

Abstract
The robustness and biocompatibility of bacterial nanocages holds promise for bio‐nanotechnologies. The propensity of these nano‐carriers to penetrate cells has been demonstrated, which calls for the development of tracking strategies, both in vitro and in vivo. Here, we label bacterial nanocages with photo‐switchable fluorophores, to facilitate their imaging by super‐resolution microscopy. We demonstrate the functionalization of the encapsulin from Brevibacterium linens with a spiropyran, which is not fluorescent, by covalent attachment to the amine residues at the outer encapsulin shell. Upon alternating irradiation with ultraviolet and visible light, the spiropyran switches forth and back to its fluorescent merocyanine photo‐isomer and thus the fluorescence can be switched on and off, reversibly. We also show that the bacterial compartments preserve their structural integrity upon covalent modification and over at least five irradiation cycles.
Keywords: molecular switches, photochromism, protein cages, self-assembly, spiropyrans
Encapsulins are protein‐based nanocages that are found in bacteria such as Brevibacterium linens and hyperthermophilic Thermotoga maritima.1, 2, 3, 4 They are composed of 60 identical proteins that self‐assemble into cages, with a diameter ranging between 20 and 24 nm (Figure 1). Their structure has been compared to T=1 viral protein cages in terms of dimension and symmetry.3, 5 However, while viruses encapsulate genetic material, encapsulins enclose smaller proteins in their inner cavity, for instance a dye‐decolorizing peroxidase in B. linens encapsulins and a ferritin‐like protein in T. maritima encapsulins.1, 2 Encapsulins have emerged as promising building blocks for applications in nanotechnology in view of their robustness and their biocompatibility, for example as compartments for functional proteins and as drug delivery agents.1, 6, 7 Moreover, encapsulins are robust with respect to pH and temperature,1, 4 and their symmetrical protein‐based structures allows modifying them as desired, genetically or chemically. Due to the symmetrical structures, a modification introduced within a protein unit is symmetrically distributed over the entire structure.5
Figure 1.
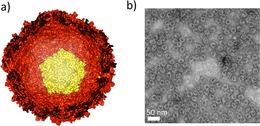
The cage‐like structure of encapsulins: a) Representation of an encapsulin nanocage made up by 12 pentamers of identical protein subunits (a pentamer is depicted in yellow). The encapsulin structure presented here is from T. maritima 2 (PDB: 3DKT) as the crystal structure for B. linens encapsulin is not yet solved. b) Image of recombinant B. linens encapsulin particles before any modification, obtained with transmission electron microscopy.
In comparison to their viral cage counterparts,5, 8 encapsulins are characterized by their robustness, which allows more freedom in introducing chemical modifications, without compromising the stability of the cage‐like structure.9 It has been proven that encapsulins can penetrate cells while carrying functional molecules,6 but further understanding of their in vivo behavior calls for the development of non‐invasive imaging and tracking strategies. Overcoming the diffraction barrier for microscopy can be achieved in live cells by using super‐resolution fluorescence imaging,10, 11, 12, 13, 14, 15, 16 by photo‐switching fluorescence on and off to statistically turn on a small fraction of fluorophores only.14, 15, 16, 17, 18, 19 The mapping of fluorescent points is achieved by activating only a subset of fluorophores stochastically at any given time while the rest are switched off.15 Super‐resolution images are then reconstructed from multiple time‐resolved fluorescence images at different areas. Moreover, the possibility to switch fluorescence on or off might reveal useful to avoid overlapping signals in the presence of various labeled structures. With this in mind, we set out to label bacterial nanocages with photo‐switchable fluorophores.
Natural fluorophores, such as the green fluorescent protein, show an on/off blinking behavior under examination at the single‐molecule level, and thus, they have been used as fluorescent probes in live‐cell imaging.20, 21 However, despite their biocompatibility, they suffer drawbacks compared to synthetic fluorophores, which are often brighter than their biological counterparts,10 allow chemical modifications on surfaces22, 23 and are less likely to interfere with protein folding because they are smaller than fluorescent proteins.18 Consequently, synthetic and photo‐switchable fluorescent probes have emerged as powerful tools for in vivo imaging,21 including diarylethenes24 and spiropyran derivatives.25 However, their moderate brightness remains an issue, which is why multiple fluorophores are often grafted at the surface of the same object.22, 26 Here, we graft multiple spiropyran switches on the surface of the encapsulin. The spiropyran switches can be converted reversibly into their fluorescent photo‐isomer upon irradiation with light, thus allowing on/off control over fluorescence.27, 28, 29, 30, 31, 32, 33, 34 In addition, the fluorescent isomer of spiropyran provides sufficient brightness for biological applications in live‐cell imaging.10, 25, 35
B. linens encapsulin is produced recombinantly in E. coli (Figure S1 of the Supporting Information, SI). B. linens encapsulin counts 240 lysine residues that are accessible on its surface, which accounts for four lysines per monomer, the amine groups of which are used as active sites for chemical modification.
A spiropyran was functionalized with a succinimide moiety (Scheme 1 a and Figure S2), which reacts with the amine groups of lysines to form an amide. All samples were kept in a neutral solution (the pH of the PBS buffer was pH 7.4) to avoid the spontaneous conversion of spiropyran into merocyanine, which occurs under acidic conditions.28 The spiropyran (closed, off‐state) is converted to the colored merocyanine form (open, on‐state) upon irradiation with UV light. This conversion is reversible upon subsequent irradiation with visible light (Scheme 1b). The merocyanine also undergoes a slow thermal relaxation into spiropyran in the dark, although the open (charged) form is more favored in a polar environment.28
Scheme 1.
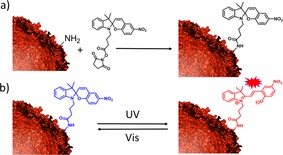
a) Coupling of encapsulin and spiropyran via amine‐succinimide reaction. b) Photo‐isomerization of non‐fluorescent spiropyran (blue) to fluorescent merocyanine (red).
Coupling of the encapsulin nanocages to spiropyran is achieved by adding a 1000‐times excess of spiropyran to a purified encapsulin solution containing 10 % DMSO. After overnight incubation, the non‐reacted spiropyran and DMSO are removed by dialysis and size‐exclusion chromatography. In this process, non‐covalently bound spiropyran molecules are removed by the combination of overnight dialysis and 48‐times dilution throughout the size‐exclusion chromatography column (a buffered saline is used as the eluent to avoid electrostatic interactions).36 The chromatogram reveals a protein peak at around V=12 mL which is characteristic for encapsulin particles (Figure 2 a).
Figure 2.
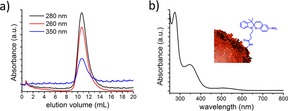
Labelling encapsulin with a spiropyran photo‐switch: a) Size‐exclusion chromatogram showing the characteristic encapsulin elution at V=12 mL as well as the characteristic spiropyran absorption at λ=350 nm at the corresponding elution volume. b) UV/Vis spectrum of encapsulin labelled with spiropyran switches.
Because of their size, which ranges between 20–24 nm, encapsulins are known to elute at volumes around V=12 mL, a fraction in which their presence can be detected by monitoring UV/Vis absorption at λ≈280 nm, that is, a range where the proteins absorb light (Figure S1a). When the encapsulins are modified with spiropyran switches, the outcome of the size‐exclusion chromatography is monitored at λ=350 nm also, in order to determine at which elution volume the spiropyran appears (Figure 2 a). We note that at λ=350 nm, only the spiropyran absorbs light, and the encapsulin does not (Figure S1b). Moreover, grafting spiropyrans on the encapsulins is not expected to modify the size of these protein cages significantly, and indeed the hybrid system still elutes at V=12 mL as demonstrated by that fact that that fraction shows absorption at λ=280 nm (Figure 2 a). Importantly, the same fraction also shows absorption at λ=350 nm, which indicates that the spiropyran is covalently bound to the encapsulin. The presence of spiropyrans is further confirmed by UV/Vis spectroscopy (Figure 2 b). The small absorption band observed at around λ=520 nm prior to any irradiation (Figure 2 b) is likely due to a small degree of spontaneous conversion from the spiropyran form to the merocyanine form, which occurs in aqueous environment despite keeping the pH at 7.4. From the absorption spectra and gel densitometry, we estimate that 116 lysines of the encapsulin surface are modified with spiropyran molecules (see Figure S1, SI).
We further examine the photo‐isomerization of the labeled encapsulin in PBS (pH 7.4) by irradiating it for 2 min with UV light (λ=365 nm) in solution. The photo‐isomerization of spiropyran into merocyanine is indicated by the appearance of an absorption band around λ=540 nm (Figure 3 a). This conversion is complete after 2 min of irradiation and prolonged irradiation does not significantly increase the merocyanine concentration. We also observe the appearance of merocyanine fluorescence (Figure 3 c) at λ=615 nm upon excitation at λ=535 nm. Excitation with λ=535 nm light for fluorescence measurement is not likely to significantly convert merocyanine back into spiropyran because it occurs within less than a minute, while the conversion back to the spiropyran form requires 7 min of continuous irradiation with visible light at 46 mW cm−2.
Figure 3.

Photo‐triggered on/off fluorescence of spiropyran‐labelled encapsulin: a) UV/Vis spectra showing the characteristic absorption band of merocyanine at λ=540 nm. b) Reversible photo‐switching of spiropyran to merocyanine monitored at λ=540 nm. c) Emission spectra showing the fluorescence of merocyanine at λ=615 nm upon excitation at λ=535 nm. d) Reversible conversion of on‐state merocyanine to off‐state spiropyran monitored at λ=615 nm.
We irradiate the encapsulin–merocyanine system with visible light (λ≥420 nm) to examine whether photo‐switching is reversible. The disappearance of the absorption band at λ=550 nm after 7 min of continuous irradiation indicates that the merocyanine form is completely reverted to the spiropyran form (Figure 3 a). The merocyanine form also undergoes thermal relaxation into the spiropyran form in the dark, which can be observed by the disappearance of its absorption band at λ=550 nm (Figure S3). However, this process is much slower in comparison to the conversion back with continuous visible light irradiation.
Similarly, we observe a decrease of the fluorescence of merocyanine at λ=615 nm after irradiation with visible light (Figure 3 c). We repeat the ultraviolet/visible light‐irradiation cycle up to five times. After the fifth cycle, the merocyanine displays fluorescence and absorbance that both amount to half their initial values. The alternating decrease and increase of the fluorescence during the cycles confirms the photo‐switching of the fluorophore (Figure 3 d).
Furthermore, we characterize the hydrodynamic size of the particles before and after irradiation by dynamic light scattering (DLS). DLS reveals a diameter of about 20 nm, which indicates that the functionalized encapsulins remain intact after irradiation with both UV and visible light (Figure 4). Transmission electron microscopy indicates that the structural integrity of the particles is preserved after irradiation (Figure S4). However, structures with sizes exceeding 100 nm are also observed after the fifth cycle (Figure S5), which are likely aggregates forming upon the protein degradation that is caused by prolonged irradiation.37
Figure 4.
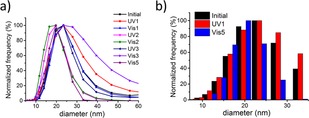
The effect of irradiation on modified encapsulin measured by DLS. a) Size distribution of encapsulin particles throughout the alternating irradiation with UV and visible light peaking at D=20–25 nm. b) Comparison of size distribution before any irradiation (black), after UV irradiation of the first cycle (red) and after visible irradiation of the fifth cycle (blue) showing that the structural integrity of the particles is preserved throughout the cycles.
In conclusion, we have demonstrated a strategy to label encapsulin nanocages isolated from Brevibacterium linens with a spiropyran photo‐switch. The fluorescence of the hybrid nano‐compartment resulting from this operation can be photo‐switched on‐command for multiple cycles. The encapsulin particles retain their structural integrity, both upon covalent modification with the dye and upon light irradiation. These results provide opportunities towards tracking protein‐based nanocages in live cells.
Experimental Section
Materials
The encapsulin was expressed and purified according to literature procedures.1, 2 All experiments were performed at pH 7.4 in a phosphate buffered saline (0.01 m PBS, Sigma–Aldrich). Chemicals were purchased from Sigma–Aldrich unless stated otherwise.
Synthesis of the Encapsulin–Siropyran Hybrid
A 1000‐fold excess of nitro‐spiropyran bearing a succinimide was added to an encapsulin solution (in 0.01 m PBS pH 7.4 containing 10 % DMSO) at room temperature and incubated overnight. The non‐reacted spiropyran was removed by size‐exclusion chromatography (preparative column Superose 6 10/100 GL, GE Healthcare FPLC Äkta purifier 900 with a 24 mL bed volume). Prior to injecting the samples into the column, all samples were dialyzed against PBS to remove the DMSO using 12–14 kDa dialysis membranes (Spectra/Por).
Photo‐switchable Fluorescence Studies
500 μL of a solution containing the encapsulin–spiropyran hybrid was irradiated with UV light (λ=365 nm, bluepoint LED Honle Technology, 40 mW cm−2) for 2 min and subsequently with visible light (λ≥420 nm, Edmund MI‐150 High‐intensity Illuminator) for 7 min to complete a cycle of irradiation. After each irradiation, the absorption spectrum was recorded with a PerkinElmer Lambda 850 UV/Vis spectrometer and the emission spectrum at λ=555–670 nm (excitation at 535 nm) was recorded using a PerkinElmer fluorescence spectrometer.
Verification of Structural Integrity
The hydrodynamic size of the encapsulins before and after irradiation in 0.01 mm PBS (pH 7.4) was determined by dynamic light scattering (Nanotrac Wave, Microtrac).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by the European Research Council (Starting Grant Phelix 307784) and the Netherlands Organization for Scientific Research (Vidi grant 700.10.423). R.M.P. acknowledges Indonesia Endowment Fund for Education (LPDP) for funding her doctoral studies. We are grateful to Prof. Nenad Ban, Dr. Markus Sutter and Dr. Rik Rurup for the donation of plasmids and we thank Federico Lancia for help with characterizing the photo‐switch and Dr. Rico Keim for help with the TEM analysis.
R. M. Putri, J. W. Fredy, J. J. L. M. Cornelissen, M. S. T. Koay, N. Katsonis, ChemPhysChem 2016, 17, 1815.
References
- 1. Rurup W. F., Snijder J., Koay M. S. T., Heck A. J. R., Cornelissen J. J. L. M., J. Am. Chem. Soc. 2014, 136, 3828–3832. [DOI] [PubMed] [Google Scholar]
- 2. Sutter M., Boehringer D., Gutmann S., Guenther S., Prangishvili D., Loessner M. J., Stetter K. O., Weber-Ban E., Ban N., Nat. Struct. Mol. Biol. 2008, 15, 939–947. [DOI] [PubMed] [Google Scholar]
- 3. McHugh C. A., Fontana J., Nemecek D., Cheng N. Q., Aksyuk A. A., Heymann J. B., Winkler D. C., Lam A. S., Wall J. S., Steven A. C., Hoiczyk E., EMBO J. 2014, 33, 1896–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rahmanpour R., Bugg T. D. H., FEBS J. 2013, 280, 2097–2104. [DOI] [PubMed] [Google Scholar]
- 5. Putri R. M., Cornelissen J. J. L. M., Koay M. S. T., ChemPhysChem 2015, 16, 911–918. [DOI] [PubMed] [Google Scholar]
- 6. Moon H., Lee J., Min J., Kang S., Biomacromolecules 2014, 15, 3794–3801. [DOI] [PubMed] [Google Scholar]
- 7. Tamura A., Fukutani Y., Takami T., Fujii M., Nakaguchi Y., Murakami Y., Noguchi K., Yohda M., Odaka M., Biotechnol. Bioeng. 2015, 112, 13–20. [DOI] [PubMed] [Google Scholar]
- 8. Lavelle L., Michel J. P., Gingery M., J. Virol. Methods 2007, 146, 311–316. [DOI] [PubMed] [Google Scholar]
- 9. Hommersom C. A., Matt B., van der Ham A., Cornelissen J. J. L. M., Katsonis N., Org. Biomol. Chem. 2014, 12, 4065–4069. [DOI] [PubMed] [Google Scholar]
- 10. MacNevin C. J., Gremyachinskiy D., Hsu C. W., Li L., Rougie M., Davis T. T., Hahn K. M., Bioconjugate Chem. 2013, 24, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van de Linde S., Heilemann M., Sauer M., Annu. Rev. Phys. Chem. 2012, 63, 519–540. [DOI] [PubMed] [Google Scholar]
- 12. Uno S. N., Kamiya M., Yoshihara T., Sugawara K., Okabe K., Tarhan M. C., Fujita H., Funatsu T., Okada Y., Tobita S., Urano Y., Nat. Chem. 2014, 6, 681–689. [DOI] [PubMed] [Google Scholar]
- 13. Hell S. W., Wichmann J., Opt. Lett. 1994, 19, 780–782. [DOI] [PubMed] [Google Scholar]
- 14. Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Science 2006, 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- 15. Rust M. J., Bates M., Zhuang X. W., Nat. Methods 2006, 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hess S. T., Girirajan T. P. K., Mason M. D., Biophys. J. 2006, 91, 4258–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bock H., Geisler C., Wurm C. A., Von Middendorff C., Jakobs S., Schonle A., Egner A., Hell S. W., Eggeling C., Appl. Phys. B 2007, 88, 161–165. [Google Scholar]
- 18. Hirabayashi K., Hanaoka K., Shimonishi M., Terai T., Komatsu T., Ueno T., Nagano T., Chem. Eur. J. 2011, 17, 14763–14771. [DOI] [PubMed] [Google Scholar]
- 19. Hell S. W., Science 2007, 316, 1153–1158. [DOI] [PubMed] [Google Scholar]
- 20. Dickson R. M., Cubitt A. B., Tsien R. Y., Moerner W. E., Nature 1997, 388, 355–358. [DOI] [PubMed] [Google Scholar]
- 21. Fernandez-Suarez M., Ting A. Y., Nat. Rev. Mol. Cell Biol. 2008, 9, 929–943. [DOI] [PubMed] [Google Scholar]
- 22.T. Udayabhaskararao, P. K. Kundu, J. Ahrens, R. Klajn, ChemPhysChem 2015, DOI: 10.1002/cphc.201500897. [DOI] [PubMed]
- 23. Katsonis N., Lubomska M., Pollard M. M., Feringa B. L., Rudolf P., Prog. Surf. Sci. 2007, 82, 407–434. [Google Scholar]
- 24. Zou Y., Yi T., Xiao S. Z., Li F. Y., Li C. Y., Gao X., Wu J. C., Yu M. X., Huang C. H., J. Am. Chem. Soc. 2008, 130, 15750–15751. [DOI] [PubMed] [Google Scholar]
- 25. Wan W., Zhu M. Q., Tian Z. Y., Li A. D. Q., J. Am. Chem. Soc. 2015, 137, 4312–4315. [DOI] [PubMed] [Google Scholar]
- 26. Fihey A., Perrier A., Browne W. R., Jacquemin D., Chem. Soc. Rev. 2015, 44, 3719–3759. [DOI] [PubMed] [Google Scholar]
- 27. Kalisky Y., Orlowski T. E., Williams D. J., J. Phys. Chem. 1983, 87, 5333–5338. [Google Scholar]
- 28. Klajn R., Chem. Soc. Rev. 2014, 43, 148–184. [DOI] [PubMed] [Google Scholar]
- 29. Zhu M. Q., Zhu L., Han J. J., Wu W., Hurst J. K., Li A. D. Q., J. Am. Chem. Soc. 2006, 128, 4303–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szymanski W., Beierle J. M., Kistemaker H. A. V., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178. [DOI] [PubMed] [Google Scholar]
- 31. Tian Z. Y., Li A. D. Q., Acc. Chem. Res. 2013, 46, 269–279. [DOI] [PubMed] [Google Scholar]
- 32. Tian Z. Y., Wu W. W., Wan W., Li A. D. Q., J. Am. Chem. Soc. 2009, 131, 4245–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deniz E., Tomasulo M., DeFazio R. A., Watson B. D., Raymo F. M., Phys. Chem. Chem. Phys. 2010, 12, 11630–11634. [DOI] [PubMed] [Google Scholar]
- 34. Tomasulo M., Deniz E., Alvarado R. J., Raymo F. M., J. Phys. Chem. C 2008, 112, 8038–8045. [Google Scholar]
- 35. Wang S. P., Deng W. J., Sun D., Yan M., Zheng H., Xu J. G., Org. Biomol. Chem. 2009, 7, 4017–4020. [DOI] [PubMed] [Google Scholar]
- 36. Hong P., Koza S., Bouvier E. S. P., J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davies M. J., Biochem. Biophys. Res. Commun. 2003, 305, 761–770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary