

Abstract
The progression of cancer from localized to invasive disease is requisite for metastasis, and is often characterized by epithelial-to-mesenchymal transition (EMT) and alterations in cellular adhesion and migration. Studies have shown that this transition is associated with an up-regulation of embryonic stem cell-associated genes, resulting in a dedifferentiated phenotype and poor patient prognosis. Nodal is an embryonic factor that plays a critical role in promoting early invasive events during development. Nodal is silenced as stem cells differentiate; however, it re-emerges in adult life during placentation and mammary gland development, and is aberrantly expressed in many cancers. Here, we show that Nodal over-expression, in poorly-invasive breast cancer and choriocarcinoma cells, causes increased invasion and migration in vitro. Furthermore, we show that Nodal over-expression in these epithelial cancer types induces an EMT-like event concomitant with the internalization of E-Cadherin. This ability of Nodal to promote cellular invasion and EMT-like phenomena is dependent upon the phosphorylation of ERK1/2. Since Nodal normally signals through SMADs, these findings lend insight into an alternative pathway that is hijacked by this protein in cancer. To evaluate the clinical implications of our results, we show that Nodal inhibition reduces liver tumor burden in a model of spontaneous breast cancer metastasis in vivo, and that Nodal loss-of-function in aggressive breast cancer lines results in a decrease in invasive phenotypes. Our results demonstrate that Nodal is involved in promoting invasion in multiple cellular contexts, and that Nodal inhibition may be useful as a therapeutic target for patients with progressive disease.
Keywords: Nodal, invasion, EMT, cancer, metastasis
INTRODUCTION
The progression of cancer from localized to invasive disease is requisite for metastasis, and is often characterized by epithelial-to-mesenchymal transition (EMT) and alterations in cellular adhesion and migration. EMT is associated with tissue remodelling during normal physiological processes such as mammary gland development and placentation, and with various cancers, including breast cancer and choriocarcinoma (1;2). While EMT has been thoroughly investigated in breast cancer, its mechanisms are less understood in the context of choriocarcinoma.
In many cancer types, cellular invasion and EMT have been linked to the over-expression of embryonic stem cell-associated genes (3;4). Moreover, this stem cell-like gene expression profile is associated with metastasis and poor prognosis. One possible mediator of this invasive cancer signature is Nodal (5;6). Nodal is a member of the transforming growth factor-beta (TGF-β) superfamily and a morphogen during early embryonic patterning. Nodal plays an important role in promoting invasive events during primitive streak formation and mammary gland development (7;8). It has been postulated that the role that Nodal plays in mediating normal invasive events may similarly manifest during cancer progression.
Indeed, recent studies have shown that Nodal promotes cellular invasion and tumorigenicity in melanoma, prostate cancer, endometrial cancer, glioma, pancreatic cancer and hepatocellular carcinoma (9–17). There is also evidence that Nodal and its receptors are present in invasive human placental choriocarcinoma cell lines and breast cancer cell lines (7;13;18–21). Furthermore, a recent study reported that Nodal is positively correlated with disease progression in breast cancer patients, such that it is expressed to a higher level in undifferentiated, invasive lesions as compared to benign and early stage disease (20). Together these findings provide evidence that Nodal is correlated with invasive breast cancer and choriocarcinoma cell types; however, the significance of this correlation has not been fully characterized.
Nodal signals through the activin-like kinase type I (ALK4/7) and type II (ActRIIB) receptor complex, and its signal is enhanced by EGF-CFC family GPI-linked Cripto co-receptor. Activation of this receptor complex causes phosphorylation of SMAD2/3, which then associates with SMAD4. This SMAD2/3/4 complex translocates to the nucleus to regulate transcription of target genes, including NODAL and its inhibitors, LEFTY1/2 (22;23). LEFTY1/2 is limited to embryonic contexts and is not expressed in most cancer cells, resulting in uncontrolled positive feedback during cancer progression (13).
Non-SMAD pathways activated by Nodal in cancer have been poorly investigated; however, non-SMAD pathway activation in embryology has been reported. For instance, Nodal-induced anterior visceral endoderm (AVE) specification during embryonic patterning is dependent on phosphorylation of p38 (24). Furthermore, phospho-p38 amplifies Nodal signaling during this process, through phosphorylation of the SMAD2 linker region leading to increased SMAD2 activation (24). In cancer, non-SMAD pathway activation by other TGF-β-family proteins is better characterized, revealing possibilities for non-SMAD Nodal targets during disease progression. For instance, the type I receptor has been shown to activate MAPK signaling through ShcA phosphorylation and subsequent interaction with the GRB2/SOS complex in response to TGF-β signaling (25). In fact, both SMAD and ERK signaling are required for TGF-β-induced EMT in keratinocytes (26). Cross-talk between these two pathways has been shown, whereby ERK substrates interact with SMADs to regulate nuclear translocation and gene expression (26). ERK1/2 phosphorylation also promotes trophoblast and choriocarcinoma cell invasion (27). Although Nodal and TGF-β share many signaling commonalities, it is unknown whether Nodal is capable of inducing non-SMAD pathways, like MAPKs, in cancer.
Accordingly, the current study investigates the role of Nodal in cancer cell invasion. We have chosen to use breast cancer and choriocarcinoma cells as models because (i) they both arise from organs where Nodal is present during remodeling events (i.e. the breast and the placenta); (ii) Nodal is expressed to a greater degree in invasive breast cancer and choriocarcinoma cell lines, as compared to their poorly invasive counterparts; and (iii) both cell types undergo EMT, thereby allowing us to explore the effects of Nodal on this phenomenon irrespective of cellular origin. Using this approach, we demonstrate that Nodal promotes cellular invasion and migration, concomitant with an EMT-like phenotype. Furthermore, we show that these Nodal-induced phenomena are mediated in part through ERK1/2 signaling. In vivo, we demonstrate that inducible Nodal inhibition causes a reduction in spontaneous metastasis of breast cancer cells to the liver in NOD/SCID/interleukin-2γ receptor null mice (NSG mice). Our study lends insight into prospective Nodal-targeted therapies for the clinical management of cancer progression.
RESULTS
Nodal promotes migration and invasion in vitro
Cellular invasion is a complex process that involves active migration and degradation of the extracellular matrix (ECM). We first examined the role of Nodal signaling in the regulation of cellular migration by performing in vitro Transwell chamber assays using breast cancer and choriocarcinoma cell lines. In agreement with previous findings (9;28), we confirmed that rhNodal could induce SMAD2 phosphorylation in T47D cells (Fig. 1A). We also validated that transfection of BeWo cells with a Nodal expression construct (BeWo+Nodal) resulted in elevated Nodal expression compared to controls (BeWo+EV) (Fig. 1B). When we performed migration assays through Transwell chambers, we found that Nodal promoted migration of T47D cells in a dose-dependent manner (n=4, p<0.05) (Fig. 1C). We also found that over-expression of Nodal in BeWo cells caused an increase in migration (n=6, p=0.002) (Fig. 1D), and that treatment of MCF-7 cells with 50–100 ng/mL rhNodal caused an increase in migration (n=4, p<0.05) (Supp. Fig. 1A) using Transwell chambers.
Figure 1. Nodal promotes invasion and migration in breast cancer and choriocarcinoma cell lines.
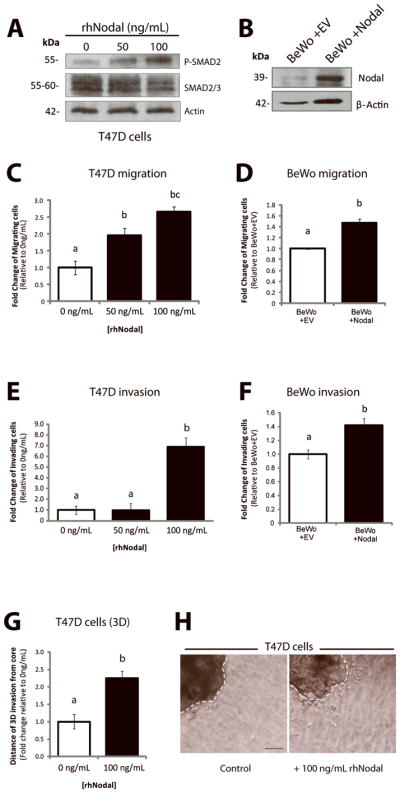
(A) Western blot validating increased P-SMAD2 in response to treatment with rhNodal in T47D cells. Total SMAD2/3 and β-Actin are used as controls. (B) Western blot validating increased Nodal expression in BeWo cells following transfection with a control versus Nodal-expression construct. The pro-Nodal (~39 kDa) band is presented and β-Actin is used as a control. (C) T47D cells were seeded in Transwell chambers and treated with 0, 50 or 100 ng/mL of rhNodal for 24 hours to assess cellular migration. Cells exhibited a significant dose-dependent up-regulation of cellular migration in response to rhNodal (n=4, p<0.05). (D) BeWo cells overexpressing Nodal (BeWo+Nodal) versus a control vector (BeWo+EV) were seeded in Transwell chambers to assess cellular migration after 24 hours. BeWo+Nodal cells exhibited elevated cellular migration compared to BeWo+EV cells (n=6, p=0.002). (E) T47D cells were seeded in Matrigel-coated Transwell chambers and treated with 0, 50 or 100 ng/mL of rhNodal for 24 hours to assess cellular invasion. Cells exhibited a significant up-regulation of cellular invasion at 100 ng/mL rhNodal (n=3, p=0.0038). (F) BeWo+Nodal cells or BeWo+EV cells were seeded in Transwell chambers coated with Matrigel to assess cellular invasion after 24 hours. BeWo+Nodal cells exhibited elevated cellular invasion compared to BeWo+EV cells (n=3, p=0.022). (G) T47D cells were grown for 3 days in 3D culture using a bioreactor, and then seeded into collagen type 1 in order to assess 3D invasion over the course of 1 week. Cells treated with 100 ng/mL displayed a significant increase in 3D cellular invasion (measured by the distance cells invaded away from the edge of the cluster) compared to controls (n=10, p=0.032). (H) Representative image showing in vitro 3D collagen invasion assay with T47D cells as described in (G). Bar equals 50 μm and hatched lines delineate the cluster boarder. For all graphs, data are presented as mean ± S.E.M for replicate values. Different letters indicate a significant difference.
In order to invade and metastasize, a cancer cell must be able to breach the basement membrane of the host tissue, and invade through the ECM. To test the effects of Nodal on cellular invasion (including ECM degradation), we used Transwell chambers coated with Matrigel, which is a protein mixture used to mimic extracellular matrix and basement membrane. Using this methodology, we found that Nodal promoted invasion of both T47D (n=3, p=0.004) and BeWo cells (n=3, p=0.022) (Fig. 1E, F). In accordance with these results, MCF-7 cells displayed a significant increase in cellular invasion through Matrigel-coated Transwell chambers in response to 100 ng/mL rhNodal compared to 0 ng/mL rhNodal (n=3, p=0.004) (Supp. Fig. 1B). Using a 3D invasion assay (29), we also found that T47D cells treated with rhNodal invaded through collagen, away from the cluster edge, more readily than control cells (n=10, p=0.032) (Fig. 1G, H).
Nodal promotes EMT-like phenomena
One of the mechanisms underlying cellular invasion in both normal and cancer contexts is EMT. EMT is characterized by a down-regulation of epithelial cell markers, such as E-Cadherin (CDH1), and an acquisition of mesenchymal cell markers, such as Vimentin (VIM), Twist (TWIST1) and N-Cadherin (CDH2) (30). In breast cancers, EMT is also associated with a reduction in Estrogen Receptor (ESR1) expression (31;32). Collectively, this phenotype allows cells to break away from the primary tumor and to invade toward secondary sites. Given that Nodal plays a role in promoting morphogenesis in early development, we opted to determine whether Nodal promotes EMT in epithelial-like breast cancer and choriocarcinoma cell lines. Using real-time RT-PCR, we found that transfection of T47D cells with a Nodal expression construct (T47D+Nodal) resulted in a significant down-regulation of ESR1 (n=3, p=0.030), and an up-regulation of the transcription factor TWIST1 (n=3, p=0.036) and the intermediate filament VIM (n=3, p=0.018) compared to T47D cells transfected with a control vector (T47D+EV) (Fig. 2A). These results were confirmed at the protein level with Western blotting (Fig. 2B). Similarly, treatment of T47D and MCF-7 cells with 100 ng/mL rhNodal caused a down-regulation ESR1 (T47D: n=4, p=0.029; MCF-7: n=5, p=0.008), and an up-regulation of TWIST1 (T47D: n=4, p=0.029; MCF-7: n=3, p=0.036) and VIM (T47D: n=4, p=0.029; MCF-7: n=5, p=0.008) compared to 0 ng/mL rhNodal vehicle controls (Supp. Fig. 2A, B). Of note, the effects observed in T47D cells with over-expression of Nodal were greater than those observed with rhNodal, likely due to differences in the length and consistency of exposure. BeWo+Nodal cells displayed a significant decrease in CDH1 expression (n=4, p<0.001), and an increase in both CDH2 (n=4, p<0.001) and VIM (n=3, p<0.001) expression compared to BeWo+EV cells (Fig. 2C). These results were confirmed at the level of protein expression using Western blot analysis (Fig. 2D).
Figure 2. Nodal promotes EMT-like phenotypes in breast cancer and choriocarcinoma cell lines.

(A) Real time RT-PCR analysis of EMT markers in T47D+EV cells versus T47D+Nodal cells. T47D+Nodal cells displayed a decrease in ESR1 expression (n=3, p=0.030), and an increase in TWIST1 (n=3, p=0.036) and VIM expression (n=3, p=0.018) compared to T47D+EV cells. CDH2 and CDH1 expression did not change (n=3, p>0.05). (B) Western blot analysis validating changes presented in (A). β-Actin is used as a loading control. (C) Real time RT-PCR analysis of EMT markers in BeWo cells transfected with a Nodal expression vector (BeWo+Nodal) versus a control vector (BeWo+EV). BeWo+Nodal cells displayed a decrease in CDH1 expression (n=4, p<0.001), and an increase in CDH2 (n=4, p<0.001) and VIM expression (n=3, p<0.001) compared to BeWo+EV cells. TWIST1 expression did not change (n=4, p>0.05). (D) Western blot analysis validating changes presented in (C). β-Actin is used as a loading control. (E) Immunofluorescence (IF) showing localization of E-Cadherin (green) in BeWo cells or (F) T47D cells transfected with a Nodal expression vector versus a control empty vector. Nuclei are stained with DAPI (blue) and bars equal 20 μm. All IF was performed ≥3 times. Data in A and C are presented as mean ± S.E.M. for replicate values. Asterisks (*) indicate a significant difference compared to control cells as specified. Expression levels are normalized to HPRT1.
In many instances, EMT is marked by changes in subcellular localization of E-cadherin protein, rather than by changes in transcript or protein production. This level of cadherin regulation is important during developmental EMT processes, such as gastrulation, that require rapid and transient alterations in cell-cell adhesions to allow for tissue remodelling (33). Accordingly, using immunofluorescence we found that over-expression of Nodal in BeWo or T47D cells caused cytoplasmic localization of E-Cadherin (even in BeWo+Nodal cells, which also express reduced E-Cadherin at the level of gene and protein expression) compared to the strong plasma membrane staining observed in corresponding control cells (Fig. 2E, F). The same effect was observed in T47D or MCF-7 cells treated with increasing concentrations of rhNodal for 48 hrs (Supp. Fig. 2C, D).
Nodal activates non-SMAD pathways to mediate invasive phenotypes
Given the emerging evidence that implicates Nodal during cancer progression, improving our understanding of both canonical and non-canonical Nodal signaling mechanisms will be imperative for developing future targeted therapies. As such, we decided to investigate whether Nodal signaling can activate the ERK pathway, as this pathway regulates cellular invasion, proliferation, migration, and differentiation, during both cancer progression and placentation. We first treated T47D cells with rhNodal (100 ng/mL) for 0, 1, 2, 5, 10, or 20 min, and found that Nodal increases P-ERK1/2 in as little as 2 min when replicates were analysed by densitometry (n=5, p<0.05) (Fig. 3A, B). Similarly, we treated BeWo cells with rhNodal (100 ng/mL) for 0, 1, 2, 5, 10 or 20 min, and found that Nodal significantly increases P-ERK1/2 by 5 min when replicates were analysed by densitometry (n=5, p<0.05) (Fig. 3C, D). In order to determine whether phosphorylation of ERK was dependent on activation of the type I receptor (similar to what has been reported in the literature for TGF-β), we examined the effects of an ALK4/7 inhibitor (SB431542; 10 μM, 1 hr) on Nodal-induced ERK1/2 phosphorylation in T47D and BeWo cells. We found that in both cell lines, SB431542 reduced Nodal-induced P-ERK1/2 (Fig. 3E, F). This was confirmed with densitometry (Supp. Fig. 3A, B). Of note, SB431542 selectively inhibits Activin, TGF-β and Nodal signaling, but not ERK (34). Hence, although our results suggest that ALK4/7 activation is required for Nodal-mediated ERK activation, we cannot exclude the possibility that indirect mechanisms (such as an up-regulation of Activin or TGF-β) are also involved. In order to determine if ERK activity may reciprocally regulate SMAD signaling, we treated T47D+Nodal and BeWo+Nodal cells with a MEK inhibitor (U0126; 10 μM, 1 hr). We determined that U0126 decreased Nodal-induced P-SMAD2 in both cases (Fig. 3G, H). Of note, using RT-PCR we found that over-expressing Nodal reduced Cripto mRNA levels in both cell lines. ALK4 and ALK7 levels were not altered in T47D cells, and over-expression of Nodal in BeWo cells resulted in an increase in ALK4, but a decrease in ALK7 (Supp. Fig. 4): Hence, in our system Nodal does not seem to mediate its effects by globally increasing receptor expression levels. Regardless, our results suggest that through the ALK4/7 receptor, Nodal induces the activation of the ERK1/2 MAPK pathway in breast cancer and choriocarcinoma cells. Moreover, Nodal-induced SMAD2 phosphorylation is in part mediated by activation of ERK1/2 MAPK.
Figure 3. Nodal activates ERK signaling.

(A) Western blot time course analysis of ERK1/2 activation in T47D cells following treatment with 100 ng/mL rhNodal for 0, 1, 2, 5, 10, or 20 minutes. P-ERK1/2 increases compared to controls after 2 minutes of rhNodal treatment. Total ERK1/2 and β-Actin are used as controls. (B) Densitometric analysis for all replicate experiments corresponding to (A). ImageJ was used to calculate band density of P-ERK1/2 relative to total ERK1/2. Data are presented as mean fold change ± S.E.M. Different letters indicate a significant difference compared to controls (n=4, p=0.029). (C) Western blot time course analysis of ERK1/2 activation in BeWo cells following treatment with 100 ng/mL rhNodal for 0, 1, 2, 5, 10, or 20 minutes. P-ERK1/2 increases compared to controls after 5 minutes of rhNodal treatment. Total ERK1/2 and β-Actin are used as controls. (D) Densitometric analysis for all replicate experiments corresponding to (C). ImageJ was used to calculate band density of P-ERK1/2 relative to total ERK1/2. Data are presented as mean fold change ± S.E.M. Different letters indicate a significant difference compared to controls (n=4, p=0.029). (E) Western blot demonstrating that P-ERK1/2 is elevated in T47D cells transfected with a Nodal expression vector (T47D+Nodal) versus a control vector (T47D+EV), and that phosphorylation is reduced when T47D+Nodal cells are treated with SB431542 (10 μM). Total ERK1/2 and β-Actin are used as controls. (F) Western blot demonstrating that P-ERK1/2 is elevated in BeWo cells transfected with a Nodal expression vector (BeWo+Nodal) versus a control vector (BeWo+EV), and that phosphorylation is reduced when BeWo+Nodal cells are treated with SB431542 (10 μM). Total ERK1/2 and β-Actin are used as controls. (G) Western blot demonstrating that P-SMAD2 is elevated in T47D+Nodal cells compared to T47D+EV cells, and that this effect is abrogated by treating T47D+Nodal cells with 10 μM U0126 (1 hr). Total SMAD2/3 and β-Actin are used as controls. (H) Western blot demonstrating that P-SMAD2 is elevated in BeWo+Nodal cells compared to BeWo+EV cells, and that this effect is abrogated by treating BeWo+Nodal cells with 10 μM U0126 (1 hr). Total SMAD2/3 and β-Actin are used as controls.
We next wanted to determine the functional significance of Nodal-induced ERK activation. We seeded T47D or BeWo cells into Transwell chambers with Matrigel (for invasion) or without Matrigel (for migration), and treated them with rhNodal alone (100 ng/mL), or with both rhNodal (100 ng/mL) and U0126 (10 μM). We found that both invasion (T47D: n=3, p=0.017; BeWo: n=3, p=0.022) and migration (T47D: n=3, p=0.010; BeWo: n=3, p=0.005) increased significantly in response to treatment with rhNodal compared to vehicle controls, and this effect was mitigated by addition of U0126 (Fig. 4A–D). Importantly, we observed no significant differences in the number of viable cells between treatments for the duration of the invasion and migration assays (24 hrs) (Fig. 4E, F). In accordance with these results, MCF-7 cells showed similar phenomena in response to rhNodal and U0126 treatments (n=3, p=0.005 for both migration and invasion) (Supp. Fig. 5A–C). Finally, using a 3D invasion assay (29) we found that treatment of T47D-derived clusters with rhNodal caused a significant increase in invasion distance compared to controls, and that this effect was prevented with U0126 (n=14, p<0.001) (Fig. 4G, H).
Figure 4. Nodal-induced cellular invasion and migration are mediated by ERK.

(A) T47D and (B) BeWo cellular invasion through a Matrigel-coated Transwell chamber increased in response to 100 ng/mL rhNodal (n=3, p≤0.022), and this effect was mitigated by 10 μM U0126. (C) T47D and (D) BeWo cellular migration through a Transwell chamber (without Matrigel) increased in response to 100 ng/mL rhNodal (n=3, p≤0.010), and this effect was mitigated by 10 μM U0126. (E) T47D viability assay, and (F) BeWo viability assay demonstrating equal cell viability after 24 hours between treatments corresponding to (A–D). (G) T47D cells grown for 3 days in 3D culture using a bioreactor, were seeded into collagen type 1 in order to assess 3D invasion over the course of 1 week. Distance of invasion was recorded as an average of 4 measurements evenly spaced around the cluster periphery (white hatched lines). Cells treated with 100 ng/mL rhNodal displayed a significant increase in 3D cellular invasion compared to controls (n=14, p<0.001), and this effect was prevented by 10 μM U0126. (H) Representative images from 3D invasion assay corresponding to data presented in (G). Bar equals 50 μm. All data are presented as mean ± S.E.M. for replicate values. Different letters indicate a significant difference as specified.
As a corollary, we determined whether ERK-activation was involved in Nodal-mediated EMT. First, we added U0126 (10 μM) to T47D or MCF-7 cells prior to treatment with rhNodal (100 ng/mL) and found that U0126 prevented Nodal-mediated induction of TWIST1 and VIM expression as quantified by real time RT-PCR, and Western blotting (Fig. 5A, B; Supp. Fig. 6A). Interestingly, ESR1 expression was still downregulated by rhNodal in the presence of U0126 (Fig. 5A, B; Supp. Fig. 6A). Additionally, using immunofluorescence, we determined that addition of U0126 to T47D or MCF-7 cells treated with rhNodal caused re-localization of E-Cadherin back to the plasma membrane (Fig. 5C; Supp. Fig. 6B). Similar to our observations in breast cancer cell lines, addition of U0126 to BeWo+Nodal cells resulted in a decrease of Nodal- induced CDH2 and VIM expression, and an increase of CDH1 expression back to control levels as measured by real time RT-PCR and Western blotting (Fig. 5D, E). Furthermore, using immunofluorescent staining and confocal analysis, we found that treatment of BeWo+Nodal cells with U0126 (10 μM) caused re-localization of E-Cadherin back to the cell membrane (Fig. 5F).
Figure 5. Nodal-induced EMT is mediated by ERK.

(A) Real time RT-PCR analysis of EMT markers in T47D cells exposed to 100 ng/mL rhNodal alone, or 100 ng/mL rhNodal + 10 μM U0126 (48 hrs). In response to 100 ng/mL of Nodal, T47D cells displayed a decrease in ESR1 (Estrogen Receptor) expression (n=4, p=0.029), and an increase in TWIST1 (n=4, p=0.029) and VIM (Vimentin) expression (n=4, p=0.029) compared to controls. CDH2 (N-Cadherin) and CDH1 (E-Cadherin) expression did not change (n=4, p>0.05). Treatment of T47D cells with U0126 prior to treatment with rhNodal rescued TWIST1 and VIM expression back to control levels, but did not rescue ESR1 Expression. (B) Western blot validating changes presented in (A). β-Actin is used as a loading control. (C) Immunofluorescence (IF) showing localization of E-Cadherin (green) in T47D cells treated with 100 ng/mL rhNodal, or 100 ng/mL rhNodal + 10 μM U0126. Nuclei are stained with DAPI (blue) and bars equal 20 μm. (D) Real time RT-PCR analysis of EMT markers in BeWo cells transfected with a Nodal expression vector (BeWo+Nodal) versus a control vector (BeWo+EV), or in BeWo+Nodal cells treated with 10 μM U0126 (48 hrs). BeWo cells expressing Nodal displayed a decrease in CDH1 expression (n=4, p<0.001), and an increase in CDH2 (n=4, p<0.001) and VIM expression (n=3, p<0.001) compared to BeWo+EV cells. Treatment of BeWo+Nodal cells with U0126 rescued CDH2 and VIM expression back to BeWo+EV levels, and significantly reverted CDH1 expression to near-control levels (n=4, p=0.029). TWIST1 expression did not change with any of the treatments. (E) Western blot analysis corresponding to changes presented in (D). β-Actin is used as a loading control. (F) IF showing localization of E-Cadherin (green) in BeWo cells transfected with a Nodal expression construct (BeWo+Nodal) or an empty vector control (BeWo+EV). BeWo+Nodal cells were also treated with U0126 (10 μM) to determine the effects of ERK signaling on Nodal-induced localization of E-Cadherin. Nuclei are stained with DAPI (blue) and bars equal 20 μm. Data are presented as mean ± S.E.M. for replicate values. Different letters indicate a significant difference compared to control treatments/genes as specified. Gene expression levels (measured by PCR) are normalized to HPRT1. All IF was performed 3–4 times.
Nodal inhibition impairs invasion in vitro in part through decreased P-ERK
In a complementary set of loss-of-function experiments, we wanted to determine if Nodal inhibition could decrease the invasiveness of more aggressive breast cancer cells, which express high levels of endogenous Nodal (13;21). Using a Transwell chamber assay with Matrigel (for invasion) or without Matrigel (for migration), we found that knocking down Nodal expression in MDA-MB-231 cells with shRNA (231+shNodal) resulted in a significant decrease in both migration (n=5, p<0.001) and invasion (n=5, p=0.016) compared to 231+shControl cells (Fig. 6A–C). Furthermore, using real time RT-PCR, we determined that 231+shNodal cells displayed reduced expression of mesenchymal markers including TWIST1 (n=3, p<0.001), CDH2 (n=3, p<0.001), and VIM (n=3, p=0.031) compared to 231+shControl cells (Fig. 6D). Western blotting validated the reduction in these mesenchymal markers at the protein level (Fig. 6E). No increase in epithelial marker expression (including CDH1 and ESR1) was observed in these cells in response to Nodal knockdown. In accordance with these results, when MDA-MB-231 cells were treated with SB431542 (0, 1, or 10 μM), a reduction in cellular invasion (n=5, p<0.05) and migration (n=6, p<0.001) was observed (Fig. 6F–H). A significant decrease in cellular invasion (n=5, p<0.05) and migration (n=5, p<0.001) was also observed in Hs578t cells treated with 1–10 μM SB431542 (Supp. Fig. 7A, B). Using real time RT-PCR, we determined that MDA-MB-231 cells treated with 10 μM SB431542 displayed reduced expression of TWIST1 (n=3, p<0.001), CDH2 (n=3, p<0.001), and VIM (n=3, p=0.0011) compared to vehicle controls (Fig. 6I). These observations were validated at the protein level by Western blotting (Fig. 6J). Similar to our shRNA results, no changes in gene expression were detected for epithelial cell markers, including CDH1 and ESR1, in response to SB431542 (Fig. 6I).
Figure 6. Nodal inhibition impairs invasion and migration in vitro.

(A) Western blot validating decreased Nodal expression in MDA-MB-231 cells transfected with a Nodal-targeted shRNA (231+shNodal) versus a scramble control shRNA (231+shControl). The pro-Nodal (~39 kDa) band is presented and β-Actin is used as a control. (B) Cellular migration and (C) invasion through Matrigel of 231+shControl versus 231+shNodal cells was quantified after 24 hrs. Cellular migration and invasion were significantly reduced in response to Nodal knockdown (n=5, p<0.02). (D) Real time RT-PCR analysis of EMT markers in MDA-MB-231 cells stably transfected with a Nodal-targeted shRNA (231+shNodal) or a scrambled Control shRNA (231+shControl). 231+Nodal cells displayed reduced expression of TWIST1 (n=3, p<0.001), CDH2 (n=3, p<0.001), and VIM (n=3, p=0.031). CDH1 and ESR1 were not significantly different between 231+shNodal cells and 231+shControl cells. (E) Western blot validating changes presented in (D). β-Actin is used as a loading control. (F) Western blot validating that treatment of MDA-MB-231 cells with SB431542 (0, 1, or 10 μM) causes a dose-dependent decrease in SMAD2 phosphorylation. SMAD2/3 and β-Actin are used as loading controls. (G) Cellular migration and (H) invasion through Matrigel of MDA-MB-231 cells treated with 0, 1 or 10 μM of SB431542. Cellular migration (n=5, p<0.001) and invasion (n=5, p=0.016) were significantly reduced in response to SB431542. (I) Real time RT-PCR analysis of EMT markers in MDA-MB-231 cells treated with SB431542 (0 or 10 μM, 48 hrs). In response to SB431542, MDA-MB-231 cells displayed reduced expression of TWIST1 (n=3, p<0.001), CDH2 (n=3, p<0.001), and VIM (n=3, p=0.001). CDH1 and ESR1 did not change in response to SB431542. (J) Western blot validating changes presented in (I). β-Actin is used as a loading control. Gene expression levels (measured by PCR) are normalized to HPRT1. All parametric data are presented as mean ± S.E.M for replicate values, and non-parametric data are presented as median ± interquartile range. Different letters indicate a significant difference compared to controls (open bars), and asterisks indicate a significant difference compared to controls (normalized gene expression).
We next investigated whether Nodal inhibition in MDA-MB-231 cells could alter phosphorylation of ERK to mediate the observed changes in cellular invasion and migration. We confirmed via Western blotting that 231+shNodal cells displayed reduced P-ERK1/2 compared to 231+shControl cells, and that addition of rhNodal (100 ng/mL) to 231+shNodal cells rescued P-ERK1/2 (Fig. 7A). In accordance with these results, we found that rhNodal (100 ng/mL) could rescue cellular invasion and migration in 231+shNodal cells to levels observed for 231+shControl cells; however, this rescue effect was mitigated when downstream ERK1/2 activation was prevented with U0126 (10 μM) (Fig. 7B–E). Furthermore, we found that inhibition of ERK1/2 phosphorylation in 231+shControl cells with U0126 resulted in a significant decrease in cellular invasion (n=3, p<0.001) and migration (n=3, p=0.002), and that these effects of U0126 were not rescued by the addition of rhNodal (Fig. 7B–E). The number of viable cells did not differ between treatments over the course of the assay (24 hours) (Fig. 7C). Of note, using RT-PCR we found that knocking down Nodal resulted in a modest increase in Cripto mRNA levels. There was no change observed in ALK4 levels and there was a small reduction in ALK7 (Supp. Fig. 4). Hence, in our system, Nodal knockdown does not seem to mediate its effects by globally decreasing receptor expression levels.
Figure 7. Nodal inhibition impairs invasion in vitro in part through decreased P-ERK1/2.

(A) Western blot analysis of MDA-MB-231 cells transfected with a Control (231+shControl) or a Nodal-targeted shRNA (231+shNodal) showing that phosphorylation of ERK1/2 decreases when Nodal is reduced. Addition of recombinant human Nodal (rhNodal; 100 ng/mL) restores ERK1/2 phosphorylation in 231+shNodal cells. Total ERK1/2 and β-Actin are used as controls. (B) Western blot analysis validating that U0126 reduces phosphorylation of ERK1/2 in parental MDA-MB-231 cells. Total ERK1/2 and β-Actin are used as controls. (C) LIVE/DEAD® Cytotoxicity/Viability assays of MDA-MB-231 cells in response to treatments and transfection conditions corresponding to D and E. Viability was constant across all MDA-MB-231 treatment conditions after 24 hours (n=8). (D) Cellular invasion (through Matrigel) and (E) cellular migration (no Matrigel) of 231+shControl or 231+shNodal cells through a Transwell chamber in the presence or absence of rhNodal (100 ng/ml) ± U0126 (10 μM) (24 hrs). Cellular invasion and migration were significantly reduced in 231+shControl cells treated with U0126, and this was not rescued with rhNodal. Cellular invasion and migration were significantly decreased in 231+shNodal cells as compared to 231+shControl cells, and the inclusion of rhNodal rescued invasion and migration in 231+shNodal cells to control levels. This ability of rhNodal to rescue the effects of shNodal on invasion and migration was mitigated by U0126 (n=3, different letters are significantly different, p<0.001). All parametric data are presented as mean ± S.E.M for replicate values.
Nodal inhibition reduces spontaneous metastasis of breast cancer to the liver
As a corollary to our in vitro assays, we performed a spontaneous metastasis assay using MDA-MB-231 breast cancer cells. MDA-MB-231 cells were transfected with a Nodal-targeted doxycyclin (Dox)-inducible shRNA construct (231+ishN), or a control Dox-inducible shRNA construct (231+ishC) (Fig. 8A, B), and injected into the mammary fat pads of 7–8 week old NSG mice. Dox was administered in mouse chow one week after injections. Tumors were palpable (>2 mm diameter) at this time. Once tumors reached ~1.3 cm in diameter, which took approximately 5 weeks, mice were sacrificed, and lungs and livers were collected.
Figure 8. Nodal knockdown reduces spontaneous metastasis to the liver in NOD/SCID/IL-2γ receptor null mice.

500,000 231+ishC or 231+ishN cells were injected into the right thoracic mammary fat pad of NSG mice. Doxycyclin was administered 1 week following injection. Mice were sacrificed when tumors reached 1.0–1.5 cm in diameter and lung and livers were analysed macrosopically or using H&E stained sections. (A) Western blot analysis demonstrating that MDA-MB-231 cells transfected with a Nodal-targeted doxycyclin (Dox)-inducible shRNA (231+ishN) express lower levels of Nodal protein upon administration of Dox compared to 231+ishN cells −Dox, and compared to MDA-MB-231 cells transfected with a scrambled control Dox-inducible shRNA (231+ishC) +/− Dox. (B) Immunohistochemical staining (brown) of Nodal in 231+ishC and 231+ishN derived tumors confirming that Nodal was knocked down in 231+ishN + Dox tumors as compared to all other groups. Bars equal 50 μm. (C) Images of lung sections stained with H&E across treatment groups. Bar equals 1 mm. (D) Quantification of tumor burden in lungs show no significant differences between treatment groups (n=10). Each point represents the average tumor burden for one animal and the filled diamond indicates the median value. (E) Bright field images of livers (top panel) or of liver sections stained with H&E (bottom panel) taken from mice injected with 231+ishC or 231+ishN cells and fed either normal or Dox-containing chow. Metastases have been outlined by a white dashed line and bar equals 1 mm. (F) Quantification of tumor burden in livers show a significant decrease in metastasis in 231+ishN +Dox mice compared to all other groups (n=10, p<0.05). Each point represents the average tumor burden for one animal and the filled diamond indicates the median value. For all graphs, different letters indicate a significant difference. (B) Immunohistochemical staining (brown) of Nodal in primary tumor, lung and liver lesions from mouse injected with 231+ishN and treated with Dox. Note that Nodal expression is regained in the liver lesion. Bars equal 50 μm.
Upon examination of lung tissue stained with H&E, we saw no significant difference in the percentage of tumor burden between treatment groups (Fig. 8C, D). Lung tumor burden ranged from 10%–95%, indicating that in many cases, lung tissue was not over-saturated with tumor. Although we found no difference in tumor burden in lung tissue, we saw robust macroscopic differences in liver metastases upon sacrificing the mice, whereby 231+ishN +Dox groups had macroscopically observable lesions in only 1/10 mice (compared to 7/11 for 231+ishC −Dox, 6/10 for 231+ishC +Dox, and 11/15 for 231+ishN −Dox) (Fig. 8E). Accordingly, analysis of H&E stained tissue sections revealed a significant decrease in extent of liver metastases in 231+ishN +Dox mice compared to all other treatment groups (Fig. 8F). Of note, we determined that Nodal was knocked down in the primary tumor and in lung metastases in 231+ishN +Dox mice; however, Nodal was expressed in the few lesions that emerged in the livers of these mice, suggesting that liver metastases originated only from cells that escaped the effects of the Nodal shRNA (Fig. 8G).
DISCUSSION
Here we show that Nodal promotes invasive phenotypes in breast cancer and choriocarcinoma cell lines, concomitant with the induction of EMT-associated events. These observations are in accordance with others that have found a link between Nodal and glioma or melanoma invasion (10;35), and with recent studies showing that Nodal is highly expressed in invasive breast cancer lesions (7;20;21). In contrast, Peng and colleagues have shown that Nodal decreases the invasiveness of trophoblast cells, which are precursors to choriocarcinoma cells (19); however, that study exposed cells to rNodal levels that greatly exceeded those used here (500 versus 100 ng/mL), and focused on non-tumorigenic cells. Hence, Nodal may differentially affect cellular invasion in a concentration or transformation-dependent manner.
We found that Nodal supports cellular invasion through altered expression of epithelial and mesenchymal cell markers in poorly-invasive, well-differentiated breast cancer and choriocarcinoma cell lines. This adds Nodal to a growing list of TGF-β family members that have been shown to regulate EMT in normal epithelial cells and during cancer progression. For example, TGF-β promotes EMT in a variety of cell types including breast cancer cells, pulmonary epithelial cells and keratinocytes (26;36;37) and differentiation factor-9 (GDF-9) has been shown to induce EMT in prostate cancer (38). Interestingly, GDF is a member of the TGF-β family that dimerizes with Nodal in the embryonic node to mediate Left-Right asymmetry (39). It is possible that Nodal may similarly work with GDFs to promote EMT in both developmental and tumorigenic contexts.
We determined that Nodal promotes cellular invasion and EMT-like phenomena via the activation of the ERK1/2 MAPK pathway in both breast cancer and choriocarcinoma cells. Prior to this study, the effects of Nodal on cellular behavior during development and in cancer were largely attributable to the induction of SMAD signaling. However, many TGF-β superfamily proteins, which share similarities between type I and type II receptor components, employ non-SMAD pathways during signal propagation. Specifically, TGF-β promotes ERK signaling, through phosphorylation of ShcA and activation of the GRB2/SOS complex via its type I receptor (25). Moreover, TGF-β induces ERK and p38 signaling in human skin keratinocyte cells (HaCaT), ras-transfected H357 keratinocytes, and malignant Il-3 keratinocytes to induce EMT (26); and inhibition of MEK with U0126 reverses TGF-β-induced EMT in H357 and Il-3 cell lines (26). Cripto, Nodal’s co-receptor, can also signal through ERK (40), and Cripto over-expression is associated with EMT in breast cancer cells (41–43). Hence, ALK4/7 and Cripto may both mediate Nodal-induced MAPK activation, leading to EMT and invasion.
Our results suggest there are multiple points of crosstalk between the SMAD2/3 and ERK1/2 pathways. First, we demonstrate that rhNodal causes an up-regulation of P-ERK1/2. Based on reports of TGF-β-induced ERK activation, this may be an indirect effect via receptor phosphorylation of an upstream mediator of the ERK cascade (such as ShcA) (25), or a direct effect via receptor phosphorylation of ERK1/2. Second, we show that MEK inhibition leads to a decrease in Nodal-induced P-SMAD2 at Ser465/467, indicating that the ERK pathway supports canonical Nodal signaling. In addition to these canonical SMAD phosphorylation sites, previous studies have shown that the SMAD2/3 linker region is receptive to phosphorylation by MAPKs; however, phosphorylation at this region tends to have an inhibitory effect of SMAD signaling (24). Future studies should investigate whether phosphorylation at this linker region by activated ERK occurs in the context of Nodal signaling, and whether this putative phosphorylation event can have differential effects on Nodal-induced phenotypes. Third, we show that MEK inhibition reduces Nodal-induced TWIST1 and VIM. In agreement with previous findings, this suggests that ERK may alter SMAD-mediated gene expression, either by regulating nuclear translocation of SMADs, or by directly interfering with transcription (44). We also found that MEK inhibition rescued Nodal-induced intracellular localization of E-Cadherin. In accordance with these observations, previous reports have shown that inhibition of the ERK pathway is required for relocalization of E-Cadherin to the plasma membrane after internalization during EMT (45;46).
An important finding in our study is that Nodal inhibition in highly invasive cells can decrease invasion, and impair breast cancer metastasis to secondary organs. These results reiterate the effects observed in our gain-of-function models, and suggest that Nodal-induced phenotypes may be reversible in advanced disease. In our NSG mouse model, we observed that Nodal knockdown reduced metastases in the liver but not in the lung, suggesting that Nodal may differentially mediate organ-specific metastasis. Differential metastasis of breast cancer to lung versus liver has been only mildly reported in the literature; however, one well-characterized mediator of this phenomenon is the tight junctional protein Claudin-2. Claudin-2 is required for the initiation and growth of breast cancer liver metastases; however, cell lines generated from aggressive breast cancer liver metastases with elevated Claudin-2 expression show decreased spontaneous metastasis to the lung (47). It is possible that differential expression of Claudin-2 may in part explain the effects of Nodal on lung versus liver metastases. The results seen in our experiments could also be due to organ-specific differences in endogenous Nodal inhibitors (ex. Lefty), such that liver metastases may require higher levels of Nodal to overcome an increased presence of inhibitor. In this situation, a partial knockdown of Nodal may leave enough ligand to sustain growth in the lung, but not in the liver. In support of this possibility, we found that liver metastases in 231+shN mice treated with Dox expressed high levels of Nodal, whilst the lung and primary lesions in the same animal showed very little expression. This suggests that metastatic growth in the liver requires higher Nodal levels than are needed for growth in the lung. Future studies shall explore the role Nodal in organ-specific metastasis, as these types of studies lend insight into how to manage disease in a patient-specific manner.
In conclusion, our results demonstrate that Nodal promotes invasive phenotypes in both breast cancer and choriocarcinoma cell lines. We found that Nodal alters the expression of epithelial and mesenchymal cell markers, and that Nodal can activate non-SMAD pathways, specifically ERK, to elicit its effects. Importantly, we show that Nodal inhibition in highly invasive cells can reduce invasion and prevent breast cancer metastasis to the liver. Our findings lend insight into recent studies that implicate Nodal in the progression of various types of cancer, and provide multiple possible targets to which clinical intervention might be effective.
METHODS
Cell lines and treatments
BeWo human choriocarcinoma cells, and MDA-MB-231, Hs578t, T47D and MCF-7 breast cancer cell lines and were obtained from and verified by the ATCC. Multiple constructs and reagents were used to knockdown or induce Nodal signaling:
Nodal gain-of-function
T47D and BeWo cells were transfected via Lipofectamine (Invitrogen) with a Nodal expression vector (abbreviated “+Nodal”), made with pcDNA™3.3-TOPO® cloning kit, or a control empty pcDNA3.3 vector (abbreviated “+EV”). H9 hESCs were used to clone the human Nodal open reading frame. Vectors were sequenced and validated. Cells were stably selected with G418. Cells were also subject to treatment with 10–100 ng/mL of recombinant human Nodal (rhNodal; R&D systems) to activate P-SMAD2 signaling as an alternative gain-of-function model.
Nodal loss-of-function
As previously described (21) MDA-MB-231 cells were transfected with a GIPZ lentiviral shRNAmir against Nodal’s 3rd exon (+shNodal; Clone: V2LHS_155453, Open Biosystems) or a scrambled shRNA Control (+shControl). As an additional loss-of-function model, MDA-MB-231 cells were transfected with a TRIPZ lentiviral shRNAmir against Nodal’s 3rd exon (+ishN; Clone: V2THS_155453) with Tet-On®/Tet-Off® inducibility (Open Biosystems) or a scrambled inducible shRNA Control (+ishC). All transfected MDA-MB-231 cells were stably selected with Puromycin. To inhibit Nodal signaling, the ALK4/5/7 inhibitor SB431542 (Sigma) was used at 1–10 μM. To inhibit MEK signaling, U0126 (Sigma) was used at 10 μM. In both cases carrier (DMSO) was used in the controls. Signaling studies were done for 0–20 min to consistently measure changes that occur directly in response to Nodal or for 60 min to establish the role of signaling molecules downstream of Nodal. Functional assays and measurement of EMT markers were done for longer periods (>24 hours) to allow for necessary transcriptional and translational alterations.
Cellular migration, invasion and viability
To assess cellular migration, 50,000 cells were seeded onto a Transwell insert, and incubated for 24 hours. Migration was quantified by staining nuclei with DAPI and manual counting. To assess cellular invasion, the migration protocol was modified by coating Transwells with Matrigel (1:10 Matrigel:Serum-free RPMI) prior to seeding cells. The 3D cell cluster invasion assay was performed as previously described (29). Briefly, one million T47D cells suspended in 1 mL of complete media (RPMI +10% FBS) were loaded into a bioreactor (Synthecon Rotary Cell Culture System™). After 3 days, clusters were selected and embedded in bovine collagen type 1 (Invitrogen). Collagen was allowed to solidify and was then overlaid with RPMI +10% FBS + vehicle control, 100 ng/mL rhNodal, or 100 ng/mL rhNodal +10 μM U0126. After 1 week, four measurements evenly spaced around each cluster periphery were averaged to generate a distance score for each cluster. Adobe Photoshop was used to measure distance (Analysis tool). To assess viability of cells in response to various treatments, the LIVE/DEAD® Viability/Cytotoxycity Kit was used as per manufacturer’s instructions (Invitrogen).
Immunofluorescence
Cells were seeded on glass coverslips, fixed with 4% paraformaldehyde, made permeable with 20 mM HEPES and 0.5% TritonX-100, and blocked with serum-free protein block (Dako, Carpinteria, CA). Cells were stained using anti-E-Cadherin antibody (Supplementary Table 1), and an Alexafluor secondary antibody (Invitrogen). DAPI was used to stain nuclei. IgG isotype controls were also included.
Real-time PCR
Total RNA was isolated using the Perfect Pure RNA cultured cell kit (5 PRIME). Reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Real-time PCR was performed using 2 μg cDNA with TaqMan® gene expression human primer/probe sets (Supplementary Table 2). All gene expression was normalized to HPRT1 or RPLPO.
Western blot analysis
Protein lysate was prepared using Mammalian Protein Extraction Reagent (Thermo Scientific), Halt™ Protease Inhibitor Cocktail (Thermo Scientific), and Phosphatase Inhibitor (Thermo Scientific). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis under reducing conditions, and transferred onto Immobilon-P membranes (Millipore). Membranes were incubated with primary antibody (Supplementary Table 1) and horseradish peroxidase-labelled secondary antibody. Bands were detected by Immun-Star™ Chemiluminescent Detection kit (Bio-Rad) or SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Images were obtained with ChemiDoc™ XRS+ System (Bio-Rad) or film. For Nodal Western blots, the 39 kDa species was used to assess protein expression as previously described (28). All densitometry was performed using ImageJ.
Spontaneous metastasis in vivo
MDA-MB-231 cells were transfected with a doxycyclin (Dox)-inducible Control or Nodal-targeted shRNA. 500,000 cells in 100 μL RPMI:Matrigel (1:1) were injected into the right thoracic mammary fat pad of 7–8 week old female NSG mice. Dox was administered in mouse chow (0.625 g Dox/1 kg chow; Harlan Laboratories) after formation of palpable tumor growth. Mice were sacrificed when tumors reached ~1.3 cm in diameter. Lung and liver were collected from each mouse, fixed with 4% formaldehyde, and stained with H&E or using immunohistochemistry for Nodal as previously described (20;21). The percent tumor burden for each animal was calculated by averaging the tumor area from 2–3 sections spaced evenly through the tissue, and dividing it by the average total square area (Adobe Photoshop). Experiments involving animals were approved by the Animal Use Subcommittee at the University of Western Ontario.
Statistical analyses
Parametric data: Statistical analyses of multiple comparisons were performed using a one-way ANOVA followed by a Tukey–Kramer Comparisons Post-Hoc test. Parametric data were expressed as mean ± S.E.M. for replicate values. Non-parametric data: An ANOVA on Ranks followed by the Mann-Whitney rank-sum test was used. Non-parametric data were expressed as median ± interquartile range. When only two items were compared, a student’s t-test was used. All statistical tests were two-sided, and data comparisons were considered statistically significant at p<0.05. Statistics were performed using SigmaStat (Dundas Software) in consultation with the biostatistical support unit at the University of Western Ontario.
Supplementary Material
Acknowledgments
GRANT SUPPORT: This work was supported by the Canadian Institutes for Health Research (MOP 89714, MOP 119589 and PLS 95381) and the Cancer Research Society to LMP. DQ is supported by fellowships from the Canadian Breast Cancer Foundation, and a CIHR training grant. LMP is the recipient of the Premier New Investigator Award from the CIHR. DAH is the recipient of a new investigator award and MacDonald Scholarship for the Canadian Heart and Stroke Foundation.
We thank LA Walsh and SD Findlay for making the Nodal expression construct, and M Jewer and A Cloutier-Bosworth for assisting with tissue scoring.
Footnotes
LMP holds a patent regarding targeting Nodal in cancer. All other authors have no conflicts of interest to declare.
References
- 1.Drasin DJ, Robin TP, Ford HL. Breast cancer epithelial-to-mesenchymal transition: examining the functional consequences of plasticity. Breast Cancer Res. 2011;13:226. doi: 10.1186/bcr3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vicovac L, Aplin JD. Epithelial-mesenchymal transition during trophoblast differentiation. Acta Anat (Basel) 1996;156:202–216. doi: 10.1159/000147847. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7:246–255. doi: 10.1038/nrc2108. [DOI] [PubMed] [Google Scholar]
- 6.Strizzi L, Hardy KM, Kirsammer GT, Gerami P, Hendrix MJ. Embryonic signaling in melanoma: potential for diagnosis and therapy. Lab Invest. 2011;91:819–24. doi: 10.1038/labinvest.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strizzi L, Postovit LM, Margaryan NV, Seftor EA, Abbott DE, Seftor RE, et al. Emerging roles of nodal and Cripto-1: from embryogenesis to breast cancer progression. Breast Dis. 2008;29:91–103. doi: 10.3233/bd-2008-29110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts HJ, Hu S, Qiu Q, Leung PC, Caniggia I, Gruslin A, et al. Identification of novel isoforms of activin receptor-like kinase 7 (ALK7) generated by alternative splicing and expression of ALK7 and its ligand, Nodal, in human placenta. Biol Reprod. 2003;68:1719–1726. doi: 10.1095/biolreprod.102.013045. [DOI] [PubMed] [Google Scholar]
- 9.Lawrence MG, Margaryan NV, Loessner D, Collins A, Kerr KM, Turner M, et al. Reactivation of embryonic nodal signaling is associated with tumor progression and promotes the growth of prostate cancer cells. Prostate. 2011;71:1198–209. doi: 10.1002/pros.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee CC, Jan HJ, Lai JH, Ma HI, Hueng DY, Gladys Lee YC, et al. Nodal promotes growth and invasion in human gliomas. Oncogene. 2010;29:3110–3223. doi: 10.1038/onc.2010.55. [DOI] [PubMed] [Google Scholar]
- 11.Topczewska JM, Postovit LM, Margaryan NV, Sam A, Hess AR, Wheaton WW, et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med. 2006;12:925–932. doi: 10.1038/nm1448. [DOI] [PubMed] [Google Scholar]
- 12.Postovit LM, Seftor EA, Seftor RE, Hendrix MJ. Targeting Nodal in malignant melanoma cells. Expert Opin Ther Targets. 2007;11:497–505. doi: 10.1517/14728222.11.4.497. [DOI] [PubMed] [Google Scholar]
- 13.Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW, et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci U S A. 2008;105:4329–4334. doi: 10.1073/pnas.0800467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strizzi L, Postovit LM, Margaryan NV, Lipavsky A, Gadiot J, Blank C, et al. Nodal as a biomarker for melanoma progression and a new therapeutic target for clinical intervention. Expert Rev Dermatol. 2009;4:67–78. doi: 10.1586/17469872.4.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papageorgiou I, Nicholls PK, Wang F, Lackmann M, Makanji Y, Salamonsen LA, et al. Expression of nodal signaling components in cycling human endometrium and in endometrial cancer. Reprod Biol Endocrinol. 2009;7:122. doi: 10.1186/1477-7827-7-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9:433–446. doi: 10.1016/j.stem.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Cavallari C, Fonsato V, Herrera MB, Bruno S, Tetta C, Camussi G. Role of Lefty in the anti tumor activity of human adult liver stem cells. Oncogene. 2012 doi: 10.1038/onc.2012.114. e-pub ahead of print 2 Apr 2012. [DOI] [PubMed] [Google Scholar]
- 18.Munir S, Xu G, Wu Y, Yang B, Lala PK, Peng C. Nodal and ALK7 inhibit proliferation and induce apoptosis in human trophoblast cells. J Biol Chem. 2004;279:31277–31286. doi: 10.1074/jbc.M400641200. [DOI] [PubMed] [Google Scholar]
- 19.Nadeem L, Munir S, Fu G, Dunk C, Baczyk D, Caniggia I, et al. Nodal signals through activin receptor-like kinase 7 to inhibit trophoblast migration and invasion: implication in the pathogenesis of preeclampsia. Am J Pathol. 2011;178:1177–1189. doi: 10.1016/j.ajpath.2010.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strizzi L, Hardy KM, Margaryan NV, Hillman DW, Seftor EA, Chen B, et al. Potential for the embryonic morphogen Nodal as a prognostic and predictive biomarker in breast cancer. Breast Cancer Res. 2012;14:R75. doi: 10.1186/bcr3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quail DF, Walsh LA, Zhang G, Findlay SD, Moreno J, Fung L, et al. Embryonic Protein Nodal Promotes Breast Cancer Vascularization. Cancer Research. 2012;72:3851–3863. doi: 10.1158/0008-5472.CAN-11-3951. [DOI] [PubMed] [Google Scholar]
- 22.Schier AF. Nodal signaling in vertebrate development. Annu Rev Cell Dev Biol. 2003;19:589–621. doi: 10.1146/annurev.cellbio.19.041603.094522. [DOI] [PubMed] [Google Scholar]
- 23.Beck S, Le Good JA, Guzman M, Ben HN, Roy K, Beermann F, et al. Extraembryonic proteases regulate Nodal signaling during gastrulation. Nat Cell Biol. 2002;4:981–985. doi: 10.1038/ncb890. [DOI] [PubMed] [Google Scholar]
- 24.Clements M, Pernaute B, Vella F, Rodriguez TA. Crosstalk between Nodal/activin and MAPK p38 signaling is essential for anterior-posterior axis specification. Curr Biol. 2011;21:1289–1295. doi: 10.1016/j.cub.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, et al. TGF-beta activates Erk MAP kinase signaling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signaling pathways. J Cell Biochem. 2005;95:918–931. doi: 10.1002/jcb.20458. [DOI] [PubMed] [Google Scholar]
- 27.Chakraborty C, Gleeson LM, McKinnon T, Lala PK. Regulation of human trophoblast migration and invasiveness. Can J Physiol Pharmacol. 2002;80:116–124. doi: 10.1139/y02-016. [DOI] [PubMed] [Google Scholar]
- 28.Quail DF, Taylor MJ, Walsh LA, Dieters-Castator D, Das P, Jewer M, et al. Low oxygen levels induce the expression of the embryonic morphogen Nodal. Mol Biol Cell. 2011;22:4809–4821. doi: 10.1091/mbc.E11-03-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quail DF, Maciel TJ, Rogers K, Postovit LM. A Unique 3D In Vitro Cellular Invasion Assay. J Biomol Screen. 2012;17:1088–1095. doi: 10.1177/1087057112449863. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Scherbakov AM, Andreeva OE, Shatskaya VA, Krasil’nikov MA. The relationships between snail1 and estrogen receptor signaling in breast cancer cells. J Cell Biochem. 2012;113:2147–2155. doi: 10.1002/jcb.24087. [DOI] [PubMed] [Google Scholar]
- 32.Dhasarathy A, Kajita M, Wade PA. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol Endocrinol. 2007;21:2907–2918. doi: 10.1210/me.2007-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 35.Postovit LM, Margaryan NV, Seftor EA, Hendrix MJ. Role of nodal signaling and the microenvironment underlying melanoma plasticity. Pigment Cell Melanoma Res. 2008;21:348–357. doi: 10.1111/j.1755-148X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- 36.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–190. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolosova I, Nethery D, Kern JA. Role of Smad2/3 and p38 MAP kinase in TGF-beta1-induced epithelial-mesenchymal transition of pulmonary epithelial cells. J Cell Physiol. 2011;226:1248–1254. doi: 10.1002/jcp.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bokobza SM, Ye L, Kynaston H, Jiang WG. Growth and differentiation factor 9 (GDF-9) induces epithelial-mesenchymal transition in prostate cancer cells. Mol Cell Biochem. 2011;349:33–40. doi: 10.1007/s11010-010-0657-5. [DOI] [PubMed] [Google Scholar]
- 39.Schier AF. Nodal morphogens. Cold Spring Harb Perspect Biol. 2009;1:a003459. doi: 10.1101/cshperspect.a003459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelber JA, Panopoulos AD, Shani G, Booker EC, Belmonte JC, Vale WW, et al. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene. 2009;28:2324–2336. doi: 10.1038/onc.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannan S, De SM, Lohmeyer M, Riese DJ, Smith GH, Hynes N, et al. Cripto enhances the tyrosine phosphorylation of Shc and activates mitogen-activated protein kinase (MAPK) in mammary epithelial cells. J Biol Chem. 1997;272:3330–3335. doi: 10.1074/jbc.272.6.3330. [DOI] [PubMed] [Google Scholar]
- 42.Strizzi L, Bianco C, Normanno N, Seno M, Wechselberger C, Wallace-Jones B, et al. Epithelial mesenchymal transition is a characteristic of hyperplasias and tumors in mammary gland from MMTV-Cripto-1 transgenic mice. J Cell Physiol. 2004;201:266–276. doi: 10.1002/jcp.20062. [DOI] [PubMed] [Google Scholar]
- 43.Rangel MC, Karasawa H, Castro NP, Nagaoka T, Salomon DS, Bianco C. Role of Cripto-1 during epithelial-to-mesenchymal transition in development and cancer. Am J Pathol. 2012;180:2188–2200. doi: 10.1016/j.ajpath.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 45.Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Q, Mattingly RR. Restoration of E-cadherin cell-cell junctions requires both expression of E-cadherin and suppression of ERK MAP kinase activation in Ras-transformed breast epithelial cells. Neoplasia. 2008;10:1444–1458. doi: 10.1593/neo.08968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tabaries S, Dong Z, Annis MG, Omeroglu A, Pepin F, Ouellet V, et al. Claudin-2 is selectively enriched in and promotes the formation of breast cancer liver metastases through engagement of integrin complexes. Oncogene. 2011;30:1318–1328. doi: 10.1038/onc.2010.518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.