

Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ) is a ligand-activated nuclear receptor that regulates glucose and lipid metabolism, endothelial function and inflammation. Rosiglitazone (RGZ) and other thiazolidinedione (TZD) synthetic ligands of PPARγ are insulin sensitizers that have been used for the treatment of type 2 diabetes. However, undesirable side effects including weight gain, fluid retention, bone loss, congestive heart failure, and a possible increased risk of myocardial infarction and bladder cancer, have limited the use of TZDs. Therefore, there is a need to better understand PPARγ signaling and to develop safer and more effective PPARγ-directed therapeutics. In addition to PPARγ itself, many PPARγ ligands including TZDs bind to and activate G protein-coupled receptor 40 (GPR40), also known as free fatty acid receptor 1. GPR40 signaling activates stress kinase pathways that ultimately regulate downstream PPARγ responses. Recent studies in human endothelial cells have demonstrated that RGZ activation of GPR40 is essential to the optimal propagation of PPARγ genomic signaling. RGZ/GPR40/p38 MAPK signaling induces and activates PPARγ co-activator-1α, and recruits E1A binding protein p300 to the promoters of target genes, markedly enhancing PPARγ-dependent transcription. Therefore in endothelium, GPR40 and PPARγ function as an integrated signaling pathway. However, GPR40 can also activate ERK1/2, a proinflammatory kinase that directly phosphorylates and inactivates PPARγ. Thus the role of GPR40 in PPARγ signaling may have important implications for drug development. Ligands that strongly activate PPARγ, but do not bind to or activate GPR40 may be safer than currently approved PPARγ agonists. Alternatively, biased GPR40 agonists might be sought that activate both p38 MAPK and PPARγ, but not ERK1/2, avoiding its harmful effects on PPARγ signaling, insulin resistance and inflammation. Such next generation drugs might be useful in treating not only type 2 diabetes, but also diverse chronic and acute forms of vascular inflammation such as atherosclerosis and septic shock.
Keywords: Peroxisome proliferator-activated receptor gamma (PPARγ), Thiazolidinediones (TZDs), G protein-coupled receptor 40 (GPR40), p38 mitogen-activated protein kinase (p38 MAPK), PPARγ co-activator-1alpha (PGC-1α), E1A binding protein p300 (EP300)
Graphical abstract
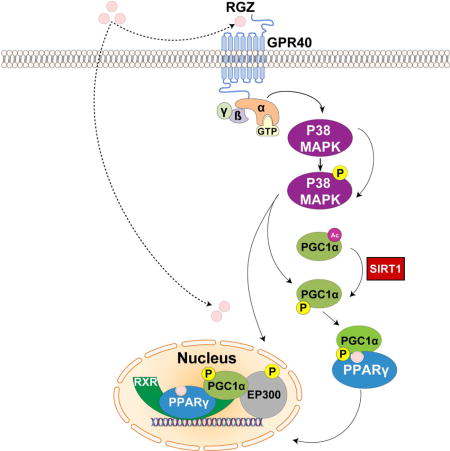
1. Introduction
1.1. Peroxisome proliferator-activated receptor gamma (PPARγ) as a target for treating type 2 diabetes and thiazolidinediones (TZDs)
In 2010, an estimated 257 million people worldwide had type 2 diabetes mellitus (T2DM) and the number is projected to rise to 395 million by 2030 (1). TZDs, commonly called glitazones, have been widely used to treat this disease (2). The anti-diabetic effects of TZDs were originally discovered in 1982 (3). Ciglitazone was the first TZD shown to normalize hyperglycemia, hyperinsulinemia, and hypertriglyceridemia in mouse models of T2DM (4). Later, troglitazone (5) and pioglitazone (6) were also shown to decrease insulin resistance by increasing insulin-stimulated glucose utilization and reducing hepatic glucose production. Rosiglitazone (RGZ), synthesized in 1988, was a more selective and potent insulin sensitizer than ciglitazone in rodent models (7, 8). In 1997, troglitazone was the first TZD approved for clinical use, but was soon withdrawn worldwide because of liver toxicity (9). The U.S. Food and Drug Administration subsequently approved RGZ and pioglitazone in 1999 for the treatment of T2DM (10). RGZ was later withdrawn in Europe and its use restricted in the United States due to an increased risk of myocardial infarction (11, 12). Pioglitazone does not appear to share this risk, but otherwise has the same adverse profile common to TZDs and has been associated with bladder cancer (13, 14).
Named for its activation by fibrates and other peroxisome proliferators (15), peroxisome proliferator-activated receptor α (PPARα/NR1C1) was first identified in 1990. Two other genes belonging to the same family, PPARβ/δ (NR1C2) and PPARγ (NR1C3), were cloned in Xenopus two years later (16). Human homologues of three PPAR isoforms, α (17), β (18) and γ (19), were soon identified. PPARγ has two isoforms, PPARγ1 and PPARγ2, with the latter harboring an additional 30 amino acids at its N-terminus (20). PPARγ1 is expressed in many tissues including leukocytes and endothelial cells, while PPARγ2 is normally restricted to adipose tissue, but can be induced elsewhere (20, 21). In 1994, PPARγ was found to be a major adipogenic transcription factor in mice (22). Long after the anti-diabetic effects of TZDs were discovered in 1982 (3), PPARγ was identified as the receptor target of TZDs in 1995 (23, 24). As noted above, this was less than two years before the first TZD was approved for clinical use (9, 25, 26). Besides synthetic TZDs, the endogenous arachidonate 15-deoxy-D12, 14-prostanglandin J2 (15d-PGJ2) and related metabolites also activate PPARγ and induce adipogenesis (24), but at concentrations above that found in cells (27). In addition some unsaturated fatty acids activate PPARγ, such as the dietary polyunsaturated eicosapentanoic acid, linolenic acids, linoleic acid, and oxidized low-density lipoprotein (28). Therefore, PPARγ may under some circumstances function as a general fatty acid sensor, with affinity KD values of 2–50 μM (29). Also two linoleic acid oxidation products detected in significant amounts in oxidized low-density lipoprotein particles, 9-HODE and 13-HODE, were previously identified as endogenous ligands and activators of PPARγ (30).
As noted above, TZDs including RGZ and pioglitazone have been associated with a number of adverse effects. These include weight gain (31), fluid retention (31, 32), and reduction in bone mineral density (33). Besides these class effects, RGZ has been associated with excess myocardial infarctions and pioglitazone with bladder cancer. These undesirable, “off-target” effects of TZDs have driven research to better understand PPARγ signaling and to develop new agents with improved efficacy and safety. TZD-induced weight gain has recently been linked to activation of PPARγ in the brain, rather than in adipose tissue (34, 35). Intraventricular TZD administration or overexpression of PPARγ in the brain of normal rats promote sustained increases in feeding and body weight (34). Conversely, mice with selected ablation of brain PPARγ consumed less food and gained less weight than controls in response to TZD treatment during high-fat feeding. These mice also showed increased physical activity and energy expenditure (35). TZD-induced fluid retention and peripheral edema has been attributed to increased sodium and water reabsorption in the distal collecting ducts of the kidney. Collecting duct-specific knockout of PPARγ blocked TZD-associated increases in plasma volume and body weight (36). How TZDs exert this action remains unclear, as findings about the role of epithelial sodium channels in this phenomenon are contradictory (36, 37). Besides weight gain, fluid retention associated with TZDs may also contribute to adverse cardiovascular events, such as congestive heart failure (20). Consistent with reductions in bone mineral density and a higher rate of fractures, TZDs caused bone loss in rodents by inhibiting osteoblastogenesis (bone formation) and enhancing osteoclastogenesis (bone resorption). TZDs were proposed to exert these effects through PPARγ-dependent induction of c-fos, β-catenin, and ERRα (38–40).
1.2. PPARγ as a therapeutic target in atherosclerosis, pulmonary arterial hypertension, adult respiratory distress syndrome, and septic shock
The direct binding of TZDs and other ligands to PPARγ activates two distinct signaling pathways. Cis-activation drives transcription through agonist-dependent conformational changes in the activation function 2 (AF-2) domain of PPARγ, recruitment of co-activators such as PPARγ co-activator-1α (PGC-1α), PPARγ dimerization with the retinoid X receptor (RXR) (41), and the binding of this complex to peroxisome proliferator response elements (PPREs) in the promoters of target genes. This signaling pathway is closely linked to the essential roles of PPARγ in adipogenesis and glucose homeostasis. Alternatively, ligand-bound PPARγ has also been shown to suppress inflammation via a mechanism called trans-repression. Trans-repression is independent of DNA binding by PPARγ as demonstrated by the PPARγ C126A/E127A mutant, which remains capable of repressing lipopolysaccharide-induced genes while being rendered incapable of cis-activation (42). This mode of repression also does not require RXR dimerization, but does require sumoylation of PPARγ2 at K395 by the small ubiquitin-like modifier and subsequent tethering of PPARγ, nuclear receptor co-repressor and histone deacetylase to NFκB and AP-1 complexes that regulate the transcription of inflammatory response genes (43). Together, these two distinct, PPARγ-dependent signaling pathways may provide benefits beyond the treatment of T2DM. Despite their adverse effects, TZDs lower blood pressure (44, 45), improve lipid profiles (46), and inhibit vascular inflammation (47). These effects in non-adipose tissue support the potential of a newer generation of PPARγ ligands in diverse conditions such as atherosclerosis, pulmonary arterial hypertension, adult respiratory distress syndrome, ulcerative colitis and septic shock. Endothelial PPARγ disruption was found to accelerate diet-induced atherosclerosis (48), while the PPARγ ligand 15d-PGJ2 reduced atherosclerotic lesion in apoE knockout mice (49). PPARγ deletion in the arterial smooth muscle cells of mice resulted in the development of pulmonary arterial hypertension (50), whereas the PPARγ ligand RGZ was beneficial in a murine model of this disease (51). Heterozygous mutation of valine to methionine at 290 (V290M) or proline to leucine at 476 (P467L) of PPARγ was linked to high blood pressure in human subjects (52). PPARγ expression and activation was found to protect animals from acute respiratory distress syndrome (53). In endotoxin challenge models of septic shock, 15d-PGJ2 reduced mortality in mice (54) and RGZ reduced organ injury and cytokine release in rats (55). Therefore, safer, more effective PPARγ activators may have broad applicability in a variety of acute and chronic inflammatory diseases.
2. Cross talk between nitric oxide (NO) and PPARγ signaling
2.1. Endogenous PPARγ ligands formed by NO nitration of unsaturated fatty acids
NO is a free radical messenger, synthesized from L-arginine via three nitric oxide synthase isoforms, that plays a central role in vascular health. The biological effects of NO are diverse and many result from the oxidization, nitrosylation, nitrosation, and/or nitration of target molecules (56). While, fatty acids are important sources of energy, covalent modifications can transform some fatty acids into potent signaling molecules (57). For example, nitro-linoleic acid is a potent PPARγ ligand with a Ki value of 133 nM, which rivals that of RGZ (Ki = 53 nM) and is much lower than that of unmodified linoleic acid (Ki >1 μM) (58). Nitro-linoleic and nitro-oleic acids, but not their unmodified forms, induced PPARγ-driven adipogenesis in preadipocytes (57, 58). In vivo administration of nitro-oleic acid, but not parental oleic acid was also shown to ameliorate diabetic symptoms in rats (59).
Nitrated fatty acids are one of the largest pools of active NO derivatives detected in human plasma (56, 57, 60). Under certain circumstances, nitrated fatty acid concentrations in human blood may reach > 1 μM (57). In vitro, PPREs reporter gene studies have shown that PPARγ is very sensitive to nitro-oleic acid with significant activation at 100 nM, while PPARα and PPARβ/δ were activated at concentrations three times higher (57). Additional evidence suggests that nitrated (61) and oxidized (62) fatty acids can covalently bind to PPARγ at C285 via Michael addition and thereby activate PPARγ genomic signaling. Importantly, nitro derivatives of unsaturated fatty acids given to leptin-deficient ob/ob mice lower insulin and glucose levels without causing the weight gain associated with RGZ (61).
While the exact identity of biologically relevant natural ligands for PPARγ remain uncertain, nitro- and nitrohydroxy-fatty acid derivatives are among the most likely candidates (56). Furthermore, these fatty acid adjuncts may also have therapeutic applications. Nitro-oleic acid at physiological concentrations in blood decreased endotoxin-induced endothelial inflammation and neutrophil transmigration in a PPARγ-dependent manner (47). Direct lung delivery of nitro-oleic acid in a mouse model of acute lung injury significantly decreased pulmonary inflammation and injury, including capillary leak, lung edema, neutrophil infiltration, oxidant stress, and plasma cytokine levels (63). In addition, nitro-oleic acid suppressed murine allergic airway disease at least partially through PPARγ activation, and unlike the steroid drug fluticasone, induced robust apoptosis and phagocytosis of neutrophils (64). Nitro-oleic acid-mediated PPARγ activation has also been shown to attenuate colitis in experimental inflammatory bowel disease (65).
2.2. NO induces p38 mitogen-activated protein kinase (MAPK) phosphorylation in endothelium, thereby activating PPARγ signaling
Besides activation by nitro-fatty acids, NO has been demonstrated to activate PPARγ via a p38 MAPK-dependent mechanism in human endothelial cells (21). In both endothelial cells and monocytes, low-dose NO caused a rapid dose-dependent increase in PPARγ binding to a consensus PPRE sequence (21, 66). NO-induced PPARγ signaling and target gene expression was directly linked to p38 MAPK phosphorylation. Blockade of p38 MAPK with a specific inhibitor or siRNA knockdown abolished the ability of NO to increase PPARγ DNA binding or to induce PPARγ target genes (21).
An extensive literature has previously connected p38 MAPK to PPARγ activation. PPARγ-dependent adipogenesis in mesenchymal cells, 3T3-L1 pre-adipocytes, and white adipocytes have all been associated with p38 MAPK activation (67–70). In brown fat, p38 MAPK has been shown to activate PGC-1α and induce the expression of PPARγ target genes including PGC-1α itself, and uncoupling protein 1 (71, 72). Notably, p38 MAPK directly phosphorylates PGC-1α (71, 73–75) and E1A binding protein p300 (EP300) (76), which facilitates co-activator recruitment to PPARγ target genes, chromatin remodeling and PPARγ-dependent gene transcription. In addition to NO, carbon monoxide, another low molecular weight, endogenous messenger that activates p38 MAPK (77, 78), has also been shown to activate PPARγ (78, 79). Moreover, TZDs have been long known to activate p38 MAPK independent of PPARγ in a variety cell types, including adipocytes (75), astrocytes (80), cardiomyocytes (81), and epithelial cells (82). The well-documented role of p38 MAPK in PPARγ signaling and the ability of TZDs to activate both p38 MAPK and PPARγ suggest that p38 MAPK is an unrecognized facilitator of TZD-mediated PPARγ activation.
3. Post-translational modifications (PTMs) that regulate or shape PPARγ genomic signaling
3.1. Obesity, insulin resistance, diabetes and PPARγ
Obesity has become a major health problem worldwide with a prevalence of 36.9% in men and 38.0% in women (83). The failure of adipose tissue in obesity to store excess energy appropriately leads to ectopic lipid deposition, insulin resistance and ultimately T2DM (84). Four different fat depots play contrasting physiological and pathophysiological metabolic roles in humans: brown (BAT), subcutaneous (SAT), and visceral white adipose tissue (VAT), and ectopic lipid (85, 86). BAT contains numerous mitochondria and expresses uncoupling protein 1, a mitochondrial protein that uncouples oxidative phosphorylation, resulting in inefficient production of ATP and release of energy as heat, thereby acting as a thermogenic organ (85, 86). SAT, the largest fat depot, stores triglycerides that can be readily released during times of energy demand. SAT also secretes adiponectin and leptin that have largely beneficial effects on lipid oxidation, energy utilization, insulin action, and inflammation (85, 86). VAT together with ectopic lipid, are harmful and associated with insulin resistance and increased cardiovascular risk (85).
T2DM is characterized by hyperglycemia due to insulin resistance. While the precise pathogenesis of insulin resistance is not completely clear, intra-abdominal VAT, deficiency of adiponectin and/or leptin, inflammation, and mitochondrial dysfunction have all been identified as important contributors (85, 87). Intra-abdominal fat leads to labile fatty acid release with direct delivery to the liver, inflammatory cell accumulation, reduced adiponectin levels, and decreased PPARγ activity (85). Lipodystrophy, syndromes are characterized by a partial or near-complete absence of SAT, a relative increase in VAT, and marked insulin resistance (85). Beneficial effects of BAT and SAT on insulin sensitivity may primarily be attributed to increased fatty acid oxidation (85). Mitochondrial dysfunction increases reactive oxygen species production, activating serine/threonine kinases including IKK, JNK, and PKCs, which phosphorylate insulin receptor substrate (IRS). Phosphorylation of IRS-1/2 inhibits signaling pathways downstream from insulin including PI3K, Akt, and PKCζ, which decreases glucose uptake, increases glucose production, and reduces vasodilation and insulin secretion (87).
PPARγ is a key regulator of insulin sensitivity that improves insulin resistance in T2DM via multiple mechanisms. Specifically, PPARγ: 1) induces adipogenesis in beneficial SAT, but not in harmful VAT (88–90); 2) enhances uncoupling protein 1 expression in BAT (71, 72); 3) elevates serum adiponectin levels (91); 4) suppresses inflammation (43); and 5) promotes mitochondrial biogenesis and reduces mitochondrial production of reactive oxygen species (92). While TZDs are full agonists of PPARγ, they have undesirable adverse effects that prevent or restrict their clinical application as discussed earlier. An emerging understanding of PPARγ signaling has the potential to create a new generation of agonists with a better safety profile and broader applicability.
3.2. PPARγ PTMs associated with obesity and insulin resistance
Similar to other nuclear receptors, PPARγ activity is tightly regulated by PTMs. In vitro assays demonstrate that serine 112 (S112) of mouse PPARγ2, corresponding to S82 of mouse and S84 of human PPARγ1, can be phosphorylated by either ERK or JNK (93–95). Phosphorylation at these sites is an inactivating event that inhibits both ligand-dependent and -independent PPARγ transcriptional activity (93, 94). Expression of PPARγ mutants (S112A) that cannot be phosphorylated increased ligand-induced adipogenesis and eliminated the ability of mitogens to inhibit differentiation (93, 94). Mice homozygous for this S112A mutation are protected from diet-induced obesity and insulin resistance, effects associated with the increased secretion of adiponectin and leptin (96). Cdk7 has also been shown to phosphorylate PPARγ2 at S112 and Cdk7 knockdown in mouse embryonic fibroblasts induces adipogenesis and adiponectin expression (97).
In the obese state, mouse PPARγ2 is also highly phosphorylated at serine 273 (S273) by both ERK and cyclin-dependent kinase Cdk5 (84, 98). This modification of PPARγ does not change its in vitro transcriptional activity and adipogenic capacity, but in vivo reshapes the transcriptional repertoire of PPARγ. Serine 273 phosphorylation of PPARγ dysregulates genes whose expression is altered in obesity, reducing the expression of adipokines and adiponectin, insulin-sensitizing hormones secreted by adipose tissue (84, 98). Pro-inflammatory cytokines produced in obesity are known to activate ERK and Cdk5. ERK inhibition or PPARγ ligands that block S273 accessibility both prevent S273 phosphorylation and reduce insulin resistance in obese wildtype and ob/ob mice (84, 98). Several high affinity PPARγ ligands, such as MRL24 and Mbx-102 that are poor PPARγ agonists, effectively inhibit S273 phosphorylation and show strong anti-diabetic activity in vivo (98). These results suggest that blockade of S273 phosphorylation alone has insulin-sensitizing effects independent of classical notions of PPARγ activation.
In addition to phosphorylation, PPARγ acetylation has been reported to regulate its function. EP300 and Tip60 acetylate, while SIRT1 de-acetylates PPARγ, respectively enhancing and repressing PPARγ transcriptional activity and its adipogenic potential in 3T3-L1 preadipocytes (99–101). Conversely, K268 and K293 of PPARγ are highly acetylated in obesity and ligand-induced de-acetylation by SIRT1 was shown to induce BAT and repress VAT genes, potentially reducing insulin resistance (102). As such, the regulation of PPARγ by acetylation is complex with many possible lysine-site combinations and as yet inadequately explored functional consequences.
Lysine site sumoylation adds another layer of control over PPARγ genomic signaling. Two functional sumoylation sites have been identified for PPARγ2, K107 and K395 (20, 43, 103). K107 sumoylation blocks the cis-transcriptional activity of PPARγ, possibly by promoting co-repressor recruitment (20, 103, 104). Recent findings in fibroblast growth factor 21-knockout mice show that this factor increases PPARγ activity in adipocytes by preventing K107 sumoylation, enhancing insulin sensitivity (105). Interestingly, K107 is close to S112 in position and together they form a consensus sumoyl-phospho motif (106). K107 sumoylation is to some extent linked to S112 phosphorylation and ablation of S112 phosphorylation significantly diminished K107 sumoylation (103). Sumoylation at K395 recruits PPARγ monomers to NF-κB and AP1 DNA-binding sites, preventing clearance of co-repressor complexes, thereby trans-repressing inflammatory response genes (20, 43). Sumoylation at this residue has also been reported to influence the ability of carbon dioxide to decrease pro-inflammatory signaling in macrophages (78). Additionally, the binding of ligands to PPARγ and interferon-γ exposure were both found to induce PPARγ ubiquitination. Although ubiquitination residues are yet to be identified, PPARγ ubiquitination is determined by the ligand binding/AF-2 domains and leads to degradation of PPARγ via the 26S proteasome (107). Regulation of PPARγ protein levels through this mechanism likely also plays an important role in obesity.
4. GPR40 and PPARγ, an integrated two-receptor signaling pathway
4.1. Biological function of GPR40
GPR40, also known as free fatty acid receptor 1 (FFAR1), was deorphanized in 2003 as a medium-to-long chain FFA (both saturated and unsaturated) receptor (108). GPR40 is highly expressed in pancreatic β cells, and also, albeit to a lesser degree, in other tissues, including the intestinal tract, bone cells, lung epithelial cells, brain, and monocytes (109, 110). A well-documented function of GPR40 is to mediate effects of FFAs, such as linoleic acid and linolenic acid, on glucose-stimulated insulin secretion in pancreatic β cells (109, 111, 112). GPR40 loss-of-function via siRNA in β cells (112), or gene deletion in Gpr40−/− mice (113) consistently resulted in a significant reduction in FFA-induced insulin secretion. Conversely, transgenic overexpression of GPR40 prevented the development of hyperglycemia in mice fed a high-fat diet, and improved insulin secretion and glucose tolerance in genetically diabetic mice (114). Furthermore, GW1100, a GPR40 antagonist, was shown to inhibit GPR40-mediated insulin secretion in the MIN6 β-cell line (115). In humans, a natural variant of GPR40 (G180S) diminishes the ability of pancreatic β cells to sense lipids and impairs FFA-induced insulin secretion (116). GPR40-mediated effects on glucose-stimulated insulin secretion were associated with increased Ca2+ influx and were not altered by inhibition of ERK, which was also activated when FFAs bound to GPR40 (112, 117). Food intake activates GPR40 expressed on enteroendocrine cells of the intestinal tract, mediating the secretion of incretin hormones (109), which also stimulate β cell insulin release (111). GPR40 full agonists increased incretin levels in a mouse model of T2DM, normalizing blood glucose levels (118). Gpr40−/− mice have reduced incretin and insulin secretion in response to fat, and are not protected from insulin resistance induced by a high-fat diet (119).
4.2. Ligands that activate both GPR40 and PPARγ
Many FFAs and their derivatives, including lauric acid, myristic acid, palmitic acid, oleic acid, linoleic acid, linolenic acid, arachidonic acid, eicosapentaenoic acid, and 9-HODE have been shown to be endogenous ligands of both GPR40 and PPARγ (28, 112, 120, 121). TZDs, including ciglitazone, troglitazone, RGZ, and pioglitazone, all bind to and activate GPR40 with subsequent signal transduction that includes stress kinase pathways (122). RGZ compared with pioglitazone causes a more robust and sustained activation of ERK (122), a stress kinase often linked to inflammation. These differences in ERK activation could possibly explain some of the efficacy and safety differences among existing synthetic TZDs. Recently, several PPARγ ligands have been found that promote insulin sensitization with fewer side effects in T2DM rodent models (123, 124). Whether these ligands can activate GPR40 or/and ERK has not been reported. Importantly, it is not entirely clear whether adverse effects associated with TZDs are attributable to the activation of GPR40 or PPARγ or are entirely independent of both receptors. TZD-induced osteoblast and osteocyte apoptosis has been directly linked to GPR40. The activation of ERK with subsequent recruitment of the pro-apoptotic factor Bax to the outer membrane of the mitochondria was posited as the cause of TZD-related bone loss (125). In opposition to this mechanism, GW9508, a GPR40 agonist that blocks osteoclast differentiation in vitro, was found to prevent ovariectomy-induced bone loss in wildtype, but not Gpr40−/− mice (126).
4.3. GPR40 and PPARγ function together as a two-receptor signaling pathway
We recently demonstrated that GPR40 and PPARγ crosstalk functions as an integrated two-receptor signal transduction pathway in human endothelium (127). RGZ was found to require GPR40 activation with downstream p38 MAPK phosphorylation to optimally propagate PPARγ nuclear signaling (Fig. 1). The role of p38 MAPK in PPARγ transcriptional activation also further explains how NO/p38 MAPK signaling, independent of GPR40, induces PPARγ target genes (21, 127). GPR40 activation of p38 MAPK induced PGC-1α expression, and directly phosphorylated both PGC-1α and EP300, thereby promoting their recruitment to PPARγ response elements. EP300, a histone acetyltransferase that docks with PGC-1α, remodels chromatin to optimize the transcription of PPARγ target genes. In human primary pulmonary artery endothelial cells, knockdown of GPR40, p38 MAPK, PGC-1α, SIRT1 (an essential PGC-1α activator) or EP300 all substantially reduced the ability of RGZ to induce PPARγ regulated genes. GPR40 and PPARγ were seen to cooperate at least additively and sometimes synergistically to initiate PPARγ genomic responses, depending on the transcriptional context (127). Collectively, this work demonstrated that p38 MAPK, PGC-1α, and EP300 link GPR40 to downstream PPARγ genomic signaling. Binding to and activating both GPR40 and PPARγ appears to be a common feature of many PPARγ agonists. This connection between GPR40 signaling and PPARγ transcriptional activation argues that the effects of TZDs on human endothelium might be best understood as a cognate two-receptor system, integrated by p38 MAPK, PGC-1α and EP300.
Figure 1.
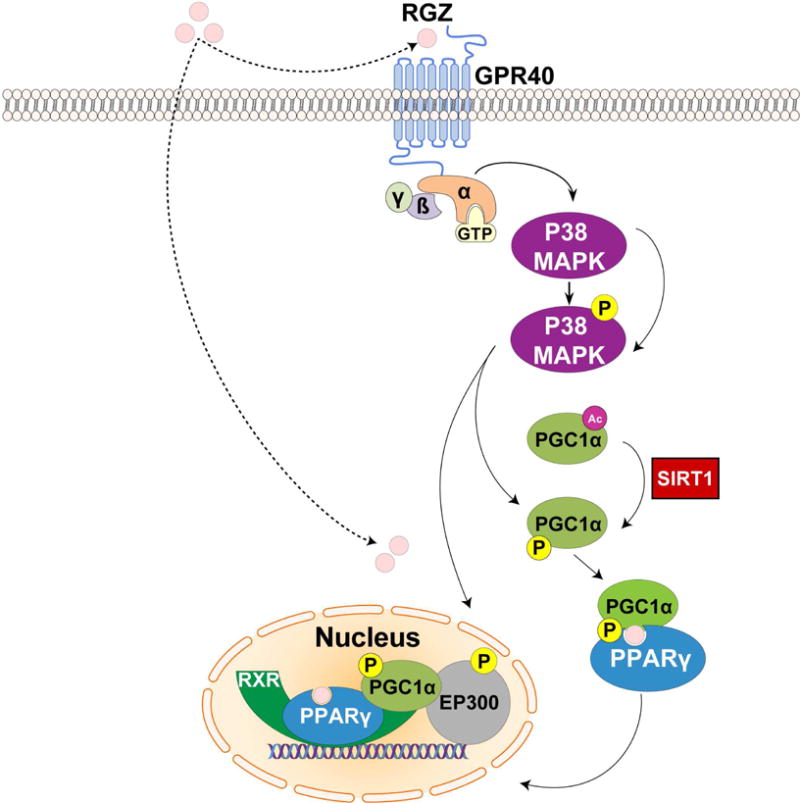
Crosstalk between GPR40 and PPARγ signaling in human endothelium.
In the classical signaling pathway, rosiglitazone (RGZ) binds directly to and activates PPARγ. In human endothelial cells, RGZ requires GPR40 activation with downstream p38 MAPK phosphorylation to optimally propagate PPARγ nuclear signaling. GPR40 activation of p38 MAPK induces PGC-1α expression, and directly phosphorylates both PGC-1α and EP300, thereby activating them. EP300, a histone acetyltransferase that docks with PGC-1α, remodels chromatin to optimize the transcription of PPARγ target genes.
4.4. PPARγ agonism without GPR40 activation or GPR40/PPARγ agonism without ERK1/2 activation
PPARγ agonists that do not bind to or activate GPR40 have not yet been characterized in a systematic fashion. The natural PPARγ ligand 15d-PGJ2 has been reported to selectively activate PPARγ, but not GPR40 in human bronchial epithelial cells (110). However, 15d-PGJ2 does appear to activate stress kinases in other cell types, which could be a signature for unrecognized GPR40 activation. Stress kinase activation by 15d-PGJ2 includes ERK in renal epithelial cells, vascular smooth muscle cells and mesangial cells (128–130), ERK and p38 MAPK in osteosarcoma cells (131), and ERK, p38 MAPK and JNK in astrocytes (80). Nonetheless, the possibility that 15d-PGJ2 or other agonists might activate PPARγ without binding to or activating GPR40 remains an open question that requires further investigation. PPARγ ligands that can strongly activate PPARγ genomic responses without interacting with GPR40 or other G-protein coupled receptors would avoid the harmful effects of concomitant stress kinase activation.
For ligands that activate both receptors, some downstream signaling from GPR40 is clearly useful (p38 MAPK) while other aspects (ERK1/2) are undesirable in regards to PPARγ activation. While GPR40/p38 MAPK activation augments the genomic effects of PPARγ, ERK1/2 phosphorylates and thereby inactivates PPARγ (93, 94), and separately has inflammatory and proliferative effects in the vasculature. Furthermore, insulin resistance has been associated with ERK1/2 phosphorylation of PPARγ at S273 (84). ERK inhibition and PPARγ ligands both block PPARγ phosphorylation at S273, providing anti-diabetic benefits in rodent models of T2DM (84, 98, 132). Importantly, GPR40-mediated enhancement of glucose-stimulated insulin secretion is not dependent on ERK1/2 activation (112). Compared to existing drugs, PPARγ ligands that demonstrate no or substantially reduced GPR40/ERK signaling might be more potent agonists of PPARγ and have a better safety profile.
5. Concluding remarks
RGZ and pioglitazone, TZD PPARγ agonists, have been widely used to treat T2DM (2, 133). In addition, PPARγ agonists also have potent anti-inflammatory effects that may be beneficial in diverse conditions including coronary artery disease (44, 46), pulmonary arterial hypertension (50), acute respiratory distress syndrome (53, 134) and septic shock (54, 55). However, adverse effects, such as weight gain (2, 133), fluid retention (31, 32), congestive heart failure (13), bone fractures (31, 32) and importantly a paradoxical increase in the risk of myocardial infarctions (11, 12) have limited the usefulness of currently available PPARγ-targeted drugs. Recent efforts have focused on PPARγ ligands that block ERK/Cdk5-mediated phosphorylation of PPARγ at S273 (123, 124, 135, 136). GPR40, a FFA and TZD receptor that stimulates insulin secretion has also been identified as a potential target for new T2DM therapeutics (109, 111). As such, GPR40 agonists with little or no PPARγ binding activity have been synthesized (137, 138), but none of these agents has yet made to the bedside. Furthermore, selective GPR40 agonists, like TZDs, are likely to activate ERK1/2 in the vasculature.
In the endothelium, RGZ and pioglitazone bind to and activate both GPR40 and PPARγ, which function together through p38 MAPK to optimally propagate PPARγ genomic responses. However, GPR40 activation by these drugs also turns on ERK1/2, a stress kinase pathway that suppresses PPARγ signaling and promotes inflammation. Knowledge of this crosstalk could be used to screen for small molecules that do not bind to GPR40, but yet strongly activate PPARγ signaling. Exclusive PPARγ agonists that do not require GPR40/p38 MAPK signaling would circumvent the counterproductive and potentially harmful effects of GPR40/ERK activation. Alternatively ligands of GPR40 and PPARγ could be sought that activate GPR40 in a biased manner. Ideal ligands would retain the ability to activate p38 MAPK, but not ERK1/2. Activating p38 MAPK would enhance downstream PPARγ signaling through effects on PGC-1α and EP300. Bypassing ERK1/2 would avoid its inactivation of PPARγ and its deleterious impact on insulin resistance and inflammation. The feasibility of either of these approaches requires further investigation.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health intramural funds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no conflicts of interest with the contents of this article.
Disclaimer:
The opinions expressed in this article are the authors’ own and do not represent any position or policy of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
References
- 1.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol. 2012;8(4):228–236. doi: 10.1038/nrendo.2011.183. [DOI] [PubMed] [Google Scholar]
- 2.Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351(11):1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 3.Sohda T, Mizuno K, Imamiya E, Sugiyama Y, Fujita T, Kawamatsu Y. Studies on antidiabetic agents. II. Synthesis of 5-[4-(1-methylcyclohexylmethoxy)-benzyl]thiazolidine-2,4-dione (ADD-3878) and its derivatives. Chem Pharm Bull (Tokyo) 1982;30(10):3580–3600. doi: 10.1248/cpb.30.3580. [DOI] [PubMed] [Google Scholar]
- 4.Fujita T, Sugiyama Y, Taketomi S, Sohda T, Kawamatsu Y, Iwatsuka H, Suzuoki Z. Reduction of insulin resistance in obese and/or diabetic animals by 5-[4-(1-methylcyclohexylmethoxy)benzyl]-thiazolidine-2,4-dione (ADD-3878, U-63,287, ciglitazone), a new antidiabetic agent. Diabetes. 1983;32(9):804–810. doi: 10.2337/diab.32.9.804. [DOI] [PubMed] [Google Scholar]
- 5.Fujiwara T, Yoshioka S, Yoshioka T, Ushiyama I, Horikoshi H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes. 1988;37(11):1549–1558. doi: 10.2337/diab.37.11.1549. [DOI] [PubMed] [Google Scholar]
- 6.Sohda T, Momose Y, Meguro K, Kawamatsu Y, Sugiyama Y, Ikeda H. Studies on antidiabetic agents. Synthesis and hypoglycemic activity of 5-[4-(pyridylalkoxy)benzyl]-2,4-thiazolidinediones. Arzneimittelforschung. 1990;40(1):37–42. [PubMed] [Google Scholar]
- 7.Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, Kraegen EW. A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes. 1994;43(10):1203–1210. doi: 10.2337/diab.43.10.1203. [DOI] [PubMed] [Google Scholar]
- 8.Cantello BC, Cawthorne MA, Cottam GP, Duff PT, Haigh D, Hindley RM, Lister CA, Smith SA, Thurlby PL. [[omega-(Heterocyclylamino)alkoxy]benzyl]-2,4-thiazolidinediones as potent antihyperglycemic agents. J Med Chem. 1994;37(23):3977–3985. doi: 10.1021/jm00049a017. [DOI] [PubMed] [Google Scholar]
- 9.Watkins PB, Whitcomb RW. Hepatic dysfunction associated with troglitazone. N Engl J Med. 1998;338(13):916–917. doi: 10.1056/NEJM199803263381314. [DOI] [PubMed] [Google Scholar]
- 10.Lalloyer F, Staels B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2010;30(5):894–899. doi: 10.1161/ATVBAHA.108.179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciudin A, Hernandez C, Simo R. Update on cardiovascular safety of PPARgamma agonists and relevance to medicinal chemistry and clinical pharmacology. Curr Top Med Chem. 2012;12(6):585–604. doi: 10.2174/156802612799436632. [DOI] [PubMed] [Google Scholar]
- 12.Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA. 2007;298(10):1189–1195. doi: 10.1001/jama.298.10.1189. [DOI] [PubMed] [Google Scholar]
- 13.Shah P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opin Drug Saf. 2010;9(2):347–354. doi: 10.1517/14740331003623218. [DOI] [PubMed] [Google Scholar]
- 14.Lewis JD, Habel LA, Quesenberry CP, Strom BL, Peng T, Hedderson MM, Ehrlich SF, Mamtani R, Bilker W, Vaughn DJ, Nessel L, Van Den Eeden SK, Ferrara A. Pioglitazone Use and Risk of Bladder Cancer and Other Common Cancers in Persons With Diabetes. JAMA. 2015;314(3):265–277. doi: 10.1001/jama.2015.7996. [DOI] [PubMed] [Google Scholar]
- 15.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 16.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68(5):879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 17.Sher T, Yi HF, McBride OW, Gonzalez FJ. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32(21):5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D, Rodan GA. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol Endocrinol. 1992;6(10):1634–1641. doi: 10.1210/mend.6.10.1333051. [DOI] [PubMed] [Google Scholar]
- 19.Qi JS, Desai-Yajnik V, Greene ME, Raaka BM, Samuels HH. The ligand-binding domains of the thyroid hormone/retinoid receptor gene subfamily function in vivo to mediate heterodimerization, gene silencing, and transactivation. Mol Cell Biol. 1995;15(3):1817–1825. doi: 10.1128/mcb.15.3.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ptasinska A, Wang S, Zhang J, Wesley RA, Danner RL. Nitric oxide activation of peroxisome proliferator-activated receptor gamma through a p38 MAPK signaling pathway. FASEB J. 2007;21(3):950–961. doi: 10.1096/fj.06-6822com. [DOI] [PubMed] [Google Scholar]
- 22.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 23.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) The Journal of biological chemistry. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 24.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz S, Raskin P, Fonseca V, Graveline JF. Effect of troglitazone in insulin-treated patients with type II diabetes mellitus. Troglitazone and Exogenous Insulin Study Group. N Engl J Med. 1998;338(13):861–866. doi: 10.1056/NEJM199803263381302. [DOI] [PubMed] [Google Scholar]
- 26.Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med. 1998;338(13):867–872. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- 27.Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPARgamma. J Clin Invest. 2003;112(6):945–955. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, Lehmann JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsen TM, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int J Obes Relat Metab Disord. 2003;27(2):147–161. doi: 10.1038/sj.ijo.802223. [DOI] [PubMed] [Google Scholar]
- 30.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93(2):229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 31.Raskin P, Rendell M, Riddle MC, Dole JF, Freed MI, Rosenstock J, Rosiglitazone Clinical Trials Study G A randomized trial of rosiglitazone therapy in patients with inadequately controlled insulin-treated type 2 diabetes. Diabetes Care. 2001;24(7):1226–1232. doi: 10.2337/diacare.24.7.1226. [DOI] [PubMed] [Google Scholar]
- 32.Aronoff S, Rosenblatt S, Braithwaite S, Egan JW, Mathisen AL, Schneider RL. Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes: a 6-month randomized placebo-controlled dose-response study. The Pioglitazone 001 Study Group. Diabetes Care. 2000;23(11):1605–1611. doi: 10.2337/diacare.23.11.1605. [DOI] [PubMed] [Google Scholar]
- 33.Glintborg D, Andersen M, Hagen C, Heickendorff L, Hermann AP. Association of pioglitazone treatment with decreased bone mineral density in obese premenopausal patients with polycystic ovary syndrome: a randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2008;93(5):1696–1701. doi: 10.1210/jc.2007-2249. [DOI] [PubMed] [Google Scholar]
- 34.Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-gamma in the regulation of energy balance. Nat Med. 2011;17(5):623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu M, Sarruf DA, Talukdar S, Sharma S, Li P, Bandyopadhyay G, Nalbandian S, Fan W, Gayen JR, Mahata SK, Webster NJ, Schwartz MW, Olefsky JM. Brain PPAR-gamma promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat Med. 2011;17(5):618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11(8):861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- 37.Borsting E, Cheng VP, Glass CK, Vallon V, Cunard R. Peroxisome proliferator-activated receptor-gamma agonists repress epithelial sodium channel expression in the kidney. Am J Physiol Renal Physiol. 2012;302(5):F540–551. doi: 10.1152/ajprenal.00306.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13(12):1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- 39.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146(3):1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 40.Wei W, Wang X, Yang M, Smith LC, Dechow PC, Sonoda J, Evans RM, Wan Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010;11(6):503–516. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47(4):507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 42.Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol. 2000;20(13):4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437(7059):759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ketsawatsomkron P, Pelham CJ, Groh S, Keen HL, Faraci FM, Sigmund CD. Does peroxisome proliferator-activated receptor-gamma (PPAR gamma) protect from hypertension directly through effects in the vasculature? The Journal of biological chemistry. 2010;285(13):9311–9316. doi: 10.1074/jbc.R109.025031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, Lascols O, Capeau J, Caron-Debarle M. Peroxisome proliferator-activated receptor-gamma mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2013;33(4):829–838. doi: 10.1161/ATVBAHA.112.300962. [DOI] [PubMed] [Google Scholar]
- 46.Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, Tan MH, Khan MA, Perez AT, Jacober SJ, Investigators GS A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. 2005;28(7):1547–1554. doi: 10.2337/diacare.28.7.1547. [DOI] [PubMed] [Google Scholar]
- 47.Reddy AT, Lakshmi SP, Kleinhenz JM, Sutliff RL, Hart CM, Reddy RC. Endothelial cell peroxisome proliferator-activated receptor gamma reduces endotoxemic pulmonary inflammation and injury. J Immunol. 2012;189(11):5411–5420. doi: 10.4049/jimmunol.1201487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qu A, Shah YM, Manna SK, Gonzalez FJ. Disruption of endothelial peroxisome proliferator-activated receptor gamma accelerates diet-induced atherogenesis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2012;32(1):65–73. doi: 10.1161/ATVBAHA.111.239137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seno T, Hamaguchi M, Ashihara E, Kohno M, Ishino H, Yamamoto A, Kadoya M, Nakamura K, Murakami K, Matoba S, Maekawa T, Kawahito Y. 15-Deoxy-Delta(1)(2),(1)(4) prostaglandin J(2) reduces the formation of atherosclerotic lesions in apolipoprotein E knockout mice. PLoS One. 2011;6(10):e25541. doi: 10.1371/journal.pone.0025541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118(5):1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Tian XY, Mao G, Fang X, Fung ML, Shyy JY, Huang Y, Wang N. Peroxisome proliferator-activated receptor-gamma ameliorates pulmonary arterial hypertension by inhibiting 5-hydroxytryptamine 2B receptor. Hypertension. 2012;60(6):1471–1478. doi: 10.1161/HYPERTENSIONAHA.112.198887. [DOI] [PubMed] [Google Scholar]
- 52.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O’Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 53.Jain M, Budinger GR, Lo A, Urich D, Rivera SE, Ghosh AK, Gonzalez A, Chiarella SE, Marks K, Donnelly HK, Soberanes S, Varga J, Radigan KA, Chandel NS, Mutlu GM. Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-gamma. Am J Respir Crit Care Med. 2011;183(11):1490–1498. doi: 10.1164/rccm.201009-1409OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-delta(12,14)-prostaglandin J(2) (15D-PGJ(2)), a peroxisome proliferator activated receptor gamma ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24(1):59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 55.Wu WT, Lee CC, Lee CJ, Subeq YM, Lee RP, Hsu BG. Rosiglitazone ameliorates endotoxin-induced organ damage in conscious rats. Biol Res Nurs. 2011;13(1):38–43. doi: 10.1177/1099800409353358. [DOI] [PubMed] [Google Scholar]
- 56.Baker PR, Schopfer FJ, O’Donnell VB, Freeman BA. Convergence of nitric oxide and lipid signaling: anti-inflammatory nitro-fatty acids. Free Radic Biol Med. 2009;46(8):989–1003. doi: 10.1016/j.freeradbiomed.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. The Journal of biological chemistry. 2005;280(51):42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H, Liu H, Jia Z, Guan G, Yang T. Effects of Endogenous PPAR Agonist Nitro-Oleic Acid on Metabolic Syndrome in Obese Zucker Rats. PPAR Res. 2010;2010:601562. doi: 10.1155/2010/601562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freeman BA, Baker PR, Schopfer FJ, Woodcock SR, Napolitano A, d’Ischia M. Nitro-fatty acid formation and signaling. The Journal of biological chemistry. 2008;283(23):15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schopfer FJ, Cole MP, Groeger AL, Chen CS, Khoo NK, Woodcock SR, Golin-Bisello F, Motanya UN, Li Y, Zhang J, Garcia-Barrio MT, Rudolph TK, Rudolph V, Bonacci G, Baker PR, Xu HE, Batthyany CI, Chen YE, Hallis TM, Freeman BA. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: selective ligand activity and anti-diabetic signaling actions. The Journal of biological chemistry. 2010;285(16):12321–12333. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, Nagy L, Yamamoto K, Schwabe JW. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol. 2008;15(9):924–931. doi: 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reddy AT, Lakshmi SP, Reddy RC. The Nitrated Fatty Acid 10-Nitro-oleate Diminishes Severity of LPS-Induced Acute Lung Injury in Mice. PPAR Res. 2012;2012:617063. doi: 10.1155/2012/617063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddy AT, Lakshmi SP, Dornadula S, Pinni S, Rampa DR, Reddy RC. The nitrated fatty acid 10-nitro-oleate attenuates allergic airway disease. J Immunol. 2013;191(5):2053–2063. doi: 10.4049/jimmunol.1300730. [DOI] [PubMed] [Google Scholar]
- 65.Borniquel S, Jansson EA, Cole MP, Freeman BA, Lundberg JO. Nitrated oleic acid up-regulates PPARgamma and attenuates experimental inflammatory bowel disease. Free Radic Biol Med. 2010;48(4):499–505. doi: 10.1016/j.freeradbiomed.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Von Knethen A, Brune B. Activation of peroxisome proliferator-activated receptor gamma by nitric oxide in monocytes/macrophages down-regulates p47phox and attenuates the respiratory burst. J Immunol. 2002;169(5):2619–2626. doi: 10.4049/jimmunol.169.5.2619. [DOI] [PubMed] [Google Scholar]
- 67.Hata K, Nishimura R, Ikeda F, Yamashita K, Matsubara T, Nokubi T, Yoneda T. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol Biol Cell. 2003;14(2):545–555. doi: 10.1091/mbc.E02-06-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maekawa T, Jin W, Ishii S. The role of ATF-2 family transcription factors in adipocyte differentiation: antiobesity effects of p38 inhibitors. Mol Cell Biol. 2010;30(3):613–625. doi: 10.1128/MCB.00685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aouadi M, Jager J, Laurent K, Gonzalez T, Cormont M, Binetruy B, Le Marchand-Brustel Y, Tanti JF, Bost F. p38MAP Kinase activity is required for human primary adipocyte differentiation. FEBS Lett. 2007;581(29):5591–5596. doi: 10.1016/j.febslet.2007.10.064. [DOI] [PubMed] [Google Scholar]
- 70.Engelman JA, Berg AH, Lewis RY, Lin A, Lisanti MP, Scherer PE. Constitutively active mitogen-activated protein kinase kinase 6 (MKK6) or salicylate induces spontaneous 3T3-L1 adipogenesis. The Journal of biological chemistry. 1999;274(50):35630–35638. doi: 10.1074/jbc.274.50.35630. [DOI] [PubMed] [Google Scholar]
- 71.Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24(7):3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Y, Guo H, Deis JA, Mashek MG, Zhao M, Ariyakumar D, Armien AG, Bernlohr DA, Mashek DG, Chen X. Lipocalin 2 regulates brown fat activation via a nonadrenergic activation mechanism. The Journal of biological chemistry. 2014;289(32):22063–22077. doi: 10.1074/jbc.M114.559104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8(5):971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 74.Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument-Bromage H, Tempst P, Spiegelman BM. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18(3):278–289. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Teruel T, Hernandez R, Benito M, Lorenzo M. Rosiglitazone and retinoic acid induce uncoupling protein-1 (UCP-1) in a p38 mitogen-activated protein kinase-dependent manner in fetal primary brown adipocytes. The Journal of biological chemistry. 2003;278(1):263–269. doi: 10.1074/jbc.M207200200. [DOI] [PubMed] [Google Scholar]
- 76.Bratton MR, Frigo DE, Vigh-Conrad KA, Fan D, Wadsworth S, McLachlan JA, Burow ME. Organochlorine-mediated potentiation of the general coactivator p300 through p38 mitogen-activated protein kinase. Carcinogenesis. 2009;30(1):106–113. doi: 10.1093/carcin/bgn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chhikara M, Wang S, Kern SJ, Ferreyra GA, Barb JJ, Munson PJ, Danner RL. Carbon monoxide blocks lipopolysaccharide-induced gene expression by interfering with proximal TLR4 to NF-kappaB signal transduction in human monocytes. PLoS One. 2009;4(12):e8139. doi: 10.1371/journal.pone.0008139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haschemi A, Chin BY, Jeitler M, Esterbauer H, Wagner O, Bilban M, Otterbein LE. Carbon monoxide induced PPARgamma SUMOylation and UCP2 block inflammatory gene expression in macrophages. PLoS One. 2011;6(10):e26376. doi: 10.1371/journal.pone.0026376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bilban M, Bach FH, Otterbein SL, Ifedigbo E, d’Avila JC, Esterbauer H, Chin BY, Usheva A, Robson SC, Wagner O, Otterbein LE. Carbon monoxide orchestrates a protective response through PPARgamma. Immunity. 2006;24(5):601–610. doi: 10.1016/j.immuni.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 80.Lennon AM, Ramauge M, Dessouroux A, Pierre M. MAP kinase cascades are activated in astrocytes and preadipocytes by 15-deoxy-Delta(12–14)-prostaglandin J(2) and the thiazolidinedione ciglitazone through peroxisome proliferator activator receptor gamma-independent mechanisms involving reactive oxygenated species. The Journal of biological chemistry. 2002;277(33):29681–29685. doi: 10.1074/jbc.M201517200. [DOI] [PubMed] [Google Scholar]
- 81.Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97(4):372–379. doi: 10.1161/01.RES.0000179226.34112.6d. [DOI] [PubMed] [Google Scholar]
- 82.Gardner OS, Shiau CW, Chen CS, Graves LM. Peroxisome proliferator-activated receptor gamma-independent activation of p38 MAPK by thiazolidinediones involves calcium/calmodulin-dependent protein kinase II and protein kinase R: correlation with endoplasmic reticulum stress. The Journal of biological chemistry. 2005;280(11):10109–10118. doi: 10.1074/jbc.M410445200. [DOI] [PubMed] [Google Scholar]
- 83.Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi-Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez-Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ, Gakidou E. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384(9945):766–781. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2015;517(7534):391–395. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hocking S, Samocha-Bonet D, Milner KL, Greenfield JR, Chisholm DJ. Adiposity and insulin resistance in humans: the role of the different tissue and cellular lipid depots. Endocr Rev. 2013;34(4):463–500. doi: 10.1210/er.2012-1041. [DOI] [PubMed] [Google Scholar]
- 86.Ma X, Lee P, Chisholm DJ, James DE. Control of adipocyte differentiation in different fat depots; implications for pathophysiology or therapy. Front Endocrinol (Lausanne) 2015;6:1. doi: 10.3389/fendo.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102(4):401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carey DG, Cowin GJ, Galloway GJ, Jones NP, Richards JC, Biswas N, Doddrell DM. Effect of rosiglitazone on insulin sensitivity and body composition in type 2 diabetic patients [corrected] Obes Res. 2002;10(10):1008–1015. doi: 10.1038/oby.2002.137. [DOI] [PubMed] [Google Scholar]
- 89.Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87(6):2784–2791. doi: 10.1210/jcem.87.6.8567. [DOI] [PubMed] [Google Scholar]
- 90.Adams M, Montague CT, Prins JB, Holder JC, Smith SA, Sanders L, Digby JE, Sewter CP, Lazar MA, Chatterjee VK, O’Rahilly S. Activators of peroxisome proliferator-activated receptor gamma have depot-specific effects on human preadipocyte differentiation. J Clin Invest. 1997;100(12):3149–3153. doi: 10.1172/JCI119870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hirose H, Kawai T, Yamamoto Y, Taniyama M, Tomita M, Matsubara K, Okazaki Y, Ishii T, Oguma Y, Takei I, Saruta T. Effects of pioglitazone on metabolic parameters, body fat distribution, and serum adiponectin levels in Japanese male patients with type 2 diabetes. Metabolism. 2002;51(3):314–317. doi: 10.1053/meta.2002.30506. [DOI] [PubMed] [Google Scholar]
- 92.Fujisawa K, Nishikawa T, Kukidome D, Imoto K, Yamashiro T, Motoshima H, Matsumura T, Araki E. TZDs reduce mitochondrial ROS production and enhance mitochondrial biogenesis. Biochem Biophys Res Commun. 2009;379(1):43–48. doi: 10.1016/j.bbrc.2008.11.141. [DOI] [PubMed] [Google Scholar]
- 93.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. The Journal of biological chemistry. 1997;272(8):5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 94.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274(5295):2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 95.Camp HS, Tafuri SR, Leff T. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-gamma1 and negatively regulates its transcriptional activity. Endocrinology. 1999;140(1):392–397. doi: 10.1210/endo.140.1.6457. [DOI] [PubMed] [Google Scholar]
- 96.Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, Kaestner KH, Lazar MA. Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Dev Cell. 2003;5(4):657–663. doi: 10.1016/s1534-5807(03)00274-0. [DOI] [PubMed] [Google Scholar]
- 97.Helenius K, Yang Y, Alasaari J, Makela TP. Mat1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipocyte differentiation. Mol Cell Biol. 2009;29(2):315–323. doi: 10.1128/MCB.00347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466(7305):451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429(6993):771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van Beekum O, Brenkman AB, Grontved L, Hamers N, van den Broek NJ, Berger R, Mandrup S, Kalkhoven E. The adipogenic acetyltransferase Tip60 targets activation function 1 of peroxisome proliferator-activated receptor gamma. Endocrinology. 2008;149(4):1840–1849. doi: 10.1210/en.2007-0977. [DOI] [PubMed] [Google Scholar]
- 101.Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T. SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010;38(21):7458–7471. doi: 10.1093/nar/gkq609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, Accili D. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell. 2012;150(3):620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yamashita D, Yamaguchi T, Shimizu M, Nakata N, Hirose F, Osumi T. The transactivating function of peroxisome proliferator-activated receptor gamma is negatively regulated by SUMO conjugation in the amino-terminal domain. Genes Cells. 2004;9(11):1017–1029. doi: 10.1111/j.1365-2443.2004.00786.x. [DOI] [PubMed] [Google Scholar]
- 104.Floyd ZE, Stephens JM. Control of peroxisome proliferator-activated receptor gamma2 stability and activity by SUMOylation. Obes Res. 2004;12(6):921–928. doi: 10.1038/oby.2004.112. [DOI] [PubMed] [Google Scholar]
- 105.Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell. 2012;148(3):556–567. doi: 10.1016/j.cell.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang XJ, Gregoire S. A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol Cell. 2006;23(6):779–786. doi: 10.1016/j.molcel.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 107.Kilroy GE, Zhang X, Floyd ZE. PPAR-gamma AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity (Silver Spring) 2009;17(4):665–673. doi: 10.1038/oby.2008.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. The Journal of biological chemistry. 2003;278(13):11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 109.Mancini AD, Poitout V. The fatty acid receptor FFA1/GPR40 a decade later: how much do we know? Trends Endocrinol Metab. 2013;24(8):398–407. doi: 10.1016/j.tem.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 110.Gras D, Chanez P, Urbach V, Vachier I, Godard P, Bonnans C. Thiazolidinediones induce proliferation of human bronchial epithelial cells through the GPR40 receptor. Am J Physiol Lung Cell Mol Physiol. 2009;296(6):L970–978. doi: 10.1152/ajplung.90219.2008. [DOI] [PubMed] [Google Scholar]
- 111.Mohammad S. GPR40 Agonists for the Treatment of Type 2 Diabetes Mellitus: Benefits and Challenges. Curr Drug Targets. 2015 doi: 10.2174/1389450117666151209122702. [DOI] [PubMed] [Google Scholar]
- 112.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422(6928):173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 113.Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1(4):245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 114.Nagasumi K, Esaki R, Iwachidow K, Yasuhara Y, Ogi K, Tanaka H, Nakata M, Yano T, Shimakawa K, Taketomi S, Takeuchi K, Odaka H, Kaisho Y. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes. 2009;58(5):1067–1076. doi: 10.2337/db08-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, Kenakin TP, Andrews JL, Ammala C, Fornwald JA, Ignar DM, Jenkinson S. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148(5):619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vettor R, Granzotto M, De Stefani D, Trevellin E, Rossato M, Farina MG, Milan G, Pilon C, Nigro A, Federspil G, Vigneri R, Vitiello L, Rizzuto R, Baratta R, Frittitta L. Loss-of-function mutation of the GPR40 gene associates with abnormal stimulated insulin secretion by acting on intracellular calcium mobilization. J Clin Endocrinol Metab. 2008;93(9):3541–3550. doi: 10.1210/jc.2007-2680. [DOI] [PubMed] [Google Scholar]
- 117.Poitout V, Lin DC. Modulating GPR40: therapeutic promise and potential in diabetes. Drug Discov Today. 2013;18(23–24):1301–1308. doi: 10.1016/j.drudis.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 118.Luo J, Swaminath G, Brown SP, Zhang J, Guo Q, Chen M, Nguyen K, Tran T, Miao L, Dransfield PJ, Vimolratana M, Houze JB, Wong S, Toteva M, Shan B, Li F, Zhuang R, Lin DC. A potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS One. 2012;7(10):e46300. doi: 10.1371/journal.pone.0046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lan H, Hoos LM, Liu L, Tetzloff G, Hu W, Abbondanzo SJ, Vassileva G, Gustafson EL, Hedrick JA, Davis HR. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes. 2008;57(11):2999–3006. doi: 10.2337/db08-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3(3):397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 121.Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun. 2003;301(2):406–410. doi: 10.1016/s0006-291x(02)03064-4. [DOI] [PubMed] [Google Scholar]
- 122.Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. The Journal of biological chemistry. 2009;284(26):17527–17539. doi: 10.1074/jbc.M109.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Choi SS, Kim ES, Koh M, Lee SJ, Lim D, Yang YR, Jang HJ, Seo KA, Min SH, Lee IH, Park SB, Suh PG, Choi JH. A novel non-agonist peroxisome proliferator-activated receptor gamma (PPARgamma) ligand UHC1 blocks PPARgamma phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. The Journal of biological chemistry. 2014;289(38):26618–26629. doi: 10.1074/jbc.M114.566794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Amato AA, Rajagopalan S, Lin JZ, Carvalho BM, Figueira AC, Lu J, Ayers SD, Mottin M, Silveira RL, Souza PC, Mourao RH, Saad MJ, Togashi M, Simeoni LA, Abdalla DS, Skaf MS, Polikparpov I, Lima MC, Galdino SL, Brennan RG, Baxter JD, Pitta IR, Webb P, Phillips KJ, Neves FA. GQ-16, a novel peroxisome proliferator-activated receptor gamma (PPARgamma) ligand, promotes insulin sensitization without weight gain. The Journal of biological chemistry. 2012;287(33):28169–28179. doi: 10.1074/jbc.M111.332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mieczkowska A, Basle MF, Chappard D, Mabilleau G. Thiazolidinediones induce osteocyte apoptosis by a G protein-coupled receptor 40-dependent mechanism. The Journal of biological chemistry. 2012;287(28):23517–23526. doi: 10.1074/jbc.M111.324814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wauquier F, Philippe C, Leotoing L, Mercier S, Davicco MJ, Lebecque P, Guicheux J, Pilet P, Miot-Noirault E, Poitout V, Alquier T, Coxam V, Wittrant Y. The free fatty acid receptor G protein-coupled receptor 40 (GPR40) protects from bone loss through inhibition of osteoclast differentiation. The Journal of biological chemistry. 2013;288(9):6542–6551. doi: 10.1074/jbc.M112.429084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang S, Awad KS, Elinoff JM, Dougherty EJ, Ferreyra GA, Wang JY, Cai R, Sun J, Ptasinska A, Danner RL. G Protein-coupled Receptor 40 (GPR40) and Peroxisome Proliferator-activated Receptor gamma (PPARgamma): AN INTEGRATED TWO-RECEPTOR SIGNALING PATHWAY. The Journal of biological chemistry. 2015;290(32):19544–19557. doi: 10.1074/jbc.M115.638924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kang DS, Kwon CH, Park JY, Kim JH, Woo JS, Jung JS, Kim YK. 15-deoxy-Delta 12,14-prostaglandin J2 induces renal epithelial cell death through NF-kappaB-dependent and MAPK-independent mechanism. Toxicol Appl Pharmacol. 2006;216(3):426–435. doi: 10.1016/j.taap.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 129.Takeda K, Ichiki T, Tokunou T, Iino N, Takeshita A. 15-Deoxy-delta 12,14-prostaglandin J2 and thiazolidinediones activate the MEK/ERK pathway through phosphatidylinositol 3-kinase in vascular smooth muscle cells. The Journal of biological chemistry. 2001;276(52):48950–48955. doi: 10.1074/jbc.M108722200. [DOI] [PubMed] [Google Scholar]
- 130.Wilmer WA, Dixon C, Lu L, Hilbelink T, Rovin BH. A cyclopentenone prostaglandin activates mesangial MAP kinase independently of PPARgamma. Biochem Biophys Res Commun. 2001;281(1):57–62. doi: 10.1006/bbrc.2001.4301. [DOI] [PubMed] [Google Scholar]
- 131.Kitz K, Windischhofer W, Leis HJ, Huber E, Kollroser M, Malle E. 15-Deoxy-Delta12,14-prostaglandin J2 induces Cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic Biol Med. 2011;50(7):854–865. doi: 10.1016/j.freeradbiomed.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 132.Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidovic D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477(7365):477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McGuire DK, Inzucchi SE. New drugs for the treatment of diabetes mellitus: part I: Thiazolidinediones and their evolving cardiovascular implications. Circulation. 2008;117(3):440–449. doi: 10.1161/CIRCULATIONAHA.107.704080. [DOI] [PubMed] [Google Scholar]
- 134.Mittal N, Sanyal SN. Effect of exogenous surfactant on phosphatidylinositol 3-kinase-Akt pathway and peroxisome proliferator activated receptor-gamma during endotoxin induced acute respiratory distress syndrome. Mol Cell Biochem. 2012;361(1–2):135–141. doi: 10.1007/s11010-011-1097-6. [DOI] [PubMed] [Google Scholar]
- 135.Liu C, Feng T, Zhu N, Liu P, Han X, Chen M, Wang X, Li N, Li Y, Xu Y, Si S. Identification of a novel selective agonist of PPARgamma with no promotion of adipogenesis and less inhibition of osteoblastogenesis. Scientific reports. 2015;5:9530. doi: 10.1038/srep09530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zheng W, Qiu L, Wang R, Feng X, Han Y, Zhu Y, Chen D, Liu Y, Jin L, Li Y. Selective targeting of PPARgamma by the natural product chelerythrine with a unique binding mode and improved antidiabetic potency. Scientific reports. 2015;5:12222. doi: 10.1038/srep12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhou C, Tang C, Chang E, Ge M, Lin S, Cline E, Tan CP, Feng Y, Zhou YP, Eiermann GJ, Petrov A, Salituro G, Meinke P, Mosley R, Akiyama TE, Einstein M, Kumar S, Berger J, Howard AD, Thornberry N, Mills SG, Yang L. Discovery of 5-aryloxy-2,4-thiazolidinediones as potent GPR40 agonists. Bioorg Med Chem Lett. 2010;20(3):1298–1301. doi: 10.1016/j.bmcl.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 138.Tan CP, Feng Y, Zhou YP, Eiermann GJ, Petrov A, Zhou C, Lin S, Salituro G, Meinke P, Mosley R, Akiyama TE, Einstein M, Kumar S, Berger JP, Mills SG, Thornberry NA, Yang L, Howard AD. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabsetes. 2008;57(8):2211–2219. doi: 10.2337/db08-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]