

Abstract
The aggregation of alpha synuclein (α‐syn) is a neuropathological feature that defines a spectrum of disorders collectively termed synucleinopathies, and of these, Parkinson's disease (PD) is arguably the best characterized. Aggregated α‐syn is the primary component of Lewy bodies, the defining pathological feature of PD, while mutations or multiplications in the α‐syn gene result in familial PD. The high correlation between α‐syn burden and PD has led to the hypothesis that α‐syn aggregation produces toxicity through a gain‐of‐function mechanism. However, α‐syn has been implicated to function in a diverse range of essential cellular processes such as the regulation of neurotransmission and response to cellular stress. As such, an alternative hypothesis with equal explanatory power is that the aggregation of α‐syn results in toxicity because of a toxic loss of necessary α‐syn function, following sequestration of functional forms α‐syn into insoluble protein aggregates. Within this review, we will provide an overview of the literature linking α‐syn to PD and the knowledge gained from current α‐syn‐based animal models of PD. We will then interpret these data from the viewpoint of the α‐syn loss‐of‐function hypothesis and provide a potential mechanistic model by which loss of α‐syn function could result in at least some of the neurodegeneration observed in PD. By providing an alternative perspective on the etiopathogenesis of PD and synucleinopathies, this may reveal alternative avenues of research in order to identify potential novel therapeutic targets for disease modifying strategies.
The correlation between α‐synuclein burden and Parkinson's disease pathology has led to the hypothesis that α‐synuclein aggregation produces toxicity through a gain‐of‐function mechanism. However, in this review, we discuss data supporting the alternative hypothesis that the aggregation of α‐synuclein results in toxicity because of loss of necessary α‐synuclein function at the presynaptic terminal, following sequestration of functional forms of α‐synuclein into aggregates.
Keywords: alpha‐synuclein, dopamine, Lewy body, Parkinson's disease, substantia nigra, synucleinopathy
Abbreviations used
- AAV
adeno‐associated virus
- CMV
cytomegalovirus
- DAT
dopamine transporter
- MSA
multiple system atrophy
- PFFs
preformed fibrils
- VMAT
vesicular monoamine transporter
Research examining the role of the protein alpha synuclein (α‐syn) in the etiology of Parkinson's disease (PD) began in 1997 when two seminal discoveries provided conclusive evidence that α‐syn is intimately linked to PD pathogenesis. The first report described a missense mutation in the gene encoding α‐syn that causes familial PD (Polymeropoulos et al. 1997). Shortly thereafter, aggregated α‐syn was discovered as one of the primary components of Lewy bodies (Spillantini et al. 1997), the neuropathological hallmark of PD (Goedert et al. 2013). Since these definitive links between α‐syn and PD were identified, research on α‐syn has grown exponentially. Within this vast body of work, a preponderance of research is conducted under the prevailing hypothesis that aberrantly expressed or aggregated α‐syn produces neurotoxicity through a gain‐of‐function mechanism. A less discussed, though equally valid hypothesis, is that the aggregation of α‐syn results in neurotoxicity through a loss‐of‐function mechanism, following sequestration of functional α‐syn protein into aggregates. Indeed, as our understanding of both the biology and pathology of α‐syn expands, so too does the evidence supporting the idea that loss of α‐syn function may be a critical event in PD.
Within this review, we will present a comprehensive overview of the data linking α‐syn to PD, as well as the lessons learned from α‐syn‐based animal models of PD. Next, we will highlight how the existing data linking α‐syn to PD pathology might be explained by toxicity originating from loss of necessary α‐syn function, as well as providing some potential mechanisms by which loss of α‐syn function could cause neurodegeneration. We will then highlight the discrepant reports in the literature that seem irreconcilable with this hypothesis. Finally, we will draw corollaries to other proteins associated with neurodegenerative disease, which were once believed to elicit toxicity directly, but are now accepted as important for neuronal survival.
α‐syn: structure and aggregation
α‐syn is a small protein that, along with β and γ synuclein, comprise the synuclein family of proteins (Surguchov 2008). α‐syn is encoded by the SNCA gene, located on the long arm of chromosome 4 in humans (4q21.3–q22)(Chen et al. 1995). Full‐length α‐syn is a 140 amino acid protein, however, alternative splicing in exons 3 and 5 can result in 126, 112, or 98 amino acid isoforms (Uéda et al. 1994; Beyer et al. 2006). Similar to full‐length α‐syn, the splice variants are expressed in a region‐specific manner throughout the brain, albeit at lower levels than the full‐length protein (McLean et al. 2012). The structure of α‐syn can be subdivided into a basic N‐terminus (amino acids 1–60), a central (‘non‐amyloidogenic component’) hydrophobic core (amino acids 61–95), and an acidic C‐terminal tail (amino acids 96–140) (Surguchov 2008).
The N‐terminus of α‐syn is characterized by seven 11 amino acid repeats containing a KTKEGV consensus sequence (Jakes et al. 1994). This sequence is highly conserved between species and within the synuclein family itself (Surguchov 2008) and predicts an alpha helix secondary structure (Bussell and Eliezer 2003). Confirming these predictions, α‐syn forms either two anti‐parallel alpha helices (Chandra et al. 2003), or one contiguous alpha helix upon interaction with acidic lipid membranes (Davidson et al. 1998; Bussell and Eliezer 2003; Jao et al. 2004). The central portion of α‐syn is highly hydrophobic and is thought to underlie the aggregate prone nature of the protein. Interestingly, the first association between α‐syn and neurodegenerative disease followed the identification of a peptide fragment within plaques isolated from the brains of patients with Alzheimer's Disease (Uéda et al. 1993). This cleaved peptide was eventually mapped to the central hydrophobic core of α‐syn, giving rise to the descriptor ‘non‐amyloidogenic component’, or NAC region, of α‐syn (Uéda et al. 1993). Finally, the C‐terminal domain of α‐syn displays the most variability within the synuclein family and between species (George 2001). The C‐terminus of α‐syn contains a large number of charged residues and is subject to significant post‐translational modification (Hasegawa et al. 2002; Oueslati et al. 2010; Krumova et al. 2011), suggesting that this region may play a role in regulating α‐syn function and conformation. Beyond regulation of α‐syn itself, the C‐terminal tail may also have a functional role in mediating interactions between α‐syn and soluble NSF attachment protein receptor (SNARE) complex proteins (Burré et al. 2010, 2012).
α‐syn is now widely considered to behave as a ‘natively unfolded’ protein, showing dynamic changes in conformation depending on the environment (Weinreb et al. 1996). Natively unfolded, or intrinsically disorder proteins, are sometimes considered a unique class of proteins in themselves, characterized by lack of a uniform tertiary structure (Uversky 2002). Recent work has suggested that natively unfolded proteins, such as α‐syn, require the presence of molecular interacting partners in order take on a specific tertiary conformation (Uversky 2002; Dyson and Wright 2005). α‐syn is highly soluble and intrinsically disordered under normal conditions (Weinreb et al. 1996), however, in the presence of acidic lipid membranes (Davidson et al. 1998; Bussell and Eliezer 2003; Chandra et al. 2003; Jao et al. 2004), or membranes with high curvature (Middleton and Rhoades 2010; Jensen et al. 2011), the N‐terminus of α‐syn folds into an alpha‐helix that interacts with membranes. Thus, it seems as if α‐syn structure may conform to its physiological structure, and thus physiological function, only in the presence of molecular interacting partners. Currently, the list of α‐syn's potential interacting partners is extremely large and growing. α‐syn directly interacts with lipid membranes (Davidson et al. 1998; Fortin et al. 2004), synaptic vesicles (Maroteaux et al. 1988), SNARE complex proteins (Chandra et al. 2005; Woods et al. 2007; Burré et al. 2010), proteins involved in dopamine (DA) homeostasis (Lee et al. 2001a; Dauer et al. 2002; Perez et al. 2002; Wersinger and Sidhu 2003a; Wersinger et al. 2003; Yu et al. 2004; Tehranian et al. 2006; Fountaine and Wade‐Martins 2007; Fountaine et al. 2008), proteins involved in calcium regulation (Martinez et al. 2003), and the catalytic subunit of protein phosphatase 2A (PP2A) (Peng et al. 2005), among others too numerous to list [reviewed in (Kanaan and Manfredsson 2012)].
Beyond interacting with other proteins, α‐syn can also interact with itself to form multimers, and there is evidence that physiological α‐syn exists as a tetramer (Bartels et al. 2011; Dettmer et al. 2013, 2015). Moreover, under certain conditions, α‐syn can self‐assemble, resulting in the formation of β‐pleated sheets (Serpell et al. 2000), followed by the formation of insoluble α‐syn aggregates (Conway et al. 1998). Conditions that promote α‐syn aggregation include genetic mutations (Conway et al. 1998; Narhi et al. 1999; Fredenburg et al. 2007), molecular crowding induced by high concentrations of macromolecules (Shtilerman et al. 2002; Uversky et al. 2002) or increased α‐syn protein levels (Conway et al. 1998; Uversky 2007), post‐translational modifications [reviewed in (Uversky 2007; Stefanis 2012)], low pH (Ahmad et al. 2012), and oxidative conditions (Hashimoto et al. 1999). Under such conditions, the formation of insoluble aggregates proceeds in stereotypic manner following first‐order kinetics. For example, monomeric α‐syn will undertake a partial fold amenable to the formation of oligomeric species (dimers, trimers, etc.) which then progress to protofibrils, and finally to mature, insoluble fibrils (Uversky et al. 2001; Kanaan and Manfredsson 2012). Throughout this series of reactions, each product is more stable than the reactants, suggesting that this process is largely irreversible (Uversky and Eliezer 2009). Importantly, the progressive conversion from soluble monomeric α‐syn to insoluble fibrils proceeds in a feed‐forward mechanism, in which oligomers or protofibrils can act as seeds in order to accelerate the conversion of physiological α‐syn into aggregates (Wood et al. 1999; Luk et al. 2009).
Although the conditions that favor α‐syn aggregation have been characterized, the precise mechanism(s) that promote aggregation are unknown. It may be that simply shifting the structure of α‐syn to an unfolded, or partially folded state is sufficient to result in aggregate formation (Uversky 2007). For example, manipulating the temperature or pH of the environment slows the intramolecular diffusion rate of α‐syn, resulting in a partially folded conformation that increases the propensity to aggregate (Uversky et al. 2001; Ahmad et al. 2012). Mutations in the hydrophobic core of α‐syn that reduce the alpha helical content also exacerbate aggregation (Burré et al. 2012). Further, folding induced by intramolecular interactions between the N‐ and C‐termini are important in maintaining α‐syn in a monomeric state, such that enhancing these interactions can inhibit aggregation (Bertoncini et al. 2005; Koo et al. 2008; Hong et al. 2011), while abolishing them by truncating the C‐terminus exacerbates aggregation (Li et al. 2005; Burré et al. 2012). Thus, inhibiting the folding of α‐syn, or promoting its partial folding, result in increased exposure of the amyloidogenic NAC core of α‐syn, thereby providing a seed for templating and initiating the feed‐forward aggregation process.
α‐syn: localization and function
Consistent with its role in multiple neurodegenerative diseases, α‐syn is predominantly considered a ‘neuronal’ protein. α‐syn is expressed in high concentrations within neural tissues where it primarily localizes to the presynaptic terminal (Iwai et al. 1995) (Fig. 1a). Some analyses estimate that α‐syn may comprise up to 1% of total cytosolic protein in the central nervous system (CNS) (Iwai et al. 1995). The original identification of α‐syn described the presynaptic localization of the protein in the electric organ of the pacific electric ray (Torpedo californica) (Maroteaux et al. 1988). α‐syn was cloned and identified using antisera raised against cholinergic synaptic vesicles isolated from neural tissue within the ray's electric organ. In addition to its strong presynaptic localization, Maroteaux et al., also identified α‐syn on the nuclear envelope, thus accounting for the moniker ‘synuclein’ (SYNapse + NUCLEus, synuclein) (Maroteaux et al. 1988). Despite some nuclear localization (Fig. 1a), α‐syn is primarily a pre‐synaptic protein, with relatively low levels of expression in the somatodendritic compartment or axon (Iwai et al. 1995). Further, although α‐syn is present in some non‐neural tissue, such as red blood cells (Barbour et al. 2008), α‐syn is widely expressed throughout most neuronal tissues within both the central and peripheral nervous system, suggesting that α‐syn plays a role in synaptic transmission in general.
Figure 1.
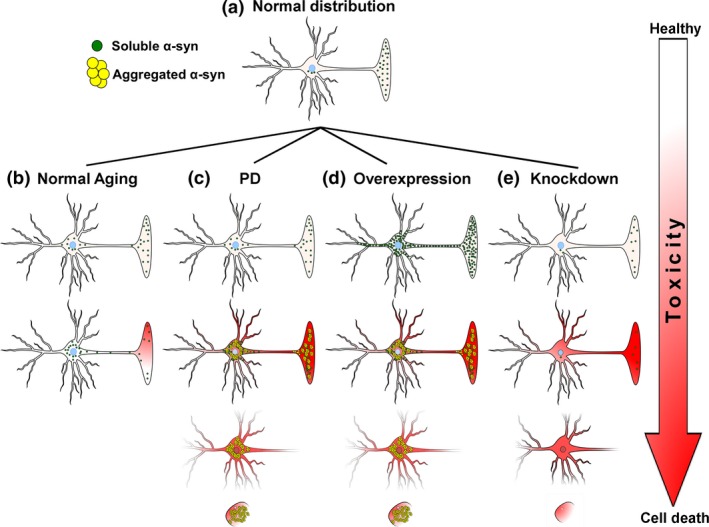
Normal and pathological subcellular distribution of α‐syn. (a) In healthy neurons, α‐syn is highly enriched within the presynaptic terminal. (b) As humans age, there is a distribution of α‐syn from the presynaptic terminal to the soma. This may predispose neurons to subsequent toxicity. (c) In PD, an initial insult (genetic mutation, oxidative stress, multiplications of SNCA gene, etc.,) induces the aggregation of α‐syn, resulting in loss of α‐syn function and subsequent toxicity leading to cell death. (d) Over‐expression of α‐syn results in increased α‐syn protein throughout the entire cell. Molecular crowding induces aggregation of α‐syn resulting in loss of α‐syn function and subsequent toxicity leading to cell death. (e) Knockdown of α‐syn decreases protein concentrations until a critical threshold is reached, below which loss of α‐syn function results in toxicity and cell death.
Currently, the exact biological function of α‐syn remains unclear. However, this is not to imply that there is a paucity of suggested physiological functions for α‐syn. α‐syn has been implicated in diverse physiological processes ranging from regulation of synaptic transmission, to calcium regulation, mitochondrial homeostasis, gene expression, protein phosphorylation, or even fatty acid binding (Sharon et al. 2001; Ellis et al. 2005). The function of α‐syn has been thoroughly reviewed elsewhere [see (Kanaan and Manfredsson 2012; Stefanis 2012)] and an exhaustive description of the potential biological functions of α‐syn is neither the purpose, nor within the scope of the current review. However, some understanding of the potential function(s) of α‐syn is necessary in order to appreciate how a loss of α‐syn function could result in neurotoxicity, and as such will be discussed briefly with a focus on neurotransmission.
The largest and most consistent body of evidence suggests that α‐syn acts as a negative regulator of synaptic transmission (Fig. 2a and b). As previously mentioned, α‐syn is highly enriched in presynaptic terminals throughout the CNS (Iwai et al. 1995). Further, α‐syn interacts with synaptic vesicles and SNARE complex proteins, potentially mediating vesicular trafficking to‐, and docking with, the presynaptic membrane, as well as vesicular endocytosis (Maroteaux et al. 1988; Burré et al. 2010, 2012; Vargas et al. 2014) (Fig. 2a and b). Ectopic expression of α‐syn results in a decrease in synaptic vesicle trafficking and docking, and a corresponding decrease in neurotransmitter release (Larsen et al. 2006; Gaugler et al. 2012; Lundblad et al. 2012; Scott and Roy 2012). In line with these observations, α‐syn null mice show decreased DA stores and increased evoked DA release (Abeliovich et al. 2000; Cabin et al. 2002), as well as decreased synaptic vesicle endocytosis (Vargas et al. 2014). α‐syn may also have a specialized role in the dopaminergic synapse; α‐syn interacts with virtually every major protein involved in DA biosynthesis and handling. α‐syn interacts with and inhibits tyrosine hydroxylase (TH) (Perez et al. 2002; Perez and Hastings 2004; Yu et al. 2004) by directly decreasing TH phosphorylation, by increasing PP2A activity (the major phosphatase regulating TH dephosphorylation), or by altering the binding of 14‐3‐3ξ protein to TH (Peng et al. 2005; Wang et al. 2009; Lou et al. 2010). α‐syn also interacts with, and inhibits, aromatic amino acid decarboxylase (AADC) (Tehranian et al. 2006) (Fig. 2a and b). α‐syn interacts with the dopamine transporter (DAT), increases DAT insertion into the presynaptic membrane, and modulates DAT activity (though there is a discrepancy as to whether α‐syn increases or decreases DAT activity), and increases the amount of vesicular monoamine transporter (VMAT) on vesicles (Lee et al. 2001a; Dauer et al. 2002; Wersinger and Sidhu 2003b; Wersinger et al. 2003; Fountaine and Wade‐Martins 2007; Fountaine et al. 2008) (Fig. 2a and b). Further, α‐syn is a highly mobile protein within the presynaptic terminal, demonstrating high concentrations within the axon terminal at rest, and rapidly dispersing away from the synapse in response to neural activity (Fortin et al. 2005; Unni et al. 2010) (Fig. 2a and b).
Figure 2.
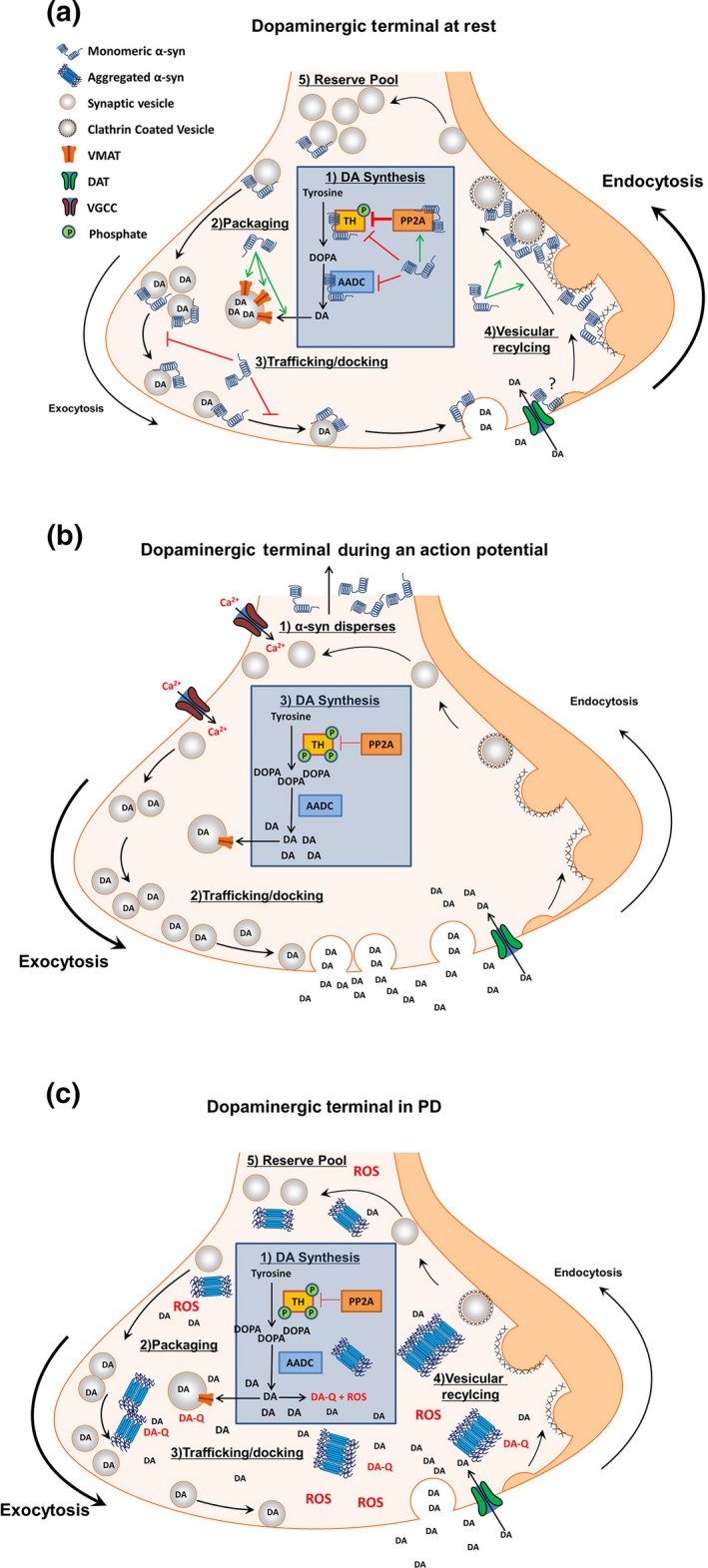
Proposed model of α‐syn function in the dopaminergic terminal. (a) In the absence of an action potential, α‐syn concentrations are high in the presynaptic terminal where it acts as a brake on chemical neurotransmission. A1) α‐syn inhibits DA synthesis by decreasing TH phosphorylation directly or by increasing PP2A activity. α‐syn also interacts with, and inhibits, AADC. A2) α‐syn aids in the sequestration of cytosolic DA by increasing the amount of VMAT on vesicles. A3) α‐syn prevents neurotransmitter release through interactions with synaptic vesicles and SNARE complex proteins to prevent trafficking and docking of vesicles with the presynaptic membrane. (A4) α‐syn facilitates the recycling of synaptic vesicles by mediating membrane bending during endocytosis, in order to (A5) maintain numbers of vesicles in the reserve (and possibly the readily releasable) vesicle pool. B1) Following neuronal stimulation and calcium influx, α‐syn rapidly disperses from the presynaptic terminal (B2) providing unimpeded vesicular trafficking and exocytosis for efficient neurotransmitter release. (B3) The absence of α‐syn disinhibits TH and AADC, allowing DA synthesis to replenish DA released during synaptic transmission. Upon repolarization, α‐syn repopulates the terminal to perform the actions listed in panel A to terminate chemical neurotransmission. (c) The aggregation of α‐syn in PD results in a loss of α‐syn function and subsequent increased cytosolic DA. (C1) Loss of α‐syn disinhibits TH and AADC, resulting in increased DA synthesis with a corresponding (C2) decrease in VMAT levels, and (C3) unregulated trafficking of synaptic vesicles. (C4) Loss of α‐syn function impairs endocytic vesicular recycling, (C5) decreasing the size of the vesicular pool. The net result is increased cytosolic DA, with a concomitant inability to efficiently sequester DA into synaptic vesicles. Increased cytosolic DA auto‐oxidizes to produce ROS and DA quinones. DA quinones react with sulfhydryl groups in proteins, forming DA‐cysteinyl adducts that covalently modify proteins, impairing enzymatic function. ROS oxidize proteins and lipids. DA‐cysteinyl adducts and ROS inhibit the electron transport chain, resulting in increased oxidative stress and opening of the mitochondrial permeability pore, as well as decreasing enzymatic break down of DA to DOPAC, further increasing cytosolic DA. Increased ROS and the formation of DA‐α‐syn adducts promote α‐syn aggregation. Together, an initial loss of α‐syn function can initiate a vicious cycle of toxicity ultimately resulting in cell death. Abbreviations: tyrosine hydroxylase (TH), aromatic amino acid decarboxylase (AADC), dopamine (DA), dihydroxyphenylalanine (DOPA), protein phosphatase 2A (PP2A), vesicular monoamine transporter (VMAT), dopamine transporter (DAT), voltage gated calcium channel (VGCC), reactive oxygen species (ROS), dopamine quinone (DA‐Q).
With this scenario, α‐syn may act as a brake on dopaminergic neurotransmission. For example, following neuronal stimulation, α‐syn rapidly disperses from the presynaptic terminal (Fortin et al. 2005); allowing unimpeded vesicular trafficking and exocytosis for efficient neurotransmitter release (Fig. 2b). Upon termination of synaptic transmission, α‐syn rapidly repopulates the presynaptic terminal (Fortin et al. 2005), presumably acting to impede the trafficking and docking of synaptic vesicles with the presynaptic membrane, thus halting chemical transmission (Fig. 2a). Further, within the dopaminergic terminal, the dispersion of α‐syn from the presynaptic terminal during neuronal firing would effectively disinhibit TH and AADC, allowing de novo DA synthesis to replenish DA for release during synaptic transmission (Fig. 2b). Finally, as α‐syn repopulates the presynaptic terminal following the termination of neuronal stimulation, α‐syn could increase the rate of synaptic vesicle recycling through endocytosis and also activate DAT and increase VMAT, resulting in more efficient DA reuptake and packaging, thereby enabling a continuing level of high‐fidelity neuronal signaling (Fig. 2a). With α‐syn potentially performing so many critical functions within the dopaminergic synapse alone, it is easy to envision how a loss of α‐syn function could result in neurotoxicity.
α‐syn in PD
Although α‐syn pathology is most commonly associated with PD, aggregation of α‐syn is a common thread linking a spectrum of neurodegenerative disorders collectively referred to as synucleinopathies. Disorders classified as synucleinopathies include PD (Spillantini et al. 1997), dementia with Lewy bodies (DLB) (Kosaka 1978; Spillantini et al. 1998a), multiple system atrophy (MSA) (H et al. 1998), pure autonomic failure (Arai et al. 2000), Lewy body variant of Alzheimer's disease (Lippa et al. 1998), and neurodegeneration with brain iron accumulation (Arawaka et al. 1998; Wakabayashi et al. 1999). Synucleinopathies are clinically differentiated by the symptoms that patients manifest; however, there are overlapping symptoms that are shared by these disorders (Marti et al. 2003). For example, both MSA and DLB are associated with clinical Parkinsonism; yet, they differ from classical PD by the presence of additional symptoms such as autonomic failure and cerebellar ataxia in MSA, or early cognitive impairment in DLB (Marti et al. 2003). The differential clinical presentations of synucleinopathies are also accompanied by differences in the neuroanatomical location of pathological α‐syn deposits. PD and DLB are characterized by aggregated α‐syn within the soma and neurites of neurons (Spillantini et al. 1997, 1998a), while MSA is characterized by α‐syn deposition primarily within myelinating glial cells (H et al. 1998). The fact that aggregation of α‐syn can have deleterious effects in many distinct cell populations strengthens the argument that α‐syn likely plays a crucial role in neurophysiology in general. Further, the location of α‐syn deposits directly correlates with the symptomatology observed, lending support to the idea that α‐syn or rather the loss of α‐syn, is directly responsible for neurotoxicity.
α‐syn genetics and PD
The first link between α‐syn and PD was the identification of a missense mutation on chromosome 4 that segregated with an autosomal‐dominant inherited form of PD (Polymeropoulos et al. 1997). This mutation was discovered in an Italian kindred as well as in four (ostensibly) unrelated Greek kindreds (Polymeropoulos et al. 1997). The actual mutation was a single base‐pair change at position 209 from G to A, resulting in an alanine to threonine substitution at amino acid 53 (A53T) (Polymeropoulos et al. 1997). Since the seminal discovery of the A53T mutation, seven additional missense mutations in the SNCA gene have been identified to result familial PD. In the following year, the A30P mutation was described, and in 2004, the E46K mutation (Kruger et al. 1998; Zarranz et al. 2004). Most recently, the H50Q, G51D, A53E, A18T, and A29S mutations were reported (Appel‐Cresswell et al. 2013; Hoffman‐Zacharska et al. 2013; Lesage et al. 2013; Pasanen et al. 2014). Beyond missense mutations, genome‐wide association studies have identified several single‐nucleotide polymorphisms in the SNCA gene that segregate with typical PD (Satake et al. 2009; Simón‐Sánchez et al. 2009; Edwards et al. 2010; Li et al. 2013). Interestingly, all currently identified mutations in the SNCA gene that result in PD occur within the N‐terminus of the protein. Functional studies examining the lipid‐binding characteristic or aggregation kinetics of these mutants confirm that familial mutations either disrupt membrane binding or increase the aggregation kinetics of α‐syn (Conway et al. 1998; Fredenburg et al. 2007; Burré et al. 2012; Lesage et al. 2013; Fares et al. 2014), resulting in impaired α‐syn function.
Multiplications of the SNCA gene also result in familial PD. Both duplications and triplications of the SNCA locus result in PD (Singleton et al. 2003; Chartier‐Harlin et al. 2004; Farrer et al. 2004; Ibanez et al. 2004). Patients with a duplication of the SNCA locus present at a similar age as idiopathic PD (Chartier‐Harlin et al. 2004; Ibanez et al. 2004). In contrast, triplication of the SNCA locus results in an early onset and very rapidly progressing disease phenotype (Singleton et al. 2003; Farrer et al. 2004). In line with increased copies of the SNCA gene, triplications patients have twice the amount of protein within the blood (Miller et al. 2004). Interestingly, although mRNA is doubled in the brains of SNCA triplication patients, there is no increase in the amount of soluble α‐syn in the brain. However, there is an increase in aggregated α‐syn within the brain, suggesting that increased SNCA protein product becomes sequestered into intracellular aggregates (Fig. 1c and d) (Miller et al. 2004). Multiplications in the SNCA gene have been identified in several other kindreds with familial PD (Nishioka et al. 2006; Fuchs et al. 2007; Ikeuchi et al. 2008), and even in patients with sporadic PD (Ahn et al. 2008).
Mutations in the SNCA promoter are also linked to PD. Genome‐wide association studies have identified several polymorphisms in the SNCA gene and promoter that are risk factors for developing PD (Farrer et al. 2001; Satake et al. 2009; Simón‐Sánchez et al. 2009; Edwards et al. 2010; Li et al. 2013). In particular, repeats in the REP1 allele within the 5′ untranslated region of SNCA are linked to PD. REP1‐SNCA is a region approximately 10kb upstream of the SNCA gene that is necessary for the proper expression of α‐syn (Chiba‐Falek and Nussbaum 2001). This microsatellite contains natural repeats, which are expanded in some patients with PD (Chiba‐Falek and Nussbaum 2001). Longer REP1 allele lengths (263 base pairs) result in increased SNCA expression in cultured cells, transgenic animals, and in human blood and brain (Chiba‐Falek and Nussbaum 2001; Fuchs et al. 2008; Cronin et al. 2009; Linnertz et al. 2009). In contrast, shorter REP1 allele lengths (259 base pairs) are associated with decreased SNCA expression (Maraganore et al. 2006). REP1 allele lengths that correlate with increased SNCA expression are risk factors for developing PD. This is in line with data from patients with multiplications in the SNCA locus, in which there seems to be a gene dosage effect, wherein increased expression of SNCA is directly correlated with disease severity.
Finally, altered α‐syn expression also occurs in sporadic PD patients. There have been several studies examining the expression of the SNCA gene in the midbrain and other brain regions of patients with idiopathic PD (Neystat et al. 1999; Beyer et al. 2004; Kingsbury et al. 2004; Tan et al. 2005; Chiba‐Falek et al. 2006; Dächsel et al. 2007; Gründemann et al. 2008). Altogether, the results from these studies are fairly inconsistent, with some results showing increased SNCA expression (Chiba‐Falek and Nussbaum 2001; Gründemann et al. 2008), and others showing decreased expression (Kingsbury et al. 2004; Dächsel et al. 2007) or no change in SNCA expression (Tan et al. 2005; Quinn et al. 2012). These inconsistent results have been attributed to normalizing SNCA expression levels to housekeeping genes that change during disease progression. Examination of SNCA expression using more appropriate control genes provides a consensus that α‐syn mRNA is decreased in the substantia nigra pars compacta (SNc) of PD patients (Dächsel et al. 2007). However, whether these reductions in expression represent an initial maladaptive change in SNCA expression that triggers toxicity, or merely a compensatory reduction in SNCA expression because of increased α‐syn protein accumulation in the soma, is not currently clear. For example, as healthy humans age, there is an age‐related redistribution of α‐syn protein from the pre‐synaptic terminal to the somatodendritic compartment in neuromelanin‐containing neurons of SNc (Fig. 1b) (Chu and Kordower 2007; Xuan et al. 2011). Further, PD is characterized by perinuclear accumulation of aggregated α‐syn (Fig. 1c). These increases in protein could serve as negative feedback to decrease SNCA gene expression. Alternatively, a reduction in SNCA gene expression could simply be a normal process of aging. α‐syn mRNA levels are reduced in normally aged humans and rodents (Galvin et al. 2001; Adamczyk et al. 2005; Mak et al. 2009). Accordingly, as aging is the primary risk factor for PD (Collier et al. 2011), decreased SNCA expression could represent an independent comorbidity that does not actually contribute to the pathogenesis of sporadic PD. That being said, while changes in SNCA gene expression may not directly contribute to the neurotoxicity of sporadic PD, it is clear that a shift in the levels of soluble to insoluble α‐syn protein is directly related to PD pathogenesis.
α‐syn aggregation in PD
Along with the loss of midbrain nigrostriatal DA neurons, PD is histopathologically defined by the presence of Lewy bodies (LB) and Lewy neurites (LN) (Braak et al. 2003a). LB are large cytoplasmic protein inclusions that were originally identified in the brains of PD patients by Frederic Heinrich Lewy in 1912 (Goedert et al. 2012). For over a century, Lewy pathology has been the defining neuropathological hallmark used for postmortem confirmation of PD (Goedert et al. 2012). However, it was not until the advent of more sophisticated histological techniques that LB were discovered to contain aggregated α‐syn (Spillantini et al. 1997). Shortly after the discovery that mutations in the SNCA gene result in familial PD, Spillantini et al., used affinity purified anti‐α‐syn antiserum to label LB and LN in brain tissue from patients with PD and DLB (Spillantini et al. 1997). In the following years, these more sensitive histological techniques identified widespread α‐syn aggregation throughout both the central and peripheral nervous systems of PD patients (Spillantini et al. 1998a; Galvin et al. 1999; Bendor et al. 2013). Further, extensive comparisons of normal and Parkinson diseased brains has revealed that the neural loci affected by α‐syn aggregation proceeds in a relatively stereotypic manner, progressing in a caudal to rostral pattern from the brainstem to the cortex over the course of the disease (Braak et al. 2003b). This revelation has led to implementation of various staging schema to diagnose disease severity based on the neuroanatomical nuclei displaying α‐syn aggregates (Marui et al. 2002; Braak et al. 2003a). The initial sites displaying α‐syn aggregates in the CNS are the dorsal motor nucleus of the vagus in the brainstem and the olfactory bulb (Braak et al. 2003a). As the disease progresses, aggregated α‐syn next appears in the pontine tegmentum, followed by the amygdala and SNc, and eventually reaching the temporal cortex and neocortex in the terminal stages of the disease (Braak et al. 2003b). Although this staging schema has been useful in segregating disease progression into quantal stages, not all patients follow the staging schema, and as such the utility of this classification system has been called into question (Halliday et al. 2006; Jellinger 2008; Beach et al. 2009), with new attempts at a unified staging scheme being put forth (Beach et al. 2009).
In addition to synuclein pathology in the brains of PD patients, there is also widespread synuclein pathology in the peripheral nervous system (Braak et al. 2006; Beach et al. 2010). The first LBs identified outside of the brain were found in the enteric nervous system (Wakabayashi et al. 1988, 1990, 1993). More recently, an extensive analysis identified aggregated α‐syn in the gastrointestinal system, the spinal cord, sympathetic ganglia, as well as in the sciatic and vagus nerves (Beach et al. 2010). The presence of aggregated α‐syn in cells outside of the basal ganglia provides a potential underlying mechanism for the myriad of non‐motor symptoms associated with PD (Chaudhuri et al. 2006). For example, GI dysfunction is one of the most common non‐motor symptoms associated with PD, and GI symptoms appear years prior to the onset of motor behavior (Pfeiffer 2003). In line with this, the most consistent location of aggregated α‐syn outside the brain is the enteric nervous system, with PD patients presenting α‐syn aggregates throughout the entirety of the enteric nervous system, from the esophagus to the rectum (Braak et al. 2006; Beach et al. 2010). Further, aggregated α‐syn appears in the enteric nervous system years prior to CNS pathology and dysfunction in PD as do many GI symptoms (Wakabayashi et al. 1988; Braak et al. 2006; Beach et al. 2010; Shannon et al. 2012a,b), suggesting that aggregated α‐syn within neurons of the enteric nervous system is likely the source of this GI dysfunction. This scenario is not limited to the GI tract; other non‐motor symptoms of PD include hyposmia, autonomic disturbances, impaired micturation, rapid eye movement (REM) sleep disorder, as well as dementia and depression (Chaudhuri et al. 2006). In line with these symptoms, aggregated α‐syn is observed in the olfactory bulb, autonomic ganglia, the dorsal horn of the sacral spinal cord, glossopharyngeal nerves, other brainstem nuclei, and cortical areas, potentially providing a pathophysiological substrate for all non‐motor symptoms associated with PD (Dickson et al. 2009; VanderHorst et al. 2015).
Propagation of aggregated α‐syn
The fact that the appearance of α‐syn pathology proceeds throughout the PD‐affected brain in a spatially and temporally stereotypic pattern has led to the suggestion that misfolded α‐syn may spread from cell‐to‐cell in a prion‐like manner (Olanow and Prusiner 2009; Lee et al. 2014). This ‘prion hypothesis’ for α‐syn has been supported by several observations and experimental findings. First and foremost, two independent publications report that fetal mesencephalic DA neurons engrafted into the striatum (ST) of PD patients displayed LB‐like structures (Kordower et al. 2008; Li et al. 2008). These LB‐like structures were composed of aggregated α‐syn, suggesting that misfolded or aggregated α‐syn from the patient's brain had infiltrated the grafts, potentially serving as templates to initiate aberrant folding and accumulation of normal, endogenous α‐syn expressed by the engrafted neurons (Li et al. 2008; Kordower et al. 2013). This host‐to‐graft transmission of pathological α‐syn was replicated in vitro and in a transgenic mouse model of PD pathology (Desplats et al. 2009). These observations led to significant interest in the route by which α‐syn could potentially be released by ‘donor’ cells and subsequently taken up by ‘recipient’ cells. To this end, both monomeric and oligomeric α‐syn are secreted from cultured neurons through unconventional exocytosis (Lee et al. 2005, 2014; Jang et al. 2010) and the released α‐syn can in turn be taken up by recipient neurons and glia through endocytosis (Lee et al. 2008, 2014). Within cultured cells, aggregated α‐syn can transmit from neuron‐to‐neuron via these exocytosis and endocytosis events, and can propagate aggregates by serving as a seeding template to initiate the aggregation of endogenous α‐syn in recipient cells (Desplats et al. 2009; Luk et al. 2009, 2012a; Volpicelli‐Daley et al. 2011). These results have been mirrored in vivo; brain homogenates from old, symptomatic transgenic (α‐syn over‐expressing) mice exhibiting α‐syn pathology were injected into young transgenic mice, which accelerated the formation of LB‐like α‐syn aggregates within the young mice (Luk et al. 2012a). Moreover, injection of preformed α‐syn fibrils into the rodent brain leads to the formation of α‐syn aggregates that appear distal to the injection site months after the injection. It remains to be proven, however, whether misfolded α‐syn can propagate/transmit throughout the CNS over time, akin to that seen in prion disorders, or whether these findings are the result of diffusion and transport of the ectopic synuclein seeds to these distal areas (Luk et al. 2009, 2012a; Paumier et al. 2015). Finally, the presence of aggregated α‐syn within oligodendrocytes (which do not normally express α‐syn) of patients with MSA has been proposed to occur through a prion‐like propagation from neurons to glia (Spillantini et al. 1998b; Miller et al. 2005) Although there is considerable evidence supporting a prion‐like transfer of α‐syn, this area of research remains contentious and not all researchers concur with a model by which cell‐to‐cell transfer of α‐syn causes disease.
Animal models of PD
Because of the prominent role that α‐syn plays in the pathology of PD and other synucleinopathies, many animal models have been developed in an attempt to elucidate both the normal function of α‐syn, and the role α‐syn plays in the pathology of synucleinopathies. To date, the vast majority of work on α‐syn animal models has been performed in rodents (mice and rats), however, there has been research performed in larger mammals such as non‐human primates, as well as in invertebrates (Drosophila and Caenorhabditis elegans). Across species, animal models centered on α‐syn can be subdivided into α‐syn over‐expression models, and α‐syn knockdown or knockout models. The α‐syn over‐expression models can then be further subdivided into viral vector‐mediated α‐syn over‐expression and α‐syn transgenic over‐expressing mouse lines. This section will briefly describe the pathology observed following manipulation of α‐syn expression within animal models, and some insights gained therein.
Over‐expression: transgenic mice
The list of transgenic human α‐syn (WT or mutant) over‐expressing mouse lines is currently quite large and seems to be growing (Hashimoto et al. 1999; Kahle et al. 2000; Masliah et al. 2000; van der Putten et al. 2000; Lee et al. 2001b, 2002; Matsuoka et al. 2001; Giasson et al. 2002; Richfield et al. 2002; Rockenstein et al. 2002; Gispert et al. 2003; Gomez‐Isla et al. 2003; Thiruchelvam et al. 2004; Tofaris et al. 2006; Wakamatsu et al. 2008; Lin et al. 2012). The majority of transgenic lines over‐expressing mutant α‐syn encode either the A53T or A30P familial mutations (Kahle et al. 2000; van der Putten et al. 2000; Matsuoka et al. 2001; Giasson et al. 2002; Lee et al. 2002; Rockenstein et al. 2002; Gispert et al. 2003; Gomez‐Isla et al. 2003; Lin et al. 2012), however, there are also double mutant transgenic lines that express both A53T and A30P mutations (Richfield et al. 2002; Thiruchelvam et al. 2004), as well as C‐terminal truncated mutants (Tofaris et al. 2006), or even a combination of familial mutations with C‐ terminal truncation (Wakamatsu et al. 2008). One major difference between existing α‐syn transgenic mice is the promoter used to control transgene expression. Existing promoters used include the Thy1 promoter (Kahle et al. 2000; van der Putten et al. 2000; Rockenstein et al. 2002), the human platelet‐derived growth factor β promoter (Masliah et al. 2000; Rockenstein et al. 2002), the TH promoter (Richfield et al. 2002; Thiruchelvam et al. 2004; Tofaris et al. 2006; Wakamatsu et al. 2008), and the prion promoter (Giasson et al. 2002; Lee et al. 2002; Gispert et al. 2003; Gomez‐Isla et al. 2003). Additionally, a tetracycline inducible α‐syn expression system was recently described (Lin et al. 2012). A notable exception to the use of artificial promoters to guide transgenic α‐syn expression are mice lacking murine α‐syn gene (mSNCA−/−) and carrying a p1 artificial chromosome expressing the entire human SNCA locus, including the endogenous SNCA promoter (Kuo et al. 2010).
The type of promoter used will determine the type of cells that the α‐syn transgene will be expressed within. For example, many of the α‐syn transgenic lines show widespread expression through the entire animal, however, some lines show more restricted expression, such as those utilizing the TH promoter (Matsuoka et al. 2001; Richfield et al. 2002; Thiruchelvam et al. 2004; Tofaris et al. 2006). Beyond determining the type of cells that the transgene will be expressed in, the choice of promoter will also determine the temporal onset of expression as well as the level of transgene expression. Currently, the range of α‐syn over‐expression observed in transgenic mouse lines is very large, varying from 0.3 to more than 30 fold increased expression over that of endogenous α‐syn levels (Fleming et al. 2005). Finally, although most current transgenic mouse lines are converging toward a consensus of using the C57Bl6 strain background, not all α‐syn transgenic mice are of the same strain, and this can affect the pattern and severity of pathology observed. Although this will not be discussed further, it is an important caveat to consider when interpreting data from various transgenic animals, and should thus be kept in mind.
The ectopic over‐expression of α‐syn in transgenic mice can result in the formation of aggregated α‐syn (Fig. 1d) (Kahle et al. 2000; Masliah et al. 2000; van der Putten et al. 2000; Giasson et al. 2002; Lee et al. 2002; Rockenstein et al. 2002; Tofaris et al. 2006; Lin et al. 2012), however, this is not always the case (Matsuoka et al. 2001; Richfield et al. 2002; Gispert et al. 2003; Gomez‐Isla et al. 2003; Thiruchelvam et al. 2004). Differences in the aggregation profiles observed in the varying transgenic lines likely reflect the varying levels of expression achieved, as well as the type of α‐syn mutation expressed. When aggregates are observed, the extent to which these aggregates recapitulate the aggregation of α‐syn in PD is uncertain, however, many α‐syn transgenic lines do show markers associated with LB‐like pathology, such as positive staining with thioflavin or silver stain, co‐labeling with ubiquitin or α‐syn phosphorylated at serine 129, (Masliah et al. 2000; van der Putten et al. 2000; Giasson et al. 2002; Lee et al. 2002; Rockenstein et al. 2002; Tofaris et al. 2006). Further, in many instances in which aggregates are observed, they are not necessarily observed within the nigrostriatal system. For example, the Thy1 or prion promoter lines demonstrate extensive α‐syn aggregation in the spinal cord and/or neuromuscular junction (van der Putten et al. 2000; Giasson et al. 2002; Lee et al. 2002; Gomez‐Isla et al. 2003; Cabin et al. 2005). Other transgenic lines show extensive α‐syn aggregation throughout the brain (e.g. cortex, hippocampus, cerebellum, and brainstem) with little or no aggregates found in the SNc (Masliah et al. 2000; Giasson et al. 2002; Rockenstein et al. 2002). Accordingly, it is likely that although many of these mice do develop some motor impairments, those impairments are not the result of degeneration of the nigrostriatal system, but more likely represent pyramidal or motor neuron degeneration (van der Putten et al. 2000; Giasson et al. 2002; Cabin et al. 2005). Although the motor dysfunction in some α‐syn transgenic lines may not recapitulate the motor impairment of PD with perfect fidelity, the models are still valuable for studying the effects of synucleinopathy outside the nigrostriatal system, which also occurs in PD (Beach et al. 2010). For example, some α‐syn transgenic mice develop olfactory, GI, and autonomic dysfunction reminiscent of changes observed in PD (Fleming et al. 2008; Wang et al. 2008; Kuo et al. 2010; Hallett et al. 2012; Noorian et al. 2012; Farrell et al. 2014).
In general, a major shortcoming of current α‐syn transgenic mice is the lack of degeneration of the nigrostriatal DA system (van der Putten et al. 2000; Giasson et al. 2002; Gispert et al. 2003; Gomez‐Isla et al. 2003). The loss of midbrain nigrostriatal neurons and the corresponding loss of DA innervation to the ST underlies the motor impairment which characterize parkinsonism, and as such any reliable model used to study PD pathology should recapitulate this defining feature. There are notable exceptions to this criticism. Mice expressing WT α‐syn under control of the platelet‐derived growth factor β promoter display decreased ST DA along with decreased TH terminal density, TH protein, and TH activity in the ST (Masliah et al. 2000; Rockenstein et al. 2002). ST pathology correlates with the presence of α‐syn inclusions within neurons and glia throughout the brain, and result in motor impairment (Masliah et al. 2000; Rockenstein et al. 2002; Hashimoto et al. 2003). Interestingly, although there was a significant loss of TH and DA levels in the ST of these animals, the majority of α‐syn inclusions were located outside the midbrain, with only occasional aggregates found in THir neurons of the SNc (Masliah et al. 2000). Further, there was no loss of THir neurons of the SNc, suggesting that the pathology observed reflects a loss of the TH phenotype within the nerve terminals in the absence of any nigrostriatal neuron loss (Masliah et al. 2000). Another commonly used α‐syn transgenic mouse is the Thy1 α‐syn mouse. There have been several iterations of this particular transgenic mouse with varying degrees of pathology (Kahle et al. 2000; van der Putten et al. 2000; Rockenstein et al. 2002). For instance, Thy‐1 A53T α‐syn mice display prominent α‐syn inclusions that are particularly enriched in the spinal cord and neuromuscular junction, but absent in the SNc (van der Putten et al. 2000). These animals manifest a severe motor phenotype at an early age, which likely reflects pyramidal or motor neuron degeneration (van der Putten et al. 2000). In contrast, WT or A30P α‐syn expressed by the Thy‐1 promoter produces α‐syn aggregation in the SNc and ST, as well as decreased ST DAT and VMAT, with no reported cell loss (Kahle et al. 2000; Rockenstein et al. 2002; Fleming et al. 2005). Expression of human α‐syn under the control of the catecholaminergic specific TH promoter results in a more robust pathological phenotype within the nigrostriatal system producing a loss of TH neurons with age (Richfield et al. 2002; Thiruchelvam et al. 2004). However, loss of TH neurons was not observed in an alternative report, despite high levels of expression and the accumulation of α‐syn in the SNc. (Matsuoka et al. 2001) Expression of a double mutant A53T+A30P α‐syn under the TH promoter results in a loss of neurons in the SNc and a corresponding loss of ST DA and DA metabolites, along with reduced locomotor activity (Richfield et al. 2002; Thiruchelvam et al. 2004). Finally, TH promoter‐mediated expression of C‐terminally truncated human α‐syn carrying the A53T mutation causes a loss of midbrain DA neurons in the SNc and a loss of DA in the ST (Wakamatsu et al. 2008).
In contrast to the mice described above, in which α‐syn expression is initiated during development, post‐developmental expression of α‐syn achieved through a tet‐inducible system produces a pattern of pathology that more faithfully recapitulates PD (Lin et al. 2012). Following the activation of transgene α‐syn expression, there is a 2–4 fold increase in human α‐syn expression that is largely restricted to midbrain DA neurons (Lin et al. 2012). Increased α‐syn expression results in the formation of α‐syn aggregates in the somata and terminals of TH neurons (Lin et al. 2012). This conditional A53T human α‐syn mouse also develops a progressive loss of THir neurons in the SNc (Lin et al. 2012) concomitant with ST denervation and impaired DA release (Lin et al. 2012). Finally, this pathology results in profound motor impairment that is reversed by inactivating the transgene (Lin et al. 2012). The more robust and consistent pathology seen when α‐syn is altered within neurons of adult animals, as opposed to germline manipulation of α‐syn, highlights the confounding variable of genetic compensation observed when animals harbor a genetic aberration during development. As will be discussed in detail in later sections, with particular emphasis on α‐syn null mice, the issue of genetic compensation often diminishes the conclusions that can be drawn from germline transgenic animals.
Over‐expression: transgenic invertebrates
Transgenic over‐expression of α‐syn has also been modeled in Drosophila and C. elegans, neither of which normally express the α‐syn gene. In Drosophila, transgenic flies expressing either WT, A30P, or A53T α‐syn downstream of multiple binding sites for the yeast transcriptional activator protein GAL4 were crossed with flies expressing GAL4 under control of a pan neuronal promoter, or the DA neuron specific promoter DOPA‐decarboxylase, resulting in an age‐dependent loss of THir neurons in the dorsomedial cluster (Feany and Bender 2000). The over‐expression of the α‐syn transgene resulted in formation of aggregates that paralleled the temporal progression of toxicity (Feany and Bender 2000). Synucleinopathy and loss of DA neurons resulted in a motor phenotype, in the form of a reduction in negative geotactic motor behavior (Feany and Bender 2000). Interestingly, as has been observed in transgenic mice, this pathology could be completely rescued by the co‐expression of the chaperone, Hsp‐70 (Auluck et al. 2002). Co‐expression of Hsp‐70 did not affect the number, size, or location of the α‐syn aggregates, yet prevented the neurodegeneration caused by the α‐syn transgene (Auluck et al. 2002). Finally, either WT or A53T α‐syn over‐expression in the nematode, C. elegans, results in rare formation of α‐syn aggregates and a loss of DA neurons (Lakso et al. 2003).
Over‐expression: viral vectors
Though transgenic mouse models may improve our understanding of α‐syn biology, to date, no mouse model accurately recapitulates the progressive neurodegenerative changes that are the hallmark of PD. In this regard, viral over‐expression of α‐syn may be considered a more accurate model of PD pathology. The majority of reports on virally mediated over‐expression of α‐syn utilize adeno‐associated virus (AAV) (Kirik et al. 2002), however, there have also been reports of lentivirus (LV)‐mediated over‐expression (Bianco et al. 2002). As in the case of transgenic animals, the major differences between viral over‐expression models is the type of promoter used to control transgene expression, as well as the use of either WT or mutant forms of α‐syn. Virally mediated over‐expression of α‐syn is commonly driven by the highly active and ubiquitous chicken β‐actin promoter (CBA; aka CAG), or the slightly less active cytomegalovirus (CMV) promoter. However, other promoters are now being utilized, such as the neuron specific synapsin promoter. Transgene expression following viral delivery is not only dependent on the promoter used, but also on the number of viral particles injected (Gombash et al. 2013), as well as the pseudotype of virus utilized. Accordingly, virally mediated over‐expression of α‐syn offers several added levels of control, which can act to spatially and temporally guide transgene expression within phenotypically distinct cells of the brain.
LV has been used to deliver both WT and mutant forms of α‐syn to the SNc of mice and rats (Bianco et al. 2002; Lauwers et al. 2003). Injection of vesicular stomatitis virus glycoprotein pseudotyped LV expressing either WT, A53T, or A30P α‐syn under control of the phosphoglycerate kinase (PKG) promoter into the rat SNc results in aggregation of α‐syn and a corresponding degeneration of TH immunoreactive (THir) nigrostriatal soma (24–35% loss) and terminals (Bianco et al. 2002). The degeneration of THir neurons in the SNc is slightly progressive, increasing from 3 weeks, to peak at the 6 week time point (Bianco et al. 2002). In contrast, LV‐mediated expression of WT or A30P α‐syn in the mouse SNc produces comparably less pathology (Lauwers et al. 2003). Using the CMV promoter, there is a 10–25% loss of THir neurons in the SNc, with no loss of striatal DA, that requires up to 12 months of transgene expression to manifest (Lauwers et al. 2003). It is interesting to note that LV‐mediated α‐syn over‐expression in TH neurons of α‐syn null mice results in α‐syn aggregation and loss of THir in dopaminergic neurons that are still present in CNS (Alerte et al. 2008). This raises the possibility that such effects may lead to erroneous interpretation of a loss of dopamine neurons if quantification of THir cells is the sole outcome measured.
AAV is the most commonly used viral vector system to over‐express α‐syn. The original description of the AAV‐α‐syn model utilized AAV serotype 2 to express either WT α‐syn or the A53T mutant under control of the CBA promoter (Kirik et al. 2002). Surprisingly, there is no difference in the pathology observed following injection of WT versus the A53T mutant form of α‐syn. In both treatments, over‐expression results in the appearance of aggregated α‐syn, dystrophic α‐syn‐immunoreactive neurites, and shrunken pyknotic soma (Kirik et al. 2002). Pathology is slightly progressive, beginning at approximately 3 weeks, and reaching a plateau at 6 weeks post‐transduction (Kirik et al. 2002). Peak pathology reported was highly variable, 30–80% loss of THir neurons in the SNc, and a corresponding 50% loss of TH fibers and DA in the ST (Kirik et al. 2002). Further, striatal TH enzymatic activity was also reduced (Kirik et al. 2002). Finally, AAV2‐α‐syn over‐expression produces a variable loss of motor behavior, likely resulting from the inconsistent amount of neurodegeneration observed in the SNc (Kirik et al. 2002). Interestingly, by 6 months post‐surgery, striatal TH and THir neuron numbers in the SNc appear to increase, demonstrating some recovery from the neuropathology (e.g. loss of TH phenotype) induced by AAV‐α‐syn expression (Kirik et al. 2002). Other laboratories replicating this model report an increase in microglial activation, B‐ and T‐cell infiltration (Theodore et al. 2008), and changes in proteins involved in axonal trafficking with a corresponding loss of proteins involved in synaptic transmission (Chung et al. 2009), pathological changes that are observed in PD.
A similar pattern of pathology occurs with nigral over‐expression of the A30P mutant α‐syn, again producing α‐syn aggregates and an approximate 50% loss of SNc neurons (Klein et al. 2002). AAV2 expressing WT α‐syn under control of the CMV promoter injected into the SNc produces a similar loss of THir nigral neurons, as well as accumulation of α‐syn phosphorylated at serine 129 (Yamada et al. 2004). Finally, as was observed using LV, injection of the same expression cassette in mice does not elicit the same degree of pathology as compared to rats: Mice injected with AAV2 expressing WT α‐syn under control of the CBA promoter do not develop obvious α‐syn inclusions, with a mere 25% neurodegeneration of nigral neurons that does not appear until 24 weeks post‐transduction (St Martin et al. 2007) [as compared with a 40–50% loss of nigral neurons by 6 weeks in rats (Kirik et al. 2002)]. In this report, the proportion of THir nigral neurons transduced was also highly variable, (St Martin et al. 2007), and accordingly, the reduced pathology in mice may result from mistargeted injections or decreased viral expression.
Perhaps, the best‐characterized and most accurate recapitulation of PD pathology reported using virally mediated over‐expression is achieved using AAV2/6 expressing WT α‐syn under control of the neuron‐specific synapsin promoter (Decressac et al. 2012). This model produces robust degeneration of the nigrostriatal system that is highly specific and progressive. For example, there is a progressive loss of THir neurons of the SNc that proceeds from 50% at 3 weeks post‐surgery to 80% at 8 weeks post‐surgery (Decressac et al. 2012). The progressive loss of THir neurons of the SNc is mirrored by a progressive decline in TH fibers, TH activity, and DA concentration in the ST (Decressac et al. 2012). The authors also reported a progressive decline in DA release and reuptake, along with a progressive decline in motor function that could be rescued by L‐DOPA administration (Decressac et al. 2012). The progressive nature of the SNc pathology by these investigators very nicely models human PD.
The patterns of neuropathology and behavioral deficits observed in viral rodent models have been replicated in monkeys, albeit over a much more protracted time course. Both AAV2 and AAV 2/5 expressing WT or A53T α‐syn under control of the CBA promoter have been injected into the SNc of adult marmosets (Kirik et al. 2003; Eslamboli et al. 2007). AAV2 expressing α‐syn produces a significant loss of neurons of the SNc and TH fibers in ST at 16 weeks post‐surgery. This degeneration is accompanied by the presence of dystrophic neurites, swollen axons, pyknotic soma, and the appearance of granular α‐syn aggregates (Kirik et al. 2003). Using AAV2/5 and a longer time course, Eslamboli et al. 2007 described a large amount of α‐syn aggregation, loss of nigrostriatal THir neurons and fibers, as well as marked behavioral abnormalities, including impaired contralateral forelimb use and a bias in the head position test (Eslamboli et al. 2007).
Taken together, most pathological changes observed using either transgenic α‐syn mouse lines or virally mediated over‐expression of α‐syn replicate changes observed in the PD brain, lending support to the construct validity of these models. However, although some aspects of PD pathology may be replicated in one particular model, the same model often lacks other pathological changes observed in PD, and currently there is no single model that replicates every aspect of PD neuropathology. For instance, virally mediated over‐expression of α‐syn results in a distinct pattern of neuropathology that, unlike that seen in transgenic over‐expressing animals, is highly selective to the nigrostriatal system. Viewed as a whole, the use of a viral vector to express an α‐syn transgene results in the aggregation of α‐syn, followed by a progressive loss of THir neurons of the SNc, as well as a corresponding loss of nigrostriatal fibers and DA concentrations in the ST. However, the extent to which these models are truly progressive could be argued, as all virally mediated α‐syn lesions plateau within a relatively short time (Bianco et al. 2002; Kirik et al. 2002; Klein et al. 2002; Yamada et al. 2004; Decressac et al. 2012), and in some instances, even seem to recover (Kirik et al. 2002), perhaps reflecting a transient loss of the TH phenotype induced by α‐syn aggregation (Alerte et al. 2008).
In contrast, the neurodegeneration observed in PD is truly progressive, in that it proceeds in an inexorable sequence ultimately leading to death (Hely et al. 1999). Neuropathology with viral vector‐mediated over‐expression appears very quickly, sometimes as early as 2–4 weeks (Bianco et al. 2002; Kirik et al. 2002; Decressac et al. 2012), which is in stark contrast to pathology seen in transgenic animals that require months or years to achieve neurodegeneration (Masliah et al. 2000; Lee et al. 2001b; Giasson et al. 2002; Rockenstein et al. 2002). In this sense, the presentation of neuropathology in transgenic animals, in which α‐syn accumulation and subsequent neurodegenerative changes occur over the lifetime of an animal, may better model the slow progression of the human disease. Because of the fact that virus is often injected directly into the SNc, the pathology observed in viral over‐expression models is usually restricted to the nigrostriatal system. This localized injection can achieve very high concentrations of transgene product, somewhat exclusively within the nigrostriatal system, which presumably accounts for the much more consistent and robust nigrostriatal degeneration observed by viral over‐expression (Bianco et al. 2002; Kirik et al. 2002, 2003; Klein et al. 2002; Yamada et al. 2004; Eslamboli et al. 2007; St Martin et al. 2007; Decressac et al. 2012), as compared to transgenic animals in which nigrostriatal degeneration is often completely absent (van der Putten et al. 2000; Giasson et al. 2002; Gispert et al. 2003; Gomez‐Isla et al. 2003). Although virus is beneficial to study degeneration of the nigrostriatal system, PD is a systemic disease that affects many regions of the brain as well as the periphery (Beach et al. 2010), and in this sense, the α‐syn expression achieved throughout the entire organism in α‐syn over‐expressing mice may be a more useful PD model. Finally, in many of the viral over‐expression models, deficits in motor behavior are either variable or completely lacking (Bianco et al. 2002; Kirik et al. 2002; Klein et al. 2002; Yamada et al. 2004; St Martin et al. 2007). However, it is likely that the lack of motor deficits result from a partial lesion of the nigrostriatal system (Kirik et al. 2002). A critical threshold of nigrostriatal neurodegeneration is known to be necessary to induce the onset of motor deficits in humans and certain PD models (Bezard et al. 2001; Kirik et al. 2002), suggesting that most PD models fail to reach this critical threshold. This idea is further supported by a clear gene‐dosage effect, whereby increased α‐syn expression and accumulation cause a correspondingly increased toxicity (Masliah et al. 2000; Lee et al. 2001b; Giasson et al. 2002; Gispert et al. 2003; Decressac et al. 2012; Gombash et al. 2013).
Silencing α‐syn expression: α‐syn null mice
In addition to transgenic mice over‐expressing human α‐syn, efforts to elucidate the biological function(s) of α‐syn have also focused on removing the protein from the entire animal. To this end, there have been several independent α‐syn knockout (KO) mouse lines created (Abeliovich et al. 2000; Dauer et al. 2002; Schlüter et al. 2003; Cabin et al. 2005). Additionally, a spontaneous deletion of the SNCA locus was identified in a subpopulation of C57bl/6j mice from Harlan Laboratories (Specht and Schoepfer 2001; Schlüter et al. 2003). Overall, the α‐syn null mice do not display overt neuropathological or behavioral phenotypes, however, in depth analyses reveal abnormalities that largely localize to the presynaptic terminal. α‐syn null mice evaluated by Abeliovich et al. (2000). display increased DA release following paired electrical stimulus, a modest (18%) reduction in striatal DA content, reduced rearing in the open‐field test, and an attenuation of amphetamine‐induced locomotor response [the latter of these findings failed to replicate in an alternate α‐syn KO model (Cabin et al. 2002)]. Ultrastructural analyses of the neurons of α‐syn null mice generated by Cabin et al., show a 50% reduction in the reserve pool of synaptic vesicles in primary hippocampal cultures and hippocampal sections from 2‐month‐old mice (Cabin et al. 2002). Some reports show that α‐syn null mice have a slightly reduced number of THir neurons in SNc (Robertson et al. 2004), however, others failed to replicate this (Abeliovich et al. 2000; Cabin et al. 2002). Finally, still others have shown that aged (2 years) α‐syn null mice develop denervated ST as reflected by decreased DA, TH, DAT, and synaptotagmin concentrations (Al‐Wandi et al. 2010).
It was originally believed that functional redundancy between α and β synuclein could have masked some of the results in α‐syn null mice. There is a high degree of homology in the primary amino acid sequences of α and β synuclein, which tend to co‐localize in presynaptic terminals throughout much of the brain, suggesting that the two proteins may be functionally redundant (Surguchov 2008). In contrast, γ synuclein is highly enriched within neural tissue of the periphery, and largely absent from the brain (Surguchov 2008). Accordingly, there have also been double synuclein KO mice created, in which either α and β (Chandra et al. 2004) or α and γ synuclein (Robertson et al. 2004) were deleted, as well as triple KOs in which all three synucleins were deleted (Greten‐Harrison et al. 2010). However, double and triple KO mice do not have a substantially different neuropathological profile from α‐syn null mice. The α/β double KO mice have reduced striatal DA that is approximately equal to that reported in the α‐syn null mouse (Chandra et al. 2004). Interestingly, despite a largely peripheral localization in normal animals, KO of γ synuclein alone results in a 15–20% loss of THir neurons in the SNc, and this effect is not significantly altered in the α and γ synuclein double KO mice (Robertson et al. 2004). The idea of functional redundancy and genetic compensation in germ line transgenic animals is further highlighted by the finding that all of the double KO synuclein animals show a compensatory increase in the remaining synuclein family member. For instance, the α/β KO mouse displays increased γ synuclein (Chandra et al. 2004), while the α/γ KO mice have increased β synuclein (Robertson et al. 2004). Some groups have reported a small, region‐specific compensatory increase in β synuclein in α‐syn null mice (Schlüter et al. 2003), however, the majority of reports saw no difference in β synuclein expression after deletion of the SNCA gene (Abeliovich et al. 2000; Cabin et al. 2002; Dauer et al. 2002). Finally, removal of all synucleins significantly increases the mortality of mice (Greten‐Harrison et al. 2010). The triple α/β/γ synuclein KO mouse also develop an age‐dependent reduction in synapse size and altered synaptic transmission (Greten‐Harrison et al. 2010). The reduction in synapse size can be rescued by crossing the triple KO with the Thy1 α‐syn over‐expressing mouse in order to restore α‐syn function, demonstrating a specific effect of synuclein (Greten‐Harrison et al. 2010).
In several independent reports, α‐syn null mice exhibit either a complete or partial resistance to 1‐methyl, 4‐phenyl‐1, 2, 3, 6, tetrahydropyradine (MPTP) (Dauer et al. 2002; Schlüter et al. 2003; Drolet et al. 2004; Robertson et al. 2004). MPTP is a pro‐toxicant that, once metabolized to 1‐meth, 4‐phenylpyradinium (MPP+), is transported into dopaminergic terminals and synaptic vesicles through direct interactions with DAT and VMAT, respectively (Dauer and Przedborski 2003). Interestingly, α‐syn null mice are more sensitive to the neurotoxicant rotenone (Dauer et al. 2002), which is lipophilic and does not require active transport via DAT or VMAT for entering dopaminergic terminals. The differential susceptibility of α‐syn null mice to MPTP and rotenone suggests that the decreased MPTP sensitivity of the mice is likely mediated through changes in the dopaminergic terminal. Supporting this, α‐syn null mice also display reduced MPTP‐induced DA efflux (Dauer et al. 2002), a phenomenon that occurs as a result of reversal of DA flow through DAT, in response to large increases in cytosolic DA following DA displacement from synaptic vesicles by MPP+(Lotharius and O'Malley 2000). It is likely that changes in synaptic vesicle mobility and concentrations underlie the altered responses. For example, the reduced size of the reserve vesicular pool could potentially account for the lack of MPTP‐induced DA efflux. Further, if α‐syn acts as a negative regulator of vesicular trafficking and exocytosis, it is possible the MPP+ within synaptic vesicles could be purged more rapidly in α‐syn null than WT mice, preventing or diminishing the amount of MPP+ available to interact with Complex I.
Silencing α‐syn expression: α‐syn Knock Down
In contrast to germline deletion of α‐syn, knock down of endogenous α‐syn in post‐mitotic neurons of adult animals causes severe neurodegeneration (Fig. 1e) (Gorbatyuk et al. 2010; Khodr et al. 2011; Kanaan and Manfredsson 2012; Collier et al. 2016). For example, delivery of AAV expressing α‐syn shRNA into the SNc of rats results in a rapid and robust loss of neurons in the SNc (Gorbatyuk et al. 2010). Here, there is a dose‐dependent loss of nigral neurons, in which the level of knockdown of endogenous α‐syn correlates with the amount of nigral neuron loss (Gorbatyuk et al. 2010). Importantly, the neuropathology and behavioral deficits observed are rescued by supplementation of rat α‐syn (rendered insensitive to the shRNA), demonstrating that the toxicity is explicitly because of the loss of endogenous α‐syn (Gorbatyuk et al. 2010). We have repeated this approach in mice (Fig. 3) and non‐human primates (Collier et al. 2016), as well as by using a microRNA approach in rats (Fig. 3). In every instance, regardless of the method or species used, a removal of endogenous α‐syn beyond a critical threshold produces SNc neurodegeneration. Other laboratories have also reported a pronounced loss of nigral neurons following AAV‐mediated delivery of an shRNA targeting α‐syn (Khodr et al. 2011), while an alternative report found that low level α‐syn knock down results in a 20% reduction in ST DA (Zharikov et al. 2015). Further, knock down of endogenous α‐syn in primary hippocampal neurons using antisense oligonucleotides decreases the size of the presynaptic vesicle pool (Murphy et al. 2000). However, not all studies show that reduction of endogenous α‐syn results in neuronal loss in vivo. For example, no toxicity was reported after knockdown of endogenous α‐syn in mouse hippocampus using naked siRNA (Lewis et al. 2008), however, there was no detailed analysis of cell loss or toxicity in that report. Additionally, delivery of siRNA targeting endogenous α‐syn to the midbrain of squirrel monkeys does not result in appreciable nigrostriatal degeneration (McCormack et al. 2010). These discrepant findings likely reflect differing levels of knockdown of endogenous α‐syn.
Figure 3.
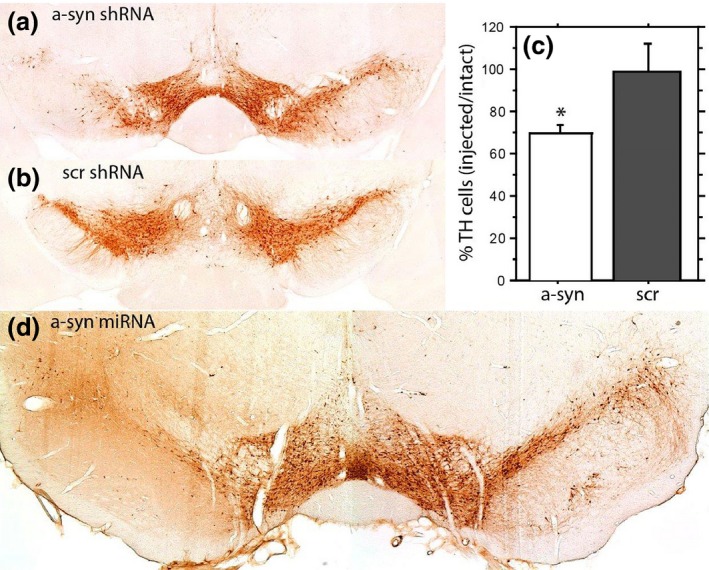
Nigral neurodegeneration due to loss of α‐syn translates across species and knockdown approaches. (a and b) Adult mice were injected in the left substantia nigra (SNc) with adeno‐associated virus (rAAV) 2/5 expressing a shRNA targeting mouse α‐syn (A) or a scrambled control shRNA(Scr) (B) (1.5 μL of 2.6 × 1012 vector genomes/mL). (c) 28‐days following vector injection animals were sacrificed and numbers of tyrosine hydroxylase neurons in the SNc were quantified using unbiased stereology. rAAV‐mediated expression of α‐syn shRNA results in an approximate 30% loss of neurons of the SNc (expressed as percent of intact hemisphere). * Indicates significantly different than scrambled shRNA control (p < 0.05). (d) Several microRNA sequences targeting rat α‐syn were evaluated in vitro. The microRNA achieving the most efficient knockdown of α‐syn in vitro was packaged into rAAV2/5 and injected in to the left SNc of adult rats (1.5 μL of 1 × 1012 vector genomes/mL). MicroRNA‐mediated knockdown of α‐syn results in a robust reduction of tyrosine hydroxylase neurons within the injected SNc.
There appears to be a critical threshold of α‐syn knockdown required in order to produce toxicity, and the severity of toxicity appears to be inversely proportional to the amount of endogenous α‐syn remaining (Gorbatyuk et al. 2010; Kanaan and Manfredsson 2012). In this sense, it seems likely that alternative reports did not observe toxicity because the level of α‐syn knockdown was either insufficient or not sustained for a requisite amount of time. For example, the maximum reduction in α‐syn protein reported following delivery of α‐syn siRNA to the squirrel monkey was 40% (McCormack et al. 2010). In contrast, toxicity in the rat was initially observed when α‐syn protein was decreased by ~ 70% following delivery of an α‐syn shRNA (Gorbatyuk et al. 2010). Further, although there was a 55% reduction in α‐syn protein following the direct injection of siRNA into the mouse hippocampus, knockdown persisted for only 1 week, after which time endogenous α‐syn rebounded to control levels (Lewis et al. 2008). As is the case in viral over‐expression models, mice seem to require longer periods of time to develop a corresponding level of toxicity observed in a rat (Fig. 3). Finally, as is evident with the neurodegeneration observed in PD, there is a differential susceptibility of central neuronal populations to α‐syn associated toxicity. It is likely that nigrostriatal neurons are particularly sensitive to loss of α‐syn, possibly for reasons that will be elaborated below.
Although there are not many studies reporting neurodegeneration following knockdown of endogenous α‐syn in vivo, there have been several descriptions of toxicity and cell loss after removal of endogenous α‐syn in vitro. For example, knockdown of α‐syn significantly decreases viability of SH‐SY5Y or MN9D DA cells (Liu et al. 2008; Han et al. 2011). Knockdown of endogenous α‐syn in SK‐MEL 28 cells increases levels of the pro‐apoptotic protein Bax, and reduces levels of the anti‐apoptotic proteins Bcl2 and Bcl‐xl, causing significantly reduced proliferative indices and increased sensitivity to staurosporine‐induced apoptosis (Choong and Say 2011). Knockdown of endogenous α‐syn in HeLa cells impairs mitochondrial calcium uptake, and results in an increase in fragmented mitochondria (Cali et al. 2012). Within that study, an increase in α‐syn expression was induced, resulting in the formation of α‐syn aggregates, presumably sequestering soluble α‐syn and reducing functional α‐syn levels within the cell (Cali et al. 2012). When α‐syn was sequestered in aggregates, the ability of mitochondria to buffer calcium was decreased, replicating the pathological effects observed with α‐syn knockdown (Cali et al. 2012). Finally, other studies have demonstrated a beneficial effect of α‐syn knockdown in cultured cells, which replicated studies in α‐syn null mice that demonstrate a resistance to MPP+ (Fountaine and Wade‐Martins 2007; Wu et al. 2009).
Seeding α‐syn aggregation; α‐syn Preformed Fibrils
In addition to genetic models of PD pathology, newer studies demonstrate that inoculating animals with preformed fibrils (PFFs) of recombinant α‐syn induces a progressive pattern of pathology that strongly resembles the synucleinopathy in the PD brain. As previously mentioned regarding the prion hypothesis of α‐syn, recombinant α‐syn can enter cultured cells via endocytosis to induce aggregation of endogenous α‐syn. Following seeding and aggregation, aberrant conformers of α‐syn are then released from cells. In theory, the released, misfolded/aggregated α‐syn may then initiate a new cycle of endocytosis and seeding of endogenous α‐syn in recipient cells, and through this series of events, propagate misfolded/aggregated α‐syn from cell to cell. α‐syn aggregates induced by treatment with recombinant PFFs display many of the same phenotypic markers of LB, such as co‐localization with ubiquitin, α‐syn phosphorylated at serine 129, and positive staining with Thioflavin S (Desplats et al. 2009; Luk et al. 2009, 2012a,b; Volpicelli‐Daley et al. 2011; Paumier et al. 2015). Additionally, similar to PD, in which the spatial and temporal pattern of toxicity is paralleled by α‐syn deposition into LB, aggregation of endogenous α‐syn induced by PFFs results in toxicity in experimental models. For instance, treatment of primary hippocampal neurons with PFFs results in recruitment of endogenous α‐syn into aggregates, and a subsequent decrease in SNARE proteins (Volpicelli‐Daley et al. 2011). Primary neurons exposed to PFFs also display a disruption of signaling and increased cell death (Volpicelli‐Daley et al. 2011). Similarly, α‐syn PFFs injected into either transgenic or WT mice or rats produce LB‐like α‐syn aggregates throughout the brain over time (Luk et al. 2009, 2012a,b; Paumier et al. 2015). Although the progressive accumulation of aggregated α‐syn in different brain regions over time may reflect a prion‐like spread, the proposed cell‐to‐cell transfer of α‐syn remains contentious, an alternative hypothesis is that the injected protein travels to distant neuronal nuclei in white matter tracts, thus inducing aggregation in the distant brain regions in the absence of cell‐to‐cell transfer. Nevertheless, as observed in PD, the presence of aggregated α‐syn throughout the brain preceeds pathology, with deposition of α‐syn aggregates in nigrostriatal neurons preceeding loss of striatal DA, TH, and DAT, subsequent death of SNc neurons, and a corresponding loss in motor performance (Luk et al. 2012a,b; Paumier et al. 2015). Interestingly, the toxicity induced by α‐syn PFFs, both in vitro and in vivo, is completely absent in the isolated neurons and brains of α‐syn null mice, reinforcing the concept that sequestration of endogenous α‐syn into aggregates appears to be a critical event in cell death associated with synucleinopathy.
Synucleinopathy in toxicant models of PD
Lastly, there have been reports of α‐syn aggregation in toxicant models. Specifically, chronic administration of rotenone (Betarbet et al. 2000), paraquat (Manning‐Bog et al. 2002), or MPTP (Fornai et al. 2005a) results in the formation of α‐syn aggregates. It is likely that the dramatic increase in oxidative stress observed after toxicant exposure produces a redox environment that favors α‐syn aggregation. However, results pertaining to the toxicity of these aggregates in view of the loss‐of‐function hypothesis are confounded by a myriad of other effects produced by these compounds. Yet, it should be noted that in some of these reports, α‐syn expression was up‐regulated following chronic toxicant exposure (Manning‐Bog et al. 2002), adding to a fairly large body of literature (discussed below) that suggest a novel role for α‐syn in response to cellular stress.
The α‐syn loss‐of‐function hypothesis
Overall, the lessons learned from PD patients and animal models of synucleinopathy demonstrate discrepancies. Arguably the most consistent trend throughout all α‐syn‐based research is a gene‐dosage effect between α‐syn expression and pathology. In SNCA multiplication patients as well as in transgenic and virally mediated over‐expression models, increasing the expression of α‐syn increases the aggregation profile of the protein, resulting in a dose‐dependent toxicity (Fig. 1d) (Masliah et al. 2000; Singleton et al. 2003; Chartier‐Harlin et al. 2004; Decressac et al. 2012; Gombash et al. 2013). In contrast, removal of endogenous α‐syn produces a similar pattern of toxicity (Fig. 1e). Here, we find that there is a dose‐dependent toxicity that is inversely proportional to the amount of endogenous α‐syn remaining (Gorbatyuk et al. 2010). While these findings may at first seem discordant, it is possible that the similar pathological profiles observed following either α‐syn over‐expression or knockdown reflect a single common underlying mechanism of toxicity, which is the loss of α‐syn function. This mechanism is obvious in the case of the toxicity associated with removal of α‐syn from neurons. However, toxicity originating from loss of necessary α‐syn function may appear somewhat counterintuitive in the context of synucleinopathy or α‐syn over‐expression models, where α‐syn seems to be abundantly present in affected cells. However, here one must distinguish between functional α‐syn versus misfolded or mislocalized α‐syn.
The α‐syn loss‐of‐function hypothesis posits that as α‐syn aggregation proceeds, endogenous functional α‐syn becomes sequestered into inclusions. The incorporation of functional α‐syn into inclusions; be they oligomers, fibrillar species, or mature LB or LN, would impede the ability of α‐syn to perform normal cellular functions, producing a loss of function form of toxicity (Fig. 1) (Perez et al. 2002; Perez and Hastings 2004; Cookson 2006; Kanaan and Manfredsson 2012; Wu et al. 2012). Hypothetically, α‐syn loss of function toxicity could result solely from sequestration into aggregates; however, changes in the subcellular localization of α‐syn could exacerbate this process. α‐syn is highly enriched in presynaptic terminals (Fig. 1a) (Iwai et al. 1995), while aggregated α‐syn is largely present in LB located in the soma (Fig. 1c and d). As such, incorporation of α‐syn into aggregates would decrease α‐syn in its normal subcellular compartment, further preventing soluble α‐syn from performing its normal functions. Mislocalization of α‐syn from the synapse to the soma is likely exacerbated with age, as there is a redistribution of α‐syn from the presynaptic terminal to soma in aging humans and monkeys (Fig. 1b) (Chu and Kordower 2007). This is in line with epidemiological studies that identify age as the primary risk factor for developing PD (Bennett et al. 1996; Tanner and Goldman 1996). Finally, loss of α‐syn function and subsequent toxicity would be expedited by mutations or multiplications of the SNCA gene, which increase the rate of aggregation, or directly impair function, depriving the cell of normal α‐syn functions (discussed more below).
Although the α‐syn loss‐of‐function hypothesis may seem counterintuitive at first glance, there is abundant evidence from the literature to support this theory. In particular, the pattern of toxicity and changes in the localization of endogenous α‐syn observed following exposure to α‐syn PFFs provides incontrovertible evidence that sequestration of endogenous α‐syn into aggregates is a critical event in toxicity. Indeed, as aggregation following PFF administration proceeds, a decline in cytoplasmic, soluble α‐syn is observed (Volpicelli‐Daley et al. 2011; Osterberg et al. 2015). An important aspect of all PFF studies reported thus far, is that the aggregates formed by treatment with PFFs, both in vitro and in vivo, are primarily composed of endogenous α‐syn, with very little of the initial PFF seed comprising the resulting inclusions. When Myc‐tagged α‐syn PFFs are used to seed aggregation, there is a small amount myc immunoreactivity in the center of the observed aggregates, however, the majority of α‐syn forming the aggregates is composed of untagged endogenous α‐syn (Luk et al. 2009). This point is further highlighted by the fact that the formation of aggregates, the propagation of aggregates, and the toxicity produced by recombinant α‐syn PFFs is completely absent in primary neurons or in the brains of α‐syn null mice (that do not have endogenous α‐syn to sequester) (Volpicelli‐Daley et al. 2011; Luk et al. 2012a). Alternatively, hemizygous α‐syn ± mice display an intermediate level of degeneration, demonstrating that the recruitment of endogenous α‐syn into aggregates is an essential component of aggregate formation and toxicity (Luk et al. 2012a). Further highlighting the sequestration of endogenous α‐syn: treatment of primary neuronal cultures with α‐syn PFFs results in a relocalization of endogenous α‐syn away from the synapse into perinuclear aggregates within the soma (Volpicelli‐Daley et al. 2011). This recruitment of endogenous α‐syn into aggregates results in an 80% decrease in soluble endogenous α‐syn in primary neurons (Volpicelli‐Daley et al. 2011). This result was replicated in vivo in an elegant study using multiphoton microscopy to image single neurons over time in living mice after they received injections of recombinant α‐syn PFFs. Inclusions undergo a maturation process in which early aggregates appear alongside soluble α‐syn, however, as inclusions become more compacted the normal homogenous immunoreactivity of soluble α‐syn is lost (Osterberg et al. 2015). The loss of soluble α‐syn into compact perinuclear aggregates coincides with the eventual death of the inclusion‐bearing cells (Osterberg et al. 2015). These results demonstrate the potential toxicity that can be elicited by reducing endogenous α‐syn below a critical threshold, and further supporting the α‐syn loss‐of‐function hypothesis.
Although there is ample evidence to support the α‐syn loss‐of‐function hypothesis, this theory is largely overlooked in favor of those ascribing α‐syn toxicity to a gain‐of‐function mechanism. Arguments supporting a direct toxicity resulting from α‐syn, and α‐syn aggregates are as follow: (i) Missense mutations in the SNCA gene result in an autosomal dominant pattern of inheritance. (ii) SNCA multiplication patients show a gene‐dosage effect in the severity of the PD phenotype. (iii) The pathological features of PD can be mimicked by the over‐expression of WT α‐syn. (iv) The brains of synucleinopathy patients show accumulation of excess α‐syn. (v) Most α‐syn null mice do not show overt neuropathological or behavioral phenotypes. These points provide a compelling argument for a direct mechanism of toxicity induced by α‐syn and associated aggregates; however, each individual argument can also be explained by the loss‐of‐function hypothesis as addressed below.
mutations in the SNCA gene result in an autosomal‐dominant pattern of inheritance.
Mutations in the SNCA gene could result in loss of functional α‐syn through two distinct means: First, mutations could impair the ability of α‐syn to form alpha helices and thus promote β sheet formation, thereby promoting aggregation. For example, the A53T, E46K, and H50Q mutations show drastically increased aggregation kinetics (Conway et al. 1998; Burré et al. 2010; Ghosh et al. 2013). In contrast, the A30P mutation does not increase the formation of mature fibrils over WT α‐syn, however, initial oligomerization of α‐syn is still dramatically increased (Conway et al. 1998; Li et al. 2001). In either scenario, the increased propensity of mutant α‐syn to self‐assemble into either mature fibrils or oligomers, could sequester soluble α‐syn into aggregates (including WT α‐syn from the unaffected allele), resulting in a depletion of functional α‐syn (Fig. 1c). Thus, an initial impetus promoting α‐syn aggregation could act in a dominant negative fashion to decrease both mutant and WT α‐syn protein levels below a critical threshold, resulting in toxicity by loss of α‐syn function, which would also produce a dominant pattern of inheritance. Second, the mutation itself could directly impair the ability of mutant α‐syn protein to perform its normal function. For example, the A30P, A53E, and G51D mutations impair the ability of α‐syn to interact with lipid membranes or localize to presynaptic membranes, directly resulting in a loss of α‐syn function (Conway et al. 1998; Fortin et al. 2004; Burré et al. 2010; Fares et al. 2014; Ghosh et al. 2014). Because of the myriad of vital functions of α‐syn performs, as well as the severe consequences observed following partial removal of α‐syn function from neurons of adult animals (Gorbatyuk et al. 2010), haploinsufficiency may be adequate to produce the dominant pattern of neurodegeneration observed in familial PD. This could particularly hold true for loss of α‐syn function within dopaminergic neurons (discussed below).
multiplication patients show a gene‐dosage effect in the severity of the PD phenotype, and,
pathological features of PD can be mimicked by over‐expression of WT α‐syn.
If PD pathology is mediated by a loss‐of‐function mechanism, then how can one account for the fact that increased SNCA expression causes toxicity? As mentioned above, data from patients with SNCA gene multiplications present a strong case for a gene‐dosage effect in which increased SNCA expression and α‐syn protein produces a more aggressive disease phenotype (Singleton et al. 2003; Chartier‐Harlin et al. 2004; Ibanez et al. 2004; Han et al. 2011). Further, lessons learned from α‐syn animal models show that ectopic over‐expression of α‐syn produces neurotoxicity in a dose‐dependent fashion (Masliah et al. 2000; Giasson et al. 2002; Gomez‐Isla et al. 2003; Fleming et al. 2005; Decressac et al. 2012; Gombash et al. 2013). As previously mentioned, increasing α‐syn protein concentrations, as well as general increases in molecular crowding, can both increase the aggregation kinetics of α‐syn (Conway et al. 1998; Uversky 2007). SNCA expression in multiplication patients has been confirmed to produce increased α‐syn protein (Miller et al. 2004), while α‐syn over‐expression models range from approximately 0.3–30 fold increases in α‐syn protein (Fleming et al. 2005). Accordingly, it is very likely that the increased α‐syn expression in multiplication patients, or α‐syn over‐expression in animal models, increases the aggregation kinetics of α‐syn, thus impairing the ability of α‐syn to perform its physiological function after sequestration into aggregates (Fig. 1c and d). Supporting this, the brains of SNCA triplication patients do not show any increase in soluble α‐syn, but exhibit a large increase in α‐syn aggregates (Miller et al. 2004). Further, levels of soluble α‐syn protein are decreased in the brains of patients with idiopathic PD (Baba et al. 1998; Quinn et al. 2012). This could also account for the much more aggressive disease phenotype seen in triplication carriers over duplication carriers, wherein increased α‐syn expression would accelerate α‐syn aggregation kinetics over that associated with SNCA duplication. Finally, ectopic over‐expression of α‐syn in animal models causes a progressive decline in soluble α‐syn protein levels with a concomitant increase in insoluble α‐syn, providing further support for the loss of function hypothesis (Volpicelli‐Daley et al. 2011; Osterberg et al. 2015). Thus, it appears that α‐syn exists in a state of tightly controlled homeostatic maintenance (Fig. 1). α‐syn is one of the most abundant proteins in the brain, yet neurons are exquisitely sensitive to changes in expression levels of α‐syn, with a small (fold) increase in expression able to produce widespread neurodegeneration and an aggressive disease phenotype. At the same time, small decreases in SNCA expression are also associated with disease severity. Supporting this is a recent study examining the relationship between SNCA expression and disease outcome in over 1000 well‐characterized PD patients. This study demonstrates that patients with the low repeat REP1 allele (259 base pairs; resulting in decreased SNCA expression), are associated with worse motor and cognitive outcomes, while the high‐repeat REP1 allele (263 base pairs; increases SNCA expression) results in better motor and cognitive outcomes (Markopoulou et al. 2014). Taken together, these data imply that α‐syn homeostasis is constantly balanced on a knife‐edge, where small perturbations that increase or decrease its expression produce neurotoxicity that is likely mediated through a common mechanism, the loss of normal α‐syn function.
The brains of synucleinopathy patients show accumulation of excess α‐syn.
It is well documented that although the brains of synucleinopathy patients do show accumulation of α‐syn, the majority of that α‐syn is aberrantly folded and/or contained within aggregates (Miller et al. 2004; Kramer and Schulz‐Schaeffer 2007; Quinn et al. 2012). Thus, despite the excess α‐syn in the brain, there are actually reduced levels of soluble, functional α‐syn available to perform its physiological functions.
A more difficult issue to resolve is whether α‐syn aggregates are directly toxic. If α‐syn aggregates are directly toxic, one would expect to see cell loss occurring with the initial presentation of the inclusions. If, however, aggregates produce toxicity by progressively sequestering soluble α‐syn until a critical threshold is lost, toxicity would appear following a necessary lag time needed to achieve a significant depletion of the soluble α‐syn pool. Indeed, there is widespread LB pathology in brain and spinal cords of presymptomatic PD patients, yet it is not until much later that symptoms and widespread cell loss are observed (Braak et al. 2003a; Halliday et al. 2006; Jellinger 2008). Next, as many as 8–12% of clinically normal individuals over the age of 60 have extensive LB pathology in the CNS, without a corresponding cell loss or PD symptoms (Forno 1969; Dickson et al. 2008). Further, in PD itself, there is a differential susceptibility of neuronal populations to the presence of α‐syn aggregates. Although late‐stage PD patients develop α‐syn containing LB throughout much of the body (Braak and Braak 2000; Braak et al. 2003b, 2006; Beach et al. 2010), only a particular subpopulation of neurons with aggregated α‐syn display neurodegeneration and cell death. For example, the formation of α‐syn inclusion within neurons of the enteric nervous system correlates with GI dysfunction in PD, however, there has never been any documentation of neuronal cell loss in the enteric nervous system. This is also observed in animal models where over‐expression of rat α‐syn in rats results in α‐syn aggregate formation but no cell loss (Bianco et al. 2002), presumably because continued expression of rat α‐syn maintains synuclein function despite the presence of aggregates. Moreover, there are LB in many surviving neurons in the brains of PD patients, suggesting that mature LB may actually represent a protective effort of the cell to sequester aggregate prone α‐syn away from the site (s) of action where α‐syn function is needed, i.e. the presynaptic terminal. Here, it becomes important to distinguish between mature highly processed and compacted LB, and self‐assembled oligomeric or fibrillar α‐syn. The latter would likely perpetuate a cycle of aggregation by serving as templating seeds within the neuron. Supporting this idea is evidence demonstrating that presynaptic α‐syn aggregates, not LB, cause neurodegeneration in models of synucleinopathy (Kramer and Schulz‐Schaeffer 2007). Further, some studies have even reported that neurons containing LB in late‐stage PD appear morphologically normal as compared to neurons without LB (Gertz et al. 1994; Tompkins et al. 1997). Finally, abundant literature describes α‐syn‐mediated toxicity associated with a number of key cellular processes such as mitochondrial function, calcium buffering, and protein clearance. Here, it seems possible that a loss‐of‐function toxicity is misinterpreted as a direct toxicity induced by α‐syn. For example, as mentioned above, both aggregation and knockdown of α‐syn impair the ability of mitochondria to buffer calcium, implying a common toxicity induced by loss of α‐syn function (Cali et al. 2012). However, many of these pathological changes could actually represent downstream toxic effects caused by a loss of α‐syn function. As will be discussed in the following section, loss of α‐syn could also cause DA‐associated toxicity, which would negatively affect virtually every organelle in a dopaminergic neuron.
α‐syn null mice do not show overt neuropathological or behavioral phenotypes.
Although α‐syn null mice show deficits in vesicular dynamics and synaptic transmission, admittedly the pathological changes observed in α‐syn null mice are modest, and do not reflect the proposed consequences resulting from loss of crucial α‐syn function. However, this could result from an effect of developmental genetic compensation resulting from germline removal of the SNCA gene. This notion is supported by several studies. First, unlike germline knockout of SNCA, removal of endogenous α‐syn from mature neurons in adult animals results in severe neurodegeneration (Gorbatyuk et al. 2010; Khodr et al. 2011; Zharikov et al. 2015; Collier et al. 2016) (Fig. 3). Second, genetic compensation resulting from germline removal of the endogenous SNCA gene is well documented. As compared to WT littermates, α‐syn null mice have 369 transcripts differentially expressed (Kuhn et al. 2007). These include increases in 14‐3‐3 and TH expression (proteins that both interact with α‐syn) (Ostrerova et al. 1999; Perez et al. 2002; Wang et al. 2009), decreases in the expression of apoptotic genes, altered expression of neurotrophic factors like Brain derived neurotrophic factor (BDNF) (Yuan et al. 2010), as well as differential expression of genes involved in vesicular function (Kuhn et al. 2007; Ubhi et al. 2010). Further, as was mentioned above, there is also a compensatory increase in other synuclein family members, which suggests functional redundancy could mask the effects of silencing α‐syn expression (Schlüter et al. 2003; Chandra et al. 2004; Robertson et al. 2004). As a result of the plethora of differentially regulated genes in α‐syn null mice, and direct evidence that manipulating α‐syn expression in adult animals produces more severe consequences than those observed by germline manipulations, drawing conclusions based solely from α‐syn null animals could lead to gross underestimation of the impact of loss of α‐syn function.
Potential mechanisms of α‐syn Loss‐of‐function toxicity
Though the precise physiological function of α‐syn has not been established, α‐syn does play a key role in many functions involved in synaptic transmission, with a potentially specialized role in the dopaminergic presynaptic terminal. Based on a survey of the literature, α‐syn plays a role in regulating the size of the vesicular pool, vesicular trafficking to‐ and docking with the presynaptic membrane, as well as subsequent clathrin‐associated formation of synaptic vesicles. In addition, α‐syn appears to play a crucial role in the regulation of DA biosynthesis and handling (see α‐syn function section above). Based on our model of α‐syn acting as a regulatory brake on dopaminergic synaptic transmission (Fig. 2), a loss of α‐syn function would produce dysregulated DA handling and subsequent DA associated toxicity (Fig. 2c).
Loss of α‐syn function has been proposed to result in increased cytosolic DA through several distinct mechanisms (Perez et al. 2002; Perez and Hastings 2004; Wu et al. 2012) (Fig. 2): First, loss of α‐syn from the dopaminergic terminal can disinhibit TH and AADC, resulting in increased de novo DA synthesis (Perez et al. 2002; Perez and Hastings 2004; Yu et al. 2004; Tehranian et al. 2006; Alerte et al. 2008; Lou et al. 2010). Second, loss of α‐syn decreases the size of the vesicular pool (Murphy et al. 2000; Cabin et al. 2002), as well as levels of VMAT, and causes dysregulation of synaptic vesicle trafficking and recycling (Maroteaux et al. 1988; Fountaine et al. 2008; Burré et al. 2010, 2012; Vargas et al. 2014). The net result of these effects would be increased free cytosolic DA, with a concomitant inability to sequester the excess DA in synaptic vesicles. Cytosolic DA readily auto‐oxidizes to produce reactive oxygen species as well as the highly reactive DA quinone, ultimately resulting in neurotoxicity (Fig. 2c) (Graham et al. 1978; Hermida‐Ameijeiras et al. 2004). Further, DA oxidation and associated changes in the cellular redox environment could inhibit the electron transport chain, resulting in further oxidative stress by opening the mitochondrial permeability pore (Berman and Hastings 1999; Khan et al. 2005), as well as causing a decreased ability of the mitochondrial enzymes MAO‐B and aldehyde dehydrogenase to convert DA to Dihydroxyphenylacetic acid (DOPAC). Finally, increased reactive oxygen species as well as direct interactions between DA‐α‐syn adducts are able to promote α‐syn aggregation (Hashimoto et al. 1999; Conway et al. 2001), further depleting the cell of functional α‐syn. Taken together such changes initiated by a loss of α‐syn function would create a feed‐forward cycle of neurotoxicity, ultimately resulting in DA cell loss. Importantly, this increase in oxidative stress can produce phenomena such as mitochondrial inhibition and dysfunction of the protein clearance machinery (Höglinger et al. 2003; Porras and Perez 2014), many of which have been attributed to an α‐syn toxic gain of function (Wang et al. 2009).
Our proposed mechanism of α‐syn loss of function toxicity is further supported by studies demonstrating that apoptosis associated with α‐syn over‐expression depends on endogenous DA production, while α‐syn over‐expression in DA‐depleted cells is not toxic, and that α‐syn may actually be neuroprotective in non‐dopaminergic cells (Xu et al. 2002). In addition, the activity of the TH‐modulating protein phosphatase, PP2A, is decreased in the brains of α‐syn triplication patients and patients with DLB in regions with robust α‐syn aggregation (Wu et al. 2012), lending further support to the idea that aggregation of α‐syn may cause unregulated DA production. DA associated toxicity following the loss of α‐syn function could also account for the differential susceptibility of central DA neurons to synucleinopathy.
Again, aggregated α‐syn is observed throughout many neuronal populations, yet only certain pools of neurons, primarily catecholaminergic nuclei, show significant cell loss. It is thus likely that the neurotransmitter used within a given neuron directly contributes to its susceptibility caused by loss of α‐syn function. The associated increased cytosolic DA results in reactive oxygen species that would not occur in neuronal subpopulations that utilize other, less volatile neurotransmitters. This mechanism of toxicity is also supported by studies documenting the temporal pattern of PD pathology in which dopaminergic fiber loss precedes the actual loss of nigrostriatal neurons (Kordower et al. 2013). Within years of manifestating motor symptoms, there is an approximate 70–90% loss of nigrostriatal DA fibers in the ST, while at this same time, point up to 40–70% of SNc neuronal cell bodies remain intact (Kordower et al. 2013). This ‘dying back’ neurotoxicity in PD could be associated with a loss of α‐syn function and subsequent DA toxicity originating in presynaptic DA terminals.
An additional mechanism whereby loss of α‐syn function produces neurodegeneration is by a diminished ability to handle cellular stress. Many studies have shown that α‐syn is up‐regulated following cellular stress, suggesting a neuroprotective role for α‐syn. α‐syn is up‐regulated in neuroblastoma cells in response to endoplasmic reticulum or proteasomal stress (Häbig et al. 2009), exposure to mitochondrial toxins in vitro and in vivo (Manning‐Bog et al. 2002; Fountaine and Wade‐Martins 2007), or drugs that specifically target catecholaminergic terminals (Fornai et al. 2005b). Indeed, increased α‐syn expression protects against a variety of cellular stressors, including DA toxicity, oxidative stress, staurosporine‐induced apoptosis, and serum deprivation (Alves da Costa et al. 2002; Wersinger and Sidhu 2003b; Zourlidou et al. 2003; Quilty et al. 2006). Moreover, α‐syn expression drastically lowers the apoptotic response of neuronal cells by decreasing both p53 expression and p53 transcriptional activity, and this effect is lost when soluble α‐syn becomes sequestered in aggregates (Alves da Costa et al. 2002). Taken together, loss of α‐syn function likely results in an increase in DA‐mediated cellular stressors, along with a concomitant decrease in the ability of neuronal cells to rebound from the same toxicity.
Discrepancies and lessons learned from other Proteins
Although the α‐syn loss‐of‐function hypothesis can reconcile much of the conflicting published data, there are some findings that seem irreconcilable with toxicity caused by loss of α‐syn function. Many of these come from data in α‐syn animal models. For example, several α‐syn over‐expressing transgenic mouse lines develop pathology in the absence of large α‐syn aggregates (Matsuoka et al. 2001; Richfield et al. 2002; Thiruchelvam et al. 2004). Alternatively, in one α‐syn over‐expressing transgenic line, large aggregates of human α‐syn are detected, however, they are devoid of endogenous murine α‐syn (Masliah et al. 2000). Aggregates containing endogenous α‐syn would seem to be a prerequisite if toxicity resulted following the sequestration of functional α‐syn into aggregates. However, these findings are rare and may reflect either; 1) the use of insufficiently sensitive techniques to identify species‐specific α‐syn or detect microaggregates or, 2) endogenous α‐syn oligomers or immature aggregate species that inhibit normal α‐syn function but are undetectable via histological techniques.
Another potential flaw of the loss‐of‐function hypothesis is studies reporting toxicity as a result of the aggregation of human α‐syn in animals lacking endogenous α‐syn. For example, Drosophila and C. elegans do not express endogenous α‐syn, nor is there currently a known homolog of α‐syn in these animals. Nonetheless, transgenic Drosophila or C. elegans expressing human α‐syn develop α‐syn aggregates and subsequent toxicity (Feany and Bender 2000; Lakso et al. 2003). Further, over‐expression of mutant A53T human α‐syn in α‐syn null mice results in toxicity that exceeds the effects of A53T human α‐syn over‐expression in WT mice (Cabin et al. 2005). Because of the fact that these animals do not express endogenous α‐syn, there is no endogenous α‐syn to be lost to aggregation. Here, it is possible that an as of yet unidentified homolog of α‐syn, or some other essential protein, gets sequestered into aggregates, resulting in similar pattern of loss‐of‐function toxicity to that proposed herein with α‐syn. Supporting this possibility, a potential α‐syn homolog has been identified in Drosophila (Shin et al. 2000). Further, co‐expression of a chaperone protein such as Hsp70, completely prevents toxicity induced by human α‐syn over‐expression in Drosophila (Auluck et al. 2002). Importantly, although co‐expression of Hsp70 prevents toxicity, it does not alter the size, number, or morphology of the human α‐syn aggregates, suggesting that it is preventing toxicity by maintaining the proper concentrations and conformation of an endogenous Droshophila protein that, when lost, produces toxicity (Auluck et al. 2002).
Oligomeric or protofibrillar α‐syn is thought to have the ability to puncture lipid membranes. Protofibrillar α‐syn also has enhanced affinity for lipid membranes and forms pore‐like structures in membranes (Volles et al. 2001; Caughey and Lansbury 2003). Further, mature fibrils of α‐syn permeabilize membranes (Pieri et al. 2012), while familial mutations in α‐syn enhance its tendency to form pores in membranes (Volles et al. 2001) Permeabilization of membranes has been hypothesized to cause membrane leakiness, allowing unregulated passage of ions and small (potentially toxic) macromolecules between the intra‐ and extracellular space, and/or between subcellular compartments, ultimately resulting in toxicity. Although the vast majority of work demonstrating α‐syn‐induced membrane leakiness has been performed in vitro with supraphysiological concentrations of α‐syn, this could represent a potential toxic gain of function.
Finally, inhibition of the autophagy–lysosomal pathway by mutant forms of α‐syn may also represent a potential gain of function toxicity. WT α‐syn is selectively translocated to the lysosome for active degradation by chaperone‐mediated autophagy (Cuervo et al. 2004). However, both the A53T and the A30P α‐syn mutations appear to inhibit their own degradation, as well as the degradation of other substrates, by this pathway (Cuervo et al. 2004). Accordingly, the subsequent accumulation of α‐syn, as well as other damaged or potentially toxic proteins, has been proposed as a possible gain of function mechanism underlying the toxicity of these α‐syn mutants (Cuervo et al. 2004).
Taken together, these findings suggest that α‐syn may have a two‐pronged mechanism of toxicity developing independently and over many years during disease pathogenesis. We believe that it is likely that a significant amount of toxicity results from loss of α‐syn function; however, it is also likely that a pathological cascade of protofibrillar or fibrillar α‐syn exerts a degree of toxicity as well. Importantly, the idea that a single protein could cause toxicity by both loss‐ and gain of function is not unique to α‐syn.
Other proteins associated with the pathogenies of neurological disorders have demonstrated a multifaceted mechanism of toxicity. For example, it has been hypothesized that accumulation of hyperphosphorylated tau directly results in the pathological sequellae observed in AD. Strikingly similar to the story of α‐syn described above, the tau hypothesis is supported by the fact that 1) neurofibrillary tangle burden correlates with cognitive loss and disease progression (Braak and Braak 1991), 2) mutations in the gene encoding tau result in disease‐related tauopathies (Hutton et al. 1998), 3) tau knockout animals do not exhibit premature mortality or major neurological deficits (Morris et al. 2011), and 4) the induction of neurofibrillary tangles in worms, flies, and rodents mimic the neurodegeneration observed in human disease (Trojanowski and Lee 2005; Götz et al. 2007). However, there is now significant evidence to suggest that loss of tau function, following sequestration into Neurofibrillary Tangle (NFTs) or by inhibition following its hyperphosphorylation, contributes significantly to AD pathology (Morris et al. 2011; Bin Zhang et al. 2005; Trojanowski and Lee 2005; Santacruz et al. 2005). Indeed, the current debate in the tau field is centered on what function(s) of tau is lost in disease (e.g. microtubule stability or associated axonal transport) and what toxic gain of function(s) occur (e.g. impairing protein degradation machinery or signaling dysregulation) that lead to neurodegeneration. Because of these remarkable similarities between tau and α‐syn, it is somewhat surprising that the contribution of decreased α‐syn function to PD pathogenesis is not more widely accepted.
Conclusions
It is clear that α‐syn plays an integral role in the pathology of PD; however, the mechanism(s) whereby aberrant α‐syn homeostasis causes neurodegeneration remain unclear. Here, we have summarized the existing data that demonstrate how a loss of function may represent a major mechanism underlying α‐syn toxicity. As science has only recently begun to understand some of the biological functions of α‐syn, research examining the contribution of α‐syn to PD‐associated pathology has understandably been undertaken with a rather myopic focus on gain‐of‐function mechanisms following α‐syn aggregation. However, as our understanding of the normal biology of α‐syn matures, it is increasingly evident that α‐syn is a multifunctional chaperone‐like protein that contributes to essential cellular processes. Because of the complexity of α‐syn biology and pathology, it is essential to further elucidate and validate the normal functions of α‐syn in order to truly appreciate how those mechanisms could underly α‐syn toxicity. In doing so, we can work to develop novel therapeutics aimed at preventing α‐syn aggregation while also maintaining α‐syn function (Vargas‐Medrano et al. 2014), in order to provide the most efficacious disease modifying therapies possible.
Acknowledgments and conflict of interest disclosure
This work was supported by Mercy Health Saint Mary's (FPM), the Parkinson's Disease Foundation (MJB), and funds from the Texas Tech University Health Sciences Center El Paso (RGP).
The authors have no conflict of interest to declare.
Work was completed in Grand Rapids MI, USA.
References
- Abeliovich A. A., Schmitz Y. Y., Fariñas I. I. et al (2000) Mice lacking alpha‐synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 14–14. [DOI] [PubMed] [Google Scholar]
- Adamczyk A., Solecka J. and Strosznajder J. B. (2005) Expression of alpha‐synuclein in different brain parts of adult and aged rats. J. Physiol. Pharmacol. 56, 29–37. [PubMed] [Google Scholar]
- Ahmad B., Chen Y. and Lapidus L. J. (2012) Aggregation of alpha‐synuclein is kinetically controlled by intramolecular diffusion. PNAS 109, 2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn T. B., Kim S. Y., Kim J. Y. et al (2008) alpha‐Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70, 43–49. [DOI] [PubMed] [Google Scholar]
- Alerte T. N. M., Akinfolarin A. A., Friedrich E. E., Mader S. A., Hong C.‐S. and Perez R. G. (2008) alpha‐synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci. Lett. 435, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves da Costa C., Paitel E., Vincent B. and Checler F. (2002) Alpha‐synuclein lowers p53‐dependent apoptotic response of neuronal cells. Abolishment by 6‐hydroxydopamine and implication for Parkinson's disease. J. Biol. Chem. 277, 50980–50984. [DOI] [PubMed] [Google Scholar]
- Al‐Wandi A., Ninkina N., Millership S., Williamson S. J. M., Jones P. A. and Buchman V. L. (2010) Absence of α‐synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 31, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel‐Cresswell S., Vilarino‐Guell C., Encarnacion M. et al (2013) Alpha‐synuclein p. H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 28, 811–813. [DOI] [PubMed] [Google Scholar]
- Arai K., Kato N., Kashiwado K. and Hattori T. (2000) Pure autonomic failure in association with human alpha‐synucleinopathy. Neurosci. Lett. 296, 171–173. [DOI] [PubMed] [Google Scholar]
- Arawaka S., Saito Y., Murayama S. and Mori H. (1998) Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha‐synuclein. Neurology 51, 887–889. [DOI] [PubMed] [Google Scholar]
- Auluck P. K., Chan H. Y. E., Trojanowski J. Q., Lee V. M. Y. and Bonini N. M. (2002) Chaperone suppression of alpha‐synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865–868. [DOI] [PubMed] [Google Scholar]
- Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q. and Iwatsubo T. (1998) Aggregation of alpha‐synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879. [PMC free article] [PubMed] [Google Scholar]
- Barbour R., Kling K., Anderson J. P. et al (2008) Red blood cells are the major source of alpha‐synuclein in blood. Neurodegener. Dis. 5, 55–59. [DOI] [PubMed] [Google Scholar]
- Bartels T., Choi J. G. and Selkoe D. J. (2011) α‐Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach T. G., Adler C. H., Lue L. et al (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 117, 613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach T. G., Adler C. H., Sue L. I. et al (2010) Multi‐organ distribution of phosphorylated alpha‐synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 119, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor J. T., Logan T. P. and Edwards R. H. (2013) The Function of α‐Synuclein. Neuron 79, 1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett D. A., Beckett L. A., Murray A. M., Shannon K. M., Goetz C. G., Pilgrim D. M. and Evans D. A. (1996) Prevalence of parkinsonian signs and associated mortality in a community population of older people. N. Engl. J. Med. 334, 71–76. [DOI] [PubMed] [Google Scholar]
- Berman S. B. and Hastings T. G. (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J. Neurochem. 73, 1127–1137. [DOI] [PubMed] [Google Scholar]
- Bertoncini C. W., Jung Y.‐S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M. and Zweckstetter M. (2005) Release of long‐range tertiary interactions potentiates aggregation of natively unstructured alpha‐synuclein. PNAS 102, 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R., Sherer T. B., MacKenzie G., Garcia‐Osuna M., Panov A. V. and Greenamyre J. T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Beyer K., Lao J. I., Carrato C., Mate J. L., Lopez D., Ferrer I. and Ariza A. (2004) Differential expression of alpha‐synuclein isoforms in dementia with Lewy bodies. Neuropathol. Appl. Neurobiol. 30, 601–607. [DOI] [PubMed] [Google Scholar]
- Beyer K., Humbert J., Ferrer A., Lao J. I., Carrato C., Lopez D., Ferrer I. and Ariza A. (2006) Low alpha‐synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. NeuroReport 17, 1327–1330. [DOI] [PubMed] [Google Scholar]
- Bezard E., Dovero S., Prunier C., Ravenscroft P., Chalon S., Guilloteau D., Crossman A. R., Bioulac B., Brotchie J. M. and Gross C. E. (2001) Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐lesioned macaque model of Parkinson's disease. J. Neurosci. 21, 6853–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco Lo. C., Ridet J.‐L., Schneider B. L., Deglon N. and Aebischer P. (2002) alpha ‐Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral‐based model of Parkinson's disease. PNAS 99, 10813–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H. and Braak E. (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H. and Braak E. (2000) Pathoanatomy of Parkinson's disease. J. Neurol. 247, II3–II10. [DOI] [PubMed] [Google Scholar]
- Braak H., Del Tredici K., Rüb U., deVos R. A. I. , Jansen Steur E. N. H. and Braak E. (2003a) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Braak H., Rüb U., Gai W. P. and Del Tredici K. (2003b) Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 110, 517–536. [DOI] [PubMed] [Google Scholar]
- Braak H., de Vos R. A. I., Bohl J. and Del Tredici K. (2006) Gastric alpha‐synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease‐related brain pathology. Neurosci. Lett. 396, 67–72. [DOI] [PubMed] [Google Scholar]
- Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M. R. and Südhof T. C. (2010) Alpha‐synuclein promotes SNARE‐complex assembly in vivo and in vitro. Science 329, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J., Sharma M. and Südhof T. C. (2012) Systematic mutagenesis of α‐synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci. 32, 15227–15242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussell R. and Eliezer D. (2003) A structural and functional role for 11‐mer repeats in alpha‐synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 329, 763–778. [DOI] [PubMed] [Google Scholar]
- Cabin D. E., Shimazu K., Murphy D. et al (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha‐synuclein. J. Neurosci. 22, 8797–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin D. E., Gispert‐Sanchez S., Murphy D., Auburger G., Myers R. R. and Nussbaum R. L. (2005) Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol. Aging 26, 25–35. [DOI] [PubMed] [Google Scholar]
- Cali T., Ottolini D., Negro A. and Brini M. (2012) α‐Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum‐mitochondria interactions. J. Biol. Chem. 287, 17914–17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B. and Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. [DOI] [PubMed] [Google Scholar]
- Chandra S. S., Chen X. X., Rizo J. J., Jahn R. R. and Südhof T. C. T. (2003) A broken alpha ‐helix in folded alpha ‐Synuclein. J. Biol. Chem. 278, 15313–15318. [DOI] [PubMed] [Google Scholar]
- Chandra S., Fornai F., Kwon H.‐B. et al (2004) Double‐knockout mice for alpha‐ and beta‐synucleins: effect on synaptic functions. PNAS 101, 14966–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S., Gallardo G., Fernández‐Chacón R., Schlüter O. M. and Südhof T. C. (2005) Alpha‐synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. [DOI] [PubMed] [Google Scholar]
- Chartier‐Harlin M.‐C., Kachergus J., Roumier C. et al (2004) Alpha‐synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Chaudhuri K. R., Healy D. G. and Schapira A. (2006) Non‐motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. 3, 235–245. [DOI] [PubMed] [Google Scholar]
- Chen X. H., Desilva H., Pettenati M. J., Rao P. N., Stgeorgehyslop P., Roses A. D., Xia Y., Horsburgh K., Uéda K. and Saitoh T. (1995) The human Nacp/alpha‐synuclein gene ‐ chromosome assignment to 4q21.3‐Q22 and Taqi Rflp analysis. Genomics 26, 425–427. [DOI] [PubMed] [Google Scholar]
- Chiba‐Falek O. and Nussbaum R. L. (2001) Effect of allelic variation at the NACP‐Rep1 repeat upstream of the alpha‐synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 10, 3101–3109. [DOI] [PubMed] [Google Scholar]
- Chiba‐Falek O., Lopez G. J. and Nussbaum R. L. (2006) Levels of alpha‐synuclein mRNA in sporadic Parkinson disease patients. Mov. Disord. 21, 1703–1708. [DOI] [PubMed] [Google Scholar]
- Choong C.‐J. and Say Y.‐H. (2011) Knockdown of alpha‐synuclein enhances susceptibility to staurosporine‐induced apoptosis in human melanoma SK‐MEL 28 cells. J. Biol. Sci. 11, 135–145. [Google Scholar]
- Chu Y. and Kordower J. H. (2007) Age‐associated increases of alpha‐synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson's disease? Neurobiol. Dis. 25, 134–149. [DOI] [PubMed] [Google Scholar]
- Chung C. Y., Koprich J. B., Siddiqi H. and Isacson O. (2009) Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha‐synucleinopathy. J. Neurosci. 29, 3365–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier T. J., Kanaan N. M. and Kordower J. H. (2011) Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non‐human primates. Nat. Rev. Neurosci. 12, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier T. J., redmond D. E., Steece‐Collier K., lipton J. W. and Manfredsson F. P. (2016) Is alpha‐synuclein loss‐of‐function a contributor to parkinsonian pathology? Evidence from non‐human primates. Front. Neurosci. 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway K. A., Harper J. D. and Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant alpha‐synuclein linked to early‐onset Parkinson disease. Nat. Med. 4, 1318–1320. [DOI] [PubMed] [Google Scholar]
- Conway K. A., Rochet J. C., Bieganski R. M. and Lansbury P. T. (2001) Kinetic stabilization of the alpha‐synuclein protofibril by a dopamine‐alpha‐synuclein adduct. Science 294, 1346–1349. [DOI] [PubMed] [Google Scholar]
- Cookson M. R. (2006) Hero versus antihero: the multiple roles of alpha‐synuclein in neurodegeneration. Exp. Neurol. 199, 238–242. [DOI] [PubMed] [Google Scholar]
- Cronin K. D., Ge D., Manninger P., Linnertz C. et al (2009) Expansion of the Parkinson disease‐associated SNCA‐Rep1 allele upregulates human alpha‐synuclein in transgenic mouse brain. Hum. Mol. Genet. 18, 3274–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T. and Sulzer D. (2004) Impaired degradation of mutant alpha‐synuclein by chaperone‐mediated autophagy. Science 305, 1292–1295. [DOI] [PubMed] [Google Scholar]
- Dächsel J. C., Lincoln S. J., Gonzalez J., Ross O. A., Dickson D. W. and Farrer M. J. (2007) The ups and downs of alpha‐synuclein mRNA expression. Mov. Disord. 22, 293–295. [DOI] [PubMed] [Google Scholar]
- Dauer W. and Przedborski S. (2003) Parkinson's Disease: mechanisms and models. Neuron 39, 889–909. [DOI] [PubMed] [Google Scholar]
- Dauer W., Kholodilov N., Vila M. et al (2002) Resistance of alpha‐synuclein null mice to the parkinsonian neurotoxin MPTP. PNAS 99, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson W. S., Jonas A., Clayton D. F. and George J. M. (1998) Stabilization of alpha‐synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- Decressac M., Mattsson B., Lundblad M., Weikop P. and Bjorklund A. (2012) Progressive neurodegenerative and behavioural changes induced by AAV‐mediated overexpression of alpha‐synuclein in midbrain dopamine neurons. Neurobiol. Dis. 45, 939–953. [DOI] [PubMed] [Google Scholar]
- Desplats P., Lee H.‐J., Bae E.‐J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E. and Lee S.‐J. (2009) Inclusion formation and neuronal cell death through neuron‐to‐neuron transmission of alpha‐synuclein. PNAS 106, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U., Newman A. J., Luth E. S., Bartels T. and Selkoe D. (2013) In vivo cross‐linking reveals principally oligomeric forms of α‐synuclein and β‐synuclein in neurons and non‐neural cells. J. Biol. Chem. 288, 6371–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U., Newman A. J., von Saucken V. E., Bartels T. and Selkoe D. (2015) KTKEGV repeat motifs are key mediators of normal a‐synuclein tetramerization: their mutation causes excess monomers and neurotoxicity. Proc. Natl Acad. Sci. 112, 9596–9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson D. W., Fujishiro H., DelleDonne A. et al (2008) Evidence that incidental Lewy body disease is pre‐symptomatic Parkinson's disease. Acta Neuropathol. 115, 437–444. [DOI] [PubMed] [Google Scholar]
- Dickson D. W., Fujishiro H., Orr C., DelleDonne A., Josephs K. A., Frigerio R., Burnett M., Parisi J. E., Klos K. J. and Ahlskog J. E. (2009) Neuropathology of non‐motor features of Parkinson disease. Parkinsonism Relat. Disord. 15(Suppl 3), S1–S5. [DOI] [PubMed] [Google Scholar]
- Drolet R. E., Behrouz B., lookingland K. J. and Goudreau J. L. (2004) Mice lacking alpha‐synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology 25, 761–769. [DOI] [PubMed] [Google Scholar]
- Dyson H. J. and Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. [DOI] [PubMed] [Google Scholar]
- Edwards T. L., Scott W. K., Almonte C. et al (2010) Genome‐wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 74, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis C. E., Murphy E. J., Mitchell D. C., Golovko M. Y., Scaglia F., Barceló‐Coblijn G. C. and Nussbaum R. L. (2005) Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha‐synuclein. Mol. Cell. Biol. 25, 10190–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslamboli A., Romero‐Ramos M., Burger C., Bjorklund T., Muzyczka N., Mandel R. J., Baker H., Ridley R. M. and Kirik D. (2007) Long‐term consequences of human alpha‐synuclein overexpression in the primate ventral midbrain. Brain 130, 799–815. [DOI] [PubMed] [Google Scholar]
- Fares M.‐B., Ait‐Bouziad N., Dikiy I. et al (2014) The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α‐synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell K. F., Krishnamachari S., Villanueva E., Lou H., Alerte T. N. M., Peet E., Drolet R. E. and Perez R. G. (2014) Non‐motor parkinsonian pathology in aging A53T alpha‐Synuclein mice is associated with progressive synucleinopathy and altered enzymatic function. J. Neurochem. 128, 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M., Maraganore D. M., Lockhart P., Singleton A., Lesnick T. G., de Andrade M., West A., de Silva R., Hardy J. and Hernandez D. (2001) alpha‐Synuclein gene haplotypes are associated with Parkinson's disease. Hum. Mol. Genet. 10, 1847–1851. [DOI] [PubMed] [Google Scholar]
- Farrer M., Kachergus J., Forno L. et al (2004) Comparison of kindreds with parkinsonism and alpha‐synuclein genomic multiplications. Ann. Neurol. 55, 174–179. [DOI] [PubMed] [Google Scholar]
- Feany M. B. and Bender W. W. (2000) A Drosophila model of Parkinson's disease. Nature 404, 394–398. [DOI] [PubMed] [Google Scholar]
- Fleming S. M., Fernagut P.‐O. and Chesselet M.‐F. (2005) Genetic mouse models of parkinsonism: strengths and limitations. NeuroRx 2, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming S. M., Tetreault N. A., Mulligan C. K., Hutson C. B., Masliah E. and Chesselet M.‐F. (2008) Olfactory deficits in mice overexpressing human wildtype alpha‐synuclein. Eur. J. Neurosci. 28, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornai F. F., Schlüter O. M. O., Lenzi P. P. et al (2005a) Parkinson‐like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin‐proteasome system and alpha‐synuclein. PNAS 102, 3413–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornai F., Lenzi P., Ferrucci M. et al (2005b) Occurrence of neuronal inclusions combined with increased nigral expression of alpha‐synuclein within dopaminergic neurons following treatment with amphetamine derivatives in mice. Brain Res. Bull. 65, 405–413. [DOI] [PubMed] [Google Scholar]
- Forno L. S. (1969) Concentric hyalin intraneuronal inclusions of Lewy type in the brains of elderly persons (50 incidental cases): relationship to parkinsonism. J. Am. Geriatr. Soc. 17, 557–575. [DOI] [PubMed] [Google Scholar]
- Fortin D. L., Troyer M. D., Nakamura K., Kubo S., Anthony M. D. and Edwards R. H. (2004) Lipid rafts mediate the synaptic localization of alpha‐synuclein. J. Neurosci. 24, 6715–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D. L., Nemani V. M., Voglmaier S. M., Anthony M. D., Ryan T. A. and Edwards R. H. (2005) Neural activity controls the synaptic accumulation of alpha‐synuclein. J. Neurosci. 25, 10913–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountaine T. M. and Wade‐Martins R. (2007) RNA interference‐mediated knockdown of alpha‐synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 85, 351–363. [DOI] [PubMed] [Google Scholar]
- Fountaine T. M., Venda L. L., Warrick N., Christian H. C., Brundin P., Channon K. M. and Wade‐Martins R. (2008) The effect of alpha‐synuclein knockdown on MPP plus toxicity in models of human neurons. Eur. J. Neurosci. 28, 2459–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredenburg R. A., Rospigliosi C., Meray R. K., Kessler J. C., Lashuel H. A., Eliezer D. and Lansbury P. T. J. (2007) The impact of the E46K mutation on the properties of alpha‐synuclein in its monomeric and oligomeric states. Biochemistry 46, 7107–7118. [DOI] [PubMed] [Google Scholar]
- Fuchs J., Nilsson C., Kachergus J. et al (2007) Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68, 916–922. [DOI] [PubMed] [Google Scholar]
- Fuchs J., Tichopad A., Golub Y., Munz M., Schweitzer K. J., Wolf B., Berg D., Mueller J. C. and Gasser T. (2008) Genetic variability in the SNCA gene influences alpha‐synuclein levels in the blood and brain. FASEB J. 22, 1327–1334. [DOI] [PubMed] [Google Scholar]
- Galvin J. E., Uryu K., Lee V. M. and Trojanowski J. Q. (1999) Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha‐, beta‐, and gamma‐synuclein. PNAS 96, 13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin J. E., Schuck T. M., Lee V. M. Y. and Trojanowski J. Q. (2001) Differential expression and distribution of α‐, β‐, and γ‐synuclein in the developing human substantia nigra. Exp. Neurol. 168, 347–355. [DOI] [PubMed] [Google Scholar]
- Gaugler M. N., Genc O., Bobela W. et al (2012) Nigrostriatal overabundance of alpha‐synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 123, 653–669. [DOI] [PubMed] [Google Scholar]
- George J. M. (2001) The synucleins. Genome Biol. 3, 3002.1–3002.6. [Google Scholar]
- Gertz H. J., Siegers A. and Kuchinke J. (1994) Stability of cell size and nucleolar size in Lewy body containing neurons of substantia nigra in Parkinson's disease. Brain Res. 637, 339–341. [DOI] [PubMed] [Google Scholar]
- Ghosh D., Mondal M., Mohite G. M., Singh P. K., Ranjan P., Anoop A., Ghosh S., Jha N. N., Kumar A. and Maji S. K. (2013) The Parkinson's disease‐associated H50Q mutation accelerates α‐Synuclein aggregation in vitro. Biochemistry 52, 6925–6927. [DOI] [PubMed] [Google Scholar]
- Ghosh D., Sahay S., Ranjan P. et al (2014) The newly discovered Parkinson's disease associated Finnish mutation (A53E) attenuates α‐synuclein aggregation and membrane binding. Biochemistry 53, 6419–6421. [DOI] [PubMed] [Google Scholar]
- Giasson B. I., Duda J. E., Quinn S. M., Zhang B., Trojanowski J. Q. and Lee V. (2002) Neuronal alpha‐synucleinopathy with severe movement disorder in mice expressing A53T human alpha‐synuclein. Neuron 34, 521–533. [DOI] [PubMed] [Google Scholar]
- Gispert S., Del Turco D., Garrett L. et al (2003) Transgenic mice expressing mutant A53T human alpha‐synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell Neurosci. 24, 419–429. [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Del Tredici K. and Braak H. (2012) 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Del Tredici K. and Braak H. (2013) 100 years of Lewy pathology. 9, 13–24. [DOI] [PubMed] [Google Scholar]
- Gombash S. E., Manfredsson F. P., Kemp C. J., Kuhn N. C., Fleming S. M., Egan A. E., Grant L. M., Ciucci M. R., MacKeigan J. P. and Sortwell C. E. (2013) Morphological and behavioral impact of AAV2/5‐mediated overexpression of human wildtype alpha‐synuclein in the rat nigrostriatal system. PLoS ONE 8, e81426–e81426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Isla T., Irizarry M. C., Mariash A. et al (2003) Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha‐synuclein A30P transgenic mice. Neurobiol. Aging 24, 245–258. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk O. S., Li S., Nash K. et al (2010) In vivo RNAi‐mediated alpha‐synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 18, 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J., Deters N., Doldissen A., Bokhari L., Ke Y., Wiesner A., Schonrock N. and Ittner L. M. (2007) A decade of tau transgenic animal models and beyond. Brain Pathol. 17, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D. G. D., Tiffany S. M. S., Bell W. R. W. and Gutknecht W. F. W. (1978) Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6‐hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 14, 644–653. [PubMed] [Google Scholar]
- Greten‐Harrison B., Polydoro M., Morimoto‐Tomita M. et al (2010) αβγ‐Synuclein triple knockout mice reveal age‐dependent neuronal dysfunction. Proc. Natl Acad. Sci. 107, 19573–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann J., Schlaudraff F., Haeckel O. and Liss B. (2008) Elevated alpha‐synuclein mRNA levels in individual UV‐laser‐microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson's disease. Nucleic Acids Res. 36, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- H T. P., Galvin J. E., baba M., Giasson B., Tomita T., Leight S., Nakajo S., Iwatsubo T., Trojanowski J. Q. and Lee V. M. (1998) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha‐synuclein. Ann. Neurol. 44, 415–422. [DOI] [PubMed] [Google Scholar]
- Häbig K., Walter M., Stappert H., Riess O. and Bonin M. (2009) Microarray expression analysis of human dopaminergic neuroblastoma cells after RNA interference of SNCA–a key player in the pathogenesis of Parkinson's disease. Brain Res. 1256, 19–33. [DOI] [PubMed] [Google Scholar]
- Hallett P. J., McLean J. R., Kartunen A., Langston J. W. and Isacson O. (2012) α‐Synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol. Dis. 47, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday G. M., Del Tredici K. and Braak H. (2006) Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson's disease. J. Neural Transm. Suppl. 70, 99–103. [DOI] [PubMed] [Google Scholar]
- Han Y., Khodr C. E., Sapru M. K., Pedapati J. and Bohn M. C. (2011) A microRNA embedded AAV α‐synuclein gene silencing vector for dopaminergic neurons. Brain Res. 1386, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Fujiwara H., Nonaka T., Wakabayashi K., Takahashi H., Lee V., Trojanowski J. Q., Mann D. and Iwatsubo T. (2002) Phosphorylated alpha‐synuclein is ubiquitinated in alpha‐synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. [DOI] [PubMed] [Google Scholar]
- Hashimoto M., Hsu L. J., Xia Y., Takeda A., Sisk A., Sundsmo M. and Masliah E. (1999) Oxidative stress induces amyloid‐like aggregate formation of NACP/alpha‐synuclein in vitro. NeuroReport 10, 717–721. [DOI] [PubMed] [Google Scholar]
- Hashimoto M., Rockenstein E. and Masliah E. (2003) Transgenic models of alpha‐synuclein pathology ‐ Past, present, and future. Ann. N. Y. Acad. Sci. 991, 171–188. [PubMed] [Google Scholar]
- Hely M. A., Morris J. G., Traficante R., Reid W. G., O'Sullivan D. J. and Williamson P. M. (1999) The sydney multicentre study of Parkinson's disease: progression and mortality at 10 years. J. Neurol. Neurosurg. Psychiatr. 67, 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida‐Ameijeiras A., Méndez‐Alvarez E., Sánchez‐Iglesias S., Sanmartín‐Suárez C. and Soto‐Otero R. (2004) Autoxidation and MAO‐mediated metabolism of dopamine as a potential cause of oxidative stress: role of ferrous and ferric ions. Neurochem. Int. 45, 103–116. [DOI] [PubMed] [Google Scholar]
- Hoffman‐Zacharska D., Koziorowski D., Ross O. A. et al (2013) Novel A18T and pA29S substitutions in α‐synuclein may be associated with sporadic Parkinson's disease. Parkinsonism Relat. Disord. 19, 1057–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger G. U., Carrard G., Michel P. P., Medja F., Lombès A., Ruberg M., Friguet B. and Hirsch E. C. (2003) Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson's disease. J. Neurochem. 86, 1297–1307. [DOI] [PubMed] [Google Scholar]
- Hong D.‐P., Xiong W., Chang J.‐Y. and Jiang C. (2011) The role of the C‐terminus of human alpha‐synuclein: intra‐disulfide bonds between the C‐terminus and other regions stabilize non‐fibrillar monomeric isomers. FEBS Lett. 585, 561–566. [DOI] [PubMed] [Google Scholar]
- Hutton M., Lendon C. L., Rizzu P. et al (1998) Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393, 702–705. [DOI] [PubMed] [Google Scholar]
- Ibanez P., Bonnet A.‐M., Debarges B., Lohmann E., Tison F., Pollak P., Agid Y., Durr A., Brice A. and Group F. P. D. S. (2004) Causal relation between a‐synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364, 1169–1171. [DOI] [PubMed] [Google Scholar]
- Ikeuchi T., Kakita A., Shiga A. et al (2008) Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch. Neurol. 65, 514–519. [DOI] [PubMed] [Google Scholar]
- Iwai A., Masliah E., Yoshimoto M., Ge N. F., Flanagan L., Desilva H., Kittel A. and Saitoh T. (1995) The precursor protein of non‐a‐beta component of Alzheimers‐disease amyloid Is a presynaptic protein of the central‐nervous‐system. Neuron 14, 467–475. [DOI] [PubMed] [Google Scholar]
- Jakes R., Spillantini M. G. and Goedert M. (1994) Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32. [DOI] [PubMed] [Google Scholar]
- Jang A., Lee H.‐J., Suk J.‐E., Jung J.‐W., Kim K.‐P. and Lee S.‐J. (2010) Non‐classical exocytosis of a‐synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 5, 1263–1274. [DOI] [PubMed] [Google Scholar]
- Jao C. C., Der‐Sarkissian A., Chen J. and Langen R. (2004) Structure of membrane‐bound alpha‐synuclein studied by site‐directed spin labeling. PNAS 101, 8331–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K. A. (2008) A critical reappraisal of current staging of Lewy‐related pathology in human brain. Acta Neuropathol. 116, 1–16. [DOI] [PubMed] [Google Scholar]
- Jensen M. B., Bhatia V. K., Jao C. C., Rasmussen J. E., Pedersen S. L., Jensen K. J., Langen R. and Stamou D. (2011) Membrane curvature sensing by amphipathic helices: a single liposome study using α‐synuclein and annexin B12. J. Biol. Chem. 286, 42603–42614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle P. J., Neumann M., Ozmen L. et al (2000) Subcellular localization of wild‐type and Parkinson's disease‐associated mutant alpha ‐synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan N. M. and Manfredsson F. P. (2012) Loss of functional alpha‐synuclein: a toxic event in Parkinson's disease? J. Parkinsons Dis. 2, 249–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan F. H., Sen T., Maiti A. K., Jana S., Chatterjee U. and Chakrabarti S. (2005) Inhibition of rat brain mitochondrial electron transport chain activity by dopamine oxidation products during extended in vitro incubation: implications for Parkinson's disease. Biochim. Biophys. Acta 1741, 65–74. [DOI] [PubMed] [Google Scholar]
- Khodr C. E., Sapru M. K., Pedapati J., Han Y., West N. C., Kells A. P., Bankiewicz K. S. and Bohn M. C. (2011) An alpha‐synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. 1395, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury A. E., Daniel S. E., Sangha H., Eisen S., Lees A. J. and Foster O. J. F. (2004) Alteration in alpha‐synuclein mRNA expression in Parkinson's disease. Mov. Disord. 19, 162–170. [DOI] [PubMed] [Google Scholar]
- Kirik D., Rosenblad C., Burger C., Lundberg C., Johansen T. E., Muzyczka N., Mandel R. J. and Björklund A. (2002) Parkinson‐like neurodegeneration induced by targeted overexpression of alpha‐synuclein in the nigrostriatal system. J. Neurosci. 22, 2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D., Annett L. E., Burger C., Muzyczka N., Mandel R. J. and Björklund A. (2003) Nigrostriatal alpha‐synucleinopathy induced by viral vector‐mediated overexpression of human alpha‐synuclein: a new primate model of Parkinson's disease. PNAS 100, 2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R. L., King M. A., Hamby M. E. and Meyer E. M. (2002) Dopaminergic cell loss induced by human A30P alpha‐synuclein gene transfer to the rat substantia nigra. Hum. Gene Ther. 13, 605–612. [DOI] [PubMed] [Google Scholar]
- Koo H.‐J., Lee H.‐J. and Im H. (2008) Sequence determinants regulating fibrillation of human α‐synuclein. Biochem. Biophys. Res. Commun. 368, 772–778. [DOI] [PubMed] [Google Scholar]
- Kordower J. H., Chu Y., Hauser R. A., Freeman T. B. and Olanow C. W. (2008) Lewy body‐like pathology in long‐term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506. [DOI] [PubMed] [Google Scholar]
- Kordower J. H., Olanow C. W., Dodiya H. B., Chu Y., Beach T. G., Adler C. H., Halliday G. M. and Bartus R. T. (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka K. (1978) Lewy bodies in cerebral cortex, report of three cases. Acta Neuropathol. 42, 127–134. [DOI] [PubMed] [Google Scholar]
- Kramer M. L. and Schulz‐Schaeffer W. J. (2007) Presynaptic alpha‐synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 27, 1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L. and Riess O. (1998) Ala30Pro mutation in the gene encoding alpha‐synuclein in Parkinson's disease. Nat. Genet. 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Krumova P., Meulmeester E., Garrido M. et al (2011) Sumoylation inhibits alpha‐synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M., Haebig K., Bonin M., Ninkina N., Buchman V. L., Poths S. and Riess O. (2007) Whole genome expression analyses of single‐ and double‐knock‐out mice implicate partially overlapping functions of alpha‐ and gamma‐synuclein. Neurogenetics 8, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y.‐M., Li Z., Jiao Y. et al (2010) Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease‐associated alpha‐synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 19, 1633–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M., Vartiainen S., Moilanen A. M., Sirvio J., Thomas J. H., Nass R., Blakely R. D. and Wong G. (2003) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha‐synuclein. J. Neurochem. 86, 165–172. [DOI] [PubMed] [Google Scholar]
- Larsen K. E., Schmitz Y., Troyer M. D. et al (2006) alpha‐synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E., Debyser Z., Van Dorpe J., De Strooper B., Nuttin B. and Baekelandt V. (2003) Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector‐mediated overexpression of alpha‐synuclein. Brain Pathol. 13, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee F., Liu F., Pristupa Z. B. and Niznik H. B. (2001a) Direct binding and functional coupling of alpha‐synuclein to the dopamine transporters accelerate dopamine‐induced apoptosis. FASEB J. 15, 916–926. [DOI] [PubMed] [Google Scholar]
- Lee M. H., Hyun D. H. and Halliwell B. (2001b) Effect of the overexpression of wild‐type or mutant a‐synuclein on cell susceptibility to insult. ‐ Lee ‐ 2001 ‐ Journal of Neurochemistry ‐ Wiley Online Library. J. Neurochem. 76, 998–1009. [DOI] [PubMed] [Google Scholar]
- Lee M. K., Stirling W., Xu Y., Xu X., Qui D., Mandir A. S., Dawson T. M., Copeland N. G., Jenkins N. A. and Price D. L. (2002) Human ‐synuclein‐harboring familial Parkinson's disease‐linked Ala‐53 ‐> Thr mutation causes neurodegenerative disease with ‐synuclein aggregation in transgenic mice. Proc. Natl Acad. Sci. 99, 8968–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J., Patel S. and Lee S. J. (2005) Intravesicular localization and exocytosis of alpha‐synuclein and its aggregates. J. Neurosci. 25, 6016–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.‐J., Suk J.‐E., Bae E.‐J., Lee J.‐H., Paik S. R. and Lee S.‐J. (2008) Assembly‐dependent endocytosis and clearance of extracellular alpha‐synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849. [DOI] [PubMed] [Google Scholar]
- Lee H.‐J., Bae E.‐J. and Lee S.‐J. (2014) Extracellular α–synuclein‐a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 10, 92–98. [DOI] [PubMed] [Google Scholar]
- Lesage S., Anheim M., Letournel F. et al (2013) G51D α‐synuclein mutation causes a novel parkinsonian‐pyramidal syndrome. Ann. Neurol. 73, 459–471. [DOI] [PubMed] [Google Scholar]
- Lewis J., Melrose H., Bumcrot D. et al (2008) In vivo silencing of alpha‐synuclein using naked siRNA. Mol. Neurodegener. 3, 19–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Uversky V. N. and Fink A. L. (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha‐synuclein. Biochemistry 40, 11604–11613. [DOI] [PubMed] [Google Scholar]
- Li W., West N., Colla E. et al (2005) Aggregation promoting C‐terminal truncation of alpha‐synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease‐linked mutations. PNAS 102, 2162–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.‐Y., Englund E., Holton J. L. et al (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host‐to‐graft disease propagation. Nat. Med. 14, 501–503. [DOI] [PubMed] [Google Scholar]
- Li N.‐N., Mao X.‐Y., Chang X.‐L., Zhao D.‐M., Zhang J.‐H., Liao Q., Yu W.‐J., Tan E.‐K. and Peng R. (2013) SNCA rs356219 variant increases risk of sporadic Parkinson's disease in ethnic Chinese. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 452–456. [DOI] [PubMed] [Google Scholar]
- Lin X., Parisiadou L., Sgobio C. et al (2012) Conditional expression of Parkinson's disease‐related mutant a‐synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 32, 9248–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnertz C., Saucier L., Ge D., Cronin K. D., Burke J. R., Browndyke J. N., Hulette C. M., Welsh‐Bohmer K. A. and Chiba‐Falek O. (2009) Genetic regulation of alpha‐synuclein mRNA expression in various human brain tissues. PLoS ONE 4, e7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa C. F., Fujiwara H., Mann D. M. et al (1998) Lewy bodies contain altered alpha‐synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 153, 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Jin L., Wang H. et al (2008) Silencing alpha‐synuclein gene expression enhances tyrosine hydroxylase activity in MN9D cells. Neurochem. Res. 33, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotharius J. and O'Malley K. L. (2000) The parkinsonism‐inducing drug 1‐methyl‐4‐phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J. Biol. Chem. 275, 38581–38588. [DOI] [PubMed] [Google Scholar]
- Lou H., Montoya S. E., Alerte T. N. M. et al (2010) Serine 129 phosphorylation reduces the ability of alpha‐synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 285, 17648–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk K. C., Song C., O'Brien P., Stieber A., Branch J. R., Brunden K. R., Trojanowski J. Q. and Lee V. M. Y. (2009) Exogenous alpha‐synuclein fibrils seed the formation of Lewy body‐like intracellular inclusions in cultured cells. Proc. Natl Acad. Sci. 106, 20051–20056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk K. C., Kehm V. M., Zhang B., O'Brien P., Trojanowski J. Q. and Lee V. M. Y. (2012a) Intracerebral inoculation of pathological alpha‐synuclein initiates a rapidly progressive neurodegenerative alpha‐synucleinopathy in mice. J. Exp. Med. 209, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk K. C., Kehm V., Carroll J., Zhang B., O'Brien P., Trojanowski J. Q. and Lee V. M. Y. (2012b) Pathological α‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad M., Decressac M., Mattsson B. and Björklund A. (2012) Impaired neurotransmission caused by overexpression of alpha‐synuclein in nigral dopamine neurons. PNAS 109, 3213–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak S. K., McCormack A. L., Langston J. W., Kordower J. H. and Di Monte D. A. (2009) Decreased alpha‐synuclein expression in the aging mouse substantia nigra. Exp. Neurol. 220, 359–365. [DOI] [PubMed] [Google Scholar]
- Manning‐Bog A. B., McCormack A. L., Li J., Uversky V. N., Fink A. L. and Di Monte D. A. (2002) The herbicide paraquat causes up‐regulation and aggregation of alpha‐synuclein in mice: paraquat and alpha‐synuclein. J. Biol. Chem. 277, 1641–1644. [DOI] [PubMed] [Google Scholar]
- Maraganore D. M., de Andrade M., Elbaz A. et al (2006) Collaborative analysis of alpha‐synuclein gene promoter variability and Parkinson disease. JAMA 296, 661–670. [DOI] [PubMed] [Google Scholar]
- Markopoulou K., Biernacka J. M., Armasu S. M. et al (2014) Does a‐synuclein have a dual and opposing effect in preclinical vs. clinical Parkinson's disease? Parkinsonism Relat. Disord. 20, 584–589– discussion 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L., Campanelli J. T. and Scheller R. H. (1988) Synuclein: a neuron‐specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M. J., Tolosa E. and Campdelacreu J. (2003) Clinical overview of the synucleinopathies. Mov. Disord. 18, S21–S27. [DOI] [PubMed] [Google Scholar]
- Martinez J., Moeller I., Erdjument‐Bromage H., Tempst P. and Lauring B. (2003) Parkinson's disease‐associated alpha‐synuclein is a calmodulin substrate. J. Biol. Chem. 278, 17379–17387. [DOI] [PubMed] [Google Scholar]
- Marui W., Iseki E., Nakai T., Miura S., Kato M., Uéda K. and Kosaka K. (2002) Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J. Neurol. Sci. 195, 153–159. [DOI] [PubMed] [Google Scholar]
- Masliah E., Rockenstein E., Veinbergs I., Mallory M., Hashimoto M., Takeda A., Sagara Y., Sisk A. and Mucke L. (2000) Dopaminergic loss and inclusion body formation in alpha‐synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y., Vila M., Lincoln S. et al (2001) Lack of nigral pathology in transgenic mice expressing human alpha‐synuclein driven by the tyrosine hydroxylase promoter. Neurobiol. Dis. 8, 535–539. [DOI] [PubMed] [Google Scholar]
- McCormack A. L., Mak S. K., Henderson J. M., Bumcrot D., Farrer M. J. and Di Monte D. A. (2010) Alpha‐synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE 5, e12122–e12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean J. R., Hallett P. J., Cooper O., Stanley M. and Isacson O. (2012) Transcript expression levels of full‐length alpha‐synuclein and its three alternatively spliced variants in Parkinson's disease brain regions and in a transgenic mouse model of alpha‐synuclein overexpression. Mol. Cell Neurosci. 49, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton E. R. and Rhoades E. (2010) Effects of curvature and composition on alpha‐synuclein binding to lipid vesicles. Biophys. J . 99, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. W., Hague S. M., Clarimon J., Baptista M., Gwinn‐Hardy K., Cookson M. R. and Singleton A. B. (2004) Alpha‐synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62, 1835–1838. [DOI] [PubMed] [Google Scholar]
- Miller D. W., Johnson J. M., Solano S. M., Hollingsworth Z. R., Standaert D. G. and Young A. B. (2005) Absence of α‐synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural. Transm. 112, 1613–1624. [DOI] [PubMed] [Google Scholar]
- Morris M., Maeda S., Vossel K. and Mucke L. (2011) The many faces of tau. Neuron 70, 410–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D. D., Rueter S. M., Trojanowski J. Q. and Lee V. M. (2000) Synucleins are developmentally expressed, and alpha‐synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi L., Wood S. J., Steavenson S. et al (1999) Both familial Parkinson's disease mutations accelerate alpha‐synuclein aggregation. J. Biol. Chem. 274, 9843–9846. [DOI] [PubMed] [Google Scholar]
- Neystat M., Lynch T., Przedborski S., Kholodilov N., Rzhetskaya M. and Burke R. E. (1999) Alpha‐synuclein expression in substantia nigra and cortex in Parkinson's disease. Mov. Disord. 14, 417–422. [DOI] [PubMed] [Google Scholar]
- Nishioka K., Hayashi S., Farrer M. J. et al (2006) Clinical heterogeneity of alpha‐synuclein gene duplication in Parkinson's disease. Ann. Neurol. 59, 298–309. [DOI] [PubMed] [Google Scholar]
- Noorian A. R., Rha J., Annerino D. M., Bernhard D., Taylor G. M. and Greene J. G. (2012) Alpha‐synuclein transgenic mice display age‐related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol. Dis. 48, 9–19. [DOI] [PubMed] [Google Scholar]
- Olanow C. W. and Prusiner S. B. (2009) Is Parkinson's disease a prion disorder? Proc. Natl Acad. Sci. 106, 12571–12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterberg V. R., Spinelli K. J., Weston L. J., Luk K. C., Woltjer R. L. and Unni V. K. (2015) Progressive aggregation of alpha‐synuclein and selective degeneration of lewy inclusion‐bearing neurons in a mouse model of parkinsonism. Cell Rep. 10, 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrerova N., Petrucelli L., Farrer M., Mehta N., Choi P., Hardy J. and Wolozin B. (1999) alpha‐Synuclein shares physical and functional homology with 14‐3‐3 proteins. J. Neurosci. 19, 5782–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A., Fournier M. and Lashuel H. A. (2010) Role of post‐translational modifications in modulating the structure, function and toxicity of alpha‐synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145. [DOI] [PubMed] [Google Scholar]
- Pasanen P., Myllykangas L., Siitonen M., Raunio A., Kaakkola S., Lyytinen J., Tienari P. J., Pöyhönen M. and Paetau A. (2014) Novel α‐synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease‐type pathology. Neurobiol. Aging 35(2180), e1–e5. [DOI] [PubMed] [Google Scholar]
- Paumier K. L., Luk K. C., Manfredsson F. P. et al (2015) Intrastriatal injection of pre‐formed mouse a‐synuclein fibrils into rats triggers a‐synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 82, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X., Tehranian R., Dietrich P., Stefanis L. and Perez R. G. (2005) alpha‐Synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 118, 3523–3530. [DOI] [PubMed] [Google Scholar]
- Perez R. G. and Hastings T. G. (2004) Could a loss of alpha‐synuclein function put dopaminergic neurons at risk? J. Neurochem. 89, 1318–1324. [DOI] [PubMed] [Google Scholar]
- Perez R. G., Waymire J. C., Lin E., Liu J. J., Guo F. L. and Zigmond M. J. (2002) A role for alpha‐synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer R. F. (2003) Gastrointestinal dysfunction in Parkinson's disease. Lancet Neurol. 2, 107–116. [DOI] [PubMed] [Google Scholar]
- Pieri L., Madiona K., Bousset L. and Melki R. (2012) Fibrillar a‐synuclein and huntingtin exon 1 assemblies are toxicto the cells. Biophys. J . 102, 2894–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos M. H., Lavedan C., Leroy E. and Ide S. E. (1997) Mutation in the a‐Synuclein Gene Identified in Families with Parkinson's Disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Porras J. L. and Perez R. G. (2014) Potential contribution of alpha‐synuclein dysregulation to Parkinson's disease pathology in Nova Publishers ‐ Alpha‐Synuclein (Kanowitz M. P. A. H. C., ed), pp. 51–70. NY: NOVA Science Publishers. [Google Scholar]
- van der Putten H., Wiederhold K. H., Probst A. et al (2000) Neuropathology in mice expressing human alpha‐synuclein. J. Neurosci. 20, 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quilty M. C., King A. E., Gai W. P., Pountney D. L., West A. K., Vickers J. C. and Dickson T. C. (2006) Alpha‐synuclein is upregulated in neurones in response to chronic oxidative stress and is associated with neuroprotection. Exp. Neurol. 199, 249–256. [DOI] [PubMed] [Google Scholar]
- Quinn J. G., Coulson D. T. R., Brockbank S., Beyer N., Ravid R., Hellemans J., Irvine G. B. and Johnston J. A. (2012) α‐Synuclein mRNA and soluble α‐synuclein protein levels in post‐mortem brain from patients with Parkinson”s disease, dementia with Lewy bodies, and Alzheimer”s disease. Brain Res. 1459, 71–80. [DOI] [PubMed] [Google Scholar]
- Richfield E. K., Thiruchelvam M. J., Cory‐Slechta D. A., Wuertzer C., Gainetdinov R. R., Caron M. G., Di Monte D. A. and Federoff H. J. (2002) Behavioral and neurochemical effects of wild‐type and mutated human alpha‐synuclein in transgenic mice. Exp. Neurol. 175, 35–48. [DOI] [PubMed] [Google Scholar]
- Robertson D. C., Schmidt O., Ninkina N., Jones P. A., Sharkey J. and Buchman V. L. (2004) Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma‐synuclein, alpha‐synuclein and double alpha/gamma‐synuclein null mutant mice. J. Neurochem. 89, 1126–1136. [DOI] [PubMed] [Google Scholar]
- Rockenstein E., Mallory M., Hashimoto M., Song D., Shults C. W., Lang I. and Masliah E. (2002) Differential neuropathological alterations in transgenic mice expressing alpha‐synuclein from the platelet‐derived growth factor and Thy‐1 promoters. J. Neurosci. Res. 68, 568–578. [DOI] [PubMed] [Google Scholar]
- Santacruz K., Lewis J., Spires T. et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W., Nakabayashi Y., Mizuta I. et al (2009) Genome‐wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Schlüter O. M., Fornai F., Alessandri M. G., Takamori S., Geppert M., Jahn R. R. and Südhof T. C. (2003) Role of a‐synuclein in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced parkinsonism in mice. Neuroscience 118, 985–1002. [DOI] [PubMed] [Google Scholar]
- Scott D. and Roy S. (2012) α‐Synuclein inhibits intersynaptic vesicle mobility and maintains recycling‐pool homeostasis. J. Neurosci. 32, 10129–10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell L. C., Berriman J., Jakes R., Goedert M. and Crowther R. A. (2000) Fiber diffraction of synthetic alpha‐synuclein filaments shows amyloid‐like cross‐beta conformation. PNAS 97, 4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon K. M., Keshavarzian A., Dodiya H. B., Jakate S. and Kordower J. H. (2012a) Is alpha‐synuclein in the colon a biomarker for premotor Parkinson's disease? Evidence from 3 cases. Mov. Disord. 27, 716–719. [DOI] [PubMed] [Google Scholar]
- Shannon K. M., Keshavarzian A., Mutlu E., Dodiya H. B., Daian D., Jaglin J. A. and Kordower J. H. (2012b) Alpha‐synuclein in colonic submucosa in early untreated Parkinson's disease. Mov. Disord. 27, 709–715. [DOI] [PubMed] [Google Scholar]
- Sharon R., Goldberg M. S., Bar‐Josef I., Betensky R. A., Shen J. and Selkoe D. J. (2001) alpha‐synuclein occurs in lipid‐rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid‐binding proteins. PNAS 98, 9110–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin E. C., Cho S. E., Lee D. K., Hur M. W., Paik S. R., Park J. H. and Kim J. (2000) Expression patterns of alpha‐synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol. Cells 10, 65–70. [DOI] [PubMed] [Google Scholar]
- Shtilerman M. D., Ding T. T. and Lansbury P. T. (2002) Molecular crowding accelerates fibrillization of alpha‐synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry 41, 3855–3860. [DOI] [PubMed] [Google Scholar]
- Simón‐Sánchez J., Schulte C., Bras J. M. et al (2009) Genome‐wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton A. B., Farrer M., Johnson J. et al (2003) alpha‐Synuclein locus triplication causes Parkinson's disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Specht C. G. and Schoepfer R. (2001) Deletion of the alpha‐synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Schmidt M. L., Lee V. and Trojanowski J. Q. (1997) [alpha]‐Synuclein in Lewy bodies: article: nature. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M. (1998a) α‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc. Natl Acad. Sci. 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Crowther R. A., Jakes R. and Cairns N. J. (1998b) Filamentous a‐synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neuroscience 251, 205–208. [DOI] [PubMed] [Google Scholar]
- St Martin J. L., Klucken J. and Outeiro T. F. (2007) Dopaminergic neuron loss and up‐regulation of chaperone protein mRNA induced by targeted over‐expression of alpha‐synuclein in mouse substantia nigra. J. Neurochem. 100, 1449–1457. [DOI] [PubMed] [Google Scholar]
- Stefanis L. (2012) Synuclein in Parkinson's Disease. Cold Spring Harb. Perspect Med. 2, a009399–a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surguchov A. (2008) Chapter 6 molecular and cellular biology of synucleins. Inter Rev. Cell Mol. Biol. 271, 227–282. [DOI] [PubMed] [Google Scholar]
- Tan E.‐K., Chandran V. R., Fook‐Chong S., Shen H., Yew K., Teoh M.‐L., Yuen Y. and Zhao Y. (2005) Alpha‐synuclein mRNA expression in sporadic Parkinson's disease. Mov. Disord. 20, 620–623. [DOI] [PubMed] [Google Scholar]
- Tanner C. M. and Goldman S. M. (1996) Tanner: epidemiology of Parkinson's disease ‐ Google Scholar. Neurol. Clin. 14, 317–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehranian R., Montoya S. E., Van Laar A. D., Hastings T. G. and Perez R. G. (2006) Alpha‐synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. J. Neurochem. 99, 1188–1196. [DOI] [PubMed] [Google Scholar]
- Theodore S., Cao S., McLean P. J. and Standaert D. G. (2008) Targeted Overexpression of Human alpha‐Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 67, 1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam M. J., Powers J. M., Cory‐Slechta D. A. and Richfield E. K. (2004) Risk factors for dopaminergic neuron loss in human alpha‐synuclein transgenic mice. Eur. J. Neurosci. 19, 845–854. [DOI] [PubMed] [Google Scholar]
- Tofaris G. K., Reitbock P. G., Humby T. et al (2006) Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha‐synuclein (1‐120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins M. M., Basgall E. J., Zamrini E. and Hill W. D. (1997) Apoptotic‐like changes in Lewy‐body‐associated disorders and normal aging in substantia nigral neurons. Am. J. Pathol. 150, 119–131. [PMC free article] [PubMed] [Google Scholar]
- Trojanowski J. Q. and Lee V. M. Y. (2005) Pathological tau: a loss of normal function or a gain in toxicity? Nat. Neurosci. 8, 1136–1137. [DOI] [PubMed] [Google Scholar]
- Ubhi K., Rockenstein E., Mante M., Inglis C., Adame A., Patrick C., Whitney K. and Masliah E. (2010) Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial‐derived neurotrophic factors. J. Neurosci. 30, 6236–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uéda K., Fukushima H., Masliah E., Xia Y., Iwai A., Yoshimoto M., Otero D. A., Kondo J., Ihara Y. and Saitoh T. (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. PNAS 90, 11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uéda K., Saitoh T. and Mori H. (1994) Tissue‐dependent alternative splicing of messenger‐Rna for Nacp, the precursor of non‐a‐beta component of Alzheimers‐disease amyloid. Biochem. Biophys. Res. Commun. 205, 1366–1372. [DOI] [PubMed] [Google Scholar]
- Unni V. K., Weissman T. A., Rockenstein E., Masliah E., McLean P. J. and Hyman B. T. (2010) In vivo imaging of alpha‐synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS ONE 5, e10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. (2002) What does it mean to be natively unfolded? Eur. J. Biochem. 269, 2–12. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. (2007) Neuropathology, biochemistry, and biophysics of alpha‐synuclein aggregation. J. Neurochem. 103, 17–37. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. and Eliezer D. (2009) Biophysics of Parkinson's disease: structure and aggregation of alpha‐synuclein. Curr. Protein Pept. Sci. 10, 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N., Li J. and Fink A. L. (2001) Evidence for a partially folded intermediate in alpha‐synuclein fibril formation. J. Biol. Chem. 276, 10737–10744. [DOI] [PubMed] [Google Scholar]
- Uversky V. N., M Cooper E., Bower K. S., Li J. and Fink A. L. (2002) Accelerated alpha‐synuclein fibrillation in crowded milieu. FEBS Lett. 515, 99–103. [DOI] [PubMed] [Google Scholar]
- VanderHorst V. G., Samardzic T., Saper C. B., Anderson M. P., Nag S., Schneider J. A., Bennett D. A. and Buchman A. S. (2015) synuclein pathology accumulates in sacral spinal visceral sensory pathways. Ann. Neurol. 78, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas K. J., Makani S., Davis T., Westphal C. H., Castillo P. E. and Chandra S. S. (2014) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 34, 9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas‐Medrano J., Krishnamachari S., Villanueva E., Godfrey W. H., Lou H., Chinnasamy R., Arterburn J. B. and Perez R. G. (2014) Novel FTY720‐based compounds stimulate neurotrophin expression and phosphatase activity in dopaminergic cells. ACS Med. Chem. Lett. 5, 782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles M. J., Lee S. J., Rochet J. C., Shtilerman M. D., Ding T. T., Kessler J. C. and Lansbury P. T. (2001) Vesicle permeabilization by protofibrillar alpha‐synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 40, 7812–7819. [DOI] [PubMed] [Google Scholar]
- Volpicelli‐Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q. and Lee V. M. Y. (2011) Exogenous α‐synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K., Takahashi H., Takeda S., Ohama E. and Ikuta F. (1988) Parkinson's disease: the presence of Lewy bodies in Auerbach's and Meissner's plexuses. Acta Neuropathol. 76, 217–221. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K., Takahashi H., Ohama E. and Ikuta F. (1990) Parkinson's disease: an immunohistochemical study of Lewy body‐containing neurons in the enteric nervous system. Acta Neuropathol. 79, 581–583. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K., Takahashi H., Ohama E., Takeda S. and Ikuta F. (1993) Lewy bodies in the visceral autonomic nervous system in Parkinson's disease. Adv. Neurol. 60, 609–612. [PubMed] [Google Scholar]
- Wakabayashi K., Yoshimoto M., Fukushima T., Koide R., Horikawa Y., Morita T. and Takahashi H. (1999) Widespread occurrence of alpha‐synuclein/NACP‐immunoreactive neuronal inclusions in juvenileand adult‐onset Hallervorden‐Spatz diseasewith Lewy bodies. Neuropathol. Appl. Neurobiol. 25, 363–368. [DOI] [PubMed] [Google Scholar]
- Wakamatsu M., Ishii A., Iwata S. et al (2008) Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha‐synuclein in mice. Neurobiol. Aging 29, 574–585. [DOI] [PubMed] [Google Scholar]
- Wang L., Fleming S. M., Chesselet M.F., Tache Y. (2008) Abnormal colonic motility in mice overexpressing human wild‐type α‐synuclein. NeuroReport 19, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Lou H., Pedersen C. J., Smith A. D. and Perez R. G. (2009) 14‐3‐3zeta contributes to tyrosine hydroxylase activity in MN9D cells: localization of dopamine regulatory proteins to mitochondria. J. Biol. Chem. 284, 14011–14019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb P. H., Zhen W., Poon A. W., Conway K. A. and Lansbury P. T. (1996) NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. [DOI] [PubMed] [Google Scholar]
- Wersinger C. and Sidhu A. (2003a) Differential cytotoxicity of dopamine and H2O2 in a human neuroblastoma divided cell line transfected with a‐synuclein and its familial Parkinson's disease‐linked mutants. Neurosci. Lett. 342, 124–128. [DOI] [PubMed] [Google Scholar]
- Wersinger C. and Sidhu A. (2003b) Attenuation of dopamine transporter activity by alpha‐synuclein. Neurosci. Lett. 340, 189–192. [DOI] [PubMed] [Google Scholar]
- Wersinger C., Prou D., Vernier P. and Sidhu A. (2003) Modulation of dopamine transporter function by alpha‐synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J. 17, 2151. [DOI] [PubMed] [Google Scholar]
- Wood S. J., Wypych J., Steavenson S., Louis J. C., Citron M. and Biere A. L. (1999) alpha‐synuclein fibrillogenesis is nucleation‐dependent. Implications for the pathogenesis of Parkinson's disease. J. Biol. Chem. 274, 19509–19512. [DOI] [PubMed] [Google Scholar]
- Woods W. S., Boettcher J. M., Zhou D. H., Kloepper K. D., Hartman K. L., Ladror D. T., Qi Z., Rienstra C. M. and George J. M. (2007) Conformation‐specific binding of alpha‐synuclein to novel protein partners detected by phage display and NMR spectroscopy. J. Biol. Chem. 282, 34555–34567. [DOI] [PubMed] [Google Scholar]
- Wu F., Poon W. S., Lu G., Wang A., Meng H., Feng L., Li Z. and Liu S. (2009) Alpha‐synuclein knockdown attenuates MPP+ induced mitochondrial dysfunction of SH‐SY5Y cells. Brain Res. 1292, 173–179. [DOI] [PubMed] [Google Scholar]
- Wu J., Lou H., Alerte T. N. M., Stachowski E. K., Chen J., Singleton A. B., Hamilton R. L. and Perez R. G. (2012) Lewy like aggregation of alphansynuclein reduces protein phosphatase 2A activity. Neuroscience 207, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Kao S.‐Y., Lee F. J. S., Song W., Jin L.‐W. and Yankner B. A. (2002) Dopamine‐dependent neurotoxicity of alpha‐synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 8, 600–606. [DOI] [PubMed] [Google Scholar]
- Xuan Q., Xu S.‐L., Lu D.‐H., Yu S., Zhou M., Uéda K., Cui Y.‐Q., Zhang B.‐Y. and Chan P. (2011) Increased expression of α‐synuclein in aged human brain associated with neuromelanin accumulation. J. Neural. Transm. 118, 1575–1583. [DOI] [PubMed] [Google Scholar]
- Yamada M., Iwatsubo T. and Mizuno Y. (2004) Overexpression of a‐synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of a‐synuclein and activation of caspase‐9: resemblance to pathogenetic changes in Parkinson's disease. J. Neurochem. 91, 451–461.‐ [DOI] [PubMed] [Google Scholar]
- Yu S., Zuo X. H., Li Y. H., Zhang C., Zhou M., Zhang Y. A., Uéda K. and Chan P. (2004) Inhibition of tyrosine hydroxylase expression in alpha‐synuclein‐transfected dopaminergic neuronal cells. Neurosci. Lett. 367, 34–39. [DOI] [PubMed] [Google Scholar]
- Yuan Y., Sun J., Zhao M., Hu J., Wang X., Du G. and Chen N. H. (2010) Overexpression of alpha‐synuclein down‐regulates BDNF expression. Cell. Mol. Neurobiol. 30, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz J. J., Alegre J., Gómez‐Esteban J. C. et al (2004) The new mutation, E46K, of alpha‐synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. [DOI] [PubMed] [Google Scholar]
- Zhang Bin., Maiti A., Shively S. et al (2005) Microtubule‐binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. PNAS 102, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharikov A. D., Cannon J. R., Tapias V., Bai Q., Horowitz M. P., Shah V., El Ayadi A., Hastings T. G., Greenamyre J. T. and Burton E. A. (2015) shRNA targeting α‐synuclein prevents neurodegeneration in a Parkinson's disease model. J. Clin. Invest. 125, 2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zourlidou A., Smith M. D. P. and Latchman D. S. (2003) Modulation of cell death by alpha‐synuclein is stimulus‐dependent in mammalian cells. Neurosci. Lett. 340, 234–238. [DOI] [PubMed] [Google Scholar]