

ABSTRACT
Exchangeable apolipoproteins (ApoA, -C, and -E) have been shown to redundantly participate in the formation of infectious hepatitis C virus (HCV) particles during the assembly process, although their precise role in the viral life cycle is not well understood. Recently, it was shown that the exogenous expression of only short sequences containing amphipathic α-helices from various apolipoproteins is sufficient to restore the formation of infectious HCV particles in ApoB and ApoE double-gene-knockout Huh7 (BE-KO) cells. In this study, through the expression of a small library of human secretory proteins containing amphipathic α-helix structures, we identified the human cathelicidin antimicrobial peptide (CAMP), the only known member of the cathelicidin family of antimicrobial peptides (AMPs) in humans and expressed mainly in bone marrow and leukocytes. We showed that CAMP is able to rescue HCV infectious particle formation in BE-KO cells. In addition, we revealed that the LL-37 domain in CAMP containing amphipathic α-helices is crucial for the compensation of infectivity in BE-KO cells, and the expression of CAMP in nonhepatic 293T cells expressing claudin 1 and microRNA miR-122 confers complete propagation of HCV. These results suggest the possibility of extrahepatic propagation of HCV in cells with low-level or no expression of apolipoproteins but expressing secretory proteins containing amphipathic α-helices such as CAMP.
IMPORTANCE Various exchangeable apolipoproteins play a pivotal role in the formation of infectious HCV during the assembly of viral particles, and amphipathic α-helix motifs in the apolipoproteins have been shown to be a key factor. To the best of our knowledge, we have identified for the first time the human cathelicidin CAMP as a cellular protein that can compensate for the role of apolipoproteins in the life cycle of HCV. We have also identified the domain in CAMP that contains amphipathic α-helices crucial for compensation and show that the expression of CAMP in nonhepatic cells expressing claudin 1 and miR-122 confers complete propagation of HCV. We speculate that low levels of HCV propagation might be possible in extrahepatic tissues expressing secretory proteins containing amphipathic α-helices without the expression of apolipoproteins.
INTRODUCTION
According to recent estimates, more than 160 million people worldwide are chronically infected with hepatitis C virus (HCV) (1), which causes liver-related pathologies that can result in life-threatening diseases such as cirrhosis and hepatocellular carcinoma (2). Chronic HCV infection is also associated with several extrahepatic manifestations, including immunological disorders such as cryoglobulinemia and B-cell non-Hodgkin lymphoma (3). A vaccine is not yet available, but recent advances in antiviral treatment, improving upon what was for many years the standard therapy of pegylated interferon alpha plus ribavirin, now include several direct-acting antivirals (DAAs) that have increased the rate of sustained virological response (SVR) up to 80 to 100% (4). The use of pseudotype particles carrying the HCV envelope proteins and RNA replicon systems facilitated the specific analysis of viral entry and RNA replication (5, 6). Later, the development of an in vitro system based on an HCV genotype 2a JFH1 strain for the complete propagation of HCV in cell culture allowed the study of the full life cycle of HCV (7, 8). One of the less understood steps in the HCV life cycle is the assembly of viral particles because of the lack of an efficient and specific system. It is known, however, that different structural and nonstructural (NS) HCV proteins are involved in this process. For instance, the recruitment of core protein on the surface of cytosolic lipid droplets (LDs) was shown to be critical for the production of infectious virus (9, 10). The HCV NS2 protein also plays a central role in the formation of infectious viral particles and has been proposed to act as an adaptor protein interacting with the viral structural proteins E1 and E2 and nonstructural proteins (11, 12). On the other hand, host factors, especially those involved in lipid metabolism, are also important for the correct assembly of HCV particles. Among them, LDs and lipoprotein-associated proteins, such as apolipoprotein B (ApoB), ApoE, and microsomal triglyceride transfer protein, are required for the efficient formation of HCV particles (9, 13–18). Recently, it was shown that various exchangeable apolipoproteins (ApoA, ApoC, and ApoE) could compensate for one another and redundantly participate in the formation of infectious HCV particles in ApoB and ApoE double-gene-knockout (BE-KO) Huh7 cells (17). Interestingly, such compensation was not dependent on the full-length expression of the exchangeable apolipoproteins but was dependent on the expression of the 11- and 22-mer tandem repeats of amphipathic α-helices present in these proteins (19–22).
Although the specific role of the exchangeable apolipoproteins in the formation of HCV particles is currently not fully understood, amphipathic α-helices are known to play a key role in viral particle formation. In the present study, through the screening of a small library of secretory proteins, we identified the human cathelicidin hCAP18/LL-37 (CAMP) as a new cellular protein that was able to facilitate viral particle formation in BE-KO cells. CAMP was able to restore the production of infectious particles through its LL-37 domain containing amphipathic α-helices and was also able to induce the production of infectious HCV particles in nonhepatic 293T cells. These results suggest that HCV particle formation can be independent of apolipoprotein expression and that low levels of HCV propagation might occur in extrahepatic compartments expressing secretory proteins containing amphipathic α-helices.
MATERIALS AND METHODS
NextBio Body Atlas.
The NextBio Body Atlas application presents an aggregated analysis of gene expression across various normal tissues and cell types as well as cancer cell lines (23). It enables us to investigate the expression of individual genes as well as gene sets. Samples for Body Atlas data are obtained from publicly available studies that are internally curated, annotated, and processed. Body Atlas measurements are generated from all available RNA expression studies that used Affymetrix U133 Plus or U133A GeneChip arrays for human studies. The results from 128 human tissue samples were incorporated from 1,067 arrays, those from 157 human cell types were incorporated from 1,474 arrays, and those from 359 human cancer cell lines were incorporated from 376 arrays. Gene queries return a list of relevant tissues or cell types rank ordered by absolute gene expression and grouped by body systems. In this study, we determined the expression levels of CAMP in all the different available tissues using an analysis protocol developed by NextBio, the details of which were described previously (23).
Cell lines.
All cell lines were cultured at 37°C under conditions of a humidified atmosphere with 5% CO2 and maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (FCS). Human hepatocellular carcinoma-derived Huh7 and human embryonic kidney-derived 293T cells were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank. The Huh7-derived cell line Huh7.5.1 was kindly provided by F. Chisari. The BE-KO Huh7 cell line was established previously in our laboratory (17).
Plasmids.
The cDNA clones of primary microRNA miR-122, ApoE, Aequorea coerulescens green fluorescent protein (AcGFP), and claudin 1 (CLDN1) as well as those from the 44 proteins that constitute the expression library were inserted between the XhoI and XbaI sites of the lentiviral vector pCSII-EF-RfA, which was kindly provided by M. Hijikata. The resulting plasmids were designated pCSII-EF-miR-122, pCSII-EF-ApoE, pCSII-EF-AcGFP, pCSII-EF-CLDN1, and pCSII-EF-protein name, respectively. The deletion mutants of CAMP were amplified by PCR and introduced into pCSII-EF-RfA. Plasmid pHH-JFH1, which encodes a full-length cDNA of the JFH1 strain, was kindly provided by T. Wakita. pHH-JFH1-E2p7NS2mt contains three adaptive mutations (24). The secreted small luciferase (nanoluciferase; Nlucsec) fragment from the pNL1.3 vector (Promega) was inserted into the self-cleaving 2A peptides of Thosea asigna virus, which generates two separated proteins (25) between p7 and NS2 of pJFH1-E2p7NS2mt, and the resulting plasmid was designated pJFH1-E2p7NS2mt-Nlucsec. pCon1/JFH1 (genotype 1b) was used for the production of chimeric virus (26). The plasmids used in this study were confirmed by sequencing with an ABI 3130 genetic analyzer (Applied Biosystems).
Antibodies.
Mouse monoclonal antibodies to β-actin and HCV core protein were purchased from Sigma-Aldrich and Thermo Fisher Scientific, respectively. Rat monoclonal antibody (3F10) to hemagglutinin (HA) was purchased from Roche. Mouse anti-ApoE antibody was obtained from Santa Cruz Biotechnology. Rabbit anti-CLDN1 and anti-CAMP antibodies were purchased from Invitrogen and Abcam, respectively. Rabbit anti-NS5A antibody was prepared as described previously (27). Alexa Fluor 488 (AF488)-conjugated anti-rabbit or anti-mouse IgG antibodies were purchased from Thermo Fisher Scientific.
Preparation of viruses.
pHH-JFH1-E2p7NS2mt or in vitro-transcribed Con1/JFH1 and JFH1Nluc RNAs were transfected into Huh7.5.1 cells, and HCV in cell culture (HCVcc) in the culture supernatant was collected after serial passages (28). Japanese encephalitis virus (JEV) (AT31 strain) and dengue virus (DENV) (NGC strain) were prepared in C6/36 cells. Infectious titers for HCV, JEV, and DENV were determined by a focus-forming assay and expressed as focus-forming units (FFUs) (7). Pseudoparticles expressing the HCV envelope glycoprotein were generated in 293T cells as previously reported (5), and infectivity was assessed by luciferase expression with a Bright-Glo luciferase assay system (Promega) according to the protocol provided by the manufacturer and expressed as relative lights units (RLUs). miR-122-independent HCV (HCV122KO) was obtained through serial passage of HCVcc (pHH-JFH1-E2p7NS2mt) in microRNA miR-122-deficient Huh7.5.1 cells.
Lipofection and lentiviral gene transduction.
The lentiviral vectors and ViraPower lentiviral packaging mix (Thermo Fisher Scientific) were cotransfected into 293T cells by using Trans IT LT-1 transfection reagent (Mirus) according to the manufacturer's protocol. The supernatants were collected at 48 h posttransfection, the lentivirus titer was determined by using the Lenti-XTM reverse transcription-quantitative PCR (qRT-PCR) titration kit (Clontech), and the expression levels of AcGFP were determined at 48 h postinoculation.
qRT-PCR.
Total RNA was extracted from cells by using a PureLink RNA minikit (Thermo Fisher Scientific), and first-strand cDNA synthesis and qRT-PCR were performed with TaqMan EZ RT-PCR core reagents and a ViiA7 system (Thermo Fisher Scientific) according to the manufacturer's protocol. The primers for TaqMan PCR targeting the noncoding region of HCV RNA were synthesized as previously reported (29). CAMP expression was assessed by using TaqMan gene expression assays (Thermo Fisher Scientific), and fluorescent signals were analyzed with the ViiA7 system.
Enzyme-linked immunosorbent assay.
Protein concentrations of ApoE and CAMP in plasma and culture supernatants were determined by using enzyme-linked immunosorbent assay (ELISA) kits from Alercheck Inc. and Hycult Biotech, respectively, according to the manufacturers' instructions.
Immunoblotting.
Cells lysed on ice in lysis buffer (20 mM Tris-HCl [pH 7.4], 135 mM NaCl, 1% Triton X-100, 10% glycerol) supplemented with protease inhibitor mix (Nacalai Tesque) were boiled in loading buffer and subjected to 5-to-20% gradient SDS-PAGE. The proteins were transferred onto polyvinylidene difluoride membranes (Millipore) and reacted with the appropriate antibodies. The immune complexes were visualized with the SuperSignal West Femto substrate (Pierce) and detected by using an LAS-3000 image analyzer system (Fuji Film).
Immunofluorescence assay.
Cells cultured on glass slides were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at room temperature for 30 min, permeabilized for 20 min at room temperature with PBS containing 0.2% Triton X-100 after being washed 3 times with PBS, and blocked with PBS containing 2% FCS for 1 h at room temperature. The cells were then incubated with PBS containing an anticore primary antibody at room temperature for 1 h, washed 3 times with PBS, and incubated with PBS containing an AF488-conjugated secondary antibody at room temperature for 1 h. For LD staining, cells incubated in medium containing 20 μg/ml boron-dipyrromethene (BODIPY) for 20 min at 37°C were washed with prewarmed fresh medium and incubated for 20 min at 37°C. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Cells were observed with a FluoView FV1000 laser scanning confocal microscope (Olympus). Immunofluorescence images were opened in ImageJ by using the LSM toolbox, the degree of colocalization was examined with the Just Another Colocalization plug-in (JACoP) (30), and the quantitation of colocalization between core and LDs was performed by using Pearson's correlation coefficient (r) (31). Pearson's coefficients range from +1 (strong correlation) to 0 (weak to no correlation) to −1 (no correlation).
In vitro transcription, RNA transfection, and luciferase assay.
Plasmids pJFH1 and pJFH1-E2p7NS2mt-Nlucsec were linearized with XbaI and treated with mung bean exonuclease. The linearized DNA was transcribed in vitro by using the MEGAscript T7 kit (Life Technologies) according to the manufacturer's protocol. The in vitro-transcribed RNA (5 μg) was electroporated into BE-KO cells at 5 × 106 cells/0.4 ml under conditions of 190 V and 975 μF by using a Gene Pulser (Bio-Rad) and plated onto DMEM containing 10% FCS. Luciferase activity was measured by using a Bright-Glo luciferase assay system (Promega) according to the protocol provided by the manufacturer and expressed as RLU.
Intracellular infectivity.
Cells were carefully washed 3 times with PBS, scraped, and pelleted by centrifugation at 1,000 × g for 5 min. Cell pellets were resuspended in 500 μl of DMEM with 10% FCS and subjected to 3 cycles of freeze-thawing using liquid nitrogen and a thermo block (TAITEC) set to 37°C. Cell lysates were centrifuged at 10,000 × g for 10 min at 4°C to remove cell debris. Cell-associated infectivity was determined by a focus-forming assay.
Buoyant density fractionation.
Culture supernatants of cells infected with HCVcc were concentrated by using Spin-X UF concentrators (Corning), applied to the top of a linear gradient formed from 10 to 40% Optiprep (Axis-Shield) in PBS, and spun at 32,000 rpm for 16 h at 4°C by using an SW41 Ti rotor (Beckman Coulter). Aliquots of 12 consecutive fractions were collected from top to bottom, and the density, infectious titer, and HCV RNA level were determined for each fraction.
Proteinase K digestion protection assay.
The proteinase K digestion protection assay was performed as described previously (13). Briefly, cells were extensively washed with PBS, scraped, and centrifuged at 10,000 × g. Cell pellets were resuspended in 500 μl of PBS and subjected to 3 cycles of freezing and thawing using liquid nitrogen and a thermo block set to 37°C. Cell lysates were then centrifuged at 10,000 × g for 10 min at 4°C to remove cell debris and incubated with 50 μg/ml proteinase K (Thermo Fisher Scientific) in the presence or absence of 5% Triton X-100 for 1 h on ice. Digestion was terminated by the addition of phenylmethylsulfonyl fluoride (PMSF) (Wako Chemical Industries).
Exosome isolation and purification.
BE-KO cells infected with HCVcc were maintained in DMEM supplemented with 10% exosome-depleted fetal bovine serum (FBS). Cell culture supernatants were collected, centrifuged at 2,000 × g for 10 min at 4°C to remove cell debris, and filtered through a 0.22-mm filter. The culture supernatants were then concentrated to a final volume of 1 ml by using Spin-X UF concentrators (Corning). The concentrated culture supernatants were mixed with the appropriate volume of ExoQuickTC reagent (catalog number EXOTC10A-1; System Biosciences) according to the manufacturer's specifications. Samples were gently mixed and incubated for 12 h at 4°C. Next, exosomes were precipitated by centrifugation at 1,500 × g for 30 min at 4°C and resuspended in PBS. Positive selection of exosomes was done by using an ExoCap CD9 for Serum Plasma kit (JSR Life Sciences).
Statistical analysis.
The data for statistical analyses were the averages of results from 3 independent experiments. Results were expressed as means ± standard deviations. The significance of differences in the means was determined by Student's t test.
RESULTS
Expression screening and identification of CAMP.
Recently, we showed the redundant role of exchangeable apolipoproteins containing amphipathic α-helices in the formation of infectious HCV particles (17). To clarify whether some other cellular proteins could compensate for the role of the exchangeable apolipoproteins in the production of HCV particles, we first determined potential candidates using the UniProt website (http://www.uniprot.org/) to compile a set of different human proteins according to two main selection criteria shared by the exchangeable apolipoproteins. First, the protein had to contain an amino-terminal signal sequence that targets it to the endoplasmic reticulum (ER) lumen. Second, the protein had to show one or more α-helices in its secondary structure. By using these two main criteria, a total of 44 proteins were selected and tested according to the screening process shown in Fig. 1A. The exogenous expression of ApoE, used as a positive control, exhibited a 10-fold increase of infectious HCV titers in the supernatants compared to the levels detected in control cells. These results were in accordance with those previously reported by our group (17) and validated our screening method and results. None of the 44 proteins tested were able to increase the HCV titers to the same extent as the expression of ApoE, but interestingly, 6 proteins were able to significantly increase the HCV titers (Fig. 1B). To further assess the effect of the expression of the identified proteins on viral production, luciferase activity in the supernatants of BE-KO cells expressing these proteins was determined at 48 h postinfection with a luciferase reporter virus, JFH1-Nluc (Fig. 1C). Moreover, the supernatants were inoculated into Huh7.5.1 cells, and luciferase activity was determined at 48 h postinfection (Fig. 1D). Although luciferase activities in the supernatants were comparable among BE-KO cells expressing each of the identified proteins upon infection with JFH1-Nluc, higher levels of luciferase activity were obtained in the supernatant of Huh7.5.1 cells inoculated with the supernatant from cells expressing apolipoprotein A4 (ApoA4), CAMP, and serum amyloid A4 (SAA4), indicating that the expression of these proteins in BE-KO cells is capable of enhancing the production of infectious HCV particles. Although ApoA4, along with all the other members of the ApoA family, was recently reported to complement HCV production in nonhepatic 293T cells (32), our result also confirms that ApoA4 is able to act as another exchangeable apolipoprotein to induce the production of infectious HCV particles, as we previously reported for ApoA1 and ApoA2 (17). SAA4 is a minor apolipoprotein present mainly on the surface of high-density lipoproteins and constitutes >90% of the total SAA present in plasma during homeostasis (33). CAMP is the only type of cathelicidin expressed in humans (34, 35). Cathelicidins are a group of small and cationic antimicrobial peptides (AMPs) (35) that are part of the innate immune system and represent one of the first chemical defense mechanisms against bacteria and other pathogens (36). The mode of action of AMPs is based mostly on their ability to bind and disrupt bacterial membranes by different mechanisms, including the formation of pores (37). Human CAMP is organized into a conserved N-terminal cathelin domain of 103 amino acid residues (38), with a signal sequence of 33 amino acids on its N terminus and a C-terminal domain of 37 amino acid residues, called LL-37, which presents antimicrobial activity (39). CAMP is first translated as a preproprotein. The signal sequence will ultimately target the preproprotein to either specific granules for storage or the cell membrane for secretion. After cleavage of the signal sequence, the resulting proprotein, with a molecular mass of 18 kDa, is still not active until the LL-37 domain is released from the cathelin domain through cleavage by proteinase 3, kallikrein, or gastricsin enzymes, depending on the cell types or tissues (40–42). Because CAMP was the only identified protein in our screening that could increase the production of infectious HCV particles and that did not belong to the family of apolipoproteins, we decided to further determine its potential role in HCV propagation.
FIG 1.

Construction of the protein library and screening. (A) Schematic diagram of the protein library screening process. cDNAs from the 44 selected proteins were individually amplified and cloned into a lentivirus expression vector (pCSII-EF-RfA) that was transfected into 293T cells to produce lentiviruses. BE-KO cells were infected with HCV at a multiplicity of infection of 1, 72 h after infection with lentiviruses. Infectious titers of HCV in the supernatants were determined by a focus-forming assay at 72 h postinfection. (B) Infectious titers in the culture supernatants from BE-KO cells expressing the library target proteins were determined by a focus-forming assay in Huh7.5.1 cells. Average titers were plotted as fold differences compared to the titers obtained from control cells. (C) BE-KO cells expressing each of the proteins identified by screening were infected with JFH1-Nluc at a multiplicity of infection of 1, and luciferase activity in the supernatant was determined at 2 days postinfection. (D) The supernatants were further inoculated into Huh7.5.1 cells, and luciferase activity in the culture supernatants was determined at 2 days postinfection.
CAMP expression in different tissues and cell lines.
CAMP is highly expressed in bone marrow and in many cell types related to the immune system, such as neutrophils, natural killer cells, monocytes, lymphocytes, and macrophages (43–49). CAMP has also been detected in epithelial cells that are in contact with the external environment in the skin, intestine, airways, and genitals (50–54). We first used the Web-based search engine NextBio Body Atlas to compare the relative CAMP mRNA expression levels in different tissues (Fig. 2A). Blood and especially bone marrow were the only two tissues where CAMP was highly expressed, while the rest of the tissues showed comparatively low expression levels. Next, we determined the expression of CAMP in isolated peripheral blood mononuclear cells (PBMCs); 293T cells; and also the hepatic cell lines Huh7, Hep3B, and HepG2. CAMP mRNA expression was detected only in PBMCs, while much lower levels or no expression was detected in the rest of the cell lines tested (Fig. 2B). We next evaluated the CAMP and ApoE protein expression levels by immunoblot analysis (Fig. 2C). ApoE was detected only in the hepatic cell lines, while the CAMP protein was detectable only in PBMCs. We also quantified the levels of both proteins in the supernatants of the different cell lines as well as in plasma samples by an ELISA. Very similar but low levels of ApoE were detected in the supernatants of the three hepatic cell lines (ranging from 21 to 27 ng/ml), while the levels in plasma were much higher (9,515 ng/ml), and no protein was detected in the supernatant of 293T cells (Fig. 2D). The CAMP ELISA, which targets the LL-37 domain, showed detectable levels of the protein in plasma, with a concentration of 13.35 ng/ml, while no protein was detected in any of the cell lines tested (Fig. 2E). Collectively, these results demonstrated that the hepatic cell lines express ApoE but not CAMP, while PBMCs express only CAMP.
FIG 2.

Expression of CAMP in various tissues and cell lines. (A) The relative mRNA expression level of CAMP in different human tissues was determined by using the NextBio Body Atlas application. The median expression level was calculated across all 128 human tissues from 1,068 arrays by using the Affymetrix GeneChip Human Genome U133 Plus 2.0 array. The mRNA expression level for each gene was log10 transformed. (B) Endogenous expression of CAMP mRNA. Total RNA was extracted from PBMCs and 293T, Huh7, Hep3B, and HepG2 cells, and the expression of CAMP mRNA was determined by qRT-PCR. ND, not determined. (C) Endogenous expression of ApoE and CAMP was determined by immunoblotting. (D and E) Expression levels of ApoE and CAMP in plasma and supernatants from 293T, Huh7, Hep3B, and HepG2 cells were determined by using ELISA kits.
CAMP compensates for the role of apolipoproteins in the formation of infectious HCV particles.
As a recent report showed that exchangeable apolipoproteins play an important role in HCV particle assembly at a postenvelopment step (17), we examined whether CAMP plays the same role as exchangeable apolipoproteins. We first confirmed the expression of the ApoE and CAMP proteins and comparable expression levels of viral proteins in transduced BE-KO cells by immunoblot analysis (Fig. 3A) and then determined the HCV RNA levels and infectious titers in cells upon infection with the HCV genotype 2a JFH1 strain. Although the expression of CAMP had no effect on intracellular HCV RNA levels (Fig. 3B), it significantly increased the extracellular and intracellular infectious titers (Fig. 3C). In addition, the expression of CAMP did not change the intracellular HCV RNA levels (Fig. 3D), but it enhanced extracellular infectious titers (Fig. 3E) in BE-KO cells infected with genotype 1b chimeric (Con1/JFH1) HCV, as seen for infection with the genotype 2a JFH1 strain. We also electroporated RNA of a luciferase reporter virus, JFH1-Nluc, into BE-KO cells, and the luciferase activities in the supernatants were then determined at 24, 48, and 72 h postelectroporation (Fig. 3F). Moreover, Huh7.5.1 cells were infected with the supernatant from BE-KO cells at 72 h postelectroporation, and the luciferase activity in the supernatant was determined at 48 h postinfection (Fig. 3G). Although the luciferase activities in the supernatants of the cells were comparable, significantly higher levels of luciferase activity were obtained in the supernatants of Huh7.5.1 cells infected with the supernatants of cells expressing CAMP or ApoE, suggesting that CAMP participates in the enhancement of HCV infectious particle production in BE-KO cells without any effect on viral replication. To rule out the possibility that CAMP participates in HCV replication in any other way than by compensating for exchangeable apolipoproteins, we examined the effect of CAMP expression on the production of infectious particles in Huh7 cells. The expression of CAMP (Fig. 3H) exhibited no effect on the production of infectious particles in the supernatants of Huh7 cells (Fig. 3I), suggesting that CAMP plays a redundant role with the exchangeable apolipoproteins in the formation of infectious HCV particles.
FIG 3.

CAMP expression increases the production of infectious HCV particles in BE-KO cells. (A) Expression levels of ApoE and CAMP were determined by immunoblotting 48 h after transduction of lentiviruses into BE-KO cells. Expression levels of HCV core and NS5A proteins were determined 72 h after infection with HCV JFH1 at a multiplicity of infection of 1. (B and C) Intracellular HCV RNA levels (B) as well as intracellular and extracellular infectious titers (C) were determined 72 h after infection with HCV JFH1 at a multiplicity of infection of 1 by qRT-PCR and a focus-forming assay, respectively. (D and E) Intracellular HCV RNA levels (D) as well as extracellular infectious titers (E) were determined 72 h after infection with HCV Con1/JFH1 at a multiplicity of infection of 2 by qRT-PCR and a focus-forming assay, respectively. (F) BE-KO cells expressing either ApoE or CAMP were electroporated with JFH1-Nluc RNA, and luciferase activities in the supernatants collected every 24 h for 3 days were determined. (G) The supernatants at 3 days postelectroporation were further inoculated into Huh7.5.1 cells, and luciferase activity in the culture supernatants was determined at 2 days postinoculation. (H) Expression levels of ApoE and CAMP were determined by immunoblotting 48 h after transduction of lentiviruses into Huh7 cells. (I) Infectious titers in the supernatant were determined 72 h after infection with HCV JFH1 at a multiplicity of infection of 1. In all cases, asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
To determine if CAMP expression has any effect in other unrelated viruses, DENV and JEV were inoculated into BE-KO cells expressing either CAMP or ApoE proteins. Viral titers in the supernatants showed no difference between control and CAMP-expressing cells upon infection with DENV (Fig. 4A) and JEV (Fig. 4B).
FIG 4.

CAMP expression does not increase production of infectious DENV or JEV particles in BE-KO cells. Infectious titers in the supernatants of BE-KO cells expressing either ApoE or CAMP were determined by a focus-forming assay 48 h after infection with either DENV (A) or JEV (B) at a multiplicity of infection of 1.
To examine whether CAMP has any effect on the entry of HCV, retrovirus pseudotype particles bearing HCV envelope protein E1/E2 (HCVpp) or the vesicular stomatitis virus G protein (VSVpp) were inoculated into BE-KO cells expressing CAMP, and luciferase activity was determined (Fig. 5A). The expression of CAMP exhibited no significant effect on the expression of luciferase, suggesting that CAMP has no effect on the entry of HCV into BE-KO cells. The localization of HCV core proteins around LDs is known to be critical for the assembly of HCV particles (9). In our previous report (17), BE-KO cells infected with HCV were shown to exhibit greater accumulation of core proteins and LDs around the perinuclear region than in those with restored ApoE expression. Therefore, we next examined the effect of CAMP on the expression and localization of the core protein in BE-KO cells upon infection with HCV. BE-KO cells expressing CAMP had no significant effect on the expression levels of core protein (Fig. 5C), and the distribution patterns of core and LDs were similar to those of BE-KO cells expressing ApoE upon infection with HCV, in contrast to the greater accumulation of core protein and LDs around the perinuclear region in BE-KO cells (Fig. 5D). Although the accumulation of LDs around the nucleus was observed only in control BE-KO cells, the degrees of core and LD colocalization were similar between CAMP-expressing cells and control BE-KO cells (Pearson's correlation coefficient [r] of 0.552 versus 0.470) (Fig. 5D). To further assess the role of CAMP in viral particle production, the specific infectivity in the supernatants was determined by comparing the infectious titers with viral RNA level (Fig. 5E). The expression of either CAMP or ApoE increased the specific infectivity in the supernatants of BE-KO cells. HCV particles in the supernatants were also analyzed by buoyant density ultracentrifugation, and infectious titers and HCV RNA levels were determined for each fraction. The highest infectious titers were detected at a density of 1.03 g/ml of the supernatants of BE-KO cells expressing either CAMP or ApoE, although high infectious titers were also detected at densities of up 1.07 g/ml in ApoE-expressing cells (Fig. 5F). Both gradients showed very similar distributions of viral RNA through the density gradient, with two clear peaks at densities of around 1.02 and 1.10 g/ml (Fig. 5G). These results suggest that CAMP has a role similar to that of ApoE in the formation of infectious HCV particles.
FIG 5.
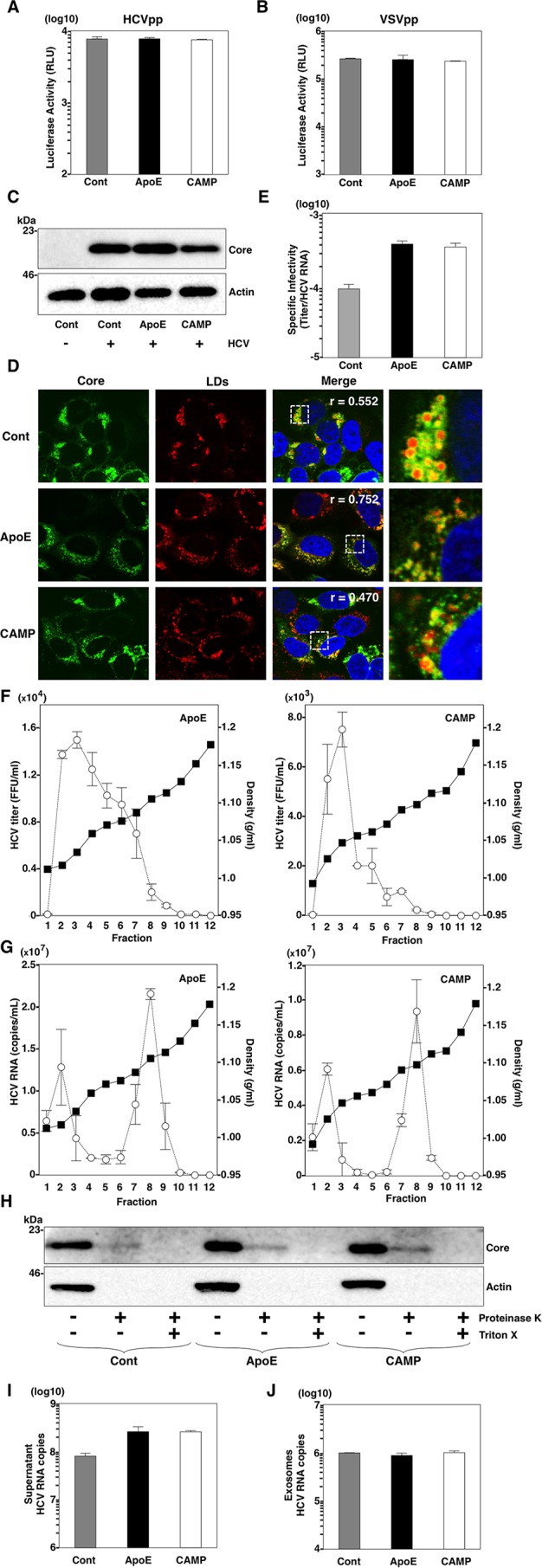
CAMP compensates for the role of apolipoproteins in BE-KO cells. (A and B) BE-KO cells expressing either ApoE or CAMP were inoculated with pseudoparticles bearing HCV envelope proteins E1 and E2 (HCVpp) (A) or VSV G protein (VSVpp) (B), and luciferase activity was determined at 24 h postinfection. (C) BE-KO cells expressing either ApoE or CAMP were infected with HCV at a multiplicity of infection of 1, and expression of the HCV core protein was determined by immunoblot analysis at 72 h postinfection. (D) Expression of core protein, lipid droplets (LDs), and cell nuclei was examined by immunofluorescence analysis after staining with anticore antibody, BODIPY, and DAPI, respectively. Pearson's correlation coefficients (r), quantifying the degree of colocalization of the HCV core protein with LDs, are presented in the merged images. The boxed areas in the merged images are magnified. (E) Specific infectivity in the supernatants of BE-KO cells expressing either ApoE or CAMP upon infection with HCV at a multiplicity of infection of 1 was determined by comparing the infectious titers with HCV RNA levels. (F and G) The supernatants of BE-KO cells expressing either ApoE or CAMP upon infection with HCV at a multiplicity of infection of 1 were subjected to density gradient fractionation, and infectious titers (G) and HCV RNA levels (F) for each fraction were determined by a focus-forming assay and qRT-PCR, respectively. (H) BE-KO cells expressing either ApoE or CAMP were lysed 72 h after infection with HCVcc and subjected to a proteinase K digestion protection assay. Cell lysates were split into 3 parts and incubated on ice for 1 h in the presence or absence of 50 μg/ml proteinase K with or without pretreatment with 5% Triton X-100. After treatment, the samples were subjected to immunoblotting using anticore antibody. (I and J) HCV RNA levels in the supernatants (I) and exosomes (J) of BE-KO cells expressing either ApoE or CAMP upon infection with HCV at a multiplicity of infection of 1 were determined by qRT-PCR.
ApoE has been shown to participate in a postenvelopment step during the process of assembly of HCV particles (14, 17). To determine whether CAMP has any effect on the envelopment of HCV particles, BE-KO cells expressing either ApoE or CAMP were infected with HCVcc, and the cell lysates were treated with proteinase K in the presence or absence of Triton X-100 (55). No significant difference in the protection of core proteins from proteinase K digestion was observed for BE-KO cells expressing either CAMP or ApoE (Fig. 5H), suggesting that neither ApoE nor CAMP participates in the envelopment step of the assembly process of HCV. Collectively, these results suggest that CAMP can compensate for the roles of ApoE in HCV particle formation at a postenvelopment step in BE-KO cells.
The exosome-mediated transmission of HCV in a hepatic cell line was recently reported (56). To test if CAMP expression had any effect on the production of exosomes containing HCV RNA, we purified exosomes from the supernatants of BE-KO cells infected with HCVcc. Although the expression of either CAMP or ApoE increased the total HCV RNA levels in the supernatants of BE-KO cells compared to those in control cells (Fig. 5I), comparable amounts of HCV RNA were detected in purified exosomes (Fig. 5J), suggesting that CAMP and ApoE have no effect on the production of exosomes containing HCV RNA.
The LL-37 domain is responsible for CAMP activity in the formation of infectious HCV particles.
Next, to determine the functional domain of CAMP responsible for HCV particle formation, an expression plasmid encoding the LL-37 domain (Fig. 6A) was expressed in BE-KO cells (Fig. 6B), and intracellular HCV RNA levels and infectious titers in the supernatants were determined upon infection with HCV. Although the expression of the LL-37 domain showed no effect on the intracellular HCV RNA levels in BE-KO cells (Fig. 6C), it significantly increased the infectious titers in the supernatants (Fig. 6D), suggesting that the LL-37 domain in CAMP participates in the production of infectious HCV particles in BE-KO cells.
FIG 6.

CAMP activity is located in the LL-37 domain. (A) Schematics of CAMP and its deletion mutant. aa, amino acids. (B) Deletion mutant with an HA tag expressed in BE-KO cells by a lentiviral vector detected by immunoblotting. (C and D) BE-KO cells expressing either ApoE, CAMP, or LL-37 were infected with HCV at a multiplicity of infection of 1, and intracellular HCV RNA levels (C) and infectious titers (D) in the supernatants at 72 h postinfection were determined by qRT-PCR and a focus-forming assay, respectively. In all cases, asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
The three-dimensional (3D) structure of the LL-37 domain in CAMP was previously determined and revealed an amphipathic helix-bend-helix conformation (57, 58) in which the lysine at position 12 (K12) is located at the breakpoint between an N-terminal α-helix and a C-terminal α-helix (Fig. 7A). To further confirm the role of amphipathic α-helices in the effect of CAMP on HCV particle formation, we constructed two expression plasmids encoding CAMP with a single deletion at each of the two α-helices (Fig. 7A), expressed them in BE-KO cells (Fig. 7B), and then determined the intracellular HCV RNA levels and infectious titers in the supernatants upon infection with HCV. Compared to control cells, the expression of the CAMP deletion mutants in BE-KO cells showed no effect on the intracellular HCV RNA levels (Fig. 7C) and infectious titers (Fig. 7D) in the supernatants upon infection with HCV. These results suggest that both α-helices in the LL-37 domain are necessary for CAMP to participate in HCV particle formation in BE-KO cells.
FIG 7.

Amphipathic α-helices in the LL-37 domain are necessary for CAMP activity. (A) Structure of LL-37 of CAMP and amino acid sequences of the LL-37 domain deletion mutants. (B) The expression of deletion mutants with HA tags in BE-KO cells by a lentiviral vector was determined by immunoblotting. (C and D) BE-KO cells expressing either ApoE, CAMP, KE-23, or LL-15 were infected with HCV at a multiplicity of infection of 1, and intracellular HCV RNA levels (C) and infectious titers (D) in the supernatants at 72 h postinfection were determined by qRT-PCR and a focus-forming assay, respectively. In all cases, asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
CAMP can induce production of infectious HCV particles in 293T cells.
Reconstitution of the complete HCV life cycle in nonhepatic cells was recently accomplished by using 293T cells (32, 59). Exogenous expression of host factors in 293T cells, including CLDN1, microRNA miR-122, and exchangeable apolipoproteins, is required for entry, viral RNA replication, and HCV particle release, respectively (27). To determine whether CAMP can compensate for the role of apolipoproteins in nonhepatic cells, we expressed CAMP in 293T cells expressing both CLDN1 and miR-122 (Fig. 8A) and then determined the intracellular viral RNA levels and infectious titers in the supernatants upon infection with HCV. Although the expression of ApoE or CAMP in 293T cells expressing both CLDN1 and miR-122 exhibited no effect on HCV RNA replication upon infection with HCV (Fig. 8B), they induced the production of infectious HCV particles in the culture supernatants (Fig. 8C). Next, we repeated the same experiment but this time in a more restricted nonhepatic environment using 293T cells expressing CLDN1 alone. We expressed ApoE or CAMP in 293T cells expressing CLDN1 (Fig. 8D) and infected them with an HCV mutant possessing a G28A substitution in the 5′ untranslated region (UTR) of the genotype 2a JFH-1 strain (HCV122KO) that facilitates viral RNA replication in miR-122-deficient Huh7.5.1 cells (60). The results showed that the expression of CAMP has no effect on the replication of HCV122KO (Fig. 8E) but induces the production of low levels of infectious particles in the culture supernatants of 293T cell expressing CLDN1 (Fig. 8F), suggesting that complete HCV propagation can be accomplished in nonhepatic cells lacking the expression of miR-122 and apolipoproteins. To confirm the bona fide production of infectious HCV particles in nonhepatic cells, JFH1 RNA was electroporated into 293T-CLDN1-miR-122 cells, and infectious titers in the supernatants were determined at 3 and 6 days posttransfection. Although no infectious titer was detected in the supernatants of 293T-CLDN1-miR-122 cells upon infection with HCVcc, low but significant infectious titers were detected in those expressing either ApoE or CAMP (Fig. 8G).
FIG 8.

CAMP expression confers production of infectious HCV particles in nonhepatic cells. (A) Expression of ApoE and CAMP in 293T-CLDN1-miR-122 cells was determined by immunoblot analysis. (B and C) Cells were infected with HCV at a multiplicity of infection of 10, and intracellular HCV RNA levels (B) and infectious titers (C) in the supernatants at 72 h postinfection were determined by qRT-PCR and a focus-forming assay, respectively. (D) Expression of ApoE and CAMP in 293T-CLDN1 cells was determined by immunoblot analysis. (E and F) Cells were infected with HCV containing a G28A mutation at a multiplicity of infection of 10, and intracellular HCV RNA levels (E) and infectious titers (F) in the supernatants at 72 h postinfection were determined by qRT-PCR and a focus-forming assay, respectively. (G) 293T-CLDN1-miR-122 cells expressing either CAMP or ApoE were electroporated with JFH1 RNA, and viral titers in the supernatant were determined by a focus-forming assay at days 3 and 6 posttransfection. In all cases, asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) versus the results for control cells.
DISCUSSION
The life cycle of HCV is tightly associated with lipid metabolism and especially with lipoprotein biogenesis (18). We previously reported the redundant roles of different exchangeable apolipoproteins in the assembly of HCV particles and identified the amphipathic α-helix motifs present in these apolipoproteins as critical factors for this process (17). These results were recently confirmed and complemented by others (32). In this study, through the screening of a small secretory protein library, we identified CAMP as a cellular protein that is able to compensate for the role of apolipoproteins in the formation of HCV particles. The expression of CAMP in BE-KO Huh7 cells increased the formation of infectious HCV particles through the amphipathic α-helices in the same way as ApoE acting at a postenvelopment step without any effect on viral RNA replication. Moreover, the expression of CAMP in Huh7 cells did not increase the production of infectious HCV particles, supporting the idea that it has a redundant role with apolipoproteins.
CAMP is the only member of the cathelicidin family of AMPs in humans and was originally identified for its broad antimicrobial activity against both Gram-positive and -negative bacteria (39). The mechanism of action of CAMP is mediated by its ability to bind parallel to the surface of the bacterial membrane and to induce curvature strain, which can result in the formation of pores disrupting the membrane (61). CAMP has also been reported to have antiviral activity against human immunodeficiency virus, influenza A virus, respiratory syncytial virus, and vaccinia virus (62) as well as several other functions in the immune system, such as chemotactic and angiogenic roles (63). Very recently, it was reported that preincubation of cells or HCV with LL-37 attenuates the infectivity of HCV (64), in contrast to our observation here that the exogenous expression of CAMP and LL-37 promotes infectious particle production in BE-KO cells. Although direct comparison is difficult, the discrepancy of the effects of LL-37 on HCV propagation might be attributable to the concentration of LL-37 used in the experiments. The amounts of LL-37 (5 to 20 μg/ml) required for a modest antiviral effect (64) were larger than normal plasma levels (25 to 250 ng/ml). Further studies are needed to clarify the roles of LL-37 in the HCV life cycle.
Virtually all activities of CAMP have been reported to be mediated through its C-terminal LL-37 domain, which consists of 37 amino acid residues with an amphipathic α-helix structure. We also revealed that both amphipathic α-helices present in the LL-37 domain of CAMP are indispensable for CAMP to compensate for infectious particle formation in BE-KO cells. While apolipoproteins play a structural role in maintaining the integrity and stability of lipoproteins, the exact mechanism by which they promote the assembly of HCV particles is currently unknown. In the same way, the exact role of CAMP in the assembly of HCV particles remains to be determined. Interestingly, CAMP has also been reported to bind to low-density lipoproteins (LDLs) and very-low-density lipoproteins (VLDLs) (65). In addition, the lipidome composition of HCV particles has been shown to resemble that of VLDLs and LDLs (66), suggesting that CAMP is able to interact with HCV particles. Moreover, some AMPs are designed based on apolipoprotein sequences (67–69). This apparent cross talk between CAMP and apolipoproteins suggests that amphipathic α-helices from both proteins probably share the ability to bind to the same types of lipid membranes and induce similar curvature effects (70, 71). Based on these observations, we speculate that exchangeable apolipoproteins and CAMP might be able to bind to the surface of HCV particles in the ER lumen and to confer some specific characteristics to the viral particles that permit maturation and release into the extracellular space through the Golgi apparatus in a postenvelopment step. In this sense, it might be interesting to examine whether other human AMPs with amphipathic α-helices can compensate for the role of exchangeable apolipoproteins in HCV particle formation. It will also be necessary to elucidate the requirements of length, charge, and hydrophobic content in the tandem repeats of amphipathic α-helices that are required for the induction of HCV assembly. Interactions of ApoE with NS5A (72) and E2 proteins (14) have been reported to be required for the assembly of infectious particles. Moreover, it has been reported that ApoE participates in the attachment of HCV to target cells through the interaction with heparan sulfate (73) or through binding to the LDL receptor (74). Further studies are needed to determine whether CAMP is capable of compensating for the role of ApoE in the interaction with NS5A and/or E2, is associated with HCV particles, and is involved in the attachment and/or entry of HCV particles in target cells.
Another important aspect of our present study is that the production of infectious HCV particles was shown to be independent of the expression of apolipoproteins in nonhepatic 293T cells. Until now, to the best of our knowledge, complete propagation of HCV in nonhepatic cells has been accomplished only either in cell lines that endogenously express some exchangeable apolipoproteins (75) or through the exogenous expression of at least one of them (59, 76, 77). Although extrahepatic propagation of HCV still remains a controversial issue, high levels of CAMP expression and possible propagation of HCV in PBMCs were previously reported (78–80). Our present results support the idea that low levels of HCV propagation might be feasible in nonhepatic cells and/or tissues expressing CAMP but not apolipoproteins, such as bone marrow or PBMCs, that could serve as a reservoir for HCV. In fact, common extrahepatic manifestations associated with HCV infection, such as cryoglobulinemia, neutropenia, or B-cell non-Hodgkin lymphoma (3, 81), are associated with dysfunctions in the above-mentioned tissues and immune cells.
In conclusion, we proved that the production of HCV particles can be independent of apolipoprotein expression through compensation by the LL-37 domain of CAMP, suggesting that low levels of HCV propagation might be possible in extrahepatic compartments with low-level or no expression of apolipoproteins but high expression levels of host proteins possessing an amphipathic α-helix structure, such as CAMP.
ACKNOWLEDGMENTS
We thank M. Tomiyama for her secretarial work and M. Ishibashi, M. Nagao, and Y. Sugiyama for their technical assistance.
This work was supported in part by health sciences research grants (research on hepatitis) from the Ministry of Health, Labor, and Welfare of Japan and AMED (Japan Agency for Medical Research and Development) and by grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology and the Takeda Science Foundation.
REFERENCES
- 1.Maasoumy B, Wedemeyer H. 2012. Natural history of acute and chronic hepatitis C. Best Pract Res Clin Gastroenterol 26:401–412. doi: 10.1016/j.bpg.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Hajarizadeh B, Grebely J, Dore GJ. 2013. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol 10:553–562. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 3.Negro F, Forton D, Craxì A, Sulkowski MS, Feld JJ, Manns MP. 2015. Extrahepatic morbidity and mortality of chronic hepatitis C. Gastroenterology 149:1345–1360. doi: 10.1053/j.gastro.2015.08.035. [DOI] [PubMed] [Google Scholar]
- 4.Elbaz T, El-Kassas M, Esmat G. 2015. New era for management of chronic hepatitis C virus using direct antiviral agents: a review. J Adv Res 6:301–310. doi: 10.1016/j.jare.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 7.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 9.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 10.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J Biol Chem 282:37158–37169. doi: 10.1074/jbc.M707329200. [DOI] [PubMed] [Google Scholar]
- 11.Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M. 2011. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J Virol 85:86–97. doi: 10.1128/JVI.01070-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol 85:1706–1717. doi: 10.1128/JVI.02268-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wölk B, Kaderali L, Pietschmann T. 2014. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol 88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JY, Acosta EG, Stoeck IK, Long G, Hiet MS, Mueller B, Fackler OT, Kallis S, Bartenschlager R. 2014. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J Virol 88:12422–12437. doi: 10.1128/JVI.01660-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuhara T, Wada M, Nakamura S, Ono C, Shiokawa M, Yamamoto S, Motomura T, Okamoto T, Okuzaki D, Yamamoto M, Saito I, Wakita T, Koike K, Matsuura Y. 2014. Amphipathic α-helices in apolipoproteins are crucial to the formation of infectious hepatitis C virus particles. PLoS Pathog 10:e1004534. doi: 10.1371/journal.ppat.1004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuhara T, Ono C, Puig-Basagoiti F, Matsuura Y. 2015. Roles of lipoproteins and apolipoproteins in particle formation of hepatitis C virus. Trends Microbiol 23:618–629. doi: 10.1016/j.tim.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. 1992. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res 33:141–166. [PubMed] [Google Scholar]
- 20.Saito H, Lund-Katz S, Phillips MC. 2004. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res 43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Hatters DM, Peters-Libeu CA, Weisgraber KH. 2006. Apolipoprotein E structure: insights into function. Trends Biochem Sci 31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Frank PG, Marcel YL. 2000. Apolipoprotein A-I: structure-function relationships. J Lipid Res 41:853–872. [PubMed] [Google Scholar]
- 23.Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, Wall GD, Wisotzkey R, Alag S, Akhtari S, Ronaghi M. 2010. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 5:e13066. doi: 10.1371/journal.pone.0013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A 105:4370–4375. doi: 10.1073/pnas.0800422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. 2001. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol 82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 26.Takebe Y, Saucedo CJ, Lund G, Uenishi R, Hase S, Tsuchiura T, Kneteman N, Ramessar K, Tyrrell DL, Shirakura M, Wakita T, McMahon JB, O'Keefe BR. 2013. Antiviral lectins from red and blue-green algae show potent in vitro and in vivo activity against hepatitis C virus. PLoS One 8:e64449. doi: 10.1371/journal.pone.0064449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuhara T, Kambara H, Shiokawa M, Ono C, Katoh H, Morita E, Okuzaki D, Maehara Y, Koike K, Matsuura Y. 2012. Expression of microRNA miR-122 facilitates an efficient replication in nonhepatic cells upon infection with hepatitis C virus. J Virol 86:7918–7933. doi: 10.1128/JVI.00567-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masaki T, Suzuki R, Saeed M, Mori K, Matsuda M, Aizaki H, Ishii K, Maki N, Miyamura T, Matsuura Y, Wakita T, Suzuki T. 2010. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. J Virol 84:5824–5835. doi: 10.1128/JVI.02397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris T, Robertson B, Gallagher M. 1996. Rapid reverse transcription-PCR detection of hepatitis C virus RNA in serum by using the TaqMan fluorogenic detection system. J Clin Microbiol 34:2933–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolte S, Cordelières FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 31.Adler J, Parmryd I. 2010. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A 77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 32.Hueging K, Weller R, Doepke M, Vieyres G, Todt D, Wölk B, Vondran FW, Geffers R, Lauber C, Kaderali L, Penin F, Pietschmann T. 2015. Several human liver cell expressed apolipoproteins complement HCV virus production with varying efficacy conferring differential specific infectivity to released viruses. PLoS One 10:e0134529. doi: 10.1371/journal.pone.0134529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Beer MC, Yuan T, Kindy MS, Asztalos BF, Roheim PS, de Beer FC. 1995. Characterization of constitutive human serum amyloid A protein (SAA4) as an apolipoprotein. J Lipid Res 36:526–534. [PubMed] [Google Scholar]
- 34.Dürr UH, Sudheendra US, Ramamoorthy A. 2006. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta 1758:1408–1425. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 35.Kościuczuk EM, Lisowski P, Jarczak J, Strzałkowska N, Jóźwik A, Horbańczuk J, Krzyżewski J, Zwierzchowski L, Bagnicka E. 2012. Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep 39:10957–10970. doi: 10.1007/s11033-012-1997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G. 2014. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel) 7:545–594. doi: 10.3390/ph7050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wimley WC. 2010. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem Biol 5:905–917. doi: 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pazgier M, Ericksen B, Ling M, Toth E, Shi J, Li X, Galliher-Beckley A, Lan L, Zou G, Zhan C, Yuan W, Pozharski E, Lu W. 2013. Structural and functional analysis of the pro-domain of human cathelicidin, LL-37. Biochemistry 52:1547–1558. doi: 10.1021/bi301008r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vandamme D, Landuyt B, Luyten W, Schoofs L. 2012. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol 280:22–35. doi: 10.1016/j.cellimm.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Sørensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. 2001. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97:3951–3959. doi: 10.1182/blood.V97.12.3951. [DOI] [PubMed] [Google Scholar]
- 41.Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, Bonnart C, Descargues P, Hovnanian A, Gallo RL. 2006. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J 20:2068–2080. doi: 10.1096/fj.06-6075com. [DOI] [PubMed] [Google Scholar]
- 42.Sørensen OE, Gram L, Johnsen AH, Andersson E, Bangsbøll S, Tjabringa GS, Hiemstra PS, Malm J, Egesten A, Borregaard N. 2003. Processing of seminal plasma hCAP-18 to ALL-38 by gastricsin: a novel mechanism of generating antimicrobial peptides in vagina. J Biol Chem 278:28540–28546. doi: 10.1074/jbc.M301608200. [DOI] [PubMed] [Google Scholar]
- 43.Büchau AS, Morizane S, Trowbridge J, Schauber J, Kotol P, Bui JD, Gallo RL. 2010. The host defense peptide cathelicidin is required for NK cell-mediated suppression of tumor growth. J Immunol 184:369–378. doi: 10.4049/jimmunol.0902110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu W, Kim CH, Liu R, Kucia M, Marlicz W, Greco N, Ratajczak J, Laughlin MJ, Ratajczak MZ. 2012. The bone marrow-expressed antimicrobial cationic peptide LL-37 enhances the responsiveness of hematopoietic stem progenitor cells to an SDF-1 gradient and accelerates their engraftment after transplantation. Leukemia 26:736–745. doi: 10.1038/leu.2011.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. 1997. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 90:2796–2803. [PubMed] [Google Scholar]
- 46.Di Nardo A, Vitiello A, Gallo RL. 2003. Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J Immunol 170:2274–2278. doi: 10.4049/jimmunol.170.5.2274. [DOI] [PubMed] [Google Scholar]
- 47.Sonawane A, Santos JC, Mishra BB, Jena P, Progida C, Sorensen OE, Gallo R, Appelberg R, Griffiths G. 2011. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol 13:1601–1617. doi: 10.1111/j.1462-5822.2011.01644.x. [DOI] [PubMed] [Google Scholar]
- 48.Rivas-Santiago B, Hernandez-Pando R, Carranza C, Juarez E, Contreras JL, Aguilar-Leon D, Torres M, Sada E. 2008. Expression of cathelicidin LL-37 during Mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect Immun 76:935–941. doi: 10.1128/IAI.01218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jörnvall H, Wigzell H, Gudmundsson GH. 2000. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood 96:3086–3093. [PubMed] [Google Scholar]
- 50.Frohm M, Agerberth B, Ahangari G, Stâhle-Bäckdahl M, Lidén S, Wigzell H, Gudmundsson GH. 1997. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem 272:15258–15263. doi: 10.1074/jbc.272.24.15258. [DOI] [PubMed] [Google Scholar]
- 51.Frohm Nilsson M, Sandstedt B, Sørensen O, Weber G, Borregaard N, Ståhle-Bäckdahl M. 1999. The human cationic antimicrobial protein (hCAP18), a peptide antibiotic, is widely expressed in human squamous epithelia and colocalizes with interleukin-6. Infect Immun 67:2561–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim BJ, Rho YK, Lee HI, Jeong MS, Li K, Seo SJ, Kim MN, Hong CK. 2009. The effect of calcipotriol on the expression of human beta defensin-2 and LL-37 in cultured human keratinocytes. Clin Dev Immunol 2009:645898. doi: 10.1155/2009/645898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF. 2002. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun 70:953–963. doi: 10.1128/IAI.70.2.953-963.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bals R, Wang X, Zasloff M, Wilson JM. 1998. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci U S A 95:9541–9546. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, Perin PM, Frentzen A, Kaderali L, Pietschmann T. 2013. Hepatitis C virus p7 is critical for capsid assembly and envelopment. PLoS Pathog 9:e1003355. doi: 10.1371/journal.ppat.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. 2013. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci U S A 110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang G. 2008. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem 283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- 58.Porcelli F, Verardi R, Shi L, Henzler-Wildman KA, Ramamoorthy A, Veglia G. 2008. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 47:5565–5572. doi: 10.1021/bi702036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Da Costa D, Turek M, Felmlee DJ, Girardi E, Pfeffer S, Long G, Bartenschlager R, Zeisel MB, Baumert TF. 2012. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J Virol 86:11919–11925. doi: 10.1128/JVI.01066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Israelow B, Mullokandov G, Agudo J, Sourisseau M, Bashir A, Maldonado AY, Dar AC, Brown BD, Evans MJ. 2014. Hepatitis C virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat Commun 5:5408. doi: 10.1038/ncomms6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Henzler Wildman KA, Lee DK, Ramamoorthy A. 2003. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 42:6545–6558. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 62.Gwyer Findlay E, Currie SM, Davidson DJ. 2013. Cationic host defence peptides: potential as antiviral therapeutics. BioDrugs 27:479–493. doi: 10.1007/s40259-013-0039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kai-Larsen Y, Agerberth B. 2008. The role of the multifunctional peptide LL-37 in host defense. Front Biosci 13:3760–3767. [DOI] [PubMed] [Google Scholar]
- 64.Matsumura T, Sugiyama N, Murayama A, Yamada N, Shiina M, Asabe S, Wakita T, Imawari M, Kato T. 26 November 2015. Antimicrobial peptide LL-37 attenuates infection of hepatitis C virus. Hepatol Res doi: 10.1111/hepr.12627. [DOI] [PubMed] [Google Scholar]
- 65.Sørensen O, Bratt T, Johnsen AH, Madsen MT, Borregaard N. 1999. The human antibacterial cathelicidin, hCAP-18, is bound to lipoproteins in plasma. J Biol Chem 274:22445–22451. doi: 10.1074/jbc.274.32.22445. [DOI] [PubMed] [Google Scholar]
- 66.Merz A, Long G, Hiet MS, Brügger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly BA, Neil SJ, McKnight A, Santos JM, Sinnis P, Jack ER, Middleton DA, Dobson CB. 2007. Apolipoprotein E-derived antimicrobial peptide analogues with altered membrane affinity and increased potency and breadth of activity. FEBS J 274:4511–4525. doi: 10.1111/j.1742-4658.2007.05981.x. [DOI] [PubMed] [Google Scholar]
- 68.Wang CQ, Yang CS, Yang Y, Pan F, He LY, Wang AM. 2013. An apolipoprotein E mimetic peptide with activities against multidrug-resistant bacteria and immunomodulatory effects. J Pept Sci 19:745–750. doi: 10.1002/psc.2570. [DOI] [PubMed] [Google Scholar]
- 69.Kelly BA, Harrison I, McKnight A, Dobson CB. 2010. Anti-infective activity of apolipoprotein domain derived peptides in vitro: identification of novel antimicrobial peptides related to apolipoprotein B with anti-HIV activity. BMC Immunol 11:13. doi: 10.1186/1471-2172-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drin G, Antonny B. 2010. Amphipathic helices and membrane curvature. FEBS Lett 584:1840–1847. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 71.Campelo F, McMahon HT, Kozlov MM. 2008. The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys J 95:2325–2339. doi: 10.1529/biophysj.108.133173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J Virol 84:11532–11541. doi: 10.1128/JVI.01021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. 2012. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol 86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Owen DM, Huang H, Ye J, Gale M. 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shiokawa M, Fukuhara T, Ono C, Yamamoto S, Okamoto T, Watanabe N, Wakita T, Matsuura Y. 2014. Novel permissive cell lines for complete propagation of hepatitis C virus. J Virol 88:5578–5594. doi: 10.1128/JVI.03839-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Narbus CM, Israelow B, Sourisseau M, Michta ML, Hopcraft SE, Zeiner GM, Evans MJ. 2011. HepG2 cells expressing microRNA miR-122 support the entire hepatitis C virus life cycle. J Virol 85:12087–12092. doi: 10.1128/JVI.05843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kambara H, Fukuhara T, Shiokawa M, Ono C, Ohara Y, Kamitani W, Matsuura Y. 2012. Establishment of a novel permissive cell line for the propagation of hepatitis C virus by expression of microRNA miR122. J Virol 86:1382–1393. doi: 10.1128/JVI.06242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Lello FA, Culasso AC, Parodi C, Baré P, Campos RH, García G. 2014. New evidence of replication of hepatitis C virus in short-term peripheral blood mononuclear cell cultures. Virus Res 191:1–9. doi: 10.1016/j.virusres.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 79.Nakai M, Seya T, Matsumoto M, Shimotohno K, Sakamoto N, Aly HH. 2014. The J6JFH1 strain of hepatitis C virus infects human B-cells with low replication efficacy. Viral Immunol 27:285–294. doi: 10.1089/vim.2013.0140. [DOI] [PubMed] [Google Scholar]
- 80.Dichamp I, Abbas W, Kumar A, Di Martino V, Herbein G. 2014. Cellular activation and intracellular HCV load in peripheral blood monocytes isolated from HCV monoinfected and HIV-HCV coinfected patients. PLoS One 9:e96907. doi: 10.1371/journal.pone.0096907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kasama Y, Sekiguchi S, Saito M, Tanaka K, Satoh M, Kuwahara K, Sakaguchi N, Takeya M, Hiasa Y, Kohara M, Tsukiyama-Kohara K. 2010. Persistent expression of the full genome of hepatitis C virus in B cells induces spontaneous development of B-cell lymphomas in vivo. Blood 116:4926–4933. doi: 10.1182/blood-2010-05-283358. [DOI] [PMC free article] [PubMed] [Google Scholar]