

Abstract
Aberrant cell cycle progression is a hallmark feature of cancer cells. Cyclin-dependent kinases 4 and 6 (CDK4/6) drive progression through the G1 stage of the cell cycle, at least in part, by inactivating the tumor suppressor, retinoblastoma (RB). CDK4/6 are targetable and the selective CDK4/6 inhibitor, palbociclib, was recently FDA approved for the treatment of estrogen receptor-positive, HER2-negative advanced breast cancer. In cutaneous melanoma, driver mutations in NRAS and BRAF promote CDK4/6 activation suggesting that inhibitors such as palbociclib are likely to provide therapeutic benefit in combination with BRAF inhibitors and/or MEK inhibitors that are FDA-approved. However, the determinants of the response to CDK4/6 inhibitors alone and in combination with other targeted inhibitors are poorly defined. Furthermore, in vivo systems to quantitatively and temporally measure the efficacy of CDK4/6 inhibitors and determine the extent that CDK activity is reactivated during acquired resistance are lacking. Here, we describe the heterogeneous effects of CDK4/6 inhibitors, the expression of anti-apoptotic proteins that associate with response to CDK4/6 and MEK inhibitors, and the development of a luciferase-based reporter system to determine the effects of CDK4/6 inhibitors alone and in combination with MEK inhibitors in melanoma xenografts. These findings are likely to inform on-going and future clinical trials utilizing CDK4/6 inhibitors in cutaneous melanoma.
Introduction
Melanoma is the most lethal form of skin cancer and the prognosis of patients with metastases remains poor. Recent FDA-approved mono-therapies including immunotherapies and mutant BRAF inhibitors have provided effective treatment options for late-stage melanoma patients. However, immune checkpoint inhibitors have 20-60% response rates and are associated with serious toxicities (1). Mutant BRAF targeting achieves higher response rates but responses are short-term (2). The combination of BRAF and MEK inhibition displays response rates of almost 80% in mutant BRAF melanoma patients; nevertheless, median progression free survival remains under 12 months (3-6). Thus, there is a clear need for additional strategies to provide long-term clinical benefit to all genetic subtypes of melanoma patients.
Aberrant cell cycle progression is a hallmark feature of cancer (7). The cell cycle consists of distinct phases:G0 (quiescence), G1 (pre-DNA synthesis), S (DNA synthesis), G2 (pre-division) and M (cell division) and is tightly regulated by a network of cyclin dependent kinases (CDKs), cyclins and CDK inhibitors (CDKI). Commitment to the cell cycle occurs in G1 phase and involves CDK4/6 in association with D-type cyclins contributing to the inactivation of the tumor suppressor, retinoblastoma (RB). Although interphase CDKs are targetable, early generation CDK inhibitors were non-selective and showed limited therapeutic value in melanoma patients (8). The clinical actions of the selective CDK4/6 inhibitor, palbociclib (IBRANCE/PD-0332991) in estrogen receptor (ER)-positive/HER2-negative breast cancer (9-11) and mantle cell lymphoma (12) has rekindled interest in targeting cell cycle progression in cancer.
In melanoma, multiple mechanisms drive aberrant progression through the cell cycle, providing a rationale for therapeutically targeting CDK4/6. Mutations in BRAF and NRAS frequently activate the MEK-ERK1/2 pathway, which upregulates cyclin D1 (13). Inactivation of RB1 also occurs through CDK4 mutation, loss of functional CDKI proteins such as p16INK4A and p14ARF, and, to a lesser degree, loss of RB1 itself. This knowledge has led to studies analyzing the effects of targeting CDK4/6 in melanoma. In vitro studies show that loss of functional p16INK4A correlated with palbociclib sensitivity (14). Mutant NRAS extinction in an inducible NRAS genetically engineered mouse model decreased cell cycle progression via effects on the expression of CDK4 and increased apoptosis following MEK-ERK1/2 pathway inhibition (15).
Given the promising development of cell cycle intervention in melanoma, it will be crucial to understand the determinants of response to CDK4/6 inhibitors alone and in combination with other targeted agents. This will identify subgroups likely to benefit from CDK4/6 inhibitors and to assist in patient selection in clinical studies. Here, we found that concurrent targeting of CDK4/6 and MEK resulted in enhanced cell death in both BRAF and NRAS mutant melanoma cells. Mechanistic investigation uncovered one potential mediator of response to CDK4/6 plus MEK inhibitors as survivin. Furthermore, we corroborated our in vitro results to demonstrate significant tumor regressions in vivo with simultaneous CDK4/6 and MEK inhibition compared to single agents alone. The efficacy of the combination was demonstrated using a novel in vivo E2F activity reporter melanoma xenograft system to temporally quantitate the effect of the inhibitors and allow for the quantitative and temporal analysis of pathway reactivation during acquired resistance.
Materials and Methods
Cell culture
CHL-1 and A375 cells (purchased from ATCC, Manassas, VA in 2013 and 2005 respectively) were cultured in DMEM with 10% FBS. WM lines, SBcl2 and 1205Lu cells (donated by Dr. Meenhard Herlyn, Wistar Institute, Philadelphia, PA in 2005) were cultured in MCDB153 with 2% FBS, 20% Leibowitz L-15 medium, 5 μg/ml insulin. BOWES cells (donated by Dr. Mark Bracke, University Hospital, Ghent, Belgium in 2013) were cultured in MEM with 10% FBS and nonessential amino acids. SKMEL207 cells (donated by Dr. David Solit, Memorial Sloan Kettering, New York, NY in 2010) were cultured in RPMI with 10% FBS. Cell lines were authenticated by sequencing at the NRAS, BRAF and CDK4 loci and by STR analysis (completed January 2015). WM1346 are a morphologically distinct subclone of WM1366.
Reagents
Palbociclib (PD0332991) was provided by Pfizer, Inc (New York, NY). Trametinib (GSK1120212) and PD0325901 were purchased from Selleck Chemicals (Houston, TX).
Western blot analysis
Protein lysates were prepared in Laemmli sample buffer, separated by SDS-PAGE and proteins transferred to PVDF membranes. Immunoreactivity was detected using horseradish protein conjugate secondary antibodies (CalBioTech, Spring Valley, CA) and chemiluminescence substrate (ThermoScientific, Waltham, MA) on a Versadoc Imaging System (Bio-Rad, Hercules, CA). Primary antibodies used are listed in Supplementary Materials and Methods.
Reverse phase protein array (RPPA) analysis
A375 cells (2×105) were plated per 33 mm well and treated with DMSO, 5 nM trametinib, 0.5 μM palbociclib or the combination for 24 hours. Cells and tumor samples were lysed and proteins analyzed, as described (16).
Cell viability assay (MTT)
Cells (2×103) were cultured in 96 well plates with indicated inhibitors. Viable cells were measured at day 4. Thiazolyl Blue Tetrazolium Bromide (Sigma) was added to growth medium, incubated for 4 hours at 37°C and then solubilized overnight with equal volume of 10% sodium dodecyl sulfate/0.1 N HCl. A 96 well plate reader, Multiskan™ Spectrum (Thermo Scientific) was used to measure absorbance at 570 nM.
Colony formation assay
Cells (4×103) were plated per 33 mm well. Inhibitors were added and replenished every 3 days. After 7 days, cells were stained with crystal violet in formalin and imaged by a scanner.
Annexin V staining
After indicated treatments, cells were resuspended in 100 μl of binding buffer, stained with annexin V-APC for 15 minutes. Apoptosis was analyzed by flow cytometry on the FACS Calibur (BD Biosciences, San Jose, CA). Data were analyzed using Flowjo software (Tree Star Inc., Ashland, OR).
Cell cycle analysis
Cells were plated at 2×105 per 33 mm dish and treated, as indicated. Adherent and floating cells were pooled, pelleted, washed twice with ice-cold PBS and fixed by drop-wise addition of ice-cold 70% ethanol. Fixed cells were washed twice and resuspended in PBS, treated with RNase A solution (Sigma) at 100 mg/ml and stained with propidium iodide (Sigma) at 10 mg/ml for 30 min. Cell-cycle analysis was performed on BD LSR II (BD Biosciences). Data were analyzed using Flowjo software.
PCR array
A375 and SBcl2 cells (2×105) were treated as indicated for 24 hours before RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA) according to manufacturer's instructions. Reverse transcription was carried out using RT2 First Strand Kit from SuperArray Biosciences. PCR reactions were performed using the human apoptosis RT2 Profiler PCR Array (PAHS-012Z) containing 84 apoptosis-related genes on the BioRad MyIQ using RT2 SYBR Green with Fluorescein master mix.
Generation of reporter cells
1205LuTR, WM1366 and SKMEL207 cells were transduced with E2F-EGFP-firefly luciferase. Reporter expressing cells were selected for with 500 μg/ml geneticin (Invitrogen, Carlsbad, CA). 1205LuTR and WM1366 reporter cells expressing high basal EGFP following transduction were enriched by cell sorting for in vivo experiments. 1205LuTR and WM1366 cells were also transduced with tdTomato fluorescent protein.
siRNA transfection
Cells were transfected for 4 hours with chemically synthesized siRNA at a final concentration of 25 nMol/L using Lipofectamine RNAiMAX (Invitrogen) transfection reagent. RNAiMAX reagent was used for knockdowns (2 μl for all cells except SBcl2 cells for which 1.5 μl was utilized). Cells were harvested after 72 hours of knockdown. Non-targeting control and Bim#2 (#D-004383-05) siRNAs were purchased from Dharmacon, Inc. (Lafayette, CO). BIRC5 (#6351S and #6546S) and Bim#1 (#6461S) siRNAs were purchased from Cell Signaling.
Immunohistochemical analysis
1205Lu xenograft samples were obtained from mice that were fed control chow for 23 days and MEK inhibitor, CDK4/6 inhibitor or MEK+CDK4/6 inhibitor chow for 51 days. Tissue were fixed in formalin and paraffin-embedded. Sections were stained with anti-Ki67 antibody (#MAI-90584) from Thermo Scientific. The intensity of staining and percentage of positive cells were evaluated by A. Goldberg, MD in a blinded manner using the Aperio ScanScope® XT slide scanning system and quantification was done with the Aperio eSlide Manager software. Entire areas of staining were analyzed.
Human patient melanoma tumor explant treatment
Tumors were collected following patient consent at Thomas Jefferson University Hospital under IRB-approved protocol (#10D.341). Less than 24 hours post-surgery, excess adipose and stromal tissue was removed and tumors were cut into 1 mm3 pieces. Vetspon absorbable hemostatic gelatin 1 cm3 sponges (Novartis, Basel Switzerland) were pre-soaked in 12-welled plates for 15 minutes at 37°C in DMEM/10% FBS containing drugs or DMSO. To avoid concerns of intra-tumoral heterogeneity, up to three 1 mm3 pieces from different locations of the original tumor were placed per sponge per treatment condition. Medium was replaced every 24 hours. Tumor pieces were homogenized in modified RPPA lysis buffer (16).
Analysis of synergy data
For in vitro drug combination studies, single agents were added simultaneously at fixed ratios (1:100). Cell viability was determined using MTT assay. Data were analyzed by Calcusyn software using the Chou-Talalay method (17).
Luciferase assay
Cells were lysed after 48 hours of treatment and firefly activity was measured using the dual luciferase assay system kit (Promega) on a Glomax Luminometer (Promega).
In vivo experiments
Animal experiments were performed in a facility at Thomas Jefferson University that is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. Studies were approved by the Institutional Animal Care and Use Committee. For all experiments, female athymic mice (NU/J, Homozygous, Jackson, 6-8 weeks, 20-25g) were used. Xenografts were allowed to reach a 50-100 mm3 volume before they were sorted into four cohorts. For the 1205Lu palbociclib experiment, mice were fed with control (AIN-76A, n=4) and CDK4/6 inhibitor (AIN-76A with 429 mg/kg palbociclib, n=8). For the 1205Lu combination experiment, mice were fed with control (AIN-76A, n=6), MEK inhibitor (AIN-76A with a diet dose of 7 mg/kg PD0325901, n=10), CDK4/6 inhibitor (n=9) and combination chow (n=9). For WM1366 xenografts, mice were fed with control (n=5), MEK inhibitor (n=5), CDK4/6 inhibitor (n=5) and combination chow (n=6). All diets were produced by Research Diets, Inc. (New Brunswick, NJ). Digital caliper measurements calculated tumor volumes using the formula: volume = length × (width2/2). In vivo bioluminescence was conducted using the Caliper IVIS Lumina-XR System (Caliper Life Sciences, Hopkinton, MA) and data acquisition was conducted using LivingImage Version 4.0 software. For firefly luciferase, mice were imaged after 10 minutes of intraperitoneal injection of D-luciferin (100 μl of 15 mg/ml stock). Pharmacokinetic studies are described in Supplementary Materials and Methods.
Statistical analysis
In vitro data were analyzed using a two-tailed t test assuming unequal variance with error bars representing SD. In vivo statistical analysis is described in Supplementary Materials and Methods.
Results
CDK4/6 inhibition induces cell cycle arrest but not apoptosis in melanoma cells
To determine the sensitivity to CDK4/6 inhibition, we examined the GI50 for palbociclib in a genetically diverse panel of melanoma cell lines (Figure 1A, Table S1). The response to palbociclib was heterogeneous with wild-type (WT) BRAF and NRAS melanoma cell lines, CHL-1 and BOWES, showing the highest sensitivity (Figure 1B). By contrast, the mutant BRAF, RB1-null cell line, SKMEL207, showed the lowest sensitivity. No clear correlation was observed between CDKN2A mutational status or INK4 family protein expression and the response to palbociclib (Figure 1A, 1B and Table S1). For example, SBcl2 cells are p16INK4A-deficient concurrent with high CDK4 and cyclin D1 expression but were relatively resistant to CDK4/6 inhibition. Phosphorylation of RB1 and cyclin A2 expression were decreased within 24 hours of exposure to palbociclib with effects more evident at low concentrations (0.05 μM) in the more sensitive cell lines (Figure S1). Irrespective of the GI50, acute treatment with 0.5 μM palbociclib resulted in decreased phosphorylation/expression of RB1 and cyclin A2 in all cell lines except SKMEL207 (Figure 1C). We further treated a panel of cells with palbociclib and performed propidium iodide (PI)-based cell cycle analysis. Twenty-four hour treatment of palbociclib induced a G0/G1 cell cycle arrest but not cell death represented by a lack of subG1 accumulation (Figure 1D). Furthermore, palbociclib as a single agent initially suppressed 1205Lu tumor growth in vivo but progressive growth ensued (Figure 1E). The mean plasma concentrations of palbociclib in mice were 690 ng/ml and 670 ng/ml after 8 and 15 days of treatment (Figure S2). Taken together, these results show that the response of melanoma cells to palbociclib is heterogeneous, may not clearly stratify to any one genotype, and leads to cytostatic effects but neither cell death nor tumor regression.
Figure 1. Differential response of melanoma cells to CDK4/6 inhibition.
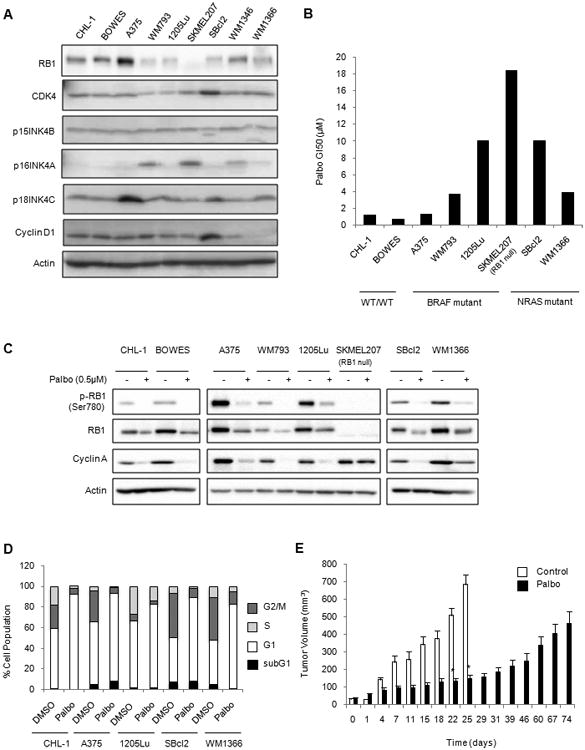
A. Western blot of potential biomarkers of response to palbociclib in melanoma lines. B. Sensitivity of melanoma cells to palbociclib. GI50 values were generated from dose-dependent curves from MTT cell viability assays. Each bar represents the average of three independent experiments. C. Palbociclib-treated melanoma cells were Western blotted for RB1 phosphorylation and expression of RB1 and cyclin A. D. Cells were treated with palbociclib for 24 hours and analyzed by PI staining. The relative distribution of cells in the subG1, G1, S and G2M phases of the cell cycle is shown. E. Mice bearing 1205Lu xenografts were treated with control chow (n=4) or palbociclib chow (n=8). (Error bars represent SEM, *p<0.05).
CDK4/6 and MEK inhibitors synergize to inhibit the growth of BRAF- and NRAS- mutant melanoma cell lines
Due to the inability of single agent palbociclib to induce cell death, we explored whether CDK4/6 targeting sensitized melanoma cells to MEK targeting. Treatment with the MEK inhibitor, trametinib, blocked the growth of all cell lines tested although SKMEL207 cells displayed decreased sensitivity (18) (Figure S3). Since the CDK4/6 plus MEK inhibitor combination represents a targeted inhibitor option that is applicable across all melanoma genotypes, cell lines were treated with palbociclib alone, trametinib alone or both inhibitors in combination over a fixed-ratio, 7-point concentration range for 96 hours. The combination of palbociclib and trametinib reduced the viability of BRAF and NRAS mutant cell lines compared to single agent treatments (Figure 2A). On the other hand, there was only a modest effect of the combinatorial treatment on CHL-1 and BOWES cells (both WT BRAF/WT NRAS) and SKMEL207. Calcusyn analysis revealed a strong synergism between the drug combination in mutant BRAF and mutant NRAS cells at median effective dose (ED50) but only moderate to slight synergism in CHL-1, BOWES and SKMEL207 cells (Figure 2B). Effective long-term responses to the combinatorial treatment in A375 and SBcl2 cells were also confirmed by colony formation assay (Figure 2C). Again, the palbociclib plus trametinib combination was less effective in CHL-1 and SKMEL207 cells compared to the other cell lines tested.
Figure 2. Enhanced effects of combined MEK and CDK4/6 inhibition in BRAF and NRAS mutant lines in vitro.
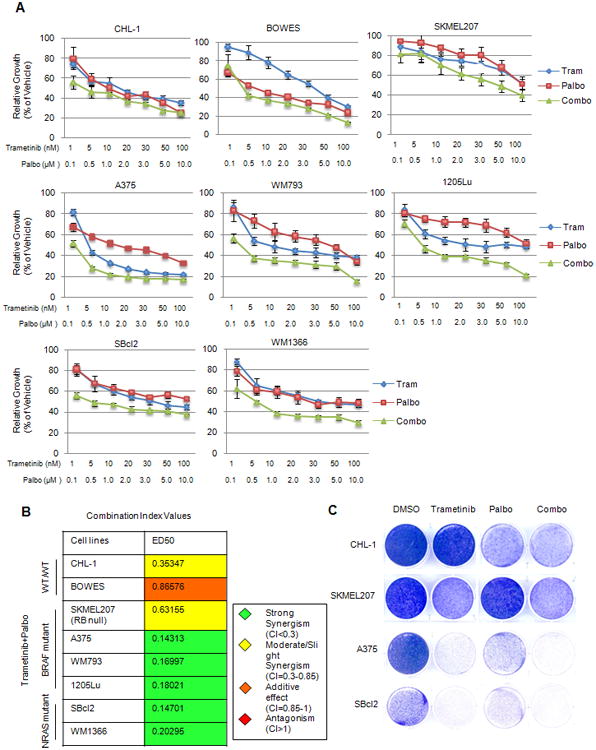
A. MTT cell viability assays of WT/WT, BRAF and NRAS mutant lines treated for four days with single agent (trametinib or palbociclib) or a fixed-ratio (1:100) combination (trametinib plus palbociclib) of both compounds (error bars represent SD). B. Calcusyn combination indices at median effective dose (ED50) generated from MTT assays in Figure A. C. Melanoma cells were plated at low density, treated with DMSO, trametinib (10 nM), palbociclib (1 μM) or the combination. After 1 week, cultures were stained with crystal violet.
Down-regulation of survivin is associated with a synergistic response to CDK4/6 plus MEK inhibitor combination
Next, we explored whether the enhanced effect seen with the combination of CDK4/6 and MEK inhibition was due to apoptosis. We observed increased annexin V staining when CDK4/6 and MEK were simultaneously inhibited in some cells (e.g. A375 and SBcl2) but not others (Figure 3A). In WM793 and 1205Lu cells, MEK inhibition alone was sufficient to induce apoptosis. Consistent with these data, trametinib increased cleaved PARP levels in cell lines that showed enhanced effects to the combination (Figure S4). To analyze effects on signaling, we performed RPPA profiling on A375 cells treated with either single agent or with the combination (Figure 3B). Phospho-RB1 and FOXM1, two established substrates of CDK4/6, were cooperatively repressed by co-inhibition of CDK4/6 and MEK. The pro-apoptotic protein, Bim-EL, was up-regulated by trametinib treatment but unaffected by palbociclib. Effects on Bim-EL levels in A375 cells were validated by Western blotting and also observed in combination inhibitor sensitive BRAF and NRAS mutant lines (Figure 3C). We detected moderate changes in Bim-EL levels in response to trametinib in the combination-resistant cell lines, CHL-1, BOWES and SKMEL207. Knockdown of Bim in 1205Lu cells partially inhibited trametinib plus palbociclib-induced apoptosis as detected by reduced annexin V staining (Figure 3D) showing a requirement of Bim.
Figure 3. Sensitivity to combined CDK4/6 and MEK inhibition is associated with survivin depletion in sensitive cells.
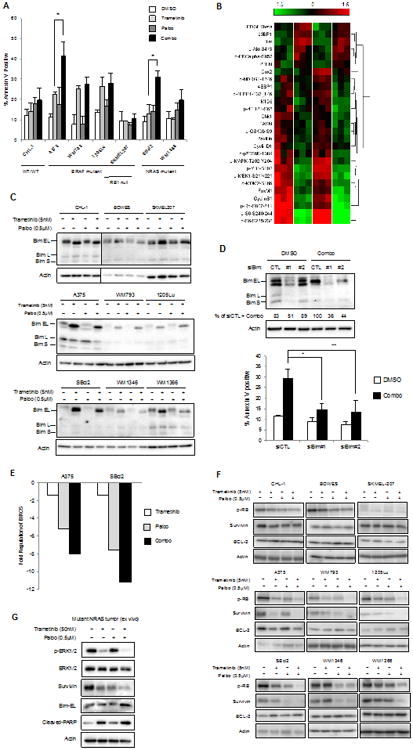
A. Representative annexin V staining of melanoma lines treated with trametinib (5 nM) and palbociclib (0.5 μM) treatment alone or a combination of both compounds for 48 hours (n=3, error bars=SD from triplicate samples, *p<0.05). B. A375 cells were treated with single agent or combination of trametinib and palbociclib for 24 hours. Lysates were analyzed by RPPA. The heatmap shows the most significantly regulated proteins (p<0.01). C. Elevated levels of Bim-EL in trametinib and combo-treated lysates. D. 1205Lu cells were transfected with siRNA to Bim in the presence or absence of trametinib and palbociclib. Knockdown of Bim rescued apoptotic phenotype elicited by trametinib and palbociclib treatments. E. Fold change in BIRC5/survivin regulation after 24 hours of treatment with indicated inhibitors. Two independent sets of tests were carried out and representative data is shown. F. Western blotting for survivin in resistant (CHL-1, Bowes, SKMEL207) and sensitive (A375, WM793, 1205Lu, SBcl2, WM1346, WM1366) cell lines in basal state as well as following treatment with trametinib and/or palbociclib for 48 hours. G. NRAS mutant melanoma tumor explant treated with DMSO, trametinib (50 nM), palbociclib (0.5 μM) or the combination.
Since Bim-EL was induced by MEK inhibition alone, we further explored the mechanistic basis of the drug synergy by testing for modulation of apoptotic genes on a quantitative PCR array. Among the 84 survival-related genes, BIRC5 exhibited the greatest change in levels in response to concurrent inhibition of MEK and CDK4/6 that was consistent between the two most sensitive cell lines, A375 and SBcl2 (Figure 3E, Figure S5). BIRC5 encodes for survivin, a protein belonging to the inhibitor of apoptosis (IAP) family (14). By Western blot, concurrent treatment with trametinib and palbociclib ablated survivin expression in sensitive cell lines, which coincided with the dephosphorylation of RB1 (Figure 3F). By contrast, the combination of trametinib and palbociclib did not affect survivin levels in less sensitive lines, CHL-1, BOWES, and SKMEL207. Although RNA expression of pro-survival protein, BCL-2, was modestly affected, we did not detect alterations at the protein level (Figure 3F, Figure S5). Finally, we utilized a primary human tumor explant culture to interrogate whether the combinatorial treatment led to an enhanced apoptotic response. Ex vivo treatment of NRAS-mutant melanoma tissue for 48 hours with single agents alone led to decreased survivin expression that was further suppressed in combination-treated tumors (Figure 3G). Bim-EL expression and cleaved PARP levels were also enhanced in combination-treated lysates. These data suggest that Bim-EL up-regulation and survivin down-regulation may be markers of the response to CDK4/6 and MEK inhibitor combinations.
Survivin is required for the survival of melanoma cells
To investigate whether survivin is required for survival, we silenced its expression in a panel of melanoma cells expressing various levels of survivin (Figure 4A, Figure S6). Knockdown of survivin led to a decrease in cell viability as compared to control transfectants in MTT assays (Figure 4B). In addition to its role in resistance to apoptosis, mitotic properties of survivin have been described (19); thus, we next examined whether depletion of survivin leads to cell cycle arrest in G2/M phase or cell death in melanoma cells by PI and annexin V staining. Survivin knockdown in a panel of melanoma cells led to accumulation in the sub-G1 phase post-knockdown indicative of apoptotic cell response but not G2/M accumulation after 72 hours (Figure 4C). Depletion of survivin at earlier time points (24 hours) did not induce an arrest in G2/M (data not shown). Similarly by annexin V staining, survivin depletion induced significant apoptosis in the majority of the cell lines we tested across different genotypes (Figure 4D). Next, we determined whether knockdown of survivin was capable of sensitizing combination-resistant CHL-1 cells to the CDK4/6-MEK inhibitor combination. Depletion of survivin in conjunction with combination treatment increased cell death in CHL-1 cells (Figure 4E). These data indicate that survivin depletion contributes to the response of melanoma cells to the combination of CDK4/6 and MEK inhibitors.
Figure 4. Survivin is essential for the survival of melanoma cells.
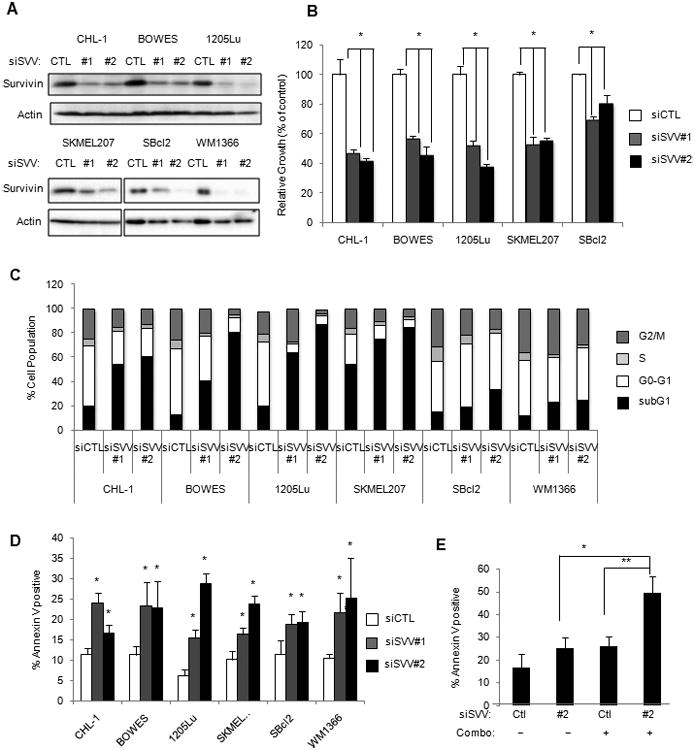
A. Cells were transfected with control or survivin-targeting siRNA #1 and #2 for 72 hours and lysates were analyzed by Western blot. B. Knockdown of survivin leads to decreased cell viability (n=4, error bars=SD *p<0.01). C. Cell cycle analysis after 72 hours of transfection with two distinct siRNA specific for survivin. Bar graphs were generated from averages of three independent knockdown experiments. D. Cells were transfected with control or two distinct survivin-targeting siRNA for 72 hours before they were analyzed for annexin V staining by flow cytometry (n=3, error bars=SD, *p<0.05). E. CHL-1 cells were transfected with control or survivin siRNA before they were treated with either DMSO or the combination of trametinib (5 nM) plus palbociclib (0.5 μM) (n=3, error bars=SD, *p<0.05 **p<0.01).
Combined CDK4/6 and MEK inhibition in vivo potently inhibits E2F reporter activity and regresses melanomas
Based on encouraging in vitro data suggesting the benefit of combined CDK4/6 and MEK inhibition, we extended our studies in vivo. We developed E2F-dependent luciferase reporter cell lines to enable temporal quantification of the effects of CDK4/6 inhibitor based treatments in a quantitative, non-invasive, and tumor-selective manner. In this reporter system, hyper-phosphorylation of RB1 leads to the uncoupling of E2F and E2F-mediated induction of firefly luciferase activity (Figure 5A). 1205Lu cells were chosen based on their ability to form tumors in vivo and their utility in a previously developed ERK1/2 reporter system (20). Tumor cells were also transduced with tdTomato fluorescent protein to selectively monitor tumor growth. In vitro, 1205Lu reporter cells showed a 60% reduction in firefly luciferase activity following 48 hours of treatment with palbociclib (Figure 5B). On the other hand, RB1-null SKMEL207 reporter cells did not show significant changes in firefly luciferase activity with palbociclib treatment suggesting that RB1 is required to modulate E2F activity (Figure 5B).
Figure 5. The combination of CDK4/6 and MEK inhibition is synergistic in vivo.
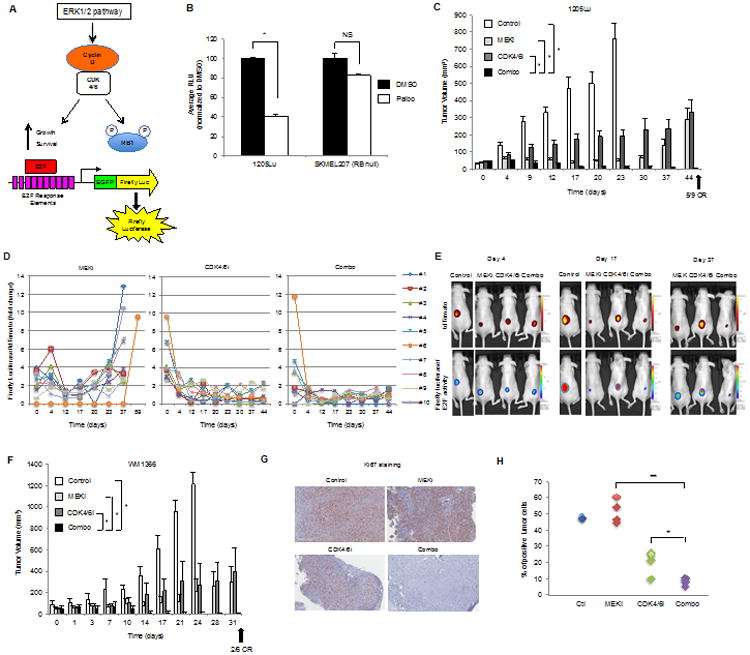
A. E2F reporter system for measuring the efficacy of CDK4/6 and MEK inhibitors in vivo. B. 1205LuTR and SKMEL207 reporter cells were treated with DMSO or palbociclib (0.5μM) for 48 hours (error bars=SD, *p<0.001, NS=not significant, n=3). C. Mice bearing1205Lu xenografts were treated with control, MEK inhibitor (PD0325901, 1 mg/kg daily dose) alone, CDK4/6 inhibitor (palbociclib, 75 mg/kg daily dose) alone, or in combination (error bars=SEM, *p<0.001 comparing combo to single agents and control). CR=complete response. D. Analysis of E2F reporter activity normalized to tdTomato fluorescent protein activity in 1205Lu xenografts. E. Images from individual mice bearing 1205Lu xenografts with tumor progression associated with high E2F reactivation in MEK inhibitor-treated mice and low E2F reactivation in CDK4/6 inhibitor and combo-treated inhibitor mice. F. Mice bearing WM1366 xenografts were treated with control, MEK inhibitor (PD0325901, 1mg/kg daily dose), CDK4/6 inhibitor (palbociclib, 75 mg/kg daily dose), or in combination (error bars=SE, *p<0.001 comparing combo to single agents and control). CR=complete response. G. Representative images of 1205Lu xenografts analyzed for Ki67. H. Quantitation of Ki67 positive cells taken from mice fed vehicle (n=2), MEK inhibitor (n=4), CDK4/6 inhibitor (n=4) and combo-laced chow (n=4). Combo versus single regiments, *p<0.05, **p<0.001.
Next, 1205Lu reporter cells were intradermally injected into immunodeficient nude mice. Tumors were allowed to form before treatment with control chow, MEK inhibitor (PD0325901) chow, CDK4/6 inhibitor (palbociclib) chow or chow containing both inhibitors. In the single agent and combination treatment cohorts, there was suppression of reporter activity that was more rapid in the CDK4/6 inhibitor-based treatments (Figures 5C, 5D and 5E). While MEK inhibitor initially maintained tumor volumes at a static level and even led to one complete regression (mouse #6), dramatic tumor regrowth was detected by treatment day 44 that was frequently preceded by strong E2F reactivation (Figures 5C, 5D, 5E and S7). Tumors from mice treated with CDK4/6 inhibitor alone showed low but continuous growth with persistent inhibition of the E2F pathway. Levels of E2F activity also remained low in tumors from mice treated with the combination of MEK plus CDK4/6 inhibitor and this regimen was the only treatment to produce significant tumor regressions in 1205Lu xenografts. Although combining inhibitors can induce toxicities, the weight of the mice in all four cohorts was comparable from the starting time to the time of termination (Figure S8).
Similar results on tumor growth were observed with a second reporter model in an NRAS-mutant melanoma background (Figure 5F, Figure S9). In contrast to the 1205Lu model, the response of WM1366 tumors to single agent treatment groups was heterogeneous; however, combination treatment resulted in uniform tumor regressions and resulted in two out of six complete responses (Figure 5F). E2F activity was diminished in the tumor (mouse #7) that showed complete response to the combination treatment (Figures S10 and S11). Fluctuations in E2F activity occurred in one tumor that did not completely regress (mouse #1) and E2F reactivation also preceded the increase in tumor volume/tdTomato activity in combination-resistant tumors (mouse #2 and #5).
Analyses of the harvested tumors from the 1205Lu reporter model showed fewer proliferating Ki67 positive cells in combination-treated samples compared to single agent treatments (Figure 5G and 5H). These data show that an E2F reporter xenograft model may be used to quantitate drug target inhibition and its association with tumor growth inhibition in vivo. Furthermore, the combination of a MEK inhibitor with palbociclib provides an effective therapeutic strategy for BRAF and NRAS-mutant melanomas.
E2F reactivation and rapid tumor regrowth from combo drug withdrawal
In the 1205Lu reporter model, five out of nine mice treated with the palbociclib/MEK inhibitor combination achieved complete response, as defined by undetectable caliper measurements (Figure 6A); however, we detected weak tdTomato signal (<0.8×1010 p/sec/cm2/sr) in position of the tumors in all five mice (Figure S12). To determine whether the complete responses were durable, these five mice were released from drug treatment (treatment day 51). Within one week off drug, a rapid reactivation of the E2F pathway and tumor regrowth were observed in all mice except one (mouse #1) (Figure 6B). The tumors re-responded rapidly to the combination drug as evident bydramatic inhibition of pathway activity. Overall there was only one complete response with no palpable tumor, residual tdTomato signal or E2F activity (mouse #2). We noticed fluctuations in E2F activity in the other four mice followed by tumor regrowth. Reactivation of the pathway was evident and predictive of tumors that would later acquire resistance to the combination therapy (e.g. mouse #6). These data suggest that residual disease remains after first-line treatment with CDK4/6 and MEK inhibitor combination and that tumors that regrow from residual disease off drug are likely to acquire resistance following re-treatment.
Figure 6. Tumor regrowth following MEK and CDK4/6 inhibitor withdrawal.
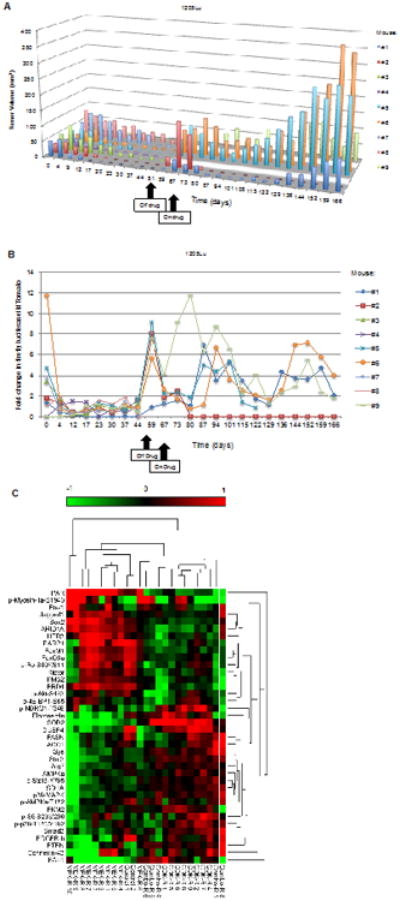
A. Mice bearing 1205Lu xenografts that showed complete response from the combination therapy were removed from treatment at day 51 and monitored for durable response. Mice were retreated with combination regimen at day 67. B. Modulation of E2F activity in combination treated mice following drug removal and retreatment. E2F reactivation is predictive of tumors that would later acquire resistance to the combination treatment. The tumor from mouse #5 exhibited necrosis by day 129 and was excluded from luciferase and tdTomato analysis. C. RPPA analysis on resistant tumors. Combination resistant (combo-R) tumors clustered with CDK inhibitor resistant (CDK-R) tumors using unsupervised hierarchical clustering. Combo-R 8 is a regressing tumor that was isolated during regression on treatment. MEK-R 9/10 was least similar to any other groups.
Finally, we utilized RPPA data to identify proteins regulated by tumor resistance. We clustered all samples based on a list of significant antibodies to reduce noise from random differences between groups. This separated the tumors into two major distinct clusters (Figure 6C). Notably, combination resistant tumors that regrew from subsequent withdrawal and retreatment of the inhibitors formed a clear cluster with CDK inhibitor resistant tumors. MEKR#9/#10 and Combo-R #8 formed their own clusters, as well as the control samples. Combo-R #8 was a sample that regressed on-treatment and was isolated during regression. Furthermore, pathway enrichment analysis resulted in Combo-R tumors versus CDK-R tumors having the fewest number of deregulated pathways whereas CDK-R tumors versus MEK-R tumors showed the largest number of deregulated pathways (Figure S13). Several significantly regulated proteins from the RPPA analysis were also validated in tumor samples. For instance, two proteins highly up-regulated in MEK-R tumors but absent in CDK-R and Combo-R tumors were stemness transcription factor, SOX2 and growth factor receptor, HER3/ERBB3 (Figure S14). Conversely, fibronectin and superoxide dismutase, SOD2 were up-regulated in CDK-R and Combo-R tumors. Taken together, these data suggest that combination-resistant tumors were molecularly similar to tumors that slowly progressed on palbociclib.
Discussion
Due to intratumoral heterogeneity and resistance mechanisms, single agent therapies frequently elicit transient effects in cutaneous melanoma patients (21, 22). It is critical to develop improved preclinical models that will monitor response and resistance to targeted agents in vivo. We describe enhanced response to combined CDK4/6 and MEK targeting in mutant BRAF and mutant NRAS melanomas both in vitro and in vivo. Combinatorial treatment-sensitive cells exhibited loss of survivin expression, indicating this may be a potential biomarker of response. Furthermore, we established a reporter system that allows the temporal quantification of changes in E2F activity in response to CDK4/6 and MEK inhibitors in melanoma cells. We show that reactivation of E2F occurs during acquired resistance in both MEK inhibitor and combination treated tumors.
Palbociclib is the first FDA-approved CDK4/6 selective inhibitor (9). Trials utilizing CDK4/6 inhibitors (palbociclib and LEE011) alone or in combination are underway in subsets of melanoma; however, to avoid their use in non-stratified clinical trials, the determinants of response must be examined. Preclinical studies in ER-positive breast cancer cells indicate that increased RB1 and cyclin D1 expression and loss of p16INK4A are associated with sensitivity to palbociclib (10). Similar alterations in patient tumors may indicate likelihood of response to CDK4/6 inhibition (14). Our studies did not show a clear correlation between genotypes or CDKN2 status and sensitivity to palbociclib; however, RB1 loss did confer resistance, as seen in other models (23).
Palbociclib likely inhibits tumor progression by inducing senescence in melanoma cells (24). Interestingly, transformed melanocytes but not normal primary melanocytes were susceptible to palbociclib-induced senescence. We evaluated the effects of palbociclib with a MEK inhibitor, a combination strategy that is broadly applicable to melanoma genotypes and may address the clinically unmet need of the lack of targeted therapies for BRAF wild-type patients. Recently, published data combining another CDK4/6 inhibitor, LY2835219 (25) with a BRAF inhibitor, vemurafenib, in BRAF mutant A375 xenografts led to additive effects in tumor growth inhibition (26). Here, we show increased cell death and tumor regression with concurrent inhibition of CDK4/6 and MEK in both BRAF and NRAS-mutant xenograft models. While Bim-EL was upregulated in MEK-inhibited cells only, the anti-apoptotic protein, survivin was further downregulated by the combination treatment. These data are consistent with survivin being an E2F-regulated gene (27). Furthermore, knockdown of survivin was sufficient to render primary resistant CHL-1 cells sensitive to the drug combination. While survivin has not been shown to interact with caspases directly, it heterodimerizes with and stabilizes XIAP, which in turn directly inhibits caspases 3, 7, and 9. Survivin functions in cytokinesis, inhibition of cell death and tumor cell invasion and metastasis (28,29). As part of the chromosomal passenger complex, survivin regulates the spindle assembly checkpoint by physically associating with inner centromere protein, aurora B and borealin. We do not rule out the possibility of cell death by mitotic catastrophe and multi-nucleation, as demonstrated by others (30). Survivin is not a traditional drug target and current targeting strategies involve interfering with survivin expression at the transcription level (31). Given the multiple functions of survivin and its association with worse outcomes in melanoma patients, survivin may not just be a determinant of response to CDK4/6 and MEK inhibitors but also a general pro-survival target.
We also describe the generation of an E2F reporter melanoma xenograft system to measure response and resistance to CDK4/6 inhibitors in vivo. This system measures pathway activity in a quantitative and tumor-specific manner. Analysis is non-invasive and, thus, permits, temporal measurements as opposed to single time point analysis provided by immunohistochemical staining. The system also determines re-activation of the pathway associated with acquired resistance. Finally, the inclusion of tdTomato allows the detection and regrowth of residual tumor, which is not completely removed by the combination treatment. Using the 1205Lu reporter model, we show differential effects on E2F firefly luciferase activity depending on the targeted agent treatment. Associated with acquired resistance to MEK inhibitor, E2F activity was dramatically induced and correlated with tumor regrowth. Under cytostatic actions of CDK4/6 inhibitor monotherapy, effective inhibition of E2F activity was sustained despite incremental but continual growth of tumors. The kinetics of tumor growth on CDK4/6 inhibitor monotherapy is distinct from the rapid regrowth associated with acquired resistance phenotype and presumably caused by a slower cell division rate.
Our data suggest that while the combination therapy was successful in leading to significant tumor regressions and removing tumor bulk, the remaining subpopulation of drug tolerant cells led to eventual resistance to the combination therapy after rounds of drug removal and retreatment. We observed fluctuations in E2F/firefly luciferase activity following retreatment of the tumors. Increases in tumor size as measured by tdTomato activity at specific time-points (e.g. day 129) could explain relatively lower E2F activity. We postulate that the tumors could have also acquired mutation(s) or signals that are E2F-independent. Detection, isolation and characterization are some of the challenges of studying dormant/drug tolerant tumor cells. Recent preclinical studies with BRAF inhibitors have suggested the use of an intermittent dosing schedule to delay the onset of resistance (32). Ongoing studies in our lab, comparing intermittent versus continuous scheduling of CDK4/6 plus MEK inhibitors will determine schedules that effectively delay the onset of resistance or reduce the population of dormant cells. Understanding the mechanisms by which these residual tumor cells survive treatment is a future priority.
Supplementary Material
Acknowledgments
We thank Neda Dadpey for technical assistance and Dr. Adam Berger for the fresh human melanoma tissue.
Grant Support: This study was supported by an Industrial Partnership Award from Melanoma Research Alliance and Pfizer, Inc., NIH R01 CA182635 and by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to A.E. Aplin. SKCC Flow Cytometry and Translational Pathology core facilities are supported by NIH/NCI Support Grant (P30 CA056036). The RPPA Core Facility at M.D Anderson is supported by a NCI Cancer Center Support Grant (CA16672).
Footnotes
Conflict of interest: This research was funded in part by Pfizer, Inc.
References
- 1.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DB, Flaherty KT, Weber JS, Infante JR, Kim KB, Kefford RF, et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697–704. doi: 10.1200/JCO.2014.57.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Burdette-Radoux S, Tozer RG, Lohmann RC, Quirt I, Ernst DS, Walsh W, et al. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest New Drugs. 2004;22:315–22. doi: 10.1023/B:DRUG.0000026258.02846.1c. [DOI] [PubMed] [Google Scholar]
- 9.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 10.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2015;373:209–19. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 12.Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119:4597–607. doi: 10.1182/blood-2011-10-388298. [DOI] [PubMed] [Google Scholar]
- 13.Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19:5320–8. doi: 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- 14.Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27:590–600. doi: 10.1111/pcmr.12228. [DOI] [PubMed] [Google Scholar]
- 15.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–10. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–21. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 17.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enz Reg. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 18.Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene. 2012;31:446–57. doi: 10.1038/onc.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altieri DC. Survivin - The inconvenient IAP. Semin Cell Dev Biol. 2015;39:91–96. doi: 10.1016/j.semcdb.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basile KJ, Abel EV, Dadpey N, Hartsough EJ, Fortina P, Aplin AE. In vivo MAPK reporting reveals the heterogeneity in tumoral selection of resistance to RAF inhibitors. Cancer Res. 2013;73:7101–10. doi: 10.1158/0008-5472.CAN-13-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–77. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- 22.Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–74. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 23.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018–32. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 24.Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–34. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32:825–37. doi: 10.1007/s10637-014-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther. 2014;13:2253–63. doi: 10.1158/1535-7163.MCT-14-0257. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y, Saavedra HI, Holloway MP, Leone G, Altura RA. Aberrant regulation of survivin by the RB/E2F family of proteins. J Biol Chem. 2004;279:40511–20. doi: 10.1074/jbc.M404496200. [DOI] [PubMed] [Google Scholar]
- 28.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenzie JA, Liu T, Goodson AG, Grossman D. Survivin enhances motility of melanoma cells by supporting Akt activation and {alpha}5 integrin upregulation. Cancer Res. 2010;70:7927–37. doi: 10.1158/0008-5472.CAN-10-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamers F, van der Ploeg I, Schild L, Ebus ME, Koster J, Hansen BR, et al. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocr Relat Cancer. 2011;18:657–68. doi: 10.1530/ERC-11-0207. [DOI] [PubMed] [Google Scholar]
- 31.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, et al. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–6. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 32.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.