

Abstract
Background
Transthyretin (TTR) pV142I (rs76992529‐A) is one of the 113 variants in the human TTR gene associated with systemic amyloidosis. It results from a G to A transition at a CG dinucleotide in the codon for amino acid 122 of the mature protein (TTR V122I). The allele frequency is 0.0173 in African Americans.
Methods
PCR‐based assays to genotype 2767 DNA samples obtained from participants in genetic studies from various African populations supplemented with sequencing data from 529 samples within the 1000 Genomes Project.
Results
The rs76992529‐A variant allele was most prevalent (allele frequency 0.0253) in the contiguous West African countries of Sierra Leone, Guinea, Ivory Coast, Burkina Faso, Ghana, and Nigeria. In other African countries, the mean allele frequency was 0.011.
Conclusions
Our data are consistent with a small number of founder carriers of the amyloidogenic TTR V122I (p.Val142Ile) allele in southern West Africa, with no apparent advantage or disadvantage of an allele carrying newborn reaching adulthood. In U.S. African Americans, the allele represents a significant risk for congestive heart failure late in life. If clinical penetrance is similar in African countries with high allele frequencies, then cardiac amyloidosis could also represent a significant cause of heart disease in the elderly in those populations.
Keywords: Africa, amyloidosis, slave trade, transthyretin
Introduction
Despite the predominant role of genomics, genome‐wide association studies, and deep sequencing technology in modern genetics, the study of well‐defined single base changes causing clinical disease in large populations remains important, particularly as therapies become available for such diseases. Furthermore, the disease‐associated variants can sometimes provide useful information regarding their evolutionary effect and the history of the carrier populations.
Transthyretin (TTR) is a homotetrameric, nonglycosylated serum and cerebrospinal fluid protein carrier of thyroxine and retinol‐binding protein charged with retinol (Buxbaum 2007). Each TTR monomer contains 127 amino acids. It is synthesized in the liver, choroid plexus, retinal epithelium, pancreas, and neurons (Buxbaum 2007; Li et al. 2011). It is a systemic carrier for thyroxine (T4) and retinol‐binding protein charged with retinol and has a critical function in transporting T4 into the central nervous system during a relatively narrow developmental window. Studies of homozygous Ttr knockout mice show significant structural disruption in the hippocampus with a quantitative deficit in neuroblast number in the supraventricular zone and neuronal loss in the CA3 region (Buxbaum et al. 2014). These are accompanied by deficits in spatial learning in adult mice (Sousa et al. 2004). It has been hypothesized that the phenotype is developmental. Neuronally synthesized TTR has been shown to behave as a stress protein, protective in transgenic murine models of human Aβ deposition with increased synthesis in human Alzheimer's disease (Wang et al. 2014).
The protein is encoded by a single‐copy gene (TTR) (NCBI reference sequence: NC_000018.10) on human chromosome 18 (Wallace et al. 1985). The gene is well conserved in humans with few variants relative to genes encoding other serum proteins and genes of the same size (Abecasis et al. 2010). A single protein polymorphism, a substitution of a serine for glycine at position 6 in the mature protein, occurs in approximately 12% of subjects of European descent (allele frequency 0.06) and to date seems unassociated with disease (Jacobson et al. 1995). Of 70 Africans screened, none were shown to carry this mutation, but it occurs in 2.5% of African Americans (Jacobson et al. 1995). The African American/White American allele frequency ratio of 0.0125/0.06 (0.21) is consistent with the mean estimate of admixture (0.17 ± 0.063) of European and African genes found in several studies across the U.S. population (Mersha and Abebe 2015). There is an additional rare coding sequence variant TTR Thr119Met (p.Thr139Met), which is kinetically very stable and has been reported to be associated with increased longevity in a large Danish population study (Hornstrup et al. 2013). The explanation for the longevity effect is unclear. Despite a significant number of large scale population surveys searching for variants, there has been no reported instance of a human completely lacking TTR.
To date there have been 123 amino acid substitutions, one deletion and one synonymous base substitution reported for the transthyretin protein and its encoding gene. Twelve have not been associated with tissue‐compromising TTR deposits, while in one instance the clinical amyloidogenicity has not been established. The amino acid variants have been shown to result in decreases in either the thermodynamic or kinetic stability of the tetramer and cause human disease (OMIM 176300.0009) (http://www.amyloidosismutations.com/mut-attr.php). The variants produce autosomal dominant diseases in which the tetramer dissociates into its constituent monomers that subsequently misfold and aggregate forming tissue oligomers and fibrils (Johnson et al. 2012). The aggregates are cytotoxic and may also compromise function on a displacement basis, depending on the target organ (Reixach et al. 2004). Most TTR variants have been described in only one or a few kindreds (Rowczenio et al. 2014). The most common variants associated with disease are pV50M (TTR V30M) and pV142I (TTRV122I). Both arose from transitions at CpG dinucleotides, a mutational “hot spot” (Cooper and Youssoufian 1988), as did the normal population variants TTR pG26S (TTR G6S) and pT139M (TTR T119M). Among the amyloidogenic TTR mutations, TTR V122I is unusual for its overwhelmingly predominant occurrence in individuals of documented African descent.
The heart is the major organ showing amyloid deposition in the pV142I (TTR V122I) carriers. It was first described in 1988 in an African American man with cardiac amyloidosis who was homozygous for the G→A transition at rs76992529 (Gorevic et al. 1989; Jacobson et al. 1990). Many other reports have confirmed the association of the variant with cardiac amyloidosis in elderly African Americans and it is now recognized as a cause of heart failure when the allele is present in either the heterozygous or homozygous state (Jacobson et al. 1990; Nichols et al. 1991). Population studies have determined that the prevalence of the TTR rs76992529‐A allele in African Americans is 0.0173 (Jacobson et al. 2015). A small number of kindreds carrying the allele without identifiable African ancestry have been reported (Gillmore et al. 1999; Asl et al. 2001; Ammirati et al. 2012) (Cappelli et al. 2015; Damy et al. 2015). The amyloidogenic allele is an infrequent but valid ancestry informative marker. It appears that in the heterozygous state the allele is clinically silent until the seventh decade of life or later. Homozygous individuals develop disease about 10 years earlier (Reddi et al. 2014). Given the late age of clinical impact, the allele is likely to be evolutionarily neutral.
One previous study reported the allele frequency in small selected African populations: it was found in 1/55 South African Zulus, 0/34 South African Xhosas, and 0/9 Nigerians (Afolabi et al. 2000). Since the African American population is largely descended from African slaves brought to the western hemisphere from West Africa in the 16th to 19th centuries (Curtin 1969), we elected to investigate its origin(s) by determining the frequency of the TTR V122I allele in various modern African cohorts.
Methods
Ethical compliance
De‐identified DNA samples from 14 modern African countries were obtained with informed consent by investigators studying the genetics of a variety of diseases not known to be related to TTR and laboratories interested in the nature of genetic differences among African populations (Table 1). The samples from the Gambia obtained specifically for this study were approved by the Gambia ethics committee of the Medical Research Council (MRC). The samples were provided to us with information about the country and, in some cases, the village and/or tribe of origin. We have also included data from 529 African DNA samples analyzed and reported in the “1000 Genomes Project” (Abecasis et al. 2010).
Table 1.
Frequency of TTR V122I in African populations
| Country/Region | Population source | V122I/Total alleles | Allele prevalence (sample) | V122I alleles | Total alleles | Allele prevalence (country) |
|---|---|---|---|---|---|---|
| Burkina Faso | Modiano et al. (1999) | 3 | 120 | 0.025 | ||
| Ghana | Total | 61 | 2424 | 0.025 | ||
| Williams et al. (2000) | 13/528 | 0.025 | ||||
| Present study | 48/1896 | 0.026 | ||||
| Guinea | Zimmerman et al. (1995) | 2 | 56 | 0.036 | ||
| Ivory Coast | Zimmerman et al. (1995) | 3 | 82 | 0.037 | ||
| Sierra Leone | Total | 31 | 1174 | 0.026 | ||
| Zimmerman et al. (1995) | 6/100 | 0.060 | ||||
| Jackson et al. (2005) | 22/904 | 0.024 | ||||
| Mende | Abecasis et al. (2010) | 3/170 | 0.018 | |||
| Nigeria | Total | 12 | 570 | 0.021 | ||
| Vulliamy et al. (1991) | 0/122 | <0.008 | ||||
| Afolabi et al. (2000) | 0/18 | <0.055 | ||||
| Esan | Abecasis et al. (2010) | 6/198 | 0.030 | |||
| Yoruba | Abecasis et al. (2010) | 6/232 | 0.026 | |||
| Total, high‐prevalence area | 112 | 4426 | 0.0253 | |||
| Gambia | Total | 8 | 636 | 0.013 | ||
| Cooke et al. (2004); Sirugo et al. (2004) | 5/410 | 0.012 | ||||
| Abecasis et al. (2010) | 3/226 | 0.013 | ||||
| Guinea‐Bissau | Zimmerman et al. (1995) | 1 | 78 | 0.013 | ||
| Mali | Kalidi et al. (1988) | 1 | 130 | 0.008 | ||
| Senegal | Zimmerman et al. (1995) | 1 | 132 | 0.008 | ||
| Tanzania | Henn et al. (2008) | 3 | 254 | 0.012 | ||
| Cameroon | Henn et al. (2008) | 3 | 230 | 0.013 | ||
| Kenya | 1 | 264 | 0.004 | |||
| Luhya (Kenya) | Abecasis et al. (2010) | 1/232 | 0.009 | |||
| Other Kenya | 0/32 | <0.030 | ||||
| South Africa | 3 | 360 | 0.008 | |||
| Xhosa | Afolabi et al. (2000) | 0/68 | <0.015 | |||
| Zulu | Afolabi et al. (2000) | 1/110 | 0.009 | |||
| Xhosa | Rousseau et al. (1985) | 2/182 | 0.011 | |||
| Total, outside high‐prevalence area | 21 | 2084 | 0.010 |
The TTR rs76992529‐A allele was identified using PCR‐based assays developed in our laboratory (Jacobson 1992) (Alexander et al. 2004) except in the data available from the 1000 Genomes Project in which complete DNA sequencing was performed (Abecasis et al. 2010).
Results
Of the roughly 2900 samples tested in our laboratories, successful results were obtained in 2767. The prevalence of the TTR allele encoding pV142I was highest in six contiguous countries of West Africa, ranging from Nigeria in the east to Guinea in the west (Table 1 and Fig. 1). The overall allele frequency in this area was 0.0253, with each of the individual countries having an allele frequency of at least 0.021. In contrast, the allele frequencies in the other 10 African countries studied ranged from 0 to 0.013, with an overall frequency in those countries of 0.011. The 0.0253 frequency in the high‐prevalence area was statistically different from the frequency in samples studied from outside this area (Table 1, Fig. 1) (P = 0.0001) (two‐tailed Z test).
Figure 1.
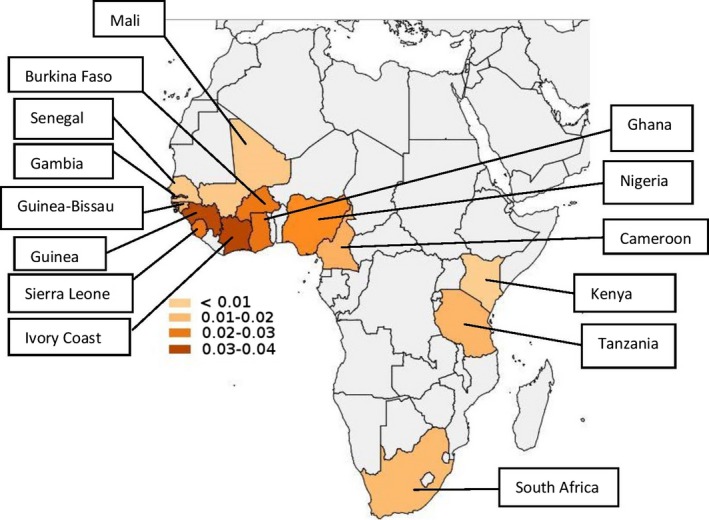
Distribution of the TTR V122I allele in Africa (see Table 1). The allele frequencies are indicated by color. Populations from the countries colored light gray were not analyzed.
Discussion
It is striking that within the African countries examined in this study, the frequency of the TTR pV142I (TTR V122I)‐encoding allele was significantly higher in one contiguous geographic area of West Africa (overall allele prevalence 0.0253, ranging from 0.021 to 0.037 in the individual countries studied) than elsewhere in Africa (allele prevalence 0.010–0.011). The overall allele prevalence of 0.0253 in the high‐prevalence area indicates that about 5.1% of the population is expected to be heterozygous for the variant allele. While the allele prevalence is significantly lower in other African regions, it was present wherever we were able to sample throughout sub‐Saharan and western Africa (Fig. 1, Table 1).
Since the Ghanaian samples were obtained from 948 newborns and 264 adults, we could compare the allele frequencies in two different age groups from the same population (as determined in a single geographic area). The allele frequency was nearly identical in the newborns (49/1896; 0.026) and the adults (13/528; 0.025) from Accra. These data, coupled with the late age of onset of the allele‐associated disease, support the conclusion that the allele does not influence survival to adulthood and is unlikely to be under positive or negative selective evolutionary pressure. The allele prevalence in African Americans is close to that in the modern West African countries that were the geographic center of the slave trade suggesting that the gene was present in similar proportions in the population ancestral to modern African Americans and had little effect on survival in North America (Curtin 1969).
A recent analysis of the African samples from the “1000 Genomes Project” showed that the G to A transition responsible for the variant amino acid occurred on five different haplotypes (Polimanti et al. 2014). Four of these differed from the haplotype containing the ancestral allele at 12 different positions within the confines of the TTR gene and were identical at all but one base, indicating a single founder haplotype that acquired noncoding mutations after the appearance of the amyloidogenic TTR V122I allele. The second potential founder haplotype differed from the ancestral haplotype at 13 positions and from the other possible founder haplotype at 12 positions, five of which it shared with the ancestral allele suggesting a second founding event or a more complex set of alterations in the gene after the initial appearance of rs76992529. In any case, despite the origin of the allele at a CpG dinucleotide, a so‐called genetic “hot spot,” the number of founder haplotypes is small. Previous haplotype analyses in African American carriers of the allele also suggested a small number of founders in the descendants of the original slave population (Jacobson et al. 1998).
Genetic, epigenetic, and environmental factors have been proposed as being responsible for variation in the clinical penetrance, that is, clinically important TTR amyloid deposition, of any TTR mutation. The first‐described and most extensively studied disease‐associated TTR variant, TTR pV50M (TTR V30M) was identified in Portugal, then Sweden, and subsequently Japan, and has since been found globally (Andrade 1952; Araki et al. 1968; Andersson 1976). The age of the allele has been estimated to be 26 generations (750 years) in Portugal, 15 generations (375 years) in Sweden (Zaros et al. 2008), and perhaps older in Italy (Iorio et al. 2014). Analysis of intronic haplotypes revealed multiple founders in Japan, but single founders in Portugal and Sweden (Yoshioka et al. 1989; Zaros et al. 2008). More recent studies have identified multiple haplotypes across Europe as well (Reilly et al. 1995; Soares et al. 2004). There are significant differences in clinical penetrance of TTRV30M among the carriers in different populations, particularly with respect to age of onset (Koike et al. 2002) (Hellman et al. 2008). We have suggested that disease penetrance is conditioned by epistatic interactions with other genes, the products of which can influence the stability of the tetramer (Soares et al. 2005). Support for that idea has recently been reported independently (Santos et al. 2015). Others have hypothesized that variation at transcription factor binding sites within populations resulting in different transcription levels presumably increased, resulting in increased serum TTR concentrations, could also be responsible for different degrees of clinical penetrance (Polimanti et al. 2014). While there are no available data regarding hepatic TTR transcription in different carriers of the same amyloidogenic allele, we think this unlikely since serum TTR concentrations tend to be lower rather than higher in carrier subjects even before there is detectable tissue deposition.
Studies of polymorphisms in a microRNA binding site in the promoter region of TTR V30M (pV50M) carriers in Sweden, while initially suggesting an explanation for the apparent late onset in Swedish carriers, could not be confirmed (Olsson et al. 2010; Norgren et al. 2012). In individuals carrying TTR V30M, it appears that the parent of inheritance (POI) has a significant effect on disease penetrance, carrier sons of carrier mothers having earlier onset, and more severe disease (Sousa et al. 1991). Some studies have suggested that the POI is related to mitochondrial genotype; however, it does not appear that the entire effect can be attributed to mitochondrial inheritance (Olsson et al. 2008; Bonaiti et al. 2010). One can assume that the same phenomena will prevail for disease related to all the amyloidogenic TTR mutations including pV142I (TTRV122I).
The mechanism of TTR amyloid formation is similar regardless of the nature of the specific mutation, that is, dissociation of the TTR tetramer due to reduced thermodynamic or kinetic stability, which allows the released monomers to misfold and aggregate (Johnson et al. 2012). It is likely that genetic or epigenetic factors that influence the development of disease operate in a similar manner regardless of the specific mutation, having a greater or lesser effect depending on the stability of the variant.
Among the samples from Sierra Leone, Cameroon, Tanzania, Burkina Faso, Nigeria, and South Africa, the variant allele was found in samples from multiple tribal origins and language groups (Tables 1 and 2). Our sample sizes were not large enough to determine whether the differences in allele frequencies among tribes were statistically significant. In the absence of larger sample sizes and haplotype analysis in those individuals we cannot determine whether these represented rare individual founders or migrants into the groups from areas in which the allele was present at higher frequencies.
Table 2.
TTR V122I allele prevalence in specific tribes in Tanzania, Cameroon, and Sierra Leone
| Tribe | TTR V122I | TTR V122V | Allele frequency |
|---|---|---|---|
| Tanzania and Cameroon | |||
| Turu | 0 | 56 | <0.018 |
| Sandawe | 1 | 61 | 0.016 |
| Burunge | 2 | 50 | 0.038 |
| Maasai | 0 | 58 | <0.017 |
| Bakola | 0 | 56 | <0.018 |
| Bamoun | 1 | 55 | 0.018 |
| Zime | 2 | 46 | 0.042 |
| Sierra Leone | |||
| Creole | 4 | 70 | 0.054 |
| Shabro | 1 | 1 | 1 |
| Mende | 9 | 236 | 0.038 |
| Temne | 3 | 195 | 0.015 |
The similar allele frequencies in newborn and adult Ghanaians suggest that the TTR V122I (pV142I) allele does not confer an advantage or disadvantage in reaching adulthood in the Ghanaian environment. Similarly, the 0.0173 allele frequency in U.S. African Americans, allowing for an average admixture of 17% with European genes, is consistent with the 0.0253 frequency in Africans in the high‐prevalence area, and suggests that there is little selection against allele carriers in the United States environment. Hence, current allele frequencies across Africa may reflect its distribution at the time of the Atlantic slave trade from the 15th to 19th centuries A.D.
The TTR V122I (pV142I) allele frequencies are highest in the non‐Bantu‐speaking Niger‐Kordofanian‐derived populations from Sierra Leone, Ghana, Cote d'Ivoire, etc. Broad genetic studies have indicated that this is the ancestry that is mostly highly represented in African Americans (Tishkoff et al. 2009; Bryc et al. 2010). Interestingly, the frequencies of the TTR V122I (pV142I) allele are low in the countries immediately adjacent to the north and west (Gambia, Guinea‐Bissau, Mali, and Senegal) as well as those to the south and east.
The major population movement as judged initially by language patterns across Africa was the so‐called Bantu expansion from 1000 B.C. to 500 A.D. During that period Bantu language‐speaking people spread, apparently in several stages, from a relatively small area in what is now the southeastern border of Nigeria and Cameroon, south and eastward, dispersing throughout the southern half of the continent. In our study, the lower allele frequency in the Cameroonian sample than in the Nigerian cohorts and the considerable variation in the frequency among the various samples from Nigeria could be consistent with a relatively indistinct western border of the Bantu region in the context of modern national boundaries. The observation that the current frequency of the TTR V122I (pV142I) allele is three times greater in coastal West Africa than in the other parts of the continent that we were able to sample is open to several interpretations. It is possible that the allele may be older than the expansion, and originated in a non‐Bantu‐speaking, western coastal population that was unaffected by the migration, and that it has persisted in the local populations because it is evolutionarily neutral or under weak purifying selection. Alternatively, the allele may have appeared after the Bantu expansion with the lower allele frequency among the Bantu speakers representing small groups of later migrating founders.
Based on studies of over 12,000 DNA samples, the prevalence of the allele encoding TTR pV142I allele in African Americans is known to be 0.0173 (Jacobson et al. 2015). Furthermore, it is estimated that modern African Americans have an average of 17% genetic admixture with groups with non‐African ancestry, although there is substantial variation among individuals and in different geographic regions in the United States (Kostrikis et al. 1998; Halder et al. 2009, 2012; Tishkoff et al. 2009; Bryc et al. 2010, 2014; Murray et al. 2010; Tandon et al. 2011; Bhatia et al. 2014; Mersha and Abebe 2015). Thus, based on an inferred population without significant admixture one can estimate that the average variant allele prevalence in the African ancestors of modern African Americans would be 0.0173/0.83 or 0.021, not significantly different from the combined observed frequency in the high‐prevalence area (Ghana, Sierra Leone, Guinea, Burkina Faso, Cote d'Ivoire, and Nigeria), that is, 0.025.
Is this calculation consistent with what is known about the Atlantic slave trade? The available historical data indicate that 50–70% of African slaves brought to North America came from areas corresponding to the modern African countries included in our designation “West Africa” plus the immediately adjacent countries of Togo and Benin, which were not represented among our samples (whereas higher percentages of slaves from other areas, such as modern‐day Congo and Angola, for which we do not have allele frequency data, may have been taken, in higher numbers, to South America) (Curtin 1969). Correlating historical records of the slave trade with modern African political boundaries is imprecise, because of gaps in the available data and the breakdown of Africa into geographic areas in the 16th to 19th centuries (e.g., Bight of Benin [including the southern coasts of Ghana, Togo, Benin, and the western coast of Nigeria], Bight of Biafra [including the presently defined southern coast of Nigeria, Cameroon, Equatorial Guinea, and the northern coast of Gabon], Windward Coasts, Gold Coast [Ghana], etc.) do not precisely correspond to modern African countries. Thus, our data and model seem consistent with the historical record of the Atlantic slave trade.
We conclude that the prevalence of the TTR V122I allele in Africa ranges from 0.037 around the area presumed to include most ancestors of African Americans to 0.010 in African countries less likely to be highly represented among the slave population brought to North America. Haplotype data from the 1000 genomes project are consistent with perhaps two ancient founder haplotypes carrying the amyloidogenic rs76992529‐A I allele (Polimanti et al. 2013). Future population‐based surveys of this allele focused on smaller regions and tribal studies within the region we have defined as “southern West Africa” plus the adjacent areas of Togo and Benin might reveal small areas with an even higher prevalence. Such studies would be of interest for both genetic purposes, and for the purposes of ultimately providing optimal care for patients from these areas. As life expectancy in the region increases, TTR V122I (pV142I) carriers may represent a cohort of individuals at substantial risk of developing clinically significant cardiac amyloidosis with aging.
Conflict of Interest
None of the authors has financial or other interests which would compromise their objectivity in either performing or interpreting the analyses reported.
Acknowledgments
We thank Rosaria Scozzari, Giorgio Sirugo, Francis J. Louis, Adrian V.S. Hill, Thomas J. Vulliamy, Peter Zimmerman, Ronald Nagel, A. S. Santachiara‐Benercetti, Ernette D. du Toit, Jeanne Rousseau, and Paul Goodfellow for contributing DNA samples for these studies. We also thank Immaculata Kane for technical assistance. The study was supported by the National Institutes of Health (NIH) grant R01AG19259 (J. N. B.).
References
- Abecasis, G. R. , Altshuler D., Auton A., Brooks L. D., Durbin R. M., Gibbs R. A., et al. 2010. A map of human genome variation from population‐scale sequencing. Nature 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afolabi, I. , Hamidi A. K., Nakamura M., Jacobs P., Hendrie H., and Benson M. D.. 2000. Transthyretin isoleucine‐122 mutation in African and American blacks. Amyloid 7:121–125. [DOI] [PubMed] [Google Scholar]
- Alexander, A. , Subramanian N., Buxbaum J. N., and Jacobson D. R.. 2004. Drop‐in, drop‐out allele‐specific PCR: a highly sensitive, single‐tube method. Mol. Biotechnol. 28:171–174. [DOI] [PubMed] [Google Scholar]
- Ammirati, E. , Marziliano N., Vittori C., Pedrotti P., Bramerio M. A., Motta V., et al. 2012. The first Caucasian patient with p.Val122Ile mutated‐transthyretin cardiac amyloidosis treated with isolated heart transplantation. Amyloid 19:113–117. [DOI] [PubMed] [Google Scholar]
- Andersson, R. 1976. Familial amyloidosis with polyneuropathy. A clinical study based on patients living in Northern Sweden. Acta Med. Scand. 590:1–64. [PubMed] [Google Scholar]
- Andrade, C. 1952. A peculiar form of peripheral neuropathy. Familial atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75:408–427. [DOI] [PubMed] [Google Scholar]
- Araki, S. , Mawatari S., Ohta M., Nakajima A., and Kuroiwa Y.. 1968. Polyneuritic amyloidosis in a Japanese family. Arch. Neurol. 18:593–602. [DOI] [PubMed] [Google Scholar]
- Asl, K. H. , Nakamura M., Yamashita T., and Benson M. D.. 2001. Cardiac amyloidosis associated with the transthyretin Ile122 mutation in a Caucasian family. Amyloid 8:263–269. [DOI] [PubMed] [Google Scholar]
- Bhatia, G. , Tandon A., Patterson N., Aldrich M. C., Ambrosone C. B., Amos C., et al. 2014. Genome‐wide scan of 29,141 African Americans finds no evidence of directional selection since admixture. Am. J. Hum. Genet. 95:437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaiti, B. , Olsson M., Hellman U., Suhr O., Bonaiti‐Pellie C., and Plante‐Bordeneuve V.. 2010. TTR familial amyloid polyneuropathy: does a mitochondrial polymorphism entirely explain the parent‐of‐origin difference in penetrance? Eur. J. Hum. Genet. 16:149–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryc, K. , Auton A., Nelson M. R., Oksenberg J. R., Hauser S. L., Williams S., et al. 2010. Genome‐wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl Acad. Sci. USA 107:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryc, K. , Durand E. Y., Macpherson J. M., Reich D., and Mountain J. L.. 2014. The Genetic Ancestry of African Americans, Latinos, and European Americans across the United States. Am. J. Hum. Genet. 96:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum, J. N. 2007. Transthyretin and the Transthyretin Amyloidoses Pp. 259–283 in Uversky V. N. and Fink A., eds. Protein misfolding, aggregation, and conformational diseases. Springer, Santa Cruz, CA. [Google Scholar]
- Buxbaum, J. N. , Roberts A. J., Adame A., and Masliah E.. 2014. Silencing of murine transthyretin and retinol binding protein genes has distinct and shared behavioral and neuropathologic effects. Neuroscience 275:352–364. [DOI] [PubMed] [Google Scholar]
- Cappelli, F. , Frusconi S., Bergesio F., Grifoni E., Fabbri A., Giuliani C., et al. 2015. The Val142Ile transthyretin cardiac amyloidosis: not only an Afro‐American pathogenic variant? A single‐centre Italian experience J. Cardiovasc. Med. (Hagerstown) 17:122–125. [DOI] [PubMed] [Google Scholar]
- Cooke, G. S. , Campbell S. J., Fielding K., Sillah J., Manneh K., Sirugo G., et al. 2004. Interleukin‐8 polymorphism is not associated with pulmonary tuberculosis in the gambia. J. Infect. Dis. 189:1545–1546. [DOI] [PubMed] [Google Scholar]
- Cooper, D. N. , and Youssoufian H.. 1988. The CpG dinucleotide and human genetic disease. Hum. Genet. 78:151–155. [DOI] [PubMed] [Google Scholar]
- Curtin, P. D. 1969. The Atlantic slave trade: a census. The University of Wisconsin Press, Madison, Milwaukee and London. [Google Scholar]
- Damy, T. , Costes B., Hagege A. A., Donal E., Eicher J. C., Slama M., et al. 2015. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur. Heart J. 37:1826–1834. [DOI] [PubMed] [Google Scholar]
- Gillmore, J. D. , Booth D. R., Pepys M. B., and Hawkins P. N.. 1999. Hereditary cardiac amyloidosis associated with the transthyretin Ile122 mutation in a white man. Heart 82:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorevic, P. D. , Prelli F. C., Wright J., Pras M., and Frangione B.. 1989. Systemic Senile Amyloidosis. Identification of a new prealbumin (transthyretin) variant in cardiac tissue: immunologic and biochemical similarity to one form of familial amyloidotic polyneuropathy. J. Clin. Invest. 83:836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder, I. , Yang B. Z., Kranzler H. R., Stein M. B., Shriver M. D., and Gelernter J.. 2009. Measurement of admixture proportions and description of admixture structure in different U.S. populations. Hum. Mutat. 30:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder, I. , Kip K. E., Mulukutla S. R., Aiyer A. N., Marroquin O. C., Huggins G. S., et al. 2012. Biogeographic ancestry, self‐identified race, and admixture‐phenotype associations in the Heart SCORE Study. Am. J. Epidemiol. 176:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman, U. , Alarcon F., Lundgren H. E., Suhr O. B., Bonaiti‐Pellie C., and Plante‐Bordeneuve V.. 2008. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid 15:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn, B. M. , Gignoux C., Lin A. A., Oefner P. J., Shen P., Scozzari R., et al. 2008. Y‐chromosomal evidence of a pastoralist migration through Tanzania to southern Africa. Proc. Natl Acad. Sci. USA 105:10693–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstrup, L. S. , Frikke‐Schmidt R., Nordestgaard B. G., and Tybjaerg‐Hansen A.. 2013. Genetic stabilization of transthyretin, cerebrovascular disease, and life expectancy. Arterioscler. Thromb. Vasc. Biol. 33:1441–1447. [DOI] [PubMed] [Google Scholar]
- Iorio, A. , De A. F., Di G. M., Luigetti M., Pradotto L., Mauro A., et al. 2014. Most recent common ancestor of TTR Val30Met mutation in Italian population and its potential role in genotype‐phenotype correlation. Amyloid 22:73–78. [DOI] [PubMed] [Google Scholar]
- Jackson, B. A. , Wilson J. L., Kirbah S., Sidney S. S., Rosenberger J., Bassie L., et al. 2005. Mitochondrial DNA genetic diversityamong four ethnic groups in Sierra Leone. Am. J. Phys. Anthropol. 128:156–163. [DOI] [PubMed] [Google Scholar]
- Jacobson, D. R. 1992. A specific test for transthyretin 122 (Val‐Ile) based on PCR‐primer introduced restriction analysis (PCR‐PIRA): confirmation of the gene frequency in Blacks. Am. J. Hum. Genet. 50:195–198. [PMC free article] [PubMed] [Google Scholar]
- Jacobson, D. R. , Gorevic P. D., and Buxbaum J. N.. 1990. A homozygous transthyretin variant associated with senile systemic amyloidosis: evidence for a late‐onset disease of genetic etiology. Am. J. Hum. Genet. 47:127–136. [PMC free article] [PubMed] [Google Scholar]
- Jacobson, D. R. , Alves I. L., Saraiva M. J., Thibodeau S. N., and Buxbaum J. N.. 1995. Transthyretin Ser 6 gene frequency in individuals without amyloidosis. Hum. Genet. 95:308–312. [DOI] [PubMed] [Google Scholar]
- Jacobson, D. R. , Schneider J., Sobel M., Yang J., Malendowicz S., Sobol J., et al. 1998. The origin of the transthyretin Ile 122 allele: haplotype analysis and population studies Pp. 227–229 in Kyle R. A., ed. Amyloid and amyloidosis. Parthenon; NY, London. [Google Scholar]
- Jacobson, D. R. , Alexander A. A., Tagoe C., and Buxbaum J. N.. 2015. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African‐Americans. Amyloid 22:171–174. [DOI] [PubMed] [Google Scholar]
- Johnson, S. M. , Connelly S., Fearns C., Powers E. T., and Kelly J. W.. 2012. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory‐agency‐approved drug. J. Mol. Biol. 42:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalidi, I. , Fofana Y., Rahly A. A., Bochu V., Dehay C., Gony J., et al. 1988. Study of HLA antigens in a population of Mali (West Africa). Tissue Antigens 31:98–102. [DOI] [PubMed] [Google Scholar]
- Koike, H. , Misu K., Ikeda S., Ando Y., Nakazato M., Ando E., et al. 2002. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early‐ versus late‐onset form. Arch. Neurol. 59:1771–1776. [DOI] [PubMed] [Google Scholar]
- Kostrikis, L. G. , Tyagi S., Mhlanga M. M., Ho D. D., and Kramer F. R.. 1998. Spectralgenotyping of human alleles. Science 279:1228–1229. [DOI] [PubMed] [Google Scholar]
- Li, X. , Masliah E., Reixach N., and Buxbaum J. N.. 2011. Neuronal production of transthyretin in human and murine Alzheimer's disease: is it protective? J. Neurosci. 31:12483–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersha, T. B. , and Abebe T.. 2015. Self‐reported race/ethnicity in the age of genomic research: its potential impact on understanding health disparities. Hum. Genomics 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modiano, D. , Petrarca V., Sirima B. S., Luoni G., Nebie I., Diallo D. A., et al. 1999. Different response to Plasmodium falciparum in west African sympatric ethnic groups: possible implications for malaria control strategies. Parassitologia 41:193–197. [PubMed] [Google Scholar]
- Murray, T. , Beaty T. H., Mathias R. A., Rafaels N., Grant A. V., Faruque M. U., et al. 2010. African and non‐African admixture components in African Americans and an African Caribbean population. Genet. Epidemiol. 34:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, W. C. , Liepnieks J. J., Snyder E. L., and Benson M. D.. 1991. Senile cardiac amyloidosis associated with homozygosity for a transthyretin variant (ILE‐122). J. Lab. Clin. Med. 117:175–180. [PubMed] [Google Scholar]
- Norgren, N. , Hellman U., Ericzon B. G., Olsson M., and Suhr O. B.. 2012. Allele specific expression of the transthyretin gene in swedish patients with hereditary transthyretin amyloidosis (ATTR V30M) is similar between the two alleles. PLoS ONE 7:e49981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson, M. , Hellman U., Plante‐Bordeneuve V., Jonasson J., Lang K., and Suhr O. B.. 2008. Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin. Genet. 11:1–7. [DOI] [PubMed] [Google Scholar]
- Olsson, M. , Norgren N., Obayashi K., Plante‐Bordeneuve V., Suhr O. B., Cederquist K., et al. 2010. A possible role for miRNA silencing in disease phenotype variation in Swedish transthyretin V30M carriers. BMC Med. Genet. 11:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polimanti, R. , Di G. M., Manfellotto D., and Fuciarelli M.. 2013. Functional variation of the transthyretin gene among human populations and its correlation with amyloidosis phenotypes. Amyloid 20:256–262. [DOI] [PubMed] [Google Scholar]
- Polimanti, R. , Di G. M., Manfellotto D., and Fuciarelli M.. 2014. In silico analysis of TTR gene (coding and non‐coding regions, and interactive network) and its implications in transthyretin‐related amyloidosis. Amyloid 21:154–162. [DOI] [PubMed] [Google Scholar]
- Reddi, H. V. , Jenkins S., Theis J., Thomas B. C., Connors L. H., van Rhee F., et al. 2014. Homozygosity for the V122I mutation in transthyretin is associated with earlier onset of cardiac amyloidosis in the African American population in the seventh decade of life. J. Mol. Diagn. 16:68–74. [DOI] [PubMed] [Google Scholar]
- Reilly, M. M. , Adams D., Davis M., Said G., and Harding A. E.. 1995. Haplotype analysis of French, British and other European patients with familial amyloid polyneuropathy (MET30 and TYR77). J. Neurol. 242:664–668. [DOI] [PubMed] [Google Scholar]
- Reixach, N. , Deechongkit S., Jiang X., Kelly J. W., and Buxbaum J. N.. 2004. Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl Acad. Sci. USA 101:2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau, J. , Mathew C. G., Rees J. S., du Toit E., Botha M. C., and Harley E. H.. 1985. Incidence of Hb Barts and alpha‐thalassaemia genotypes in a South African population. Acta Haematol. 73:159–162. [DOI] [PubMed] [Google Scholar]
- Rowczenio, D. M. , Noor I., Gillmore J. D., Lachmann H. J., Whelan C., Hawkins P. N., et al. 2014. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat. 35:E2403–E2412. [DOI] [PubMed] [Google Scholar]
- Santos, D. , Coelho T., Alves‐Ferreira M., Sequeiros J., Mendonca D., Alonso I., et al. 2015. Variants in RBP4 and AR genes modulate age at onset in familial amyloid polyneuropathy (FAP ATTRV30M). Euro. J. Hum. Gen. 24:756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirugo, G. , Sam O., Nyan O., Pinder M., Hill A. V., Kwiatkowski D., et al. 2004. A national DNA bank in The Gambia, West Africa, and genomic research in developing countries. Nat. Genet. 36:785–786. [DOI] [PubMed] [Google Scholar]
- Soares, M. L. , Coelho T., Sousa A., Holmgren G., Saraiva M. J., Kastner D. L., et al. 2004. Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype‐phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur. J. Hum. Genet. 12:225–237. [DOI] [PubMed] [Google Scholar]
- Soares, M. L. , Coelho T., Sousa A., Batalov S., Conceicao I., Sales‐Luis M. L., et al. 2005. Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: complexity in a single‐gene disease. Hum. Mol. Genet. 14:543–553. [DOI] [PubMed] [Google Scholar]
- Sousa, A. , Coelho T., and Sequeiros J.. 1991. Parental transmission and age‐of‐onset in familial amyloidotic polineuropathy (Portuguese type). Amyloid and Amyloidosis 1990. p. 691–693.
- Sousa, J. C. , Grandela C., Fernandez‐Ruiz J., de Miguel R., de Sousa L., Magalhaes A. I., et al. 2004. Transthyretin is involved in depression‐like behaviour and exploratory activity. J. Neurochem. 88:1052–1058. [DOI] [PubMed] [Google Scholar]
- Tandon, A. , Patterson N., and Reich D.. 2011. Ancestry informative marker panels for African Americans based on subsets of commercially available SNP arrays. Genet. Epidemiol. 35:80–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff, S. A. , Reed F. A., Friedlaender F. R., Ehret C., Ranciaro A., Froment A., et al. 2009. The genetic structure and history of Africans and African Americans. Science 324:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy, T. J. , Othman A., Town M., Nathwani A., Falusi A. G., Mason P. J., et al. 1991. Polymorphic sites in the African population detected by sequence analysis of the glucose‐6‐phosphate dehydrogenase gene outline the evolution of the variants A and A‐. Proc. Natl Acad. Sci. USA 88:8568–8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, M. R. , Naylor S. L., Kluve‐Beckerman B., Long G. L., McDonald L., Shows T. B., et al. 1985. Localization of the human prealbumin gene to chromosome 18. Biochem. Biophys. Res. Commun. 129:753–758. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Cattaneo F., Ryno L., Hulleman J., Reixach N., and Buxbaum J. N.. 2014. The systemic amyloid precursor transthyretin (TTR) behaves as a neuronal stress protein regulated by HSF1 in SH‐SY5Y human neuroblastoma cells and APP23 Alzheimer's disease model mice. J. Neurosci. 34:7253–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, S. M. , Addy J. H., Phillips J. A. III, Dai M., Kpodonu J., Afful J., et al. 2000. Combinations of variations in multiple genes are associated with hypertension. Hypertension 36:2–6. [DOI] [PubMed] [Google Scholar]
- Yoshioka, K. , Furuya H., Sasaki H., Saraiva M. J. M., Costa P. P., and Sakaki Y.. 1989. Haplotype analysis of familial amyloidotic polyneuropathy. Evidence for multiple origins of the Val‐Met mutation most common to the disease. Hum. Genet. 82:9–13. [DOI] [PubMed] [Google Scholar]
- Zaros, C. , Genin E., Hellman U., Saporta M., Languille L., and Wadington‐Cruz M.. 2008. On the origin of the transthyretin Val30Met familial amyloid polyneuropathy. Ann. Hum. Genet. 72:478–484. [DOI] [PubMed] [Google Scholar]
- Zimmerman, P. A. , Phadke P. M., Lee A., Elson L. H., Aruajo E., Guderian R., et al. 1995. Migration of a novel DQA1* allele (DQA1*0502) from African origin to North and South America. Hum. Immunol. 42:233–240. [DOI] [PubMed] [Google Scholar]