strong LMCT (oxo–FeIII ligand-to-metal charge transfer) absorption bands from 300 to 350 nm and much weaker bands between 400 and 700 nm.[122,151–153] The weaker bands between 400 and 700 nm may include contributions from charge-transfer bands and d–d ligand field transitions.[154–156]
Figure 7.
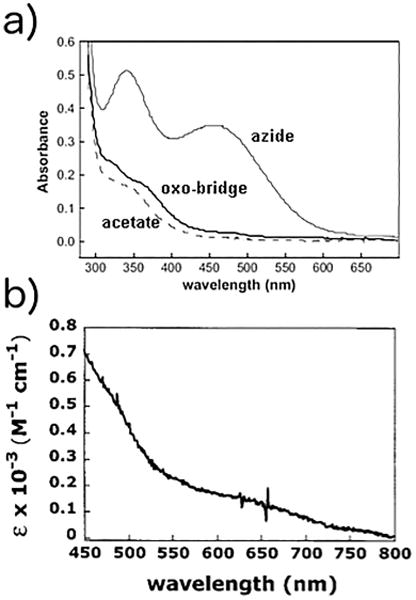
a) Spectrum of di-FeIII-DF2 and spectral changes that accompany the addition of the small molecules sodium azide and sodium acetate. b) Additional weaker di-FeIII-DF2 spectral features. (Adapted from ref.[144] Copyright 2001 American Chemical Society.).
The addition of sodium azide results in the appearance of two intense bands at 340 and 460 nm (Figure 7). The spectrum of the azide–di-FeIII-DF2 complex is nearly identical to the spectra of azide–di-FeIII-ACP Δ9-desaturase and the azide–methemerythrin complex.[122,151–153,157] The removal of the ligand by gel filtration restores the original absorption bands, which indicates that the diiron–oxo site is still intact.
Furthermore, the UV/Vis spectrum of di-FeIII-DF2 shows small changes upon the addition of acetate (see Figure 7). The intensities of the absorption bands at 330, 360, and 475 nm decrease upon sodium acetate addition. Moreover, the bands at 330 and 360 nm become less well resolved. These spectral changes suggest that acetate binds to the protein and alters the ligand field of the dimetal center. Similar spectral changes were observed upon the addition of benzoate or 4-hydroxybenzoate anions, which suggests that the carboxylate groups of these ligands interact with the metal center, similarly to natural diiron–oxo proteins.[144]
Comparison of the crystal structures and the spectroscopic data of the members of the DF1 and DF2 subsets, containing either Ala or Gly at the access site position, suggested that the water-filled, active-site pocket, observed in the crystal structure of di-MnII-L13A-DF1, is also present in di-FeIII-DF2 in aqueous solution. These experiments proved that the dinuclear metal binding site of the diferric form of the protein is accessible to azide- and carboxylate-containing molecules and established the feasibility of building an active-site cavity into the structure of DF1 scaffold.
The availability of crystal structures of the di-MnII, di-CoII, and di-ZnII complexes of several DF1 and DF2 analogues allowed an unprecedented examination of how a designed protein can accommodate different metal ions and exogenous ligands in the binding site.[142] The overall picture is of a prototypical binding site with two bridging carboxylates, two chelating carboxylates, and two monovalent His ligands. An open coordination site on both metal ions provides an attractive site for an approaching dioxygen molecule. Significant flexibility and more or less deviation from structure to structure were observed. For example, a high degree of variability was found in the bridging Glu residues, the carboxylate groups of which frequently lie in the same plane as the two metal ions, but in some structures are rotated out of this plane. Two orientations of the bridging carboxylates, as observed in the two dimers of the crystal structure of DF1, are shown in Figure 8.
Figure 8.
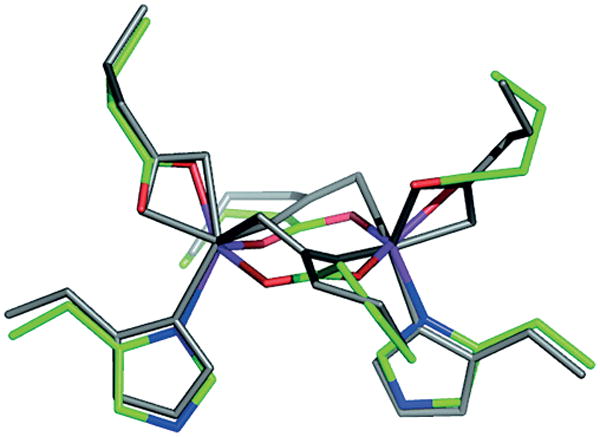
Superposition of the structure of the two independent dimers in di-ZnII-DF1. The two orientations of the bridging and chelating carboxylates observed in the two dimers of the crystal structure are shown. The side-chains of dimer 1 are in gray whereas the other side-chains of dimer 2 are green (carbon), red (oxygen), and blue (nitrogen). (Adapted from ref.[141]).
The chelating carboxylates also show a high degree of variability, and can distort towards the bond lengths expected for monovalent ligands.
All these structural findings were investigated by first principles[158] and hybrid quantum mechanical/molecular modeling (QM/MM) molecular dynamics simulations.[159] These studies indicated that a shift in the binding of the carboxylate from chelated to monodentate occurs during the simulations, at least on one of the two bridging glutamates. The calculations also showed that the second-shell interactions contribute significantly to the structural stability of the active site, and that the bulk solvent water molecules play a critical role in fine-tuning the dynamics of the system.
A further example of DF structural flexibility comes from the crystal structure of di-MnII-L13G-DF1, which crystallized with four dimers in the asymmetric unit.[145] In three of these dimers, a water molecule bridges between the metal ions (Figure 8) in a manner analogous to the di-MnII form of the R2 subunit of ribonucleotide reductase.[160] In the fourth dimer, two solvent molecules are bound trans to the His ligands. This finding was particularly interesting because the two structures resemble putative intermediates in the reaction mechanism of manganese catalases.[161] The major difference in the two sites was an increase of 0.6 Å in the metal–metal distance in the diaqua structure compared with the bridged structure. A rigid body shift of the helices accompanied this change in metal ligation. Although the C-terminal helices (2 and 2′) of the four crystal-lographic dimers are virtually invariant among the structures, the two copies of helix 1 undergo a shift in opposite directions (approximately 0.7 Å) along the z axis, away from the metal binding site in the dihydroxy structure. Indeed, this shift increases the length of the metal–metal distance. This sliding-helix mechanism may also occur in natural metalloproteins to accommodate changes in their coordination environment. However, such motions have not been observed in the parent natural metalloproteins, probably because it may be more difficult to be observed in a large complex molecule.
In conclusion, the overall structural and computational analysis performed on DF1 and DF2 analogues showed the plasticity of the fundamental DF framework, its modes of motion, and the degree of similarity of their structures to natural diiron proteins. This observed flexibility may ultimately be important for changes in response to redox shifts and organic substrate binding, towards the elaboration of catalysts capable of mediating a variety of oxygen-dependent reactions.
3.2. Expanding the Scope of the DF Family: On the Road to Functional Molecules
The lesson learned from the iterative process of design and characterization of the DF family prompted us to shift from the problem of protein folding to the design of proteins with specific catalytic functions. The results obtained suggested that it would be feasible to transfer a functional active site from a natural enzyme onto a minimal scaffold, but also to alter its chemical reactivity through specific amino acid substitutions at the active site. In the examples described below, we show that mutations of very few residues could be introduced into simple and stable four-helix bundles, as DFs, to confer catalytic functions and to perform structure–function correlation studies.
The first step on the road to converting the DF protein scaffold into functional molecules was to open up the active-site cavity, to enable the protein to accommodate substrates. The shape and accessibility of the active-site cavity are controlled by the residues at positions “g” and “d”. Residues with different steric hindrance at the above-mentioned positions influence both stability and activity. As already described, by decreasing the bulk of the residue at the “d” position with Ala or Gly variants, we were able to induce the formation of a cavity just above the dimetal site (Figure 6). The L13A-DF1[144] and L13G-DF1[145] variants, as well as the DF2 subset,[162–165] bind exogenous ligands, such as phenol and acetate, and display ferroxidase activity, as shown in the previous paragraph.
Inspection of the structures revealed that also residues at position 9 should be crucial in modulating active-site accessibility and reactivity. However, as the number of mutations increases, a very large number of combinations are generated. Thus, an exhaustive analysis would require the preparation and purification of significant quantities of hundreds to thousands of variants, and their screening for the binding of small molecules and reactivity towards a variety of substrates.
To this aim, a first screening of the active-site requirements for function were gained through the design of the DFtet subset, made up of a four-helix bundle heterotetrameric system (see Figure 9 and Table 2, entry 7),[52] consisting of four disconnected helices, which could be combinatorially assembled to create an array of the desired helical bundles.
Figure 9.
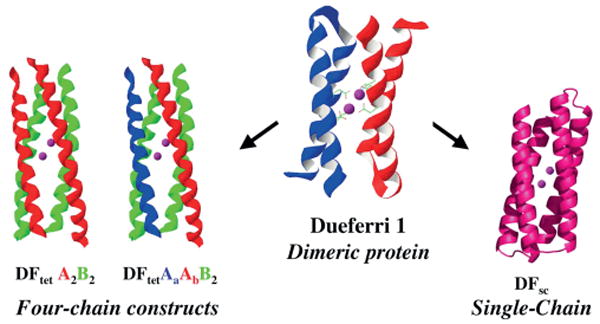
Different DF protein constructs as models of diiron proteins: Four-chain constructs (DFtet), dimeric protein (DF1), and single chain (DFsc). (Adapted from ref.[141]).
First, symmetric A2B2 and then asymmetric AaAbB2 systems[166] would give rise to a thousand different compounds starting from only 10 variants of each component. To stabilize the antiparallel coiled coil, the 24-long helices in DF1 were extended to 33 residues, offering an increased hydro-phobic interface to drive the folding (Figure 10). Next, a course-grained energy function, accounting only for electrostatic interactions, drove the design of the solvent-exposed region in a way that could stabilize only the correct topology and destabilize undesired parallel tetramerization. This design was the first to be reported in which an explicit “negative design” was accomplished.
Figure 10.
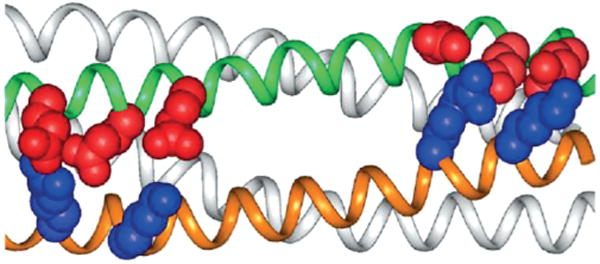
Polar side-chains to drive the correct antiparallel pairing of the coiled coil in the DFtet subset. (Reprinted with permission from ref.[166] Copyright 2002 National Academy of Sciences, USA.).
A series of asymmetrical variants were synthesized to explore the ferroxidase activity as a function of the accessibility of the metal binding site,[19] and mutations were introduced at positions 15 and 19 (corresponding to the positions 9 and 13, respectively, of DF1). The variants of DFtetA2B2 are designated G4-DFtet (in which Leu15 and Ala19 of both A chains are substituted with Gly), L2G2-DFtet (in which Leu15 is retained and Ala19 is changed to Gly), A2G2-DFtet, and G2A2-DFtet (which have Gly or Ala at the indicated positions).
All the resulting proteins were found to assemble into a helical tetramer with considerable thermodynamic stability. Furthermore, they bound CoII, ZnII, and FeII in the expected stoichiometry. All the variants were screened for their ability to react with FeII and O2 [Equation (1), Scheme 1]. To correlate the role of the bulkiness of the residues lining the active-site channel with functionality, the rate of formation of the oxo-bridged form in the ferroxidase reaction was evaluated. The variant with the fewest steric restrictions, namely G4-DFtet, showed the most rapid rate of oxidation and formation of the oxo species with no detectable intermediates. The same variant showed the greatest binding ability towards phenol, which binds to the diferric site, thus indicating a good reactivity towards dioxygen and exogenous substrates.
Scheme 1.
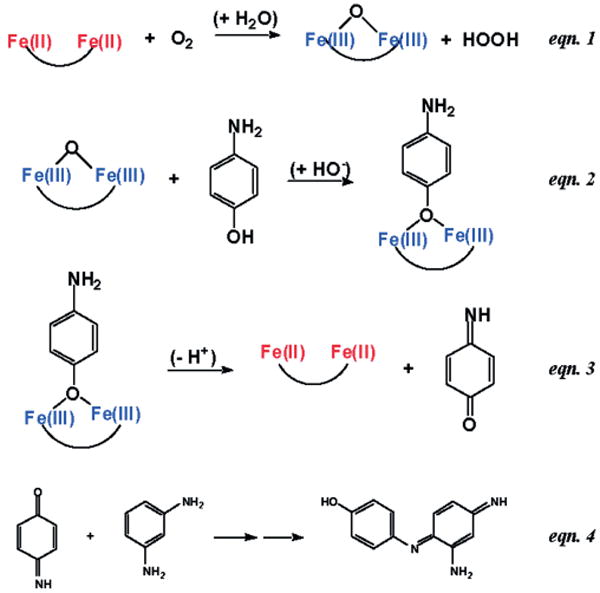
Proposed mechanism for the oxidation of 4-aminophenol. (Reprinted with permission from ref.[19] Copyright 2004 National Academy of Sciences, USA.).
Then, several analogues were screened in the two-electron oxidation of 4-aminophenol (4-AP). This reaction (Scheme 1) involves the use of O2 to oxidize the diferrous protein to a diferric species. The diferric protein then reacts with the substrate 4-AP to produce benzoquinone monoimine. The reduced diferrous form is then oxidized by O2, thereby initiating another catalytic cycle. The released quinone monoimine product is quenched and spectroscopically observed by reaction with m-phenylenediamine.[167,168]
G4-DFtet was able to enhance the reaction rate by around 1000-fold, relative to the background reaction, when the initial rate of the reaction in the presence and absence of the protein was compared. G4-DFtet catalyzed this reaction for at least 100 turnovers, and with a catalytic efficiency of kcat/KM = 1.540 m−1min−1. Changing either of the Gly residues at positions 19 or 15 to Ala gave a protein that reacted at a rate that was reduced by a factor of between 2.5 and 5.
Thus, changes as small as a methyl group had a significant effect on the catalytic activity of the protein, and the molecular basis of this effect can be deduced by inspection of a model structure in which the 4-AP has been sculpted into the DFtet active site (see Figure 11). Modeling suggested that when this molecule is inserted into the active site of the original DFtetA2B2 and modeled such that its phenolic oxygen bridges the metal ions, it makes unfavorable contacts with Leu15 and Ala19 (corresponding to positions 9 and 13, respectively, of DF1).
Figure 11.
a) Computer model of 4-aminophenol bound to the active site of DFtetA2B2. b) Solvent-accessible surface associated with G4-DFtet. The aminophenol ring in the pocket is represented in purple. (Adapted from ref.[19]).
Reducing the side-chain hindrance around the metal greatly influences the catalytic efficiency of the protein. In addition, G4-DFtet showed substrate selectivity. In agreement with the proposed mechanism of reaction, the substrate 4-methoxyaniline, in which the hydroxy group of 4-AP is converted into a methoxy, was not a substrate for the protein. Also, 4-aminoaniline, in which the phenolic hydroxy is replaced by an amino group, was oxidized at a rate only two-fold greater than the background reaction.
The screening carried out with the DFtet subset encouraged us to take more steps forward on the road to functional molecules, even though it was necessary to jump some hurdles and sometimes to change direction. Several drawbacks of the DFtet subset, such as the complex stoichiometry, marginal stability, and tendency to undergo ligand-exchange reactions, hampered attempts to fully characterize the structures and properties of these molecules.
Thus, the mutations important for functions were applied either to a single-chain protein or to the well-characterized helix-loop-helix scaffold. Both strategies aimed to obtain proteins that would have an unambiguous three-dimensional structure to investigate the structural bases for the functions.
A single-chain version of the DF proteins permitted mutations in only one position of the protein sequence, as opposed to the minimum of two for the homodimeric helix-loop-helix scaffold. A 114-residue protein, DFsc, which mimics the asymmetry of natural proteins, was designed adopting the helices of DFtet as a template for the bundle.[169] The keystone residues of the first and second coordination spheres, as well as the loop regions, were kept fixed, and SCADS[88] repacking algorithm guided the choice of the other residues. The resulting protein was highly soluble and expressed in E. coli in high yields. DFSC binds ZnII, CoII, FeII, and MnII in the intended stoichiometry with micromolar affinities. A QM/MM-refined NMR structure was solved and proved to be in good agreement with the starting model. Even though DFsc has native-like folding characteristics and is monomeric and stable, it was not able to convert any substrate, probably because of the low flexibility of the scaffold. In addition, this protein presented four Ala residues at the entrance of the metal binding site, thus limiting accessibility to the active site.
To examine the effect of Ala to Gly mutation in the single-chain scaffold, a quadruple Ala-Gly mutant (G4DFsc) was constructed by incorporating analogous Gly mutations (A10G, A14G, A43G, A47G) into the DFsc sequence.[22] As observed for the DF1 scaffold, these changes were expected to result in a large increase in the hydration and size of the active-site cavity (Figure 12).
Figure 12.
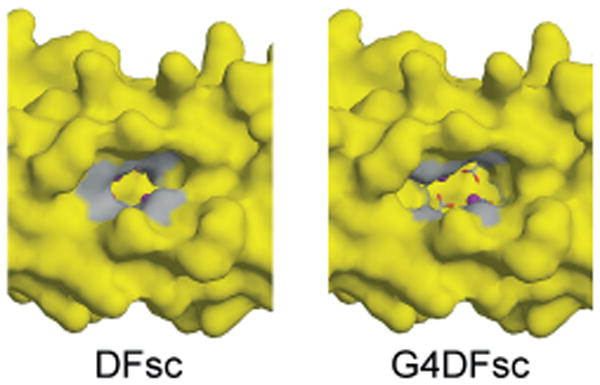
Surface models of DFsc (left; PDB ID: 2HZ8; ref.[52]) and the G4DFsc model (right) based on the initial DFsc computational design. The four Ala to Gly substitutions (shown in gray) significantly open the substrate access channel. Pictures generated with PyMOL.[52a]
The expansion of the substrate access channel into the single-chain version of the DF scaffold was effective, because, as observed for G4-DFtet, the protein showed activity towards the oxidation of 4-aminophenol to the corresponding quinone imine. Furthermore, these substitutions also minimized the formation of an off-pathway tyrosinate–iron complex that occurred in previous versions of the designed protein.[170] However, substitution of four Gly residues into the DFsc scaffold resulted in an apo form slightly less α-helical than the holo form. In addition, greater active-site accessibility increases the exposure of the iron atoms to the aqueous solvent, which renders them prone to hydrolysis. This reduces the stability of the Fe–protein complex and leads to the precipitation of iron oxides.
An impressive result was achieved through the incorporation of four mutations at different levels (one first shell, two second shell, and one third shell) in the DFsc protein, successfully switching the activity from hydroquinone oxidase to N-hydroxylase (Figure 13).
Figure 13.
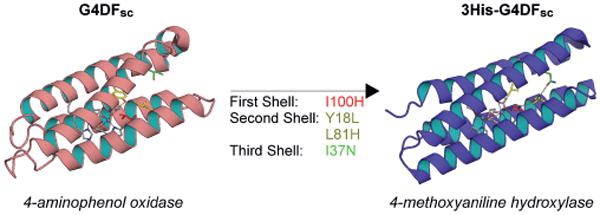
Mutations in the first, second, and third coordination spheres of the G4DFsc model (left) led to 3His-G4DFsc (right, Ala mutant PDB ID: 2LFD; ref.[22]), switching the activity from arylamine oxidase to arylamine hydroxylase. Pictures generated with PyMOL.[52a]
As reported in the previous section, AurF catalyzes N-hydroxylase activity, and is the only structurally characterized N-oxygenase known to contain a diiron catalytic center. To engineer this activity into the DFsc scaffold, a third metal-binding His residue, analogous to that found in AurF, was incorporated into the active site. The corresponding position in DFsc was that of Ile100. Additional mutations were required to stabilize this buried His ligand close to the protein core. An iterative process, alternating computer-aided design stages and expression and purification of the designed sequences, led to three crucial mutations that were able to stabilize and accommodate the mentioned I100H mutation. In particular, calculations indicated that a Y18L mutation was needed to alleviate a clash in the second shell of coordination. Furthermore, a Leu to His mutation in the second shell was needed to provide both the appropriate distance and favorable energy for the newly introduced polar metal-binding residue. In the same manner, a third-shell mutation was therefore needed to fulfil the last designed hydrogen-bond donor. Several polar residues were thus evaluated by computational design, and an I37N was finally selected as the best candidate for further studies. Interestingly, the computationally designed residues in the final 3His-G4DFsc sequence recreated part of an even more elaborate hydrogen-bonding network found in AurF (Figure 13), emphasizing both the correctness of the design process as well as the evolutionary optimized sequence of the natural counterpart.
3His-G4DFsc successfully catalyzes the N-hydroxylation of arylamines, thus showing a reactivity similar to that observed for the natural protein AurF. The addition of p-anisidine to 3His-G4DFsc in the presence of 2 equivalents of FeII resulted in the formation of 4-nitroso-4′-methoxydi-phenylamine, a diaryl product that probably arises from a nucleophilic aromatic substitution reaction between the p-nitrosoanisole intermediate and unreacted p-anisidine (Scheme 2). It is likely that the p-nitrosoanisole intermediate arises from the disproportionation of N-hydroxy-aminoanisole, which is the expected product of the initial hydroxylation reaction in the proposed AurF mechanism, but is not stable under these conditions.
Scheme 2.
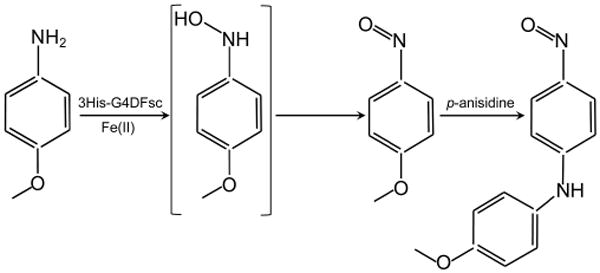
Proposed reaction scheme illustrating the oxidation of p-anisidine to p-nitrosoanisole via the N-hydroxyaminoanisole intermediate and subsequent nucleophilic aromatic substitution with unreacted p-anisidine to form 4-nitroso-4′-methoxydiphenylamine.
Interestingly, no reaction was observed in the presence of G4DFsc, and the engineered 3His-G4DFsc protein showed only background levels of activity towards arylamine oxidation. This finding demonstrates that selectivity towards substrates was achieved with the designed enzymes.
The second- and third-shell mutations needed to transform the DFsc protein from a hydroquinone oxidase into an N-hydroxylase partially disrupted the hydrophobic packing of the four-helix bundle, with a significant destabilization of the apo protein. As a consequence, apo-3His-G4DFsc is fully folded only in the presence of divalent metal ions. This finding is not unexpected given that the binding of the divalent metal ion is linked thermodynamically to protein stability in this class of proteins[169] and that 3His-G4DFsc incorporates numerous strongly helix-destabilizing substitutions as well as three apolar-to-polar substitutions within the solvent-inaccessible protein core. The addition of divalent cations provided enough driving force to promote folding, and once folded in the holo form, the protein proved sufficiently robust for characterization and reactivity studies.
It is well recognized that the requirements for protein stability and function are often diametrically opposed. The folded conformations are stabilized by maximizing the burial of hydrophobic groups, minimizing voids, and forming intramolecular hydrogen bonds. In contrast, to provide an artificial metalloprotein with functionality, the first point to be addressed is the design of a protein matrix able to accommodate all the changes occurring close to the active site, as it cycles between different functional states, without losing the native structure. Functionality requires flexibility around the metal site, ligand exchanges, and the binding of substrates. These principles are strongly maintained also in de novo designed metalloenzymes. Compelling proof is the structure–activity relationship analysis of the 2-Gly-2-Ala variant of the 3His-G4DFsc protein, namely 3His-G2A2DFsc. The structure of this variant was determined by NMR spectroscopy with ZnII. The presence of two Ala and two Gly residues along the substrate access channel profoundly affected the properties of the molecule. Even though the two Ala residues stabilized the protein, and the expected tertiary structure was confirmed, this variant showed decreased rates of N-oxygenase activity
All these excellent results highlight the delicate balance between function and stability that Nature manipulates so elegantly In the context of trying to replicate this balance in a de novo designed scaffold, a similar approach to that used for the DFsc scaffold was followed by using the helix-loop-helix version of the DF family, namely DF1 and its variants.
Before introducing into the DF1 scaffold the active-site mutations thought to be crucial for function, that is, the Gly residue at both positions 9 and 13, we tried to trace the energetic consequences of these mutations. The analysis of the thermodynamics of folding carried out on DF1, L13A, and L13G variants showed that the stability decreases as the bulk of the residues at positions “d” decreases from Leu to Ala to Gly due to a loss of the hydrophobic driving force for folding. A single mutation of Leu13 to glycine destabilizes DF1 by 10.8 kcalmol−1 dimer−1, thus precluding the introduction of a second glycine residue into this scaffold.
To increase the conformational stability of the DF1 scaffold, we modified the sequence of the interhelical turn, which adopts an αR-αL-β conformation.[77] Previous attempts to redesign the protein to accommodate different turn types[163,164] were not sufficiently successful to provide adequate stability to support the desired multiple glycine mutations. Thus, we used the database of the Protein Data Bank to derive statistical position-specific propensities for this solvent-exposed αR-αL-β turn,[163] leading us to change the original Val24-Lys25-Leu26 of DF1 to Thr24-His25-Asn26. In models, His25 appeared capable of forming stabilizing hydrogen-bonded C-capping interactions in helix 1, whereas Asn26 could either form N-capping interactions with helix 2, or with the carbonyl group of Thr24, depending on its rotamer. Introduction of this sequence, along with the two leucine-to-glycine mutations, resulted in a sequence designated DF3 (see Table 2, entry 4, and Figure 14).[20]
Figure 14.
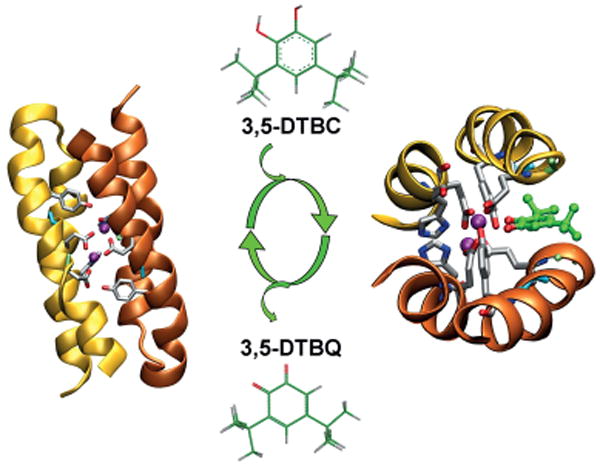
Di-ZnII-DF3 (left; PDB ID:2KIK) catalyzes the oxidation of 3,5-DTBC to the corresponding quinone (3,5-DTBQ). Model of the interaction of the substrate interaction with the DF3 active site (right). Pictures generated with VMD.[171]
The newly designed DF3 exhibited enhanced solubility (up to 3 mM in water) and an increased accessibility to the active site, as expected. DF3 was able to bind FeII, forming, upon exposure to dioxygen, a μ-oxo di-FeIII center. Notably, the di-FeIII-DF3 complex shows a long life-time, being stable under atmospheric O2 for at least 5 months at 4 °C. Only minor decomposition was observed for longer times. It can easily be reconstituted with different metal ions, and its diiron site is able to bind exogenous small molecules, such as dioxygen and azide. Compared with all DF proteins reported thus far, DF3 has the highest affinity for redox-active metals such as CoII and MnII.[172]
The thermodynamic cost of carving an active-site access channel within DF3 was largely compensated by the increased stability of the interhelical loop. As mentioned above, the substitution of Leu13/13′ with Gly in the sequence of DF1 resulted in a destabilization of 10.8 kcalmol−1 dimer−1 at pH 5.5 (ca. 5 kcal per substitution).[143] At this pH, apo-DF3 showed a ΔGuH2O value of 10.3 ± 0.6 kcalmol−1, with a destabilization of 13.2 kcalmol−1 with respect to DF1. Considering that four Gly residues were introduced into the core structure of the DF3 dimer (replacing Leu13/13′ and Leu9/9′ of DF1), a minor destabilization (ca. 3 kcal per substitution) was observed. This finding indicates that the interhelical loop has a profound influence on the DF3 stability.
Moreover, DF3 retained the intended global fold in the holo form, as confirmed by the NMR solution structural characterization of the diamagnetic di-ZnII derivative.
Finally, the stability/function tradeoff was matched with di-FeIII-DF3, because this protein behaves like a phenol oxidase, similar to the natural enzymes alternative oxidase (AOX)[126] and plastid terminal oxidase (PTOX).[173] These catalysts cycle between the di-FeII and di-FeIII states as they reduce O2, and then use the oxidizing equivalents to convert quinols into quinones.
Di-FeIII-DF3 was able to catalyze the oxidation of different substrates, such as 3,5-di-tert-butylcatechol (3,5-DTBC), 4-AP, and p-phenylenediamine (PPD). The kinetic results are summarized in Table 3. In the presence of ambient oxygen, the protein follows Michaelis–Menten kinetics for the oxidation of 4-AP to the corresponding benzoquinone monoimine with values of 1.97 ± 0.27 mm and 2.72 ± 0.19 min−1 for Km and kcat, respectively (Table 3, kcat/Km = 1380 m−1min−1, Figure 15, a). Measurement of the reaction over the course of an hour indicated that the protein was capable of at least 50 turnovers.
Table 3.
Kinetic parameters obtained for the oxidation of the different substrates by di-FeIII-DF3.[a]
| Protein | Substrate | Km [mm] | Kcat [min−1] | kcat/Km [m−1 min−1] |
|---|---|---|---|---|
| di-FeIII-DF3 | 3,5-DTBC[b] | 2.09 ± 0.31 | 13.20 ± 1.21 | 6315 |
| 4-AP[c] | 1.97 ± 0.27 | 2.72 ± 0.19 | 1380 | |
| PPD[v] | 8.87 ± 2.58 | 0.73 ± 0.03 | 83 | |
| OPD[e] | not detected | |||
| G4-DFtet | 4-AP[c] | 0.83 ± 0.06 | 1.30 ± 0.10 | 1540 |
All experiments were performed in 100 mm HEPES/100mm NaCl buffer (pH 7.0). Kinetic parameters represent mean values ± s.d., calculated by two independent measurements.
3,5-Di-tert-butylcatechol.
4-Aminophenol.
p-Phenylenediamine.
o-Phenylenediamine.
Figure 15.
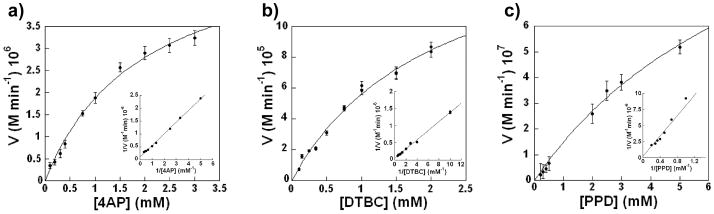
Catalytic activity of di-FeIII-DF3. Initial rate of the oxidation vs. substrate concentration for a) 4-AP, b) 3,5-DTBC, and c) PPD. The di-FeIII-DF3 concentration was 2 μm in (a) and (c), and 13 μm in (b). Kinetic parameters (kcat and Km) were determined from the Lineweaver Burk analysis (see insets). The analyzed substrates undergo a similar oxidation reaction as they bear two ortho- or para-related electron-donating groups (-OH or -NH2) in their molecular structures, giving rise to the corresponding benzoquinone monoimines, quinones, and diimines, respectively.
Kinetic investigations using other diamine and ortho-quinol substrates were consistent with the designed active-site cavity of di-FeIII-DF3. Indeed, di-FeIII-DF3 catalyzed the oxidation of 3,5-DTBC to the corresponding quinone 3,5-DTBQ (Figure 15, b), with a five-fold greater value of kcat/Km compared with 4-AP, reflecting an increase in kcat (Table 3). Amino-aniline substrates were expected to bind more weakly to the diferric center. Indeed, kcat/Km for the two-electron oxidation of PPD (Figure 15, c) was 75-fold lower than the value for 3,5-DTBC, and no catalysis was observed for o-phenylenediamine.
Enzymes are remarkable not only in their ability to catalyze reactions, but also in their ability to do so specifically. Oxygen-utilizing metalloproteins are particularly impressive in this regard because, unless single-electron processes are suppressed, O2 can react with iron ions to create radical oxygen species that engage in a wide variety of nonspecific radical reactions. Thus, it is significant that di-FeIII-DF3 did not catalyze the decomposition of other electron-rich aryl groups such as monophenols to biaryl species, catechols, and quinones. Furthermore, guaiacol was not converted into the corresponding quinone. Thus, the protein appears to catalyze the oxidation by the proposed two-electron reaction scheme while suppressing radical Fenton-like reactions.
In conclusion, catalytic diiron–oxo sites have been successfully engineered in de novo designed four-helix bundles by using both the single-chain approach and the helix-loop-helix dimerization. Each strategy holds advantages and disadvantages: for example, the single chain allows for the construction of asymmetric sites, whereas this is not possible in C2-symmetric homodimers, which, in turn, are more rigid and stable frameworks. Thus, merging the advantages of each system is highly desirable. More recently, we implemented our design strategy to obtain an asymmetric DF scaffold through the heterodimerization of two different α2 motifs, driven by selective chemical ligation.[174]
4. Conclusions
De novo metalloprotein design provides a powerful approach in many areas of research, from basic biology, for a more comprehensive understanding of reaction mechanisms, to catalysis, medicine, pharmacology, and biosensing.
The examples reported in this review show that designers can now approach with a high degree of confidence the construction of metalloenzymes in which the backbone of the protein as well as the catalytic site are elaborated from first principles.[6,9]
The development of the due ferri (DF) family of artificial proteins, based on the four-helix bundle fold, represents clear evidence that a metal binding site can be introduced into this simple scaffold. However, the principles employed during the design strategy can fail when applied to the construction of functional metalloproteins that must bind and act on substrates. In these cases, each structural element should be considered not individually, but in relation to the formation of a stable folded structure, and, most importantly, to the final desired function. Thus, the design and structure determination of metalloenzymes that fold into tertiary structures, with clefts that simultaneously bind and catalyze the transformation of organic substrates, has proved to be more challenging.
Through the development of the DF family of diiron/dimanganese proteins, we have demonstrated that the successful design of artificial metalloenzymes requires consideration of not only functional requirements, but also the careful redesign of the structural framework to compensate for stabilizing interactions lost during substrate binding-site incorporation. The introduction of catalytic activity into four-helix bundles requires engineering with Å-level precision: 1) The first- and second-shell ligands, 2) the residues that define the shape and accessibility of the active-site cavity, and 3) the thermodynamic stability, achieved by careful design of the helical packing and the interhelical turn.
In conclusion, such a highly simplified four-helix bundle structure provides attractive opportunities for determining how systematic variations in amino acid sequence and bundle geometry affect the physical and spectroscopic properties of the active site and, ultimately, its function.
Acknowledgments
The authors wish to thank all the co-workers who have contributed to the results described in this review, as well as Fabrizia Sibillo, for help with editing the manuscript. The work reported herein has been supported by the National Institutes of Health (NIH), USA, the Italian Ministry of Education and Research (MIUR), the European Union (EU) (Cost Action CM1003 – Biological Oxidation Reactions: Mechanisms and Design of New Catalysts) and the Scientific Research Department of Campania Region (BIP Project, POR FESR 2007/2013, grant number B25C13000290007, and STRAIN Project, POR FSE 2007/2013, grant number B25B0900000000 for a postdoctoral fellowship to M. C.).
Biographies

Marco Chino, 29 years old, received his BS and MS degrees in Chemistry from University of Napoli “Federico II”. In 2014, he received his Ph.D. in Chemical Sciences under the supervision of Professor Angela Lombardi, after spending six months in San Francisco in the DeGrado Lab. He is now a postdoctoral fellow in the artificial metalloenzymes group at the Department of Chemical Science in Napoli. His research interests include computational protein design, bioinorganic chemistry, and protein–protein interactions.

Ornella Maglio graduated in Industrial Chemistry, in 1989, at the University of Napoli “Federico II”, where she also received her Ph.D. in Chemical Sciences in 1992, under the supervision of professor Vincenzo Pavone. In 1996 she became Researcher at the Centro di Studio di Biocristallografia. In 1998 she was visiting fellow at the National Institute of Medical Research of London, in the research group of Dr. Annalisa Pastore. Since 2002 she is a Researcher at the Institute of Biostructures and Bioimages of the CNR in Naples. Her current research interests include the design and structural characterization, by NMR, of metalloprotein models, model systems based on peptides able to bind metal ions, and to reproduce the active site of natural metalloproteins.

Flavia Nastri is a professor of inorganic and bioinorganic chemistry at the University “Federico II” of Napoli. She graduated in Pharmaceutical Chemistry in 1990 at the University of Napoli where she received her Ph.D. in 1996 in Chemical Sciences. She was a postdoctoral fellow in the group of Vincenzo Pavone at the same University. In 1994 she joined Daniel Mansuy's group at the Université René Descartes, Paris (France). Her research interests are centered on peptide and protein chemistry, covering the fields of synthesis and structural and functional characterization of biologically active peptides and metalloprotein models.

Vincenzo Pavone is a professor of inorganic and bioinorganic chemistry at the University “Federico II” of Napoli. He graduated in Chemistry with Paolo Corradini at the University of Napoli (1976). He spent two years at the NIH (1978–1980) in the Laboratory of Chemical Biology (NIAMDD) and six months (1987) at the Mitsubishi Kasei Institute of Life Science (Japan). His scientific interests are focused on peptide/protein–metal ion interactions and include the design, synthesis, and structural characterization of “miniaturized metalloproteins”.

William (Bill) DeGrado's work focuses on the design of peptides, proteins, and peptide mimetics. He received his Ph.D. in organic chemistry from the University of Chicago (1981) working with E. T. (Tom) Kaiser. Bill was a member of DuPont Central Research and DuPont Merck Pharmaceutical Company from 1981 to 1996. In 1996, Bill moved to the Department of Biochemistry and Biophysics at the University of Pennsylvania, where he was a professor in the department of Biochemistry & Biophysics and an adjunct member of the Chemistry Department. In 2011 he moved to the Department of Pharmaceutical Chemistry at the University of California, San Francisco, where he is currently a professor and member of the Cardiovascular Research Institute. Some of Bill's research interests include: de novo design of proteins and peptide design; peptide mimetics; structure, stability, and function of membrane proteins, including integrins and viral ion channels; design of biomimetic polymers; bioinorganic chemistry; and computational approaches to small molecule and protein design. Bill is a member of the National Academy of Science and the American Academy of Arts and Sciences.

Angela Lombardi is a professor of inorganic chemistry and protein design at the University “Federico II” of Napoli. She received her Ph.D. in Chemical Sciences from the University of Napoli in 1990 working in the area of transition metal chemistry and peptide synthesis. In 1990, Angela was a Visiting Fellow at the NIH (Laboratory of Experimental Carcinogenesis, NCI), and in 1995 she spent a sabbatical in Bill DeGrado's group, at the DuPont-Merck Pharmaceutical Co and then in 1997 at the University of Pennsylvania. Angela's research interests mainly focus on the design of peptide-based metalloprotein models as tools for understanding the structural requirements for function in natural systems. New catalytically active molecules, able to promoting oxidative reactions, have been obtained, with a great potential in biosensing and diagnostics. Angela's research interests also include bioactive peptides, such as antimicrobial peptides and hormones.
References
- 1.Lu Y, Berry SM, Pfister TD. Chem Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 2.Winkler JR, Gray HB. Chem Rev. 2013 doi: 10.1021/cr4004715. [DOI] [Google Scholar]
- 3.Bertini I, Gray HB, Stiefel EI, Valentine JS. Biological Inorganic Chemistry: Structure and Reactivity. University Science Books; Sausalito, CA, USA: 2007. [Google Scholar]
- 4.Ciferri A, Perico A. Ionic Interactions in Natural and Synthetic Macromolecules. Wiley; Hoboken, NJ, USA: 2012. [Google Scholar]
- 5.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 6.De Grado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 7.Samish I, MacDermaid CM, Perez-Aguilar JM, Saven JG. Annu Rev Phys Chem. 2011;62:129–149. doi: 10.1146/annurev-physchem-032210-103509. [DOI] [PubMed] [Google Scholar]
- 8.Ueno T, Watanabe Y. Coordination Chemistry in Protein Cages: Principles, Design, and Applications. Wiley; Hoboken, NJ, USA: 2013. [Google Scholar]
- 9.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HX, O'Neil KT, DeGrado WF. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 10.Smith BA, Hecht MH. Curr Opin Chem Biol. 2011;15:421–426. doi: 10.1016/j.cbpa.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Petrik ID, Liu J, Lu Y. Curr Opin Chem Biol. 2014;19:67–75. doi: 10.1016/j.cbpa.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Chem Rev. 2014;114:3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 14.Baltzer L, Nilsson H, Nilsson J. Chem Rev. 2001;101:3153–3164. doi: 10.1021/cr0000473. [DOI] [PubMed] [Google Scholar]
- 15.Maglio O, Nastri F, Lombardi A. In: Ionic Interactions in Natural and Synthetic Macromolecules. Ciferri A, Perico A, editors. Wiley; Hoboken, NJ: 2012. pp. 361–450. [Google Scholar]
- 16.Nastri F, Bruni R, Maglio O, Lombardi A. In: Coordination Chemistry in Protein Cages: Principles, Design, and Applications. Ueno T, Watanabe Y, editors. Wiley; Hoboken, NJ, USA: 2013. pp. 43–85. [Google Scholar]
- 17.Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanda V, Koder RL. Nat Chem. 2010;2:15–24. doi: 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan J, DeGrado WF. Proc Natl Acad Sci USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat Chem Biol. 2009;5:882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Nat Chem. 2012;4:118–123. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Solomon EI, DeGrado WF. Nat Chem. 2012;4:900–906. doi: 10.1038/nchem.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohn WD, Hodges RS. Trends Biotechnol. 1998;16:379–389. [Google Scholar]
- 24.Holm RH, Solomon EI. Chem Rev. 2004;104:347–348. doi: 10.1021/cr0206364. [DOI] [PubMed] [Google Scholar]
- 25.Saven JG. Curr Opin Colloid Interface Sci. 2010;15:13–17. doi: 10.1016/j.cocis.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choma CT, Lear JD, Nelson MJ, Dutton PL, Robertson DE, DeGrado WF. J Am Chem Soc. 1994;116:856–865. [Google Scholar]
- 27.Shifman JM, Moser CC, Kalsbeck WA, Bocian DF, Dutton PL. Biochemistry. 1998;37:16815–16827. doi: 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- 28.Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, Dutton PL. Nature. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rojas NRL, Kamtekar S, Simons CT, Mclean JE, Vogel KM, Spiro TG, Farid RS, Hecht MH. Protein Sci. 1997;6:2512–2524. doi: 10.1002/pro.5560061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moffet DA, Certain LK, Smith AJ, Kessel AJ, Beckwith KA, Hecht MH. J Am Chem Soc. 2000;122:7612–7613. [Google Scholar]
- 31.Moffet DA, Case MA, House JC, Vogel K, Williams RD, Spiro TG, McLendon GL, Hecht MH. J Am Chem Soc. 2001;123:2109–2115. doi: 10.1021/ja0036007. [DOI] [PubMed] [Google Scholar]
- 32.Patel SC, Hecht MH. Protein Eng Des Sel. 2012:1–7. doi: 10.1093/protein/gzs025. [DOI] [PubMed] [Google Scholar]
- 33.Grzyb J, Xu F, Weiner L, Reijerse EJ, Lubitz W, Nanda V, Noy D. Biochim Biophys Acta (BBA) - Bioenerg. 2010;1797:406–413. doi: 10.1016/j.bbabio.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Shiga D, Nakane D, Inomata T, Funahashi Y, Masuda H, Kikuchi A, Oda M, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Tanaka T. J Am Chem Soc. 2010;132:18191–18198. doi: 10.1021/ja106263y. [DOI] [PubMed] [Google Scholar]
- 35.Kharenko OA, Ogawa MY. J Inorg Biochem. 2004;98:1971–1974. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa MY, Fan J, Fedorova A, Hong J, Kharenko OA, Kornilova AY, Lasey RC, Xie F. J Braz Chem Soc. 2006;17:1516–1521. [Google Scholar]
- 37.Kharenko OA, Kennedy DC, Demeler B, Maroney MJ, Ogawa MY. J Am Chem Soc. 2005;127:7678–7679. doi: 10.1021/ja042757m. [DOI] [PubMed] [Google Scholar]
- 38.Xie F, Sutherland DEK, Stillman MJ, Ogawa MY. J Inorg Biochem. 2010;104:261–267. doi: 10.1016/j.jinorgbio.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Rau HK, DeJonge N, Haehnel W. Angew Chem Int Ed. 2000;39:250–253. [PubMed] [Google Scholar]; Angew Chem. 2000;112:256. [Google Scholar]
- 40.Monien BH, Drepper F, Sommerhalter M, Lubitz W, Haehnel W. J Mol Biol. 2007;371:739–753. doi: 10.1016/j.jmb.2007.05.047. [DOI] [PubMed] [Google Scholar]
- 41.Schnepf R, Haehnel W, Wieghardt K, Hildebrandt P. J Am Chem Soc. 2004;126:14389–14399. doi: 10.1021/ja0484294. [DOI] [PubMed] [Google Scholar]
- 42.Khare SD, Kipnis Y, Takeuchi R, Ashani Y, Goldsmith M, Song Y, Gallaher JL, Silman I, Leader H, Sussman JL, Stoddard BL, Tawfik DS, Baker D. Nat Chem Biol. 2012;8:294–300. doi: 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Der BS, Edwards DR, Kuhlman B. Biochemistry. 2012;51:3933–3940. doi: 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeung N, Lin YW, Gao YG, Zhao X, Russell BS, Lei L, Miner KD, Robinson H, Lu Y. Nature. 2009;462:1079–1082. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu Y, Chakraborty S, Miner KD, Wilson TD, Mukherjee A, Yu Y, Liu J, Marshall NM. In: Comprehensive Inorganic Chemisty II. 2nd. Poeppelmeier JR, editor. Elsevier; Amsterdam: 2013. pp. 565–593. [Google Scholar]
- 46.Kamtekar S, Hecht MH. FASEB J. 1995;9:1013–1022. doi: 10.1096/fasebj.9.11.7649401. [DOI] [PubMed] [Google Scholar]
- 47.Yadav MK, Leman LJ, Price DJ, Brooks CL, Stout CD, Ghadiri MR. Biochemistry. 2006;45:4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 49.a) Harbury PB, Zhang T, Kim PS, Alber T. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]; b) Harbury PB, Kim PS, Alber T. Nature. 1994;371:80–83. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- 50.Deng Y, Liu J, Zheng Q, Eliezer D, Kallenbach NR, Lu M. Structure. 2006;14:247–255. doi: 10.1016/j.str.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF. J Mol Biol. 2002;321:923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 52.a) The PyMOL Molecular Graphics System, version 1.7.4. Schrödinger, LLC; [Google Scholar]; b) Grigoryan G. DrawCoil. http://www.gri-goryanlab.org/drawcoil.
- 53.Banner DW, Kokkinidis M, Tsernoglou D. J Mol Biol. 1987;196:657–675. doi: 10.1016/0022-2836(87)90039-8. [DOI] [PubMed] [Google Scholar]
- 54.Friedman AM, Fischmann TO, Steitz TA. Science. 1995;268:1721–1727. doi: 10.1126/science.7792597. [DOI] [PubMed] [Google Scholar]
- 55.Butterfoss GL, Kuhlman B. Annu Rev Biophys Biomol Struct. 2006;35:49–65. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- 56.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 57.Handel TM, Williams SA, DeGrado WF. Science. 1993;261:879. doi: 10.1126/science.8346440. [DOI] [PubMed] [Google Scholar]
- 58.Betz SF, Raleigh DP, DeGrado WF. Curr Opin Struct Biol. 1993;3:601–610. [Google Scholar]
- 59.Kuwajima K. Proteins Struct, Funct, Bioinf. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- 60.Ptitsyn OB, Pain RH, Semisotnov GV, Zerovnik E, Razgulyaev OI. FEBS Lett. 1990;262:20–24. doi: 10.1016/0014-5793(90)80143-7. [DOI] [PubMed] [Google Scholar]
- 61.Hill RB, Raleigh DP, Lombardi A, DeGrado WF. Acc Chem Res. 2000;33:745–754. doi: 10.1021/ar970004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor WR. Nature. 2002;416:657–660. doi: 10.1038/416657a. [DOI] [PubMed] [Google Scholar]
- 64.Helling R, Li H, Mélin R, Miller J, Wingreen N, Zeng C, Tang C. J Mol Graph Model. 2001;19:157–167. doi: 10.1016/s1093-3263(00)00137-6. [DOI] [PubMed] [Google Scholar]
- 65.Levitt M. Proc Natl Acad Sci USA. 2009;106:11079–11084. doi: 10.1073/pnas.0905029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grant A, Lee D, Orengo C. Genome Biol. 2004;5:107. doi: 10.1186/gb-2004-5-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Grigoryan G. Methods Enzymol. Elsevier; 2013. pp. 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moll M, Bryant DH, Kavraki LE. BMC Bioinformatics. 2010;11:555. doi: 10.1186/1471-2105-11-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 70.Johansson MU, Zoete V, Michielin O, Guex N. BMC Bioinformatics. 2012;13:173. doi: 10.1186/1471-2105-13-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johansson MU, Zoete V, Guex N. Int J Mol Sci. 2013;14:7795–7814. doi: 10.3390/ijms14047795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gonzalez G, Hannigan B, DeGrado WF. PLoS Comput Biol. 2014;10:e1003750. doi: 10.1371/journal.pcbi.1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shirvanyants D, Alexandrova AN, Dokholyan NV. Bioinformatics. 2011;27:1327–1329. doi: 10.1093/bioinformatics/btr129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adams PD, Baker D, Brunger AT, Das R, DiMaio F, Read RJ, Richardson DC, Richardson JS, Terwilliger TC. Annu Rev Biophys Bioeng. 2013;42:265–287. doi: 10.1146/annurev-biophys-083012-130253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Offredi F, Dubail F, Kischel P, Sarinski K, Stern AS, Van de Weerdt C, Hoch JC, Prosperi C, François JM, Mayo SL, Martial JA. J Mol Biol. 2003;325:163–174. doi: 10.1016/s0022-2836(02)01206-8. [DOI] [PubMed] [Google Scholar]
- 77.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, De Grado WF. Proc Natl Acad Sci USA. 2000;97:6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, Degrado WF. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lanci CJ, MacDermaid CM, Kang S, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG. Proc Natl Acad Sci USA. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Azoitei ML, Correia BE, Ban YEA, Carrico C, Kalyuzhniy O, Chen L, Schroeter A, Huang PS, McLellan JS, Kwong PD, Baker D, Strong RK, Schief WR. Science. 2011;334:373–376. doi: 10.1126/science.1209368. [DOI] [PubMed] [Google Scholar]
- 81.Bonet J, Segura J, Planas-Iglesias J, Oliva B, Fernandez-Fuentes N. Bioinformatics. 2014;30:1935–1936. doi: 10.1093/bioinformatics/btu129. [DOI] [PubMed] [Google Scholar]
- 82.Murphy GS, Sathyamoorthy B, Der BS, Machius MC, Pulavarti SV, Szyperski T, Kuhlman B. Protein Sci. 2015;24:434–445. doi: 10.1002/pro.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacDonald JT, Maksimiak K, Sadowski MI, Taylor WR. Proteins Struct, Funct, Bioinf. 2010;78:1311–1325. doi: 10.1002/prot.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grigoryan G, DeGrado WF. J Mol Biol. 2011;405:1079–1100. doi: 10.1016/j.jmb.2010.08.058. www.grigoryanlab.org/cccp/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Z, Yang Y, Zhan J, Dai L, Zhou Y. Annu Rev Biophys Bioeng. 2013;42:315–335. doi: 10.1146/annurev-biophys-083012-130315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Desmet J, Maeyer MD, Hazes B, Lasters I. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 87.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 88.Kono H, Saven JG. J Mol Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 89.Mitra P, Shultis D, Zhang Y. Nucleic Acids Res. 2013;41:W273–W280. doi: 10.1093/nar/gkt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Floudas CA, Fung HK, McAllister SR, Mönnigmann M, Rajgaria R. Chem Eng Sci. 2006;61:966–988. [Google Scholar]
- 91.Kuhlman B, Baker D. Proc Natl Acad Sci USA. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kraemer-Pecore CM, Lecomte JTJ, Desjarlais JR. Protein Sci. 2003;12:2194–2205. doi: 10.1110/ps.03190903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Larson SM, England JL, Desjarlais JR, Pande VS. Protein Sci. 2002;11:2804–2813. doi: 10.1110/ps.0203902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Havranek JJ, Harbury PB. Nat Struct Mol Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 95.Shifman JM, Mayo SL. Proc Natl Acad Sci USA. 2003;100:13274–13279. doi: 10.1073/pnas.2234277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hu X, Wang H, Ke H, Kuhlman B. Structure. 2008;16:1799–1805. doi: 10.1016/j.str.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fletcher JM, Boyle AL, Bruning M, Bartlett GJ, Vincent TL, Zaccai NR, Armstrong CT, Bromley EHC, Booth PJ, Brady RL, Thomson AR, Woolfson DN. ACS Synth Biol. 2012;1:240–250. doi: 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- 98.Thomas F, Boyle AL, Burton AJ, Woolfson DN. J Am Chem Soc. 2013;135:5161–5166. doi: 10.1021/ja312310g. [DOI] [PubMed] [Google Scholar]
- 99.Bloomer AC, Champness JN, Bricogne G, Staden R, Klug A. Nature. 1978;276:362–368. doi: 10.1038/276362a0. [DOI] [PubMed] [Google Scholar]
- 100.Stubbs G, Warren S, Holmes K. Nature. 1977;267:216–221. doi: 10.1038/267216a0. [DOI] [PubMed] [Google Scholar]
- 101.De Vos AM, Ultsch M, Kossiakoff AA. Science. 1992;255:306–312. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 102.Rozwarski DA, Gronenborn AM, Clore GM, Bazan JF, Bohm A, Wlodawer A, Hatada M, Karplus PA. Structure. 1994;2:159–173. doi: 10.1016/s0969-2126(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 103.Weber PC, Bartsch RG, Cusanovich MA, Hamlin RC, Howard A, Jordan SR, Kamen MD, Meyer TE, Weatherford DW, Xuong NH, Salemme FR. Nature. 1980;286:302–304. doi: 10.1038/286302a0. [DOI] [PubMed] [Google Scholar]
- 104.Mathews FS, Bethge PH, Czerwinski EW. J Biol Chem. 1979;254:1699–1706. [PubMed] [Google Scholar]
- 105.Kurtz DM., Jr JBIC J Biol Inorg Chem. 1997;2:159–167. [Google Scholar]
- 106.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 107.Feig AL, Lippard SJ. Chem Rev. 1994;94:759–805. [Google Scholar]
- 108.Nordlund P, Sjöberg BM, Eklund H. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 109.Lange SJ, Que L., Jr Curr Opin Chem Biol. 1998;2:159–172. doi: 10.1016/s1367-5931(98)80057-4. [DOI] [PubMed] [Google Scholar]
- 110.Andersson KK, Gräslund A. Adv Inorg Chem. 1995;43:359–408. [Google Scholar]
- 111.Harrison PM, Arosio P. Biochim Biophys Acta BBA-Bioenerg. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 112.Hempstead PD, Hudson AJ, Artymiuk PJ, Andrews SC, Banfield MJ, Guest JR, Harrison PM. FEBS Lett. 1994;350:258–262. doi: 10.1016/0014-5793(94)00781-0. [DOI] [PubMed] [Google Scholar]
- 113.Frolow F, Kalb AJ, Yariv J. Nat Struct Biol. 1994;1:453–460. doi: 10.1038/nsb0794-453. [DOI] [PubMed] [Google Scholar]
- 114.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 115.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 116.Coleman NV, Bui NB, Holmes AJ. Environ Microbiol. 2006;8:1228–1239. doi: 10.1111/j.1462-2920.2006.01015.x. [DOI] [PubMed] [Google Scholar]
- 117.Stenkamp RE. Chem Rev. 1994;94:715–726. [Google Scholar]
- 118.Nordlund P, Eklund H. J Mol Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 119.Stubbe J, van der Donk WA. Chem Biol. 1995;2:793–801. doi: 10.1016/1074-5521(95)90084-5. [DOI] [PubMed] [Google Scholar]
- 120.Logan DT, Su XD, Åberg A, Regnström K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 121.Lindqvist Y, Huang W, Schneider G, Shanklin J. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 122.Fox BG, Shanklin J, Ai J, Loehr TM, Sanders-Loehr J. Biochemistry. 1994;33:12776–12786. doi: 10.1021/bi00209a008. [DOI] [PubMed] [Google Scholar]
- 123.Zocher G, Winkler R, Hertweck C, Schulz GE. J Mol Biol. 2007;373:65–74. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 124.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci USA. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chanco E, Choi YS, Sun N, Vu M, Zhao H. Bioorg Med Chem. 2014;22:5569–5577. doi: 10.1016/j.bmc.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 126.Berthold DA, Andersson ME, Nordlund P. Biochim Biophys Acta. 2000;1460:241–254. doi: 10.1016/s0005-2728(00)00149-3. [DOI] [PubMed] [Google Scholar]
- 127.Shiba T, Kido Y, Sakamoto K, Inaoka DK, Tsuge C, Tatsumi R, Takahashi G, Balogun EO, Nara T, Aoki T, Honma T, Tanaka A, Inoue M, Matsuoka S, Saimoto H, Moore AL, Harada S, Kita K. Proc Natl Acad Sci USA. 2013;110:4580–4585. doi: 10.1073/pnas.1218386110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gomes CM, Le Gall J, Xavier AV, Teixeira M. Chem-BioChem. 2001;2:583–587. doi: 10.1002/1439-7633(20010803)2:7/8<583::AID-CBIC583>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 129.Stubbe J, Riggs-Gelasco P. Trends Biochem Sci. 1998;23:438–443. doi: 10.1016/s0968-0004(98)01296-1. [DOI] [PubMed] [Google Scholar]
- 130.Jin Y, Lipscomb JD. J Biol Inorg Chem. 2001;6:717–725. doi: 10.1007/s007750100250. [DOI] [PubMed] [Google Scholar]
- 131.Gibson KJ. Biochim Biophys Acta BBA-Lipids Lipid Me-tab. 1993;1169:231–235. doi: 10.1016/0005-2760(93)90245-5. [DOI] [PubMed] [Google Scholar]
- 132.Simurdiak M, Lee J, Zhao H. ChemBioChem. 2006;7:1169–1172. doi: 10.1002/cbic.200600136. [DOI] [PubMed] [Google Scholar]
- 133.Krebs C, Matthews ML, Jiang W, Bollinger JM. Biochemistry. 2007;46:10413–10418. doi: 10.1021/bi701060g. [DOI] [PubMed] [Google Scholar]
- 134.Holmes MA, Le Trong I, Turley S, Sieker LC, Stenkamp RE. J Mol Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 135.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew Chem Int Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2001;113:2860. [Google Scholar]
- 136.Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM. Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 137.Yang X, Chen-Barrett Y, Arosio P, Chasteen ND. Biochemistry. 1998;37:9743–9750. doi: 10.1021/bi973128a. [DOI] [PubMed] [Google Scholar]
- 138.Tinberg CE, Lippard SJ. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sazinsky MH, Lippard SJ. Acc Chem Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 140.Maglio O, Nastri F, Martín de Rosales RT, Faiella M, Pavone V, DeGrado WF, Lombardi A. C R Chim. 2007;10:703–720. [Google Scholar]
- 141.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Pept Sci. 2005;80:264–278. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 142.Geremia S, Di Costanzo L, Randaccio L, Engel DE, Lombardi A, Nastri F, DeGrado WF. J Am Chem Soc. 2005;127:17266–17276. doi: 10.1021/ja054199x. [DOI] [PubMed] [Google Scholar]
- 143.Maglio O, Nastri F, Pavone V, Lombardi A, De-Grado WF. Proc Natl Acad Sci USA. 2003;100:3772–3777. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Di Costanzo L, Wade H, Geremia S, Randaccio L, Pavone V, De Grado WF, Lombardi A. Am Chem Soc. 2001;123:12749–12757. doi: 10.1021/ja010506x. [DOI] [PubMed] [Google Scholar]
- 145.DeGrado WF, Di Costanzo L, Geremia S, Lombardi A, Pavone V, Randaccio L. Angew Chem Int Ed. 2003;42:417–420. doi: 10.1002/anie.200390127. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2003;115:433. [Google Scholar]
- 146.DeRose VJ, Liu KE, Lippard SJ, Hoffman BM. J Am Chem Soc. 1996;118:121–134. [Google Scholar]
- 147.Rosenzweig AC, Brandstetter H, Whittington DA, Nordlund P, Lippard SJ, Frederick CA. Proteins. 1997;29:141–152. [PubMed] [Google Scholar]
- 148.Andersson KK, Elgren TE, Que L, Jr, Lipscomb JD. J Am Chem Soc. 1992;114:8711–8713. [Google Scholar]
- 149.Que L. Coord Chem Rev. 1983;50:73–108. [Google Scholar]
- 150.Khangulov SV, Goldfeld MG, Gerasimenko VV, Andreeva NE, Barynin VV, Grebenko AI. J Inorg Biochem. 1990;40:279–292. [Google Scholar]
- 151.Garbett K, Darnall DW, Klotz IM, Williams RJP. Arch Biochem Biophys. 1969;135:419–434. doi: 10.1016/0003-9861(69)90559-1. [DOI] [PubMed] [Google Scholar]
- 152.Reem RC, McCormick JM, Richardson DE, Devlin FJ, Stephens PJ, Musselman RL, Solomon EI. J Am Chem Soc. 1989;111:4688–4704. [Google Scholar]
- 153.Dunn JB, Addison AW, Bruce RE, Loehr JS, Loehr TM. Biochemistry. 1977;16:1743–1749. doi: 10.1021/bi00627a035. [DOI] [PubMed] [Google Scholar]
- 154.Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]
- 155.Tolman WB, Liu S, Bentsen JG, Lippard SJ. J Am Chem Soc. 1991;113:152–164. [Google Scholar]
- 156.Kurtz DM., Jr Chem Rev. 1990;90:585–606. [Google Scholar]
- 157.Ai J, Broadwater JA, Loehr TM, Sanders-Loehr J, Fox BG. JBIC J Biol Inorg Chem. 1997;2:37–45. [Google Scholar]
- 158.Papoian GA, DeGrado WF, Klein ML. J Am Chem Soc. 2003;125:560–569. doi: 10.1021/ja028161l. [DOI] [PubMed] [Google Scholar]
- 159.Magistrato A, DeGrado WF, Laio A, Rothlisberger U, VandeVondele J, Klein ML. J Phys Chem B. 2003;107:4182–4188. [Google Scholar]
- 160.Atta M, Nordlund P, Aberg A, Eklund H, Fontecave M. J Biol Chem. 1992;267:20682–20688. [PubMed] [Google Scholar]
- 161.Law NA, Caudle MT, Pecoraro VL. Adv Inorg Chem. 1998;46:305–440. [Google Scholar]
- 162.Pasternak A, Kaplan J, Lear JD, Degrado WF. Protein Sci. 2001;10:958–969. doi: 10.1110/ps.52101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lahr SJ, Engel DE, Stayrook SE, Maglio O, North B, Geremia S, Lombardi A, Degrado WF. J Mol Biol. 2005;346:1441–1454. doi: 10.1016/j.jmb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 164.Maglio O, Nastri F, Calhoun JR, Lahr S, Wade H, Pavone V, Degrado WF, Lombardi A. JBIC J Biol Inorg Chem. 2005;10:539–549. doi: 10.1007/s00775-005-0002-8. [DOI] [PubMed] [Google Scholar]
- 165.Wade H, Stayrook SE, DeGrado WF. Angew Chem Int Ed. 2006;45:4951–4954. doi: 10.1002/anie.200600042. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:5073. [Google Scholar]
- 166.Marsh ENG, DeGrado WF. Proc Natl Acad Sci USA. 2002;99:5150–5154. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Corbett JF. J Chem Soc B. 1969:823–826. [Google Scholar]
- 168.Corbett JF, Gamson EP. J Chem Soc Perkin Trans. 1972;2:1531–1537. [Google Scholar]
- 169.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 170.Calhoun JR, Bell CB, Smith TJ, Thamann TJ, DeGrado WF, Solomon EI. J Am Chem Soc. 2008;130:9188–9189. doi: 10.1021/ja801657y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 172.Torres Martin de Rosales R, Faiella M, Farquhar E, Que L, Andreozzi C, Pavone V, Maglio O, Nastri F, Lombardi A. JBIC J Biol Inorg Chem. 2010;15:717–728. doi: 10.1007/s00775-010-0639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Josse EM, Alcaraz JP, Labouré AM, Kuntz M. Eur J Biochem. 2003;270:3787–3794. doi: 10.1046/j.1432-1033.2003.03766.x. [DOI] [PubMed] [Google Scholar]
- 174.Chino M, Leone L, Liguori D, Maglio O, Nastri F, Pavone V, Lombardi A. unpublished results, manuscript in preparation [Google Scholar]