

ABSTRACT
Currently available simian immunodeficiency virus (SIV) infectious molecular clones (IMCs) and isolates used in nonhuman primate (NHP) models of AIDS were originally derived from infected macaques during chronic infection or end stage disease and may not authentically recapitulate features of transmitted/founder (T/F) genomes that are of particular interest in transmission, pathogenesis, prevention, and treatment studies. We therefore generated and characterized T/F IMCs from genetically and biologically heterogeneous challenge stocks of SIVmac251 and SIVsmE660. Single-genome amplification (SGA) was used to identify full-length T/F genomes present in plasma during acute infection resulting from atraumatic rectal inoculation of Indian rhesus macaques with low doses of SIVmac251 or SIVsmE660. All 8 T/F clones yielded viruses that were infectious and replication competent in vitro, with replication kinetics similar to those of the widely used chronic-infection-derived IMCs SIVmac239 and SIVsmE543. Phenotypically, the new T/F virus strains exhibited a range of neutralization sensitivity profiles. Four T/F virus strains were inoculated into rhesus macaques, and each exhibited typical SIV replication kinetics. The SIVsm T/F viruses were sensitive to TRIM5α restriction. All T/F viruses were pathogenic in rhesus macaques, resulting in progressive CD4+ T cell loss in gastrointestinal tissues, peripheral blood, and lymphatic tissues. The animals developed pathological immune activation; lymphoid tissue damage, including fibrosis; and clinically significant immunodeficiency leading to AIDS-defining clinical endpoints. These T/F clones represent a new molecular platform for the analysis of virus transmission and immunopathogenesis and for the generation of novel “bar-coded” challenge viruses and next-generation simian-human immunodeficiency viruses that may advance the HIV/AIDS vaccine agenda.
IMPORTANCE Nonhuman primate research has relied on only a few infectious molecular clones for a myriad of diverse research projects, including pathogenesis, preclinical vaccine evaluations, transmission, and host-versus-pathogen interactions. With new data suggesting a selected phenotype of the virus that causes infection (i.e., the transmitted/founder virus), we sought to generate and characterize infectious molecular clones from two widely used simian immunodeficiency virus lineages (SIVmac251 and SIVsmE660). Although the exact requirements necessary to be a T/F virus are not yet fully understood, we generated cloned viruses with all the necessary characteristic of a successful T/F virus. The cloned viruses revealed typical acute and set point viral-load dynamics with pathological immune activation, lymphoid tissue damage progressing to significant immunodeficiency, and AIDS-defining clinical endpoints in some animals. These T/F clones represent a new molecular platform for studies requiring authentic T/F viruses.
INTRODUCTION
Recent studies have found that, compared with viruses that circulate during chronic infection, transmitted/founder (T/F) human immunodeficiency virus type 1 (HIV-1) has unique properties that may help establish systemic infection (1–8). However, to date, all nonhuman primate (NHP) models of AIDS employing simian immunodeficiency viruses (SIV), including vaccine evaluation and virus transmission studies, have relied on a limited number of viruses derived from infected animals during chronic or clinical endpoint stages of infection and cloned following ex vivo expansion in human cell lines. For many studies involving viral transmission and pathogenesis, or their prevention or treatment, it would be desirable to have viruses representative of those that are involved in transmission, particularly in light of data suggesting that the viruses that establish initial infection may differ from those present in the chronic phase of infection (2–4). Here, we utilized mucosal infection and the attendant genetic bottleneck to identify viral genomes that are able to cross a mucosal epithelial barrier; replicate to sufficiently high titers to evade innate and early adaptive immunity; and establish persistent, systemic, pathological infection. A priori, these viruses embody the properties required for successful transmission and establishment of initial infection, and the molecular clones representing such T/F viruses described here may serve as a useful alternative to the late-stage-infection-derived viral clones currently used in NHP studies.
Single-genome amplification (SGA), followed by direct sequencing and mathematical modeling, is now a well-established method to infer the identities and number of distinct viral genomes establishing systemic infection and to define the precise nucleotide composition of each T/F genome (9–13). We and others have used this technique to show in geographically distinct populations that productive HIV-1 infection is due to transmission of a single viral genome in ∼40% of intravenous drug users, ∼60% of men who have sex with men, and at least 80% of heterosexual transmissions (9, 12, 14–17). We have additionally used SGA analysis of viral envelope sequences to show that the titer of mucosal inoculation of SIV in rhesus macaques (Macaca mulatta) can be determined in order to recapitulate the number of T/F genomes observed in mucosal transmission of HIV-1 (18–21). Mucosal challenges whose titers have been determined and subsequent SGA-based T/F variant quantification have more recently been used to examine vaccine efficacy in preclinical NHP vaccine trials, where a reduction in the number of T/F viruses in vaccinated macaques mucosally inoculated with SIV has been proposed as a parameter to help evaluate partial vaccine efficacy in preclinical studies (22–28). Although sequencing the Env gene is typically sufficient for quantifying the T/F viruses and analyzing Env-based phenotypes, the same approach can also be used to identify and clone full-length T/F genome sequences for biological analyses (3, 29, 30). Cloning full-length T/F viral genomes directly from peripheral blood mononuclear cells (PBMCs) during acute infection allows one to capture and analyze in a reproducible fashion the genetic and biological properties of viruses that are responsible for a given clinical transmission event in a way that subsequent virus isolations do not. Here, we utilized this approach to infer and analyze the env genes and the full-length viral genomes of T/F viruses derived from SIVmac251 and SIVsmE660 swarm infections.
Compared to control HIV-1 infectious molecular clones (IMCs) derived from the chronic-phase infection, full-length T/F HIV-1 IMCs have greater infectivity and contain more envelope glycoprotein per particle; however, T/F viruses appear to replicate with kinetics comparable to those of chronic viruses in vitro (3). Importantly, in the presence of alpha interferon (IFN-α), which is present at high concentrations during primary infection, T/F HIV-1 subtype B replicated to higher titers than chronic viruses, suggesting an inherent resistance to interferon-stimulated gene products that might otherwise inhibit or prevent systemic infection (2). Recently, it was reported that although all T/F viruses replicate sufficiently to establish systemic infection, differences in the replication rates of different T/F viruses can affect immune activation and eventual disease progression (31). Here, we generated several T/F SIV IMCs and characterized their properties in vitro and in vivo.
Nonhuman primates provide unique advantages in studying virus transmission and pathogenesis, including precise manipulation of the challenge stock and the route of exposure with early and frequent blood and tissue sampling. This, combined with the ability to infect many animals with exactly the same virus, allows greater understanding of complex biological processes. Adding to the repertoire of functional SIV clones used in NHP research may be useful in assessing the differences between T/F viruses and chronic viruses in vivo. These clones also provide authentic transmitted virus clones to study the transmission process in molecular detail and to accurately model preclinical intervention studies.
Two SIV molecular clones, SIVmac239 and SIVsmE543, are widely used in nonhuman primate models of HIV-1 infection. SIVmac239 is a pathogenic, highly neutralization-resistant virus closely related to but distinct from the SIVmac251 isolate. SIVmac239 was originally derived by lambda phage cloning from DNA isolated following coculture of macaque plasma obtained during late-stage simian AIDS with the human cell line HUT-78 (32). The original SIVmac239 clone contained a premature stop codon in the nef gene and at least four additional suboptimal nucleotides (33–35). The nef mutation was subsequently corrected through site-directed mutagenesis prior to widespread use (36), but a clone with the 4 additional suboptimal mutations corrected has only recently been made available (37). Despite this, SIVmac239 has been used extensively because it was the first IMC available and it uniformly causes infection and pathogenesis in a time frame appropriate for NHP research. SIVsmE543, which is also pathogenic and neutralization resistant, is a molecular clone closely related to but distinct from the SIVsmE660 isolate (38). Similar to SIVmac239, SIVsmE543 was cloned from DNA following virus isolation on a human cell line, when PBMCs from a macaque with AIDS were cocultured with the human cell line CEMx174. For this clone, 106 clones were originally screened, but only 1, SIVsmE543, was infectious (38). This clone and the SIVsmE660 isolate were shown to be sensitive to rhesus macaque TRIM5α inhibition (39), likely reflecting the limited passage history of SIVsm in rhesus macaques. Both of the clones and their respective viral isolates originated during late-stage infection and contained nonfunctional or suboptimal mutations that were subsequently identified as or presumed to be either PCR/cloning errors, naturally occurring but rare due to the lack of effective purifying selection during clinical AIDS, or mutations that arose during ex vivo coculture. In addition, expansion in human cells prior to cloning might have altered the viral genome. Therefore, although mucosally transmissible, the SIVmac239 and SIVsmE543 clones represent chronic/AIDS-like viruses rather than viruses associated with transmission.
Here, we used a stringent mucosal-infection strategy to limit the number of genomes establishing systemic infection to identify authentic T/F viruses from the chronic SIVmac251 and SIVsmE660 isolates. We utilized the principles of SGA and identification of T/F viruses to generate genetically defined T/F molecular clones following this mucosal bottleneck. In total, eight T/F IMCs were generated, and all were fully functional in vitro. Four clones (2 from each lineage) were further characterized in vivo, where they demonstrated high replication capacity and authentic pathogenesis, as indicated by high plasma viral loads during acute and chronic infection, with CD4+ T cell decline in lymph nodes and gut, chronic immune activation, lymphoid and gut tissue damage, and clinical immunodeficiency with AIDS-defining disease endpoints. This study represents the first description of SIV-based T/F IMCs and highlights their potential utility to more clearly elucidate any unique characteristics of T/F viruses in relevant NHP models. We anticipate that these T/F IMCs will be particularly useful for transmission, natural history, and preclinical preventive-intervention studies.
MATERIALS AND METHODS
SIVmac251 and SIVsmE660 isolates used to generate T/F IMCs.
We utilized two SIVmac251 isolate stocks following mucosal infection to derive T/F IMCs. The passage history of each SIVmac251 isolate and the phylogenetic comparisons of each lineage have been described previously (40, 41). One isolate used was previously described (42) and was used to generate the SIVmacPBE and SIVmacCR53 clones. The other was a recent reexpansion in primary rhesus PBMCs from the original SIVmac251 isolate (33) and contained 64.7 ng/ml of p27 and 6.56 × 104 50% tissue culture infective doses (TCID50)/ml assayed on rhesus PBMCs. This unpublished isolate was used to derive SIVmac746 and SIVmac766 following intrarectal challenge. The SIVsmE660 isolate was initially reported (38), and infectious molecular clones were derived, from mucosally infected animals previously described (18, 43).
Viral-RNA extraction and cDNA synthesis.
At least 20,000 copies of viral RNA (vRNA) were extracted from plasma and virus inocula using the EZ1 Virus minikit v2.0 with the EZ1 Advanced XL robotic workstation (Qiagen) or manually using the QIAamp Viral RNA minikit (Qiagen). RNA was eluted and was immediately subjected to cDNA synthesis. Viral RNA, 0.5 mM each deoxynucleoside triphosphate, and 0.25 μM antisense primer were incubated at 65°C for 5 min and then moved to ice for 1 min. Then, 1× reverse transcriptase (RT) buffer, 5 mM dithiothreitol, 2 U/μl RNaseOut, and 10 U/μl of SuperScript III reverse transcription mixture (Thermo Fisher Scientific) were incubated at 50°C for 60 min, followed by an increase in temperature to 55°C for an additional 60 min. The samples were then heat inactivated at 70°C for 15 min, and 2 U of RNase H was added and incubated at 37°C for 20 min. cDNA was used immediately or frozen at −80°C.
Single-genome amplification.
For maximum sensitivity, nested-PCR amplification was performed using Platinum Taq DNA High Fidelity polymerase (Thermo Fisher Scientific) for both reactions according to the manufacturer's protocol. Briefly, 1× High Fidelity Platinum PCR buffer, 2 mM MgSO4, 0.2 mM each deoxynucleoside triphosphate, 0.2 μM each primer, and 0.025 U/μl Platinum Taq High Fidelity polymerase were combined in a 20-μl reaction mixture. cDNA was serially diluted until a concentration was found at which PCR-positive wells constituted less than 30% of the total number of reactions, as previously described (9, 18). First-round PCR mixtures were denatured at 94°C for 1 min, followed by 35 cycles of 94°C for 20 s, 55°C for 30 s, and 68°C for 1 min per kilobase and terminated with a single 10-min 68°C extension. Next, 1 μl of each reaction mixture was transferred to a second-round reaction, which was amplified under the same PCR conditions for 45 cycles. PCRs were scored as positive following gel electrophoresis. Positive wells were directly sequenced on an ABI 3730xl genetic analyzer using BigDye Terminator chemistry (Applied Biosystems). Both DNA strands were sequenced, and overlapping sequence fragments for each amplicon were assembled and edited using the Sequencher 5.0 program (Gene Codes). Chromatograms were inspected at every position for mixed bases (double peaks), which would be indicative of a Taq polymerase error in an early cycle or PCR priming from more than one template. Sequences with mixed bases were excluded from further analysis.
Sequence analysis.
All the sequences were aligned using ClustalW and then manually inspected and optimized in MacClade 4.08. Phylogenetic trees were based on nucleotide sequences and constructed using the neighbor-joining method.
IMC generation.
The generation and general description of SIVsmCG7V and SIVsmCG7G clones were reported previously (44) (accession numbers JX648291 and JX648292). SIVsmR02012 and SIVsmR95117 IMCs were constructed in two halves around a unique BstBI site. The 5′ half was amplified, using Phusion high-fidelity DNA polymerase (Invitrogen), from DNA isolated from peak infection PBMCs and subcloned into pCR-XL-TOPO (Invitrogen). To aid in the final construction, the sense primer contained a 5′ XhoI site and the antisense primer overlapped the BstB1 site. The 3′ half was de novo synthesized using the SGA sequence as the template (Blue Heron), starting at the BstB1 site, and contained a NotI site at the 3′ end. Both fragments were combined in the low-copy-number plasmid pBR-322-MCS (45) using the XhoI, BstBI, and NotI sites. SIVmacCR53 and SIVmacPBE were also constructed in halves around the same internal viral BstBI site from DNA isolated from acute-infection PBMCs. For these clones, each half was cloned into pCR-XL-TOPO, and then, the 3′ half was combined with the 5′ half using the BstBI and NotI sites. SIVmac746 and SIVmac766 were generated with a MluI site at the 5′ end, an SphI site joining the two halves, and an XhoI site at the 3′ end, again cloned into pCR-XL-TOPO. All full-length IMCs were sequenced after construction to compare them to the inferred T/F genome sequence. In one clone (SIVmac746), there was a single-nucleotide mutation from the T/F sequence, causing a G172R mutation in the RT gene.
IMC virus stocks.
Virus stocks were generated in HEK-293T cells using either Fugene 6 (Roche) or TransIt (Mirus Bio) according to the manufacturer's instructions. The culture medium was changed 24 to 48 h posttransfection. At 72 h posttransfection, virus-containing supernatant was clarified by centrifugation, sterile filtered through a 0.45-μm filter, aliquoted, and stored at −80°C. Virus titers were determined using TZM-bl reporter cells (reference no. 8129; NIH AIDS Research and Reference Reagent Program), which contain a Tat-inducible luciferase and a beta-galactosidase gene expression cassette. Infectious titers were measured by counting individual β-galactosidase-expressing cells per well in cultures infected with serial 3-fold dilutions of virus (46). Wells containing dilution-corrected blue-cell counts within the linear range of the virus dilution series were averaged to generate an infectious titer in infectious units (IU) per milliliter. Additional infection-derived stocks were generated for SIVsmCG7V and SIVsmCG7G by isolating rhesus PBMCs from whole blood by gradient centrifugation over Ficoll-Hypaque Plus (GE Healthcare). CD4+ T cells were then positively selected using magnetic nonhuman primate CD4+ microbeads with the autoMACS (Miltenyi). CD4+ T lymphocytes were activated in RPMI 1640 medium supplemented with 15% fetal bovine serum (FBS) and 3 μg/ml of staphylococcal enterotoxin B (SEB) (Sigma-Aldrich) for 48 h at 37°C. Activated CD4+ cells were infected in a small volume (<2 ml) at a multiplicity of infection (MOI) of 0.1, washed 3 times, and then suspended at a concentration of 1 × 106/ml in RPMI 1640 medium, 15% FBS, and 30 U/ml of interleukin-2 (IL-2) (Roche). On day 4 postinfection and every 2 days thereafter, cells were collected by low-speed centrifugation, and a complete medium exchange was performed and saved. Viral replication was assessed retrospectively by SIV p27 antigen enzyme-linked immunosorbent assay (Zeptometrix), and a final stock was created by combining the three time points with the highest viral loads: days 6, 8, and 10.
In vitro replication.
To evaluate the viral replication capacity of each of the newly described IMCs, CD8-depleted PBMCs were isolated from naive rhesus macaque donors. The CD8− depletion was performed using a nonhuman primate CD8 microbead kit (Miltenyi Biotec) according to the manufacturer's recommendation. The remaining mononuclear cells were stimulated with plate-bound anti-CD3 antibody and IL-2 (100 U/ml) in RPMI-Complete (RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin) for 3 days. The stimulated cells were then removed from anti-CD3-coated plates, washed once in RPMI-Complete, and then infected with each virus in independent cultures at a nominal MOI of 0.01, with cells subsequently cultured in RPMI-Complete containing IL-2 (100 U/ml). Following an overnight incubation at 37°C, the cells were washed twice with phosphate-buffered saline (PBS) and once with RPMI-Complete to remove excess virus. Viral replication was monitored using reverse transcriptase activity measured in a colorimetric detection kit (Roche). The TRIM5α genotype of each animal was determined as previously reported and indicated for each animal used (39).
Neutralization assay.
The monoclonal antibodies (MAbs) 3.11H (V3) and 1.4H (CD4i) and the newly generated SIV MAb panel have been reported previously (47, 48). SIV sera were provided by N. Letvin (Harvard University, Boston, MA) from chronically infected rhesus macaques intrarectally infected with SIVsmE660 or SIVmac251. Before use, the sera were heat inactivated at 56°C for 30 min and clarified by spinning at 3,000 × g for 5 min. Virus neutralization was assessed on TZM-bl cells as described previously (48, 49). Briefly, either the day before or the day of the experiment, TZM-bl cells were seeded and cultured in 96-well plates. Virus stock dilutions were made in Dulbecco's modified Eagle's medium (DMEM) containing 6 to 10% FBS and 40 μg/ml DEAE-dextran (Sigma-Aldrich, St. Louis, MO) to achieve 2,000 IU/well. Equal-volume virus dilutions and 5-fold serially diluted sera or MAbs were combined and incubated at 37°C for 30 to 60 min. Then, 100 μl of these mixtures was added to the TZM-bl cells in 96-well plates. All antibody dilutions were tested in triplicate. After a 48-h incubation at 37°C, the cells were lysed, and luciferase activity was recorded as a measure of residual infectivity. To prevent additional rounds of replication, the integrase inhibitor indinavir (1 μM) was added after infection. Medium-only and virus-only control wells were included as background and 100% infectivity, respectively.
Rhesus macaque infection.
Two male, Indian-origin rhesus macaques were intrarectally inoculated with 1 ml of diluted viral stock for each T/F IMC. For the transfection-produced viral clones, intrarectal challenge was performed with 2.0 × 104 IU (SIVmac746) and 2.2 × 104 IU (SIVmac766). For the infection-produced viral clones, intrarectal challenge was performed with 1.25 × 105 IU (SIVsmCG7G) and 6.8 × 104 IU (SIVsmCG7V). Following the first challenge, 4 animals, p096 (SIVmac746), p097 and p106 (SIVmac766), and p140 (SIVsmCG7V), were not productively infected. For animals p096, p097, and p106, rechallenge was performed with 100-fold more virus intravenously. For p140, rechallenge was repeated intrarectally with 10-fold more virus (6.8 × 105 IU in 2 ml). Following rechallenge, all the animals became productively infected. Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (63). For this study, the animals were maintained at the Advanced Bioscience Laboratory, an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited institution, with the approval of the Animal Care and Use Committees of the National Institutes of Health.
Plasma viral load and peripheral CD4+ T cell quantification.
Quantitative plasma SIV RNA loads were determined as previously described (50). Briefly, viral RNA was extracted from blood plasma by pelleting, guanidine HCl/proteinase K lysis, and isopropanol precipitation. Following cDNA synthesis, quantitative real-time PCR of the gag gene was performed, with an overall assay sensitivity of 30 copies/ml. Peripheral blood CD4+ T cells were monitored throughout the course of infection in all eight infected animals using TruCount (BD Biosciences).
Immunohistochemical staining.
Immunohistochemistry (IHC) for CD4+ T cells, collagen 1, Ki67, and myeloperoxidase (MPO) was performed as previously described (51) from rectal and peripheral lymph node biopsy and necropsy specimens. Briefly, IHC was performed using a biotin-free polymer approach (Golden Bridge International, Inc.) on 5-μm tissue sections mounted on glass slides, which were dewaxed and rehydrated with graded alcohols (70 to 100% ethanol) to double-distilled H2O. Heat-induced epitope retrieval was performed by heating sections in 0.01% citraconic anhydride containing 0.05% Tween 20 or 1× Diva buffer (Biocare Medical) in a pressure cooker set at 122 to 125°C for 30 s. Slides were incubated with blocking buffer (Tris-buffered saline [TBS] with 0.05% Tween 20 and 0.25% casein) for 10 min. For IHC of myeloid cells, slides were loaded on an IntelliPath autostainer (Biocare Medical), stained under optimal conditions determined empirically (consisting of a blocking step using blocking buffer for 10 min; an endogenous peroxidase block using 1.5% [vol/vol] H2O2 in TBS [pH 7.4] for 10 min; incubation with mouse anti-CD68 [1:400; clone KP1; Dako], mouse anti-CD163 [1:400; clone 10D6; Novocastra/Leica], and rabbit monoclonal anti-CD4 [1:200; clone EPR6855; Epitomics, Inc.] diluted in blocking buffer for 1 h at room temperature), washed with TBS containing 0.05% Tween 20 (TBS-Tw), and detected using a biotin-free polymer approach consisting of Mouse Polink-1 AP (Golden Bridge International, Inc.) for 30 min at room temperature followed by Rabbit Polink-1 horseradish peroxidase (HRP) (Golden Bridge International, Inc.) for 30 min at room temperature. Sections were washed and first incubated with Impact DAB (3,3′-diaminobenzidine; Vector Laboratories) to develop the CD4, washed, and developed with Warp Red (Biocare Medical, Inc.) to develop macrophage/myeloid cells and to mask the faint CD4 expressed on antigen-presenting cells (APCs) to distinctly distinguish CD4+ T cells from myeloid cells.
Accession number(s).
All the sequences used in the study were deposited in GenBank. The accession numbers are divided into 6 groups representing the Env sequence analysis: the SIVmac251 stock (KX089355 to KX089585), the acute-infection sequences for 5′ (KX089586 to KX089693) and 3′ (KX089794 to KX089925), the terminal sequence analysis for 5′ (KX089694 to KX089793) and 3′ (KX089926 to KX090026), and the IMC sequences (KU955513 to KU955518).
RESULTS
T/F Env analysis to identify candidate viruses for cloning.
We sought to generate and fully characterize T/F IMCs following mucosal infection with the two most widely used SIV isolates (SIVmac251 and SIVsmE660). We obtained T/F IMCs from these isolates by characterizing and selecting T/F viruses representative of each lineage in different starting experiments. We recently reported that viral sequences within individual laboratory expansions derived from the original SIVmac251 isolate are genetically distinct and clustered (40). Although we have identified the T/F env genes in several studies using multiple stocks of both SIVmac251 and SIVsmE660 isolates (18–25, 43, 52–54), until now, the original isolate of SIVmac251 had not yet been used in a study where T/F viruses were identified. While the original isolate itself is no longer available, we were able to analyze a 10-animal intrarectal titration study of a new expansion of the original SIVmac251 isolate that closely mirrors the diversity in the original isolate (40), with 2 animals challenged at each dilution (1:10, 1:25, 1:50, 1:100, and 1:500). Plasma vRNA was measured by real-time PCR to confirm infection. All but one animal became infected after a single challenge. At peak viremia (2 weeks postchallenge), plasma vRNA was isolated for single-genome amplification and sequence analysis to infer the number and sequence of each T/F env genome. A total of 264 sequences were obtained, with an average of 29 sequences per animal. Each animal's viral sequences were examined phylogenetically to identity low-diversity lineages containing less than 2 non-APOBEC mutated nucleotide polymorphisms from the consensus sequence across the entire env gene. For individuals with only a single T/F lineage, the inferred T/F env sequence represents the consensus of all the sequences from that animal (Fig. 1A and B). For individuals with additional low-diversity lineages, each lineage was examined independently with an inferred T/F genome for each lineage. Importantly, due to very early sampling, most sequences within each low-diversity lineage were either identical to the consensus sequence (i.e., the inferred T/F sequence) or differed by only a few nucleotides (random errors likely introduced by reverse transcription). For all the animals tested, the total number of low-diversity lineages was 33. Phylogenetic analyses of the stock and each variant identified both T/F variants that were unique to a single animal and T/F variants that were common to multiple animals (Fig. 1C). In one dominant cluster, 6 of the 33 total T/F variants can be found, representing nearly 20% of all the transmitted variants in this study. Importantly, this cluster also contains 21 of the 64 stock sequences, representing 32% of all stock sequences (Fig. 1C, red box). In addition to this overall dominant lineage, there were 6 other clusters representing selection of these variants within the stock (Fig. 1C, green boxes) or selection as transmitted/founder variants (Fig. 1C, blue boxes).
FIG 1.

(A and B) Phylogenetic trees and highlighter alignments of Env sequences from SIvmac251-infected rhesus macaques M766 (A) and M746 (B). Single nucleotide polymorphisms to the consensus sequences are denoted by colored tick marks; gaps are shown in gray and G-to-A mutations in purple. (C) Phylogenetic tree of the 33 T/F env variants (color coded by animal identifier) and sequences from the infection stock used in this study (gray circles). One large cluster (boxed in red) contains 20% of the T/F viruses and 32% of the stock sequences with identical or nearly identical envelopes. The green boxes indicate inoculum-enriched clusters, and the blue boxes indicate transmission-enriched clusters. Nucleotide differences in phylogenetic trees are shown by the scale bar.
From this SIVmac251 titration study, we identified two animals represented in Fig. 1 (M766 and M746) that were selected for IMC generation, as both animals had single-variant infections representing T/F-enriched clusters on the phylogenetic tree. Two additional animals (PBE and CR53) infected with a genetically distinct stock of SIVmac251 were also selected for full-genome analysis and IMC generation based on previously published env analysis (18). These animals were selected because each animal was infected by a single T/F variant that represented a phylogenetically distinct virus.
For the SIVsmE660 lineage, four SIVsmE660-infected animals, the envelope analyses of which were previously reported (R02012 and R95117 [43] and CG7V and CG7G [18]), were also included for full-genome analysis and IMC generation. Only one of these animals (R95117) was infected with a single variant, while R02012 was infected with two variants and CG7G and CG7V were each infected with five variants, based on env analyses.
Full-genome analysis.
While the viral envelope gene provides sufficient diversity for variant enumeration, the sequence of the entire genome was needed to generate IMCs that represented the exact transmitted/founder viral genome. We performed SGA and direct sequencing of vRNA obtained at peak viremia, amplifying overlapping 5′ and 3′ half-genomes for all 8 animals. Importantly, sequencing vRNA is sufficient to infer the entire proviral form of the SIV genome because both halves of the repeated region (R) are contained within these sequences, as well as the untranslated-5 (U5) region from the 5′ half-sequence and the untranslated-3 (U3) region from the 3′ sequence. These overlapping sequences were then concatenated in combination with the inferred Env T/F sequence to identify the full-length T/F viral genome (Fig. 2; see Fig. S1 in the supplemental material). Although three animals were infected with more than one variant (R02012, CG7G, and CG7V), only the dominant lineage was selected for viral cloning. As expected, in each case, the env-only T/F consensus was confirmed in the larger 3′ half-amplicons. For all 8 viral genomes examined, each inferred T/F viral genome contained intact open reading frames and standard start codons for all 9 SIV genes.
FIG 2.
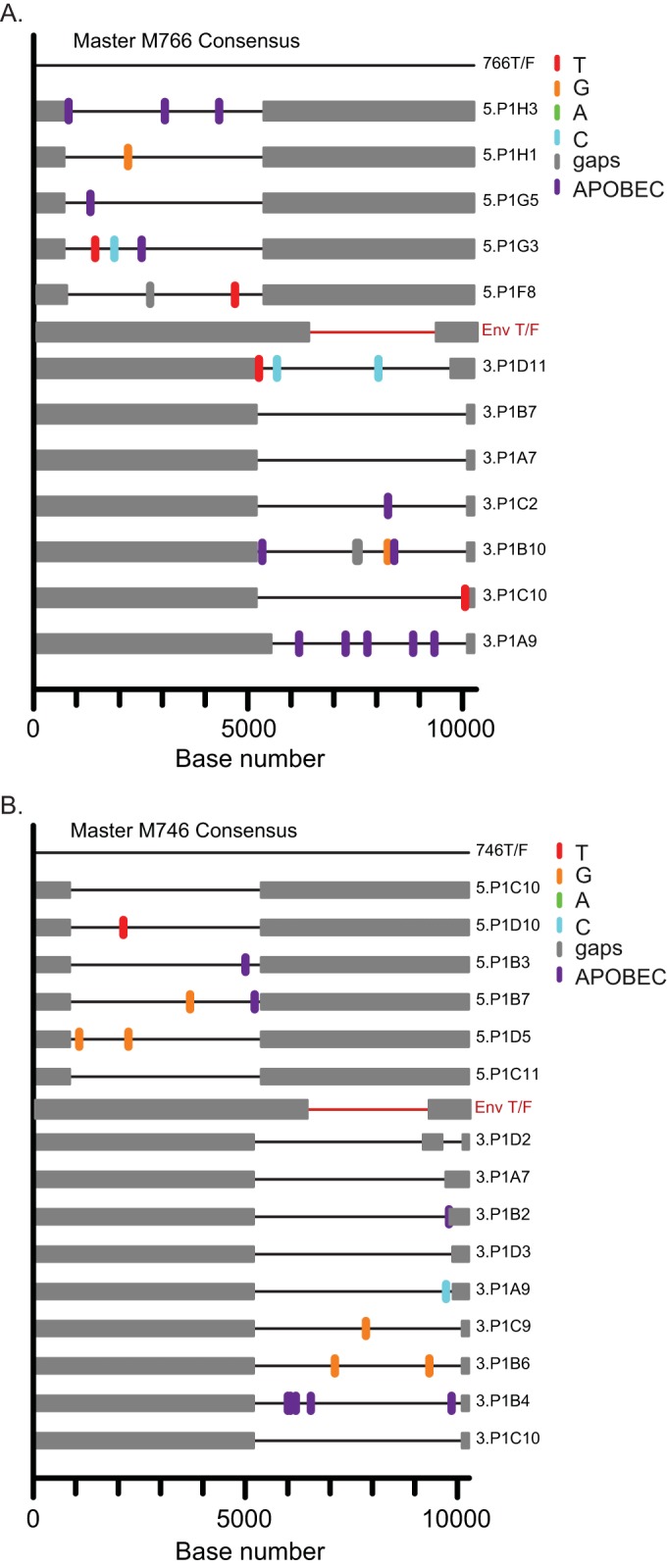
Highlighter alignments of 5′ and 3′ half-genomes from animals M766 (A) and M746 (B). Single nucleotide polymorphisms to the consensus sequences are denoted by colored tick marks; gaps are shown in gray and G-to-A mutations in purple. The two overlapping amplicons are shown in black, and the previously determined T/F env sequence is shown in red (Env T/F). The full-genome consensus sequence was identified using the overlapping half-genomes.
Generation of full-length T/F SIV molecular clones.
Although SGA was utilized to infer T/F genomes, SGA amplicons themselves are not amenable to direct cloning due to the lack of intact 5′ and 3′ long terminal repeats (LTRs) and the accumulated mutations of multiple PCR cycles necessary to amplify from a single-copy template. To overcome these challenges, we utilized a methodology originally developed for HIV-1 T/F IMC cloning (29) and SIVagm cloning (55). Two half-genome fragments were amplified from integrated PBMC DNA (or synthesized directly) to encompass the entire 5′ and 3′ LTRs and an internal unique restriction site. Each half-genome amplicon was independently cloned. Individual clones were screened by sequence analysis to match the inferred T/F genome. Final assembly was performed by combining sequence-confirmed 5′ and 3′ half-clones into one IMC using a unique internal restriction site. In total, 4 clones were generated from the SIVmac251 lineage (SIVmac766, SIVmac746, SIVmacPBE, and SIVmacCR53) and 4 from the SIVsmE660 lineage (SIVsmR02012, SIVsmR95117, SIVsmCG7V, and SIVsmCG7G).
In vitro infectivity and replication kinetics.
Viral stocks from each of the T/F IMCs were tested for infectivity on TZM-bl cells. All 8 clones were infectious, with titers for each clone greater than 1.2 × 105 IU/ml (data not shown). The viral replication capacity was then evaluated in primary rhesus macaque PBMCs depleted of CD8+ T cells and infected with equivalent numbers of infectious units of virus. To evaluate viral replication in the absence of any potential TRIM5α-mediated restriction of SIVsmE660-derived viruses, we selected PBMC donor animals with a permissive TRIM5α genotype (Q/Q) (39). As shown in Fig. 3, all 8 T/F viruses were replication competent, with high titers detected by day 3, which peaked on day 7 with titers comparable to those of SIVmac239 and SIVsmE543 controls (range, 7 × 104 to 6 × 105 pg/ml RT activity).
FIG 3.

In vitro replication curves of 8 T/F IMCs and the control clones SIVmac239 and SIVsmE543. CD8-depleted PBMCs from 2 naive, TRIM5α-permissive (Q/Q) rhesus macaques were inoculated with equivalent numbers of infectious units of each virus. Viral replication was monitored by measuring RT activity in cell-free culture supernatants at days 3, 7, 10, and 14 postinfection. (A) SIVmac lineage clones. (B) SIVsm lineage clones.
Neutralization sensitivity of T/F IMCs.
The vast majority of viruses from the SIVsmE660 lineage have been shown to be exquisitely sensitive to neutralizing antibodies (44), while viruses from the SIVmac251 lineage (including SIVmac239) are highly neutralization resistant (44, 56). Interestingly, the SIVsmE543 clone derived from the SIVsmE660 lineage is relatively neutralization resistant, while the SIVmac239 clone is completely resistant. To determine the neutralization sensitivity of these 8 new T/F IMCs, we examined each clone against 12 newly developed and characterized MAbs that target critical determinants on the viral envelope, including the CD4 binding site and variable loops 1, 2, and 3 (48). As expected, we found wide variation in neutralization sensitivities for each MAb, with the SIVmac lineage clones being overall more resistant than the SIVsm clones (Fig. 4). Importantly, none of the clones were as resistant as SIVmac239, which has a 50% inhibitory concentration (IC50) of >50 μg/ml for each MAb. We then selected 4 clones (SIVmac746, SIVmac766, SIVsmCG7G, and SIVsmCG7V), 2 from each viral lineage, and tested neutralization sensitivities using sera from chronically SIVmac251- and SIVsmE660-infected rhesus macaques. Consistent with our published results (44), the SIVsm clones were neutralization sensitive, with 50% reciprocal inhibitory dilution (ID50) titers ranging from 5 × 10−5 to 2 × 10−6 for SIVsmCG7G and 2 × 10−4 to 2 × 10−5 for SIVsmCG7V (Fig. 5A and B). The SIVmac clones again were more resistant overall to neutralization. For the SIVmac766 clone, 7 of 8 different sera from SIVmac-infected macaques (lineage autologous) were able to reach an ID50 titer ranging from 5 × 10−5 to 5 × 10−6, while only 3 of 6 SIVsm-infected macaque sera (lineage heterologous) reached an ID50 (Fig. 5C). For the SIVmac746 clone, neither autologous nor heterologous sera were able to neutralize the virus by greater than 50% at the most concentrated plasma value tested (1:500) (Fig. 5D). Two additional monoclonal antibodies recognizing a V3 epitope (3.11H) and a CD4-induced epitope (1.4H) were also utilized to test general neutralization sensitivity (Fig. 5E and F) (47). As expected, SIVmac239 was resistant even at the highest concentration (10 μg/ml) tested, while the remaining T/F clones revealed 4 distinct neutralization profiles. These neutralization data highlight the wide breadth of neutralization sensitivities of these T/F molecular clones, with evidence of several-log-unit differences in neutralization, allowing a rank order of sensitivity from the most sensitive to the most resistant: SIVsmCG7G, SIVsmCG7V, SIVmac766, and SIVmac746.
FIG 4.

Summary of neutralization profiles of the T/F clones against 12 SIV-specific MAbs. The antibodies were specific for CD4 binding site; CD4 induced; variable loops 1, 2, and 3; and sites outside the variable loops (ΔV1V2V3). Color coding indicates the ranges of IC50s, as indicated. For MAbs that did not completely neutralize, a half-maximum IC50 (Half Max) was determined based on the concentration that caused a 50% reduction of the maximal neutralization (Vmax) achieved for that antibody. Values of >50 indicate that no neutralization was detected.
FIG 5.

(A to D) Neutralization profiles of the T/F clones SIVmac746, SIVmac766, SIVsmCG7G, and SIVsmCG7V following exposure to lineage-homologous or lineage-heterologous sera from chronically infected animals. Viruses were incubated with serially diluted sera from rhesus macaques chronically infected with SIVmac251 or SIVsmE660. Residual infectivity was measured on TZM-bl cells following exposure to the sera. (E and F) Percent inhibition of virus entry was also assessed using monoclonal antibodies specific for V3-induced (E) or CD4-induced (F) epitopes.
In vivo replication.
Large infectious stocks of these four clones (SIVmac746, SIVmac766, SIVsmCG7G, and SIVsmCG7V) were generated and used to characterize the clones in vivo. For each clone, two naive Indian-origin rhesus macaques were intrarectally or intravenously infected, and disease progression was monitored by viral-load quantification and CD4+ T cell depletion in peripheral blood. For SIVmac251 clonal infection, plasma viral-load analysis showed acute replication kinetics similar to those typically seen following SIVmac251 isolate infection, with peak viral loads ranging from 2.1 × 107 to 7.4 × 107 vRNA copies/ml by days 11 to 17 postchallenge (Fig. 6A). These animals were monitored at frequent intervals for more than 600 days to assess the chronic set point viral load, peripheral blood CD4+ T cell decline, pathogenesis, and viral adaptation. The 4 macaques infected with SIVmac clones established similar viral set points irrespective of the host TRIM5α genotype (Fig. 6A). These four animals had progressive declines in peripheral blood CD4+ T cells over the study period corresponding to high plasma viral loads (Fig. 6C). Using immunohistochemistry, we evaluated the extent of acute CD4+ T cell depletion in the gastrointestinal tract—a hallmark of pathogenic SIV infection. We found substantial CD4+ T cell depletion in the gut as early as 28 days postinfection that is typical of pathogenic SIV infection (Fig. 6E). Animals p096 (SIVmac746 infected) and p097 (SIVmac766 infected) were euthanized due to simian-AIDS-defining illnesses at 1.1 and 1.3 years postinfection, respectively (see below).
FIG 6.

In vivo replication kinetics and peripheral CD4 depletion. (A and B) Plasma viral loads from animals infected with SIVmac clones (A) or SIVsm clones (B) highlight the consistency of SIVmac infections and the variable viral loads following SIVsm infection. The legend (right) indicates the infecting virus and TRIM5α genotype for each animal. †, euthanasia due to simian-AIDS-defining conditions. (C and D) Longitudinal absolute peripheral blood CD4+ T cell counts for SIVmac-infected animals (C) and SIVsm-infected animals (D). (E) Early gut CD4+ T cell depletion shown by immunohistochemistry in SIVmac766-infected animals (p106 and p097) and SIVsmCG7G-infected animals (p114 and p116) from preinfection and acute infection (day 28 postinfection). The brown stain is for CD4+ cells, and pink staining for myeloperoxidase identifies myeloid cells. Scale bars, 100 μm.
Animals infected with SIVsm clones (SIVsmCG7G and SIVsmCG7V) had similar peak viral loads ranging from 2.1 × 106 to 5.0 × 107 by days 11 to 19 postchallenge but with highly variable chronic-phase viral loads. For each SIVsm, one animal controlled viremia during the chronic phase of infection to less than 103 vRNA copies/ml (p114 and p142), while the other animal maintained viral loads of ∼105 (p140) or ∼107 (p116) through the chronic stage (Fig. 6B). The peripheral blood CD4+ T cell levels were also variable for SIVsm-infected animals but showed an overall decline during infection inversely proportional to the plasma viral load (Fig. 6D). We found substantial CD4+ T cell depletion in the gut as early as 28 days postinfection that is typical of pathogenic SIV infection (Fig. 6E). Animal p116 (SIVsmCG7G) required euthanasia at 1.1 years postinfection due to AIDS-defining illness with a sustained elevated viral load (see below).
Viral restriction.
There were two possible, but not mutually exclusive, explanations for the disparate chronic viral load outcomes following SIVsm infection: cytotoxic T lymphocyte (CTL)- or TRIM5α-mediated control. Three of the four SIVsm-infected macaques (including the two controlling animals) had a known controlling major histocompatibility complex (MHC) allele (Mamu-B*17), which has been associated with elite control in ∼20% of SIVmac239-infected animals (57). However, this seems unlikely to be the primary mechanism for viral control in these animals because both SIVsm lineage clones already contained the dominant CTL escape allele (TW9) in Nef, which has been shown to abrogate epitope processing and presentation (58). Furthermore, p106 also had the Mamu-B*017 allele, but sequence analysis revealed viral escape to PW9. To determine if differential sensitivity to TRIM5α alleles might explain the low viral load in the SIVsm infections, we sequenced the CypA binding site in gag for evidence of viral escape from the effects of TRIM5α. It has been shown that mutations in this site confer resistance to TRIM restriction (59). For SIVsmCG7V, no CypA binding site mutations were detected in the animal that controlled viral replication (p142). In contrast, in animal p140, which also had a restrictive TRIM5α genotype but high viral loads, the circulating virus contained three fixed amino acid mutations in the CypA binding site (R97S, T107N, and D109E) (Table 1). Intriguingly, for both SIVsmCG7G-infected animals, we found a V91A mutation in gag irrespective of the TRIMα genotype. For p114, an animal with a restrictive genotype (TFP/TFP), this mutation occurred very rapidly and was present in 29% of sequences by week 2 and completely fixed by week 6, whereas for p116, an animal with a permissive genotype (Q/Q), the mutation was not detected through week 12 but then was fixed by week 26. We conclude that 91A is the optimal amino acid in rhesus macaques regardless of TRIM5α genotype and that escape from TRIM5α restriction occurred in only 1 of 3 SIVsm-infected macaques (p140).
TABLE 1.
Proportions of CypA binding sitemutations over time
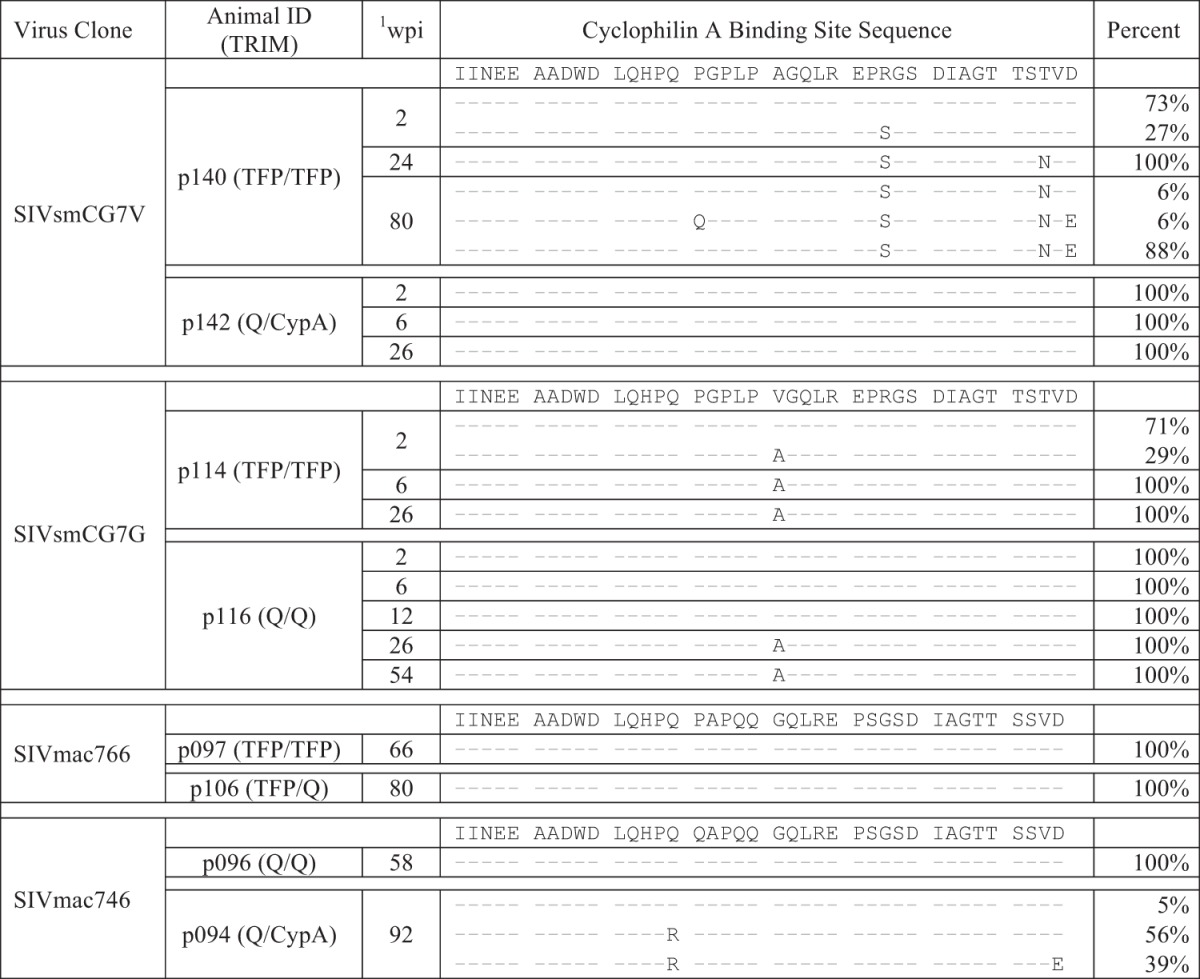
wpi, weeks postinfection.
In contrast, for animals infected with SIVmac766, there were no mutations detectable within the CypA binding site, despite restrictive (TFP/TFP) and intermediate (TFP/Q) genotypes. Of SIVmac746-infected animals, p096, with the permissive genotype (Q/Q), showed no CypA binding site mutations, but the intermediate and rare rhesus genotype (Q/CypA) in p094 revealed selective pressure and nearly complete fixation of a Q85R mutation with or without a D109E mutation, which might indicate a lack of complete adaptation of SIVmac251 to some uncommon rhesus TRIM5α alleles. Although not all of these specific mutations have been previously identified as TRIM5α escape mutations, the significant positive selection only within this region of gag in the presence of known restrictive genotypes suggests substantial selective pressure exerted by TRIM5α.
Given the fixed mutations in the CypA binding site of Gag, we sought to confirm TRIM5α restriction of SIVsm in vitro. The replication competence of each clone was tested in CD4+ T cells from animals with a known TRIM5α-restrictive genotype. Again, all of the T/F clones replicated equally well and as well as or better than the control viruses in cells with a permissive genotype (Fig. 7A), but SIVsm lineage clones replicated to levels 1 to 2 log units lower in TFP/TFP restrictive cells (Fig. 7B). Thus, we conclude that all T/F IMCs are fully functional in vivo and in vitro, but as is the case with SIVsmE543 and other SIVsmE660 lineage members, SIVsmCG7G and SIVsmCG7V are sensitive to rhesus TRIM5α restriction.
FIG 7.

In vitro replication curves of 4 T/F IMCs (SIVsmCG7G, SIVsmCG7V, SIVmac746, and SIVmac766) and the control clones SIVmac239 and SIVsmE543. Viruses from these clones were used to infect CD8-depleted PBMCs collected from 2 naive rhesus macaques. One macaque (A) possessed a permissive TRIM5α genotype (Q/Q), while the other (B) possessed a restrictive TRIM5α genotype (TFP/TFP). RT activity was measured at days 3, 7, and 10 postinfection, showing significant TRIM5α-specific restriction of SIVsm, but not SIVmac.
Sequence analysis of acute infection.
The underlying principle in using sequence analysis to identify the actual transmitted/founder virus rests on the premise that early mutations are randomly generated without selection and that the consensus genome of these early sequences represents the virus that was transmitted and became the founder of the virus population. Infection with clonal viruses allowed us to formally test this principle by examining the accumulated changes during acute infection compared to the clonal stock. We used SGA at peak viremia (2 weeks postinfection) in each infected macaque and sequenced the entire genome in overlapping 5′ and 3′ amplicons. These sequences were then compared to the T/F clonal sequence (Fig. 8). In each case, the accumulated mutations either were randomly distributed and represent unselected RT errors or were G-to-A mutations, consistent with generation by the innate restriction factor APOBEC (Fig. 8). Interestingly, there was significant variation in the numbers of APOBEC-mediated mutations seen both between animals infected with the same virus and between animals infected with different viruses. In all cases, the consensus sequence at peak viremia exactly matched the clonal viral sequence, whether or not the APOBEC mutations were included.
FIG 8.

Highlighter alignments of 5′ and 3′ half-genomes from all 8 infected animals. Plasma samples were obtained at peak viremia and used to identify the consensus genome for each infected animal. Each alignment represents two animals infected with each clone, SIVmac746 (A), SIVmac766 (B), SIVsmCG7V (C), and SIVsmCG7G (D). Single nucleotide polymorphisms to the T/F IMC sequences are denoted by colored tick marks; gaps are shown in gray and G-to-A mutations in purple. The full-genome consensus sequence (CONSENSUS) for both animals is an exact nucleotide match with that of the IMC clone used to infect each animal.
Sequence analysis of viral adaptation.
Since viruses adapt to their hosts over time, we sought to fully characterize these new clones using full-genome sequence analysis to determine the frequencies of accumulated mutations at the time of necropsy. For the two animals that controlled their infections (p114 and p142), plasma viral loads were insufficient for analysis. For the three animals necropsied due to AIDS (p116, p097, and p096), phylogenetic analysis occurred at ∼425 days postinfection, while the remaining 3 animals (p094, p140, and p106) were analyzed at ∼600 days postinfection (Fig. 9). Phylogenetic analysis of the 5′ half-genome (including gag and pol) revealed limited fixed mutations for either SIVmac-infected (Fig. 9A) or SIVsm-infected (Fig. 9C) macaques. The average overall pairwise nucleotide diversity of the 5′ half-genome for all the animals was 0.005 (range, 0.004 to 0.007), while the average nucleotide divergence from the viral clonal sequence was 0.004 (range, 0.003 to 0.005). Overall, outside the CypA binding site, the 5′ half of the genome had limited sites with fixed mutations but had accumulated modest levels of random diversity (seen as star-like pattern at the tips of the tree). This was in stark contrast to the 3′ half-genome (including Vif, Vpr, Vpx, Tat, Rev, Env, and Nef genes), where long internal branches common to all the sequences from each animal revealed significant viral adaptation and positive selection in both SIVmac-infected (Fig. 9B) and SIVsm-infected (Fig. 9D) animals. Overall, the 3′ half-genome had twice as much diversity (average diversity, 0.01; range, 0.006 to 0.012) as the 5′ half (P = 0.004). Furthermore, the 3′ half-genome had significantly higher divergence from the T/F genome (average divergence, 0.01; range, 0.007 to 0.013) than the 5′ half (P = 0.0002). Additionally, like the 5′ half-genome, the tips of the 3′ half-genome were also star-like, with a large accumulation of unique mutations. Together, these data reveal significant adaptation in the 3′ half-genome, leading to multiple fixed mutations.
FIG 9.

Unrooted phylogenetic trees of sequences from 6 of the 8 infected animals from plasma taken at necropsy. (A and B) SIVmac-infected animals, with trees representing the 5′ half-genome (A) and the 3′ half-genome (B). (C and D) SIVsm-infected animals, with trees representing the 5′ half-genome (C) and the 3′ half-genome (D). The scale bars represent 0.01-nucleotide changes. Sequences from each animal are color coded. Each TF IMC sequence is labeled in black.
We next examined the individual mutations under positive selection. For SIVsm-infected animals, the few selected mutations identified in the 5′ half-genome included the TRIM5α escape mutations described above and a mutation in Pol found in two animals, leading to an S753G substitution in animal p140 and an S753N substitution in animal p116. In the 5′ half-genome in animals infected with SIVmac766, there was a single common mutation in both animals in Gag (K404R), while SIVmac746-infected animals had no common mutations. The only other fixed mutations within Gag or Pol were single polymorphisms within individual animals, likely representing animal-specific adaptations.
The 3′ half-genome contained the most fixed mutations, with the most pronounced changes occurring within the envelope gene clustered within the V1/V2 and V4 regions, including single-nucleotide polymorphisms and potential N-linked glycosylation (PNG) mutations (see Fig. S2 in the supplemental material). Within the V1/V2 and V4 loops, we also observed substantial length variation due to insertions and deletions. One notable fixed mutation in 5 of 6 animals was the addition of a PNG near the tip of V4. In SIVmac-infected animals, the mutation was D415N, while for SIVsm-infected animals, the change was K420N. We identified several additional mutations in the 3′ half of the genome common to both animals infected with the same virus or viral lineage. We identified 4 Env mutations (I215V, W345R, D415N, and H831Q/L/N) and 2 Nef mutations (S161L and Q196H) in SIVmac766-infected animals. For SIVmac746-infected animals, there were 4 total shared mutations, including a Vif mutation (H74N), an Env mutation (T628S), and 2 Nef mutations (M144I/V and Q196H). Furthermore, we identified 7 mutations common to SIVsmCG7G- and SIVsmCG7V-infected animals, including a Vpx mutation (R50Q); a Vpr mutation (R63K), which also includes an overlapping Tat1 mutation (E13K); an Env mutation (T473I); a Rev2 mutation (N62D); and 3 Nef mutations (V4A, K7R, and Y170F/C/H). Overall, the frequent changes in V1/V2 and V4, with length polymorphisms and important glycosylation changes, are indicative of antibody-mediated selective pressure with additional virus-specific polymorphisms.
Viral pathogenesis.
All infected animals showed typical signs of progressive immunodeficiency associated with SIV infection, including acute and sustained gut CD4+ T cell depletion (Fig. 6E) and progressive declines in circulating CD4+ T cells (Fig. 6C and D). Macaques p096 (SIVmac746) and p097 (SIVmac766) reached simian-AIDS-defining clinical endpoints at 1.1 and 1.3 years postinfection, with peripheral blood CD4+ T cell counts of <200 cells/mm3, severe chronic nonresponsive diarrhea with inappetence, cachexia, and pneumonia. Animal p116 (SIVsmCG7G) was also euthanized after reaching a clinical endpoint at 1.1 years postinfection, with cachexia and a large abdominal mass. At necropsy, a 12-cm expansile, mesenteric, multifocal, extranodal B cell lymphoma characterized by random mitotic figures and large, random areas of necrosis was found, representing a classic end stage, AIDS-defining pathology.
To further characterize immunopathology, lymph nodes were collected prior to infection, throughout the study, and at necropsy for all of the animals. Representative longitudinally sampled lymph node sections from p096 and p116 showed a substantial population of CD4+ cells in the paracortical region (T cell zone) prior to infection that were severely depleted at the time of necropsy (Fig. 10A, brown). Myeloid lineage cells (pink) were largely unchanged over the course of infection. Immune activation (Ki67 staining) was largely confined to germinal centers in preinfection section lymph nodes but had progressed to generalized, diffuse activation, beginning with primary infection, that was sustained through to AIDS (Fig. 10B). In addition, collagen deposition led to extensive remodeling of the cortical lymph node architecture, a characteristic response to chronic inflammation in pathogenic lentiviral infection (Fig. 10C). Therefore, in animals in which viral replication was not substantially restricted by TRIM5α, we identified typical signs of SIV-associated immunopathology in early and late tissue samples, consistent with a progressive lentiviral infection typical of SIVmac or SIVsm infection of rhesus macaques.
FIG 10.

Immunopathology in the lymph nodes for 2 representative animals, p096 (SIVmac746) and p116 (SIVsmCG7G). Biopsy specimens obtained preinfection (Pre.) are compared to samples obtained postinfection, with the animal manifesting an AIDS-defining condition (AIDS). (A) Decrease in CD4+ T cells (brown) with unchanged myeloid lineage cells (pink) at AIDS. (B and C) Increased immune activation at AIDS as measured by Ki67 staining (brown), largely restricted to B cell follicles prior to infection but generalized throughout the lymph node when simian AIDS was diagnosed (B), and increased collagen deposition (brown) (C).
DISCUSSION
Nonhuman primate models of HIV have been instrumental in studying viral transmission, pathogenesis, and intervention strategies. Virtually all of the numerous HIV/AIDS-related studies involving SIV infection in NHP models that have been published have been conducted using a limited number of viral-swarm stocks or related molecular clones derived from late stages of infection. Here, we sought to generate and provide to the field well-characterized, infectious molecular clones representing authentic transmitted/founder viruses that a priori possess all the properties necessary and sufficient to establish systemic infection and cause classical AIDS-defining pathology. In addition to being sequence-confirmed transmitted/founder viruses, the clones were selected based on good replicative dynamics in the parental animals from which they were derived, their genetic relatedness to other clones, and a greater than average representation among transmitted variants (Fig. 1). The average intralineage diversity for the T/F IMCs was 0.5% for SIVsm clones and 1% for SIVmac clones, while the average interlineage diversity was 15%, approximating within- and between-individual diversities for HIV-infected subjects (22), making these clones ideal for use in vaccine studies testing homologous versus heterologous protection.
As predicted by our model of early T/F replication and evolution, the analysis of the envelope gene only was confirmed, when using full-length genome sequencing, in both the number of identifiable T/F variants and the inferred sequence. These data confirm that there is not a primer bias toward any particular variant and suggest that any region of the genome can be used to infer the number of T/F variants as long as there is sufficient diversity within that region. As expected from our model, all 8 T/F clones contained intact reading frames and were infectious and replication competent. Furthermore, it is clear that these T/F viruses replicated in vitro at least as well as the SIVmac239 and the SIVsmE543 clones. In fact, all the clones replicated more robustly than SIVmac239 as long as the cells were derived from a TRIM5α-permissive donor. This may be due to the suboptimal nucleotides still present in the parental SIVmac239 (35, 37). Consistent with this idea, we recently reported that correcting these suboptimal changes increased in vitro replication (37). Regardless of the reason for the poorer performance of SIVmac239, these new clones behaved precisely as hypothesized for authentic T/F viruses.
Downselection of clones for detailed characterization in vivo was in part based on neutralization sensitivity. We sought to develop IMCs representing the extensive range of neutralization sensitivities found in SIV. Based on serum neutralization assays, the SIVmac746 clone was highly neutralization resistant and similar to SIVmac239. However, there are indications that SIVmac766 has some sensitivity to neutralization and might provide a much needed alternative to the extremely neutralization-resistant SIVmac239 clone. While SIVsm clones in general are more sensitive to neutralization, the SIVsmCG7G clone is more resistant than SIVsmCG7V, providing both a highly sensitive and modestly resistant virus.
The T/F IMCs from the SIVsmE660 and SIVmac251 lineages were replication competent in vitro and in vivo but showed markedly different lineage-specific susceptibilities to TRIM5α restriction. Upon initiation of infection, plasma viremia results showed a rapid and robust primary infection with high peak viral loads, indicating a fully functional genome and excellent replicative capacity. For animals infected with SIVmac251-derived IMCs, acute and set point viral loads were consistent between animals infected with the same virus and remarkably similar to results for animals M766 and M746, from which the IMCs were derived (data not shown). Importantly, replication of the SIVsm clones was not consistent between animals infected with the same virus but varied with the TRIM5α genotype. For this study, animals were not partitioned into TRIM5α genotypes prior to infection. Of the four animals infected with SIVsm lineage viruses, only one expressed the permissive genotype Q/Q (p116). Of the remaining three animals, two were able to control viremia and the third showed evidence of viral escape from TRIM5α-mediated restriction. Interestingly, SIVsmCG7G contains a valine in a capsid at position 91 instead of the more common alanine found in SIVsmE660 lineage viruses (60), which corresponds to the virus most sensitive to TRIM5α restriction in vitro. Interestingly, this 91V also mutated to 91A by week 26 in animal p116 with a putatively nonrestrictive genotype (Q/Q), suggesting TRIM5α-mediated restriction. For consistency, in experiments other than those studying TRIM5α restriction, the use of these SIVsm lineage viruses should be restricted to TRIM5α-permissive animals or the viruses should be generated with CypA binding site modifications, as described in Table 1. Consistent with previous reports (52), TRIM5α-restrictive alleles appeared to have limited or no negative impact on viruses derived from the SIVmac251 lineage. The one possible exception to this accepted view was the selection of a Q85R mutation in capsids with or without a D109E mutation in animal p094, which possessed the rare Q/CypA genotype and was infected with SIVmac746. Although this mutation was not detected until 92 weeks postinfection, it raises the possibility that SIVmac lineage viruses are not fully resistant to minor TRIM5α genotypes due to limited exposure during their passage history prior to isolation.
While the premise of our approach of using sequence analysis to infer the identities of T/F viruses in clonally initiated infections is widely accepted (9, 12, 14–17), the model has not been subjected to direct experimental testing in rhesus macaques. We amplified the entire viral genome (in halves) at peak viremia and determined that the consensus sequence exactly matched that of the clonal T/F virus inoculum. This conclusion confirms our hypothesis that local replication prior to systemic dissemination with measurable plasma viremia is not sufficiently long or error prone to alter the clonal sequence of the incoming viral genome from the infecting inoculum. This result held for both intrarectal and intravenous infections. The approach to generating functional clones described here is a significant improvement over the original methods for isolating biologically active clones (SIVmac239 and SIVsmE543).
While at peak viremia the consensus genome matched the T/F sequence, at all later times, random mutations and selection generated a more diverse viral population. While some of the mutations seen over time were known to be associated with overcoming immune- or TRIM5α-mediated restriction, other mutations might be undefined immune selection or simply polymorphisms within a quasispecies. The frequent and clear changes in the V1/V2 and V4 regions of envelope highlight the plasticity of those domains in the face of dynamic host-specific autologous antibody responses. One other possible reason for accumulated mutations is that a particular viral clone may have suboptimal mutations that reduce overall fitness, leading to reversion in any host or in vitro. Although we generated our clones using a methodology to limit such mutations, it is possible that some fixed changes are the result of correction of suboptimal nucleotides, as has been well described for SIVmac239 (35, 37). Although we detected fewer than 7 common mutations between infected animals, additional infected animals will be needed to assess if any of these mutations are suboptimal overall or if they reflect naturally occurring polymorphisms. It is important to note that all 8 T/F IMCs have the canonical primer binding site sequence, as well as the optimal version of the other 3 mutations found in the Pol and Env genes of SIVmac239 (37). Additionally, we recently described a viral genotype in SIVsmE660 that is enriched in neutralization-sensitive clones (22, 61). In 6 of 8 new clones reported here, there is an A/K at positions 45/47 of envelope, which has been reported as a neutralization-resistant genotype in SIVsmE660 lineage viruses (22). In two clones not tested in vivo (SIVsmR02012 and SIVsmR95117), the more neutralization-sensitive genotype (T/R) was found.
All the macaques infected with the new T/F virus clones yielded characteristic signs of pathogenic SIV infection. We found declines in CD4+ T cells in peripheral blood, lymph nodes, and the gut; inflammatory polymorphonuclear leukocyte (PMN) infiltrates; chronic immune activation; and pathological collagen deposition in lymphoid tissues, with some animals progressing to AIDS-defining clinical endpoints during the study follow-up period, with disease progression associated with the degree of viral replication. Chronic-phase viremia levels and time to AIDS were consistent with pathogenic SIV infections using other clonal or swarm challenge viruses. Overall, these new IMCs representing T/F viruses from within the widely used SIVmac251 and SIVsmE660 swarms recapitulate key features of these viruses and, a priori, are also representative of the properties of transmitted viruses that represent the most authentic targets for studies of preventive interventions and viral transmission, arguably making them challenge viruses of choice for studies employing a clonal virus challenge. The ability to generate isogenic, biologically equivalent, but sequence-tagged variants of these viruses (62) and to generate similar T/F IMCs from other SIV or simian/human immunodeficiency virus (SHIV) swarms of interest should provide improved options for selecting more optimal challenge viruses for various AIDS-related studies in NHP models.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Nancy Miller and Alan Schultz of the Preclinical Research and Development Branch, NIAID, for support and thoughtful discussions. We acknowledge the staffs of the Specimen Support Core, the Viral Evolution Core, the Tissue Analysis Core, and the Quantitative Molecular Diagnostics Core from the AIDS and Cancer Virus Program, Frederick National Laboratory, for sample processing and analysis. We are grateful for MAbs from Rosemarie Mason and Mario Roederer (Vaccine Research Center, National Institutes of Health, Bethesda, MD) and James Robinson (Tulane University Medical Center, New Orleans, LA). The TZM-bl cell line was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, donated by J. C. Kappes and F. Wu.
This work was supported in part with federal funds from NIAID grant AI087383 and the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Funding Statement
This work was supported in part with federal funds from NIAID grant AI087383 and the National Cancer Institute; National Institutes of Health under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00718-16.
REFERENCES
- 1.Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S, Hunter E. 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- 2.Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, Pellegrino P, Williams I, Cohen MS, Gao F, Shaw GM, Hahn BH, Ochsenbauer C, Kappes JC, Borrow P. 2013. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 10:146. doi: 10.1186/1742-4690-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP Jr, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parrish NF, Wilen CB, Banks LB, Iyer SS, Pfaff JM, Salazar-Gonzalez JF, Salazar MG, Decker JM, Parrish EH, Berg A, Hopper J, Hora B, Kumar A, Mahlokozera T, Yuan S, Coleman C, Vermeulen M, Ding H, Ochsenbauer C, Tilton JC, Permar SR, Kappes JC, Betts MR, Busch MP, Gao F, Montefiori D, Haynes BF, Shaw GM, Hahn BH, Doms RW. 2012. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin alpha4beta7. PLoS Pathog 8:e1002686. doi: 10.1371/journal.ppat.1002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander M, Lynch R, Mulenga J, Allen S, Derdeyn CA, Hunter E. 2010. Donor and recipient envs from heterosexual human immunodeficiency virus subtype C transmission pairs require high receptor levels for entry. J Virol 84:4100–4104. doi: 10.1128/JVI.02068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlson JM, Schaefer M, Monaco DC, Batorsky R, Claiborne DT, Prince J, Deymier MJ, Ende ZS, Klatt NR, DeZiel CE, Lin TH, Peng J, Seese AM, Shapiro R, Frater J, Ndung'u T, Tang J, Goepfert P, Gilmour J, Price MA, Kilembe W, Heckerman D, Goulder PJ, Allen TM, Allen S, Hunter E. 2014. HIV transmission. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 345:1254031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deymier MJ, Claiborne DT, Ende Z, Ratner HK, Kilembe W, Allen S, Hunter E. 2014. Particle infectivity of HIV-1 full-length genome infectious molecular clones in a subtype C heterosexual transmission pair following high fidelity amplification and unbiased cloning. Virology 468-470:454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yue L, Pfafferott KJ, Baalwa J, Conrod K, Dong CC, Chui C, Rong R, Claiborne DT, Prince JL, Tang J, Ribeiro RM, Cormier E, Hahn BH, Perelson AS, Shaw GM, Karita E, Gilmour J, Goepfert P, Derdeyn CA, Allen SA, Borrow P, Hunter E. 2015. Transmitted virus fitness and host T cell responses collectively define divergent infection outcomes in two HIV-1 recipients. PLoS Pathog 11:e1004565. doi: 10.1371/journal.ppat.1004565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HY, Giorgi EE, Keele BF, Gaschen B, Athreya GS, Salazar-Gonzalez JF, Pham KT, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Hahn BH, Shaw GM, Korber BT, Bhattacharya T, Perelson AS. 2009. Modeling sequence evolution in acute HIV-1 infection. J Theor Biol 261:341–360. doi: 10.1016/j.jtbi.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baalwa J, Wang S, Parrish NF, Decker JM, Keele BF, Learn GH, Yue L, Ruzagira E, Ssemwanga D, Kamali A, Amornkul PN, Price MA, Kappes JC, Karita E, Kaleebu P, Sanders E, Gilmour J, Allen S, Hunter E, Montefiori DC, Haynes BF, Cormier E, Hahn BH, Shaw GM. 2013. Molecular identification, cloning and characterization of transmitted/founder HIV-1 subtype A, D and A/D infectious molecular clones. Virology 436:33–48. doi: 10.1016/j.virol.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bar KJ, Li H, Chamberland A, Tremblay C, Routy JP, Grayson T, Sun C, Wang S, Learn GH, Morgan CJ, Schumacher JE, Haynes BF, Keele BF, Hahn BH, Shaw GM. 2010. Wide variation in the multiplicity of HIV-1 infection among injection drug users. J Virol 84:6241–6247. doi: 10.1128/JVI.00077-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C, CAPRISA Acute Infection Study Team, Center for HIV-AIDS Vaccine Immunology Consortium. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J Virol 83:3556–3567. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haaland RE, Hawkins PA, Salazar-Gonzalez J, Johnson A, Tichacek A, Karita E, Manigart O, Mulenga J, Keele BF, Shaw GM, Hahn BH, Allen SA, Derdeyn CA, Hunter E. 2009. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog 5:e1000274. doi: 10.1371/journal.ppat.1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Bar KJ, Wang S, Decker JM, Chen Y, Sun C, Salazar-Gonzalez JF, Salazar MG, Learn GH, Morgan CJ, Schumacher JE, Hraber P, Giorgi EE, Bhattacharya T, Korber BT, Perelson AS, Eron JJ, Cohen MS, Hicks CB, Haynes BF, Markowitz M, Keele BF, Hahn BH, Shaw GM. 2010. High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathog 6:e1000890. doi: 10.1371/journal.ppat.1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw GM, Hunter E. 2012. HIV transmission. Cold Spring Harb Perspect Med 2:a006965. doi: 10.1101/cshperspect.a006965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keele BF, Li H, Learn GH, Hraber P, Giorgi EE, Grayson T, Sun C, Chen Y, Yeh WW, Letvin NL, Mascola JR, Nabel GJ, Haynes BF, Bhattacharya T, Perelson AS, Korber BT, Hahn BH, Shaw GM. 2009. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J Exp Med 206:1117–1134. doi: 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma ZM, Keele BF, Qureshi H, Stone M, Desilva V, Fritts L, Lifson JD, Miller CJ. 2011. SIVmac251 is inefficiently transmitted to rhesus macaques by penile inoculation with a single SIVenv variant found in ramp-up phase plasma. AIDS Res Hum Retroviruses 27:1259–1269. doi: 10.1089/aid.2011.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stone M, Keele BF, Ma ZM, Bailes E, Dutra J, Hahn BH, Shaw GM, Miller CJ. 2010. A limited number of simian immunodeficiency virus (SIV) env variants are transmitted to rhesus macaques vaginally inoculated with SIVmac251. J Virol 84:7083–7095. doi: 10.1128/JVI.00481-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Keele BF, Li H, Keating S, Norris PJ, Carville A, Mansfield KG, Tomaras GD, Haynes BF, Kolodkin-Gal D, Letvin NL, Hahn BH, Shaw GM, Barouch DH. 2010. Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J Virol 84:10406–10412. doi: 10.1128/JVI.01155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roederer M, Keele BF, Schmidt SD, Mason RD, Welles HC, Fischer W, Labranche C, Foulds KE, Louder MK, Yang ZY, Todd JP, Buzby AP, Mach LV, Shen L, Seaton KE, Ward BM, Bailer RT, Gottardo R, Gu W, Ferrari G, Alam SM, Denny TN, Montefiori DC, Tomaras GD, Korber BT, Nason MC, Seder RA, Koup RA, Letvin NL, Rao SS, Nabel GJ, Mascola JR. 2014. Immunological and virological mechanisms of vaccine-mediated protection against SIV and HIV. Nature 505:502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaccari M, Keele BF, Bosinger SE, Doster MN, Ma ZM, Pollara J, Hryniewicz A, Ferrari G, Guan Y, Forthal DN, Venzon D, Fenizia C, Morgan T, Montefiori D, Lifson JD, Miller CJ, Silvestri G, Rosati M, Felber BK, Pavlakis GN, Tartaglia J, Franchini G. 2013. Protection afforded by an HIV vaccine candidate in macaques depends on the dose of SIVmac251 at challenge exposure. J Virol 87:3538–3548. doi: 10.1128/JVI.02863-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel V, Jalah R, Kulkarni V, Valentin A, Rosati M, Alicea C, von Gegerfelt A, Huang W, Guan Y, Keele BF, Bess JW Jr, Piatak M Jr, Lifson JD, Williams WT, Shen X, Tomaras GD, Amara RR, Robinson HL, Johnson W, Broderick KE, Sardesai NY, Venzon DJ, Hirsch VM, Felber BK, Pavlakis GN. 2013. DNA and virus particle vaccination protects against acquisition and confers control of viremia upon heterologous simian immunodeficiency virus challenge. Proc Natl Acad Sci U S A 110:2975–2980. doi: 10.1073/pnas.1215393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pegu P, Vaccari M, Gordon S, Keele BF, Doster M, Guan Y, Ferrari G, Pal R, Ferrari MG, Whitney S, Hudacik L, Billings E, Rao M, Montefiori D, Tomaras G, Alam SM, Fenizia C, Lifson JD, Stablein D, Tartaglia J, Michael N, Kim J, Venzon D, Franchini G. 2013. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIV(mac251) acquisition in an immunization regimen that mimics the RV-144 Thai trial. J Virol 87:1708–1719. doi: 10.1128/JVI.02544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sui Y, Hogg A, Wang Y, Frey B, Yu H, Xia Z, Venzon D, McKinnon K, Smedley J, Gathuka M, Klinman D, Keele BF, Langermann S, Liu L, Franchini G, Berzofsky JA. 2014. Vaccine-induced myeloid cell population dampens protective immunity to SIV. J Clin Invest 124:2538–2549. doi: 10.1172/JCI73518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao P, Patterson LJ, Kuate S, Brocca-Cofano E, Thomas MA, Venzon D, Zhao J, DiPasquale J, Fenizia C, Lee EM, Kalisz I, Kalyanaraman VS, Pal R, Montefiori D, Keele BF, Robert-Guroff M. 2012. Replicating adenovirus-simian immunodeficiency virus (SIV) recombinant priming and envelope protein boosting elicits localized, mucosal IgA immunity in rhesus macaques correlated with delayed acquisition following a repeated low-dose rectal SIV(mac251) challenge. J Virol 86:4644–4657. doi: 10.1128/JVI.06812-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gambhira R, Keele BF, Schell JB, Hunter MJ, Dufour JP, Montefiori DC, Tang H, Rose JK, Rose N, Marx PA. 2014. Transmitted/founder simian immunodeficiency virus envelope sequences in vesicular stomatitis and Semliki forest virus vector immunized rhesus macaques. PLoS One 9:e109678. doi: 10.1371/journal.pone.0109678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. 2012. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol 86:2715–2728. doi: 10.1128/JVI.06157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med 206:1273–1289. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claiborne DT, Prince JL, Scully E, Macharia G, Micci L, Lawson B, Kopycinski J, Deymier MJ, Vanderford TH, Nganou-Makamdop K, Ende Z, Brooks K, Tang J, Yu T, Lakhi S, Kilembe W, Silvestri G, Douek D, Goepfert PA, Price MA, Allen SA, Paiardini M, Altfeld M, Gilmour J, Hunter E. 2015. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc Natl Acad Sci U S A 112:E1480–E1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naidu YM, Kestler HW III, Li Y, Butler CV, Silva DP, Schmidt DK, Troup CD, Sehgal PK, Sonigo P, Daniel MD. 1988. Characterization of infectious molecular clones of simian immunodeficiency virus (SIVmac) and human immunodeficiency virus type 2: persistent infection of rhesus monkeys with molecularly cloned SIVmac. J Virol 62:4691–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daniel MD, Letvin NL, King NW, Kannagi M, Sehgal PK, Hunt RD, Kanki PJ, Essex M, Desrosiers RC. 1985. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science 228:1201–1204. doi: 10.1126/science.3159089. [DOI] [PubMed] [Google Scholar]
- 34.Regier DA, Desrosiers RC. 1990. The complete nucleotide sequence of a pathogenic molecular clone of simian immunodeficiency virus. AIDS Res Hum Retroviruses 6:1221–1231. [DOI] [PubMed] [Google Scholar]
- 35.Alexander L, Denekamp L, Czajak S, Desrosiers RC. 2001. Suboptimal nucleotides in the infectious, pathogenic simian immunodeficiency virus clone SIVmac239. J Virol 75:4019–4022. doi: 10.1128/JVI.75.8.4019-4022.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kestler HW III, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. 1991. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65:651–662. doi: 10.1016/0092-8674(91)90097-I. [DOI] [PubMed] [Google Scholar]
- 37.Fennessey CM, Reid C, Lipkey L, Newman L, Oswald K, Piatak M Jr, Roser JD, Chertova E, Smedley J, Gregory Alvord W, Del Prete GQ, Estes JD, Lifson JD, Keele BF. 2015. Generation and characterization of a SIVmac239 clone corrected at four suboptimal nucleotides. Retrovirology 12:49. doi: 10.1186/s12977-015-0175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirsch V, Adger-Johnson D, Campbell B, Goldstein S, Brown C, Elkins WR, Montefiori DC. 1997. A molecularly cloned, pathogenic, neutralization-resistant simian immunodeficiency virus, SIVsmE543-3. J Virol 71:1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol 8:e1000462. doi: 10.1371/journal.pbio.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Prete GQ, Scarlotta M, Newman L, Reid C, Parodi LM, Roser JD, Oswald K, Marx PA, Miller CJ, Desrosiers RC, Barouch DH, Pal R, Piatak M Jr, Chertova E, Giavedoni LD, O'Connor DH, Lifson JD, Keele BF. 2013. Comparative characterization of transfection- and infection-derived simian immunodeficiency virus challenge stocks for in vivo nonhuman primate studies. J Virol 87:4584–4595. doi: 10.1128/JVI.03507-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Apetrei C, Kaur A, Lerche NW, Metzger M, Pandrea I, Hardcastle J, Falkenstein S, Bohm R, Koehler J, Traina-Dorge V, Williams T, Staprans S, Plauche G, Veazey RS, McClure H, Lackner AA, Gormus B, Robertson DL, Marx PA. 2005. Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J Virol 79:8991–9005. doi: 10.1128/JVI.79.14.8991-9005.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Letvin NL, Rao SS, Dang V, Buzby AP, Korioth-Schmitz B, Dombagoda D, Parvani JG, Clarke RH, Bar L, Carlson KR, Kozlowski PA, Hirsch VM, Mascola JR, Nabel GJ. 2007. No evidence for consistent virus-specific immunity in simian immunodeficiency virus-exposed, uninfected rhesus monkeys. J Virol 81:12368–12374. doi: 10.1128/JVI.00822-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson NA, Keele BF, Reed JS, Piaskowski SM, MacNair CE, Bett AJ, Liang X, Wang F, Thoryk E, Heidecker GJ, Citron MP, Huang L, Lin J, Vitelli S, Ahn CD, Kaizu M, Maness NJ, Reynolds MR, Friedrich TC, Loffredo JT, Rakasz EG, Erickson S, Allison DB, Piatak M Jr, Lifson JD, Shiver JW, Casimiro DR, Shaw GM, Hahn BH, Watkins DI. 2009. Vaccine-induced cellular responses control simian immunodeficiency virus replication after heterologous challenge. J Virol 83:6508–6521. doi: 10.1128/JVI.00272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopker M, Easlick J, Sterrett S, Decker JM, Barbian H, Learn G, Keele BF, Robinson JE, Li H, Hahn BH, Shaw GM, Bar KJ. 2013. Heterogeneity in neutralization sensitivities of viruses comprising the simian immunodeficiency virus SIVsmE660 isolate and vaccine challenge stock. J Virol 87:5477–5492. doi: 10.1128/JVI.03419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takehisa J, Kraus MH, Decker JM, Li Y, Keele BF, Bibollet-Ruche F, Zammit KP, Weng Z, Santiago ML, Kamenya S, Wilson ML, Pusey AE, Bailes E, Sharp PM, Shaw GM, Hahn BH. 2007. Generation of infectious molecular clones of simian immunodeficiency virus from fecal consensus sequences of wild chimpanzees. J Virol 81:7463–7475. doi: 10.1128/JVI.00551-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morcock DR, Thomas JA, Sowder RC II, Henderson LE, Crise BJ, Gorelick RJ. 2008. HIV-1 inactivation by 4-vinylpyridine is enhanced by dissociating Zn(2+) from nucleocapsid protein. Virology 375:148–158. doi: 10.1016/j.virol.2008.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong R, Li H, Georgiev I, Changela A, Bibollet-Ruche F, Decker JM, Rowland-Jones SL, Jaye A, Guan Y, Lewis GK, Langedijk JP, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2012. Epitope mapping of broadly neutralizing HIV-2 human monoclonal antibodies. J Virol 86:12115–12128. doi: 10.1128/JVI.01632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mason RD, Welles HC, Adams C, Chakrabarti BK, Gorman J, Zhou T, Nguyen R, O'Dell S, Lusvarghi S, Bewley CA, Li H, Shaw GM, Sheng Z, Shapiro L, Wyatt R, Kwong PD, Mascola JR, Roederer M. 2016. Targeted isolation of antibodies directed against major sites of SIV Env vulnerability. PLoS Pathog 12:e1005537. doi: 10.1371/journal.ppat.1005537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 50.Cline AN, Bess JW, Piatak M Jr, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol 34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 51.Hao XP, Lucero CM, Turkbey B, Bernardo ML, Morcock DR, Deleage C, Trubey CM, Smedley J, Klatt NR, Giavedoni LD, Kristoff J, Xu A, Del Prete GQ, Keele BF, Rao SS, Alvord WG, Choyke PL, Lifson JD, Brenchley JM, Apetrei C, Pandrea I, Estes JD. 2015. Experimental colitis in SIV-uninfected rhesus macaques recapitulates important features of pathogenic SIV infection. Nat Commun 6:8020. doi: 10.1038/ncomms9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fenizia C, Keele BF, Nichols D, Cornara S, Binello N, Vaccari M, Pegu P, Robert-Guroff M, Ma ZM, Miller CJ, Venzon D, Hirsch V, Franchini G. 2011. TRIM5alpha does not affect simian immunodeficiency virus SIV(mac251) replication in vaccinated or unvaccinated Indian rhesus macaques following intrarectal challenge exposure. J Virol 85:12399–12409. doi: 10.1128/JVI.05707-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolton DL, Song K, Wilson RL, Kozlowski PA, Tomaras GD, Keele BF, Lovingood RV, Rao S, Roederer M. 2012. Comparison of systemic and mucosal vaccination: impact on intravenous and rectal SIV challenge. Mucosal Immunol 5:41–52. doi: 10.1038/mi.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu M, Keele BF, Aravantinou M, Krawczyk N, Seidor S, Abraham CJ, Zhang S, Rodriguez A, Kizima L, Derby N, Jean-Pierre N, Mizenina O, Gettie A, Grasperge B, Blanchard J, Piatak MJ Jr, Lifson JD, Fernandez-Romero JA, Zydowsky TM, Robbiani M. 2014. Exposure to MIV-150 from a high-dose intravaginal ring results in limited emergence of drug resistance mutations in SHIV-RT infected rhesus macaques. PLoS One 9:e89300. doi: 10.1371/journal.pone.0089300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gnanadurai CW, Pandrea I, Parrish NF, Kraus MH, Learn GH, Salazar MG, Sauermann U, Topfer K, Gautam R, Munch J, Stahl-Hennig C, Apetrei C, Hahn BH, Kirchhoff F. 2010. Genetic identity and biological phenotype of a transmitted/founder virus representative of nonpathogenic simian immunodeficiency virus infection in African green monkeys. J Virol 84:12245–12254. doi: 10.1128/JVI.01603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edinger AL, Ahuja M, Sung T, Baxter KC, Haggarty B, Doms RW, Hoxie JA. 2000. Characterization and epitope mapping of neutralizing monoclonal antibodies produced by immunization with oligomeric simian immunodeficiency virus envelope protein. J Virol 74:7922–7935. doi: 10.1128/JVI.74.17.7922-7935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 80:5074–5077. doi: 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minang JT, Trivett MT, Coren LV, Barsov EV, Piatak M Jr, Chertov O, Chertova E, Ott DE, Ohlen C. 2008. The Mamu B 17-restricted SIV Nef IW9 to TW9 mutation abrogates correct epitope processing and presentation without loss of replicative fitness. Virology 375:307–314. doi: 10.1016/j.virol.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuda K, Dang Q, Brown CR, Keele BF, Wu F, Ourmanov I, Goeken R, Whitted S, Riddick NE, Buckler-White A, Hirsch VM. 2014. Characterization of simian immunodeficiency virus (SIV) that induces SIV encephalitis in rhesus macaques with high frequency: role of TRIM5 and major histocompatibility complex genotypes and early entry to the brain. J Virol 88:13201–13211. doi: 10.1128/JVI.01996-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reynolds MR, Sacha JB, Weiler AM, Borchardt GJ, Glidden CE, Sheppard NC, Norante FA, Castrovinci PA, Harris JJ, Robertson HT, Friedrich TC, McDermott AB, Wilson NA, Allison DB, Koff WC, Johnson WE, Watkins DI. 2011. The TRIM5{alpha} genotype of rhesus macaques affects acquisition of simian immunodeficiency virus SIVsmE660 infection after repeated limiting-dose intrarectal challenge. J Virol 85:9637–9640. doi: 10.1128/JVI.05074-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee FH, Mason R, Welles H, Learn GH, Keele BF, Roederer M, Bar KJ. 2015. Breakthrough virus neutralization resistance as a correlate of protection in a nonhuman primate heterologous simian immunodeficiency virus vaccine challenge study. J Virol 89:12388–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Del Prete GQ, Park H, Fennessey CM, Reid C, Lipkey L, Newman L, Oswald K, Kahl C, Piatak M Jr, Quinones OA, Alvord WG, Smedley J, Estes JD, Lifson JD, Picker LJ, Keele BF. 2014. Molecularly tagged simian immunodeficiency virus SIVmac239 synthetic swarm for tracking independent infection events. J Virol 88:8077–8090. doi: 10.1128/JVI.01026-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.