

Abstract
Meiotic cells possess surveillance mechanisms that monitor critical events such as recombination and chromosome synapsis. Meiotic defects resulting from the absence of the synaptonemal complex component Zip1 activate a meiosis-specific checkpoint network resulting in delayed or arrested meiotic progression. Pch2 is an evolutionarily conserved AAA+ ATPase required for the checkpoint-induced meiotic block in the zip1 mutant, where Pch2 is only detectable at the ribosomal DNA array (nucleolus). We describe here that high levels of the Hop1 protein, a checkpoint adaptor that localizes to chromosome axes, suppress the checkpoint defect of a zip1 pch2 mutant restoring Mek1 activity and meiotic cell cycle delay. We demonstrate that the critical role of Pch2 in this synapsis checkpoint is to sustain Mec1-dependent phosphorylation of Hop1 at threonine 318. We also show that the ATPase activity of Pch2 is essential for its checkpoint function and that ATP binding to Pch2 is required for its localization. Previous work has shown that Pch2 negatively regulates Hop1 chromosome abundance during unchallenged meiosis. Based on our results, we propose that, under checkpoint-inducing conditions, Pch2 also possesses a positive action on Hop1 promoting its phosphorylation and its proper distribution on unsynapsed chromosome axes.
INTRODUCTION
During meiosis, accurate distribution of chromosomes to the gametes is ensured by the action of meiosis-specific surveillance mechanisms commonly known as the meiotic recombination checkpoint or pachytene checkpoint (1,2) and, more recently, broadly referred to as the meiotic checkpoint network (3). This checkpoint monitors those meiotic events, such as chromosome synapsis and meiotic recombination, which are important to establish the adequate number and distribution of interhomolog connections essential for proper chromosome segregation. The meiotic checkpoint network reinforces the adequate order of events during normal meiotic prophase and, in addition, it is crucial to react to meiotic failures. In response to defects in synapsis and/or recombination, the pachytene checkpoint blocks or delays entry into meiosis I, thus preventing the formation of gametes harboring aneuploidy and other kinds of genetic abnormalities.
Chromosome synapsis is mediated by the synaptonemal complex (SC), an evolutionarily-conserved tripartite structure that holds homologous chromosomes together during the pachytene stage of meiotic prophase I. Meiotic recombination initiates with the generation of programmed DNA double-strand breaks (DSBs), which undergo strictly regulated repair during prophase, preferentially with a non-sister chromatid (4). A fraction of DSBs are repaired to yield crossovers that, together with sister chromatid cohesion, give rise to physical links between homologs –chiasmata– promoting proper chromosome distribution. In some organisms, including budding yeast and mouse, chromosome synapsis is tightly linked to and depends on meiotic recombination.
In Saccharomyces cerevisiae, the coiled-coil Zip1 protein is the major component of the central region of the SC. Mutants lacking Zip1 fail to synapse and undergo pachytene checkpoint-dependent meiotic arrest or delay in prophase (5). Importantly, the Red1 and Hop1 structural components of the SC lateral elements (LEs) are also involved in the checkpoint; they function as adaptors to support activation of the meiosis-specific Mek1 effector kinase. In particular, phosphorylation of Hop1 at Thr318 by the upstream sensor kinases Mec1/Tel1 is necessary for Mek1 activation by phosphorylation (6,7). Full Mek1 activation involves sequential phosphorylation events that can be genetically separated and biochemically differentiated in phos-tag gels (8). Mec1/Tel1 dependent phosphorylation of Mek1 is followed by in-trans autophosphorylation at particular sites in its activation loop (Thr327 and Thr331) (9). Active Mek1 promotes two major meiotic responses: it reinforces interhomolog (IH) recombination bias (10,11), at least in part, through the inhibitory phosphorylation of Rad54 at Thr132 (12) and, on the other hand, it prevents exit from prophase and entry into meiosis I. Several crucial cell-cycle regulators, such as Swe1, Ndt80 and Cdc5, are targeted by the checkpoint to impose the cell cycle delay in response to defective recombination/synapsis; whether they are direct targets of Mek1 activity remains to be determined. The Swe1 kinase carries out the inhibitory phosphorylation of the main budding yeast cyclin-dependent kinase Cdc28 at Tyr19. In addition, inhibition and nuclear exclusion of the meiosis-specific transcription factor Ndt80 results in transcriptional down-regulation of a number of genes including those encoding B-type cyclins and the Cdc5 polo-like kinase that, together with inactive Cdc28, lead to meiotic cell cycle arrest (13–16).
Besides the Mec1-Ddc2/Tel1 sensors, the meiotic recombination checkpoint also shares other upstream components with the canonical DNA damage checkpoint, including Rad24 and the ‘9-1-1’ (Rad17-Mec3-Ddc1) module, which interacts with Red1 (17). In addition, epigenetic regulators, such as the Sir2 histone deacetylase and the Dot1 histone methyltransferase, also operate in the meiotic checkpoint response, at least in part, by regulating the chromosomal distribution of the meiosis-specific Pch2 protein (8,18,19).
Pch2 (also known as TRIP13 in mammals) is an evolutionarily conserved AAA+ ATPase involved in various aspects of meiotic chromosome metabolism in an ample range of organisms, including budding yeast, plants, worms, flies and mice. Pch2 was initially discovered in S. cerevisiae as a component of the checkpoint responding to the meiotic defects of the synapsis-deficient zip1 mutant lacking the central region of the SC (18,20). On the other hand, functional analyses of Pch2 in wild-type meiosis, has revealed that this ATPase negatively regulates the accumulation of the Hop1 protein at chromosome axes (18,21–23); in addition, functions for Pch2Trip13 in DSB generation, crossover interference, IH recombination bias, and regulation of rDNA recombination have been also described in both yeast and mouse (24–28). Intriguingly, whereas the absence of Pch2 bypasses the meiotic delay of the synapsis-deficient zip1 mutant, it has little effect on the checkpoint response to unrepaired resected DSBs in dmc1. In contrast, Pch2 acting through Xrs2 and Tel1 (but not Mec1) signals unprocessed DSBs in sae2 strains; however, Tel1 is not required for the zip1-induced synapsis checkpoint (29). All these pieces of evidence suggest that, depending on the initiating event, Pch2 may perform specific functions in relaying the checkpoint signal generated by different meiotic perturbations.
Whereas in budding yeast Pch2 appears to be dedicated exclusively to meiotic functions, the Trip13Pch2 homolog in metazoa is also involved in the mitotic spindle assembly checkpoint regulating the Mad2 protein, which, like Hop1, contains a HORMA domain (30–32). Underscoring the relevance of Trip13Pch2 function in somatic cells, the expression levels of Trip13Pch2 in certain types of cancer define the chemotherapy resistance and prognosis (33,34). Recent work has revealed the structural properties of this conserved ATPase and the conformational changes induced in some of its substrates (35).
In order to gain further insight into the functional impact of Pch2 during SC-deficient meiosis, we first describe here a genetic overexpression screen aimed to discover factors involved in the checkpoint role of Pch2. Despite the fact that Pch2 somehow promotes the turnover of Hop1 from meiotic chromosome axes; that is, Hop1 is more abundant and more continuous on pch2 chromosomes, we surprisingly found that Hop1 overproduction suppresses the checkpoint defect of the zip1 pch2 mutant. Then, we show that, in the zip1 mutant, Pch2 is required for Mek1 auto-phosphorylation and formation of chromosomal Mek1 foci, and that HOP1 overexpression restores full Mek1 activation in zip1 pch2. We also demonstrate that, in contrast with the pch2 single mutant, Hop1 is not more abundant on zip1 pch2 chromosomes; furthermore, in response to zip1 defects, Pch2 specifically promotes high levels of Hop1 phosphorylation at Thr318 required to sustain checkpoint function. Finally, we show that pch2 mutants carrying mutations in the Walker A or Walker B motifs phenocopy the defects of the pch2 null-mutant, indicating that the ATPase activity of Pch2 is essential for its synapsis checkpoint function. Moreover, the Walker A-deficient Pch2 version fails to localize to the chromosomes and to form a stable complex suggesting that adenosine triphosphate (ATP) binding is required for formation or integrity of the Pch2 hexameric complex. We conclude that the critical function of the Pch2 ATPase in a zip1-deficient situation is to facilitate Mec1-dependent phosphorylation of Hop1 at Thr318.
MATERIALS AND METHODS
Yeast strains and meiotic time courses
The genotypes of yeast strains are listed in Supplementary Table S1. All strains are in the BR2495 or the BR1919 background (5,36). The zip1::LEU2, zip1::LYS2, zip1::URA3, zip3::URA3, ecm11::kanMX6, ndt80::LEU2, ddc1::ADE2, rad17::LEU2, rad24::TRP1, dot1::TRP1, pch2::TRP1, sir2::LEU2, hop1::hphMX4 and rad54::LEU2 gene deletions were previously described (5,8,15,18,19,37–39). The rad51::natMX4 deletion was made following a PCR (polymerase chain reaction)-based approach (40). The ndt80::kanMX3 construct was generated by marker swapping in ndt80::LEU2 strains using the M3926 plasmid (41). The pph3::kanMX6 cassette was amplified from genomic DNA of a W303-based strain containing that deletion (a gift from R. Bermejo; CIB-CSIC) and used to transform the appropriate BR1919 strains. For substitution of the PCH2 gene for the pch2Δ-lacZ::TRP1 construct, strains were transformed with the pSS67 plasmid (see below) digested with XhoI-SphI. N-terminal tagging of PCH2 with three copies of the HA epitope was previously described (18). PCH2 was also tagged with three copies of the MYC epitope at the same position using identical procedures (42). The MEK1-GFP and DDC2-GFP constructs were also previously described (8,43). The pch2-K320A and pch2-E399Q mutations were introduced at the PCH2 genomic locus using the delitto perfetto technique (44); they generate a NarI and a BstNI site, respectively, utilized for genotyping purposes during genetic crosses. The sequences of the primers used are available upon request. All strains were made by direct transformation or by genetic crosses always in an isogenic background. Sporulation conditions for meiotic time courses have been described (8). To score meiotic nuclear divisions, samples were taken at different time points, fixed in 70% Ethanol, washed in phosphate buffered saline (PBS) and stained with 1 μg/μl DAPI for 15 min. At least 300 cells were counted at each time point. Meiotic time courses were repeated several times; representative experiments are shown.
Plasmids
The plasmids used are listed in Supplementary Table S2. The pSS51 plasmid expressing a pch2-lacZ fusion was constructed by cloning a 1.3-kb XhoI-BamHI fragment containing the PCH2 promoter and the N-terminal ORF region encoding the first 90 amino acids into the SalI-BamHI sites of the YCp50-derivative R1566 plasmid harboring the E. coli lacZ gene with a BamHI site at the 5′ end to generate an in-frame fusion. pSS67 contains a 4.3-kb BglII-NheI fragment from pSS51 harboring pch2-lacZ cloned into BglII-SphI of pSS53 (18). The R1692 plasmid overexpressing HOP1 has been previously described (45). pSS54 contains a 3.2-kb KpnI-PstI PCH2 fragment in the high-copy YEp352 vector. The pSS316 and pSS317 plasmids were constructed as follows: a 2.7-kb fragment containing HOP1 ORF and the 5′ (648 bp) and 3′ (241 bp) UTR regions was first amplified by PCR from genomic DNA of a BR1919 strain using oligos HOP1-EcoRI(Fw) and HOP1-SalI(Rv) and blunt-cloned into the pJET1.2 vector (ThermoFisher Scientific) to originate pSS314. This EcoRI-SalI fragment was then subcloned into pRS426 to generate the pSS316 plasmid. A 1.6-kb fragment spanning the hop1-T318A mutation was amplified from the JCY565 strain (6) (a gift from J. Carballo; CIB-CSIC), digested with BamHI-SacI and used to replace the same 1.0-kb fragment in pSS314 to generate pSS315. The 2.7-kb EcoRI-SalI fragment of pSS315 was then transferred to pRS426 to originate the pSS317 plasmid overexpressing hop1-T318A.
Genetic screen for high-copy suppressors of zip1 pch2
The zip1 pch2-lacZ mutant (DP221) was transformed with a yeast genomic library constructed in the multicopy vector YEp24 (46). Transformants were selected on SC-Ura and replica-plated to SPO plates containing a sterile Whatman 3MM filter paper on top. As positive controls, two colonies of DP221 transformed with pSS54 were placed at known positions on every plate. After incubation at 30°C for 3 days, the filters were removed, exposed to chloroform fumes for 10 min and assayed for β-galactosidase activity by incubating them at 30°C, colony side up, in empty dishes with a solution of 500 μl of Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 40 mM β-mercatoethanol, pH 7.0) containing 100 μl of 20 mg/ml X-Gal. About 23 000 transformants were scored and 21 displayed blue color; plasmids were recovered for further analysis. The presence of PCH2 in the plasmids was discarded by PCR using internal PCH2 primers. Fifteen of the plasmids recovered fail to reproduce the phenotype when reintroduced into DP221, indicating that the suppression phenotype was not linked to the plasmids. The remaining six plasmids were analyzed by restriction mapping and sequencing of the insert ends and were grouped into two Pch-Two-Suppressors: PTS10 containing RPS9A and a truncated form of MOT1, and PTS11 containing HOP1, PCI8, MAM33, RPS24B and SEC6.
Western blotting and immunoprecipitation
Total cell extracts were prepared by trichloroacetic acid (TCA) precipitation from 5-ml aliquots of sporulation cultures as previously described (13). Analysis of Mek1 phosphorylation using Phos-tag gels was performed as reported (8). The antibodies used are listed in Supplementary Table S3. The ECL or ECL2 reagents (ThermoFisher Scientific) were used for detection. The signal was captured on films and/or with a ChemiDoc XRS system (Bio-Rad) and quantified with the Quantity One software (Bio-Rad).
For immunoprecipitation of Pch2, 5-ml aliquots from 16 h meiotic cultures were crosslinked with 1% formaldehyde for 10 min at 30°C. The reaction was quenched by adding glycine to 250 mM and incubating for 5 min on ice. Cells were collected, washed and broken with glass beads in lysis buffer (150 mM NaCl, 1% Triton X-100, 50 mM Tris HCl pH 8.0) containing protease inhibitors (Complete Ultra Tablets, Roche). Clarified extracts were immunoprecipitated with anti-HA antibodies conjugated with magnetic MicroBeads using the μMACS Epitope Tag Protein Isolation Kit (Miltenyi Biotec) following the manufacturer's protocol.
Cytology
Immunofluorescence of chromosome spreads and whole cells was performed essentially as described in (47) and (48), respectively. The antibodies used are listed in Supplementary Table S3. Images of spreads were captured with a Nikon Eclipse 90i fluorescence microscope controlled with MetaMorph software and equipped with a Hammamatsu Orca-AG CCD camera and a PlanApo VC 100 × 1.4 NA objective. Ddc2-GFP foci images were captured with an Olympus IX71 fluorescence microscope equipped with a personal DeltaVision system, a CoolSnap HQ2 (Photometrics) camera and 100x UPLSAPO 1.4 NA objective. Stacks of 10 planes at 0.4 μm intervals with 500 ms exposure time were captured. Maximum intensity projections of deconvolved images were generated using the SoftWorRx 5.0 software (Applied Precisions). Quantification of the fluorescence signal in individual spread nuclei or cells was performed with the Image J software. Background signal was subtracted using the Otsu's threshold method of Image J. DAPI images were collected using a Leica DMRXA fluorescence microscope equipped with a Hammamatsu Orca-AG CCD camera and a 63 × 1.4 NA objective.
Dityrosine fluorescence assay, sporulation efficiency and spore viability
To examine dityrosine fluorescence as an indicator of the formation of mature asci, patches of cells grown on YPDA plates were replica-plated to sporulation plates overlaid with a nitrocellulose filter (Protran BA85, Whatman). After 3-days incubation at 30°C, fluorescence was visualized by illuminating the open plates from the top with a hand-held 302 nm UV lamp. Images were taken using a Gel Doc XR system (Bio-Rad). Sporulation efficiency was quantitated by microscopic examination of asci formation after 3 days on sporulation plates. Both mature and immature asci were scored. At least 300 cells were counted for every strain. Spore viability was assessed by tetrad dissection. At least 144 spores were scored for every strain.
Statistics
To determine the statistical significance of differences a two-tailed Student t-test was used. P-Values were calculated with the GraphPad Prism 5.0 software.
RESULTS
A genetic screen for high-copy suppressors of zip1 pch2 identifies HOP1
The absence of Pch2 alleviates the checkpoint-induced meiotic arrest of the zip1 mutant. To gain insight into the function of Pch2 in this checkpoint we devised a genetic screen to identify genes that, when overexpressed, restored the meiotic block in a zip1 pch2 double mutant. This screen could potentially identify novel components of the checkpoint pathway and/or reveal additional functional interactions of Pch2 with previously known checkpoint factors. We took advantage of the fact that Pch2 is transiently produced during meiotic prophase in the wild type, but it accumulates in the prophase-arrested zip1 mutant (18). To follow PCH2 expression, we generated a construct containing the PCH2 coding sequence corresponding to the first 90 amino acids (pch2Δ90-564) fused the E. coli lacZ gene and expressed from the PCH2 promoter in a centromeric plasmid (Figure 1A). β-galactosidase activity was used as a readout for PCH2 expression and dityrosine fluorescence as an indicator of sporulation on plates. Consistent with the previously described PCH2 expression pattern, meiotic-proficient wild-type cells containing this plasmid exhibited no or low levels of β-galactosidase activity, but the prophase-arrested zip1 mutant accumulated high levels of β-galactosidase activity, manifested as blue color on sporulation plate assays (Figure 1B and C). Alleviation of the prophase arrest in a zip1 dot1 mutant (8,19) eliminated β-galactosidase production (Figure 1B), whereas the checkpoint-independent meiotic block imposed by deletion of NDT80 (49) resulted in β-galactosidase accumulation (Figure 1B). Thus, the pch2Δ90-564-lacZ fusion gene (hereafter, pch2Δ-lacZ) serves as a useful reporter for meiotic prophase arrest.
Figure 1.

Identification of HOP1 in a genetic screen for high-copy suppressors of the zip1 pch2 checkpoint defect using a pch2-lacZ construct as a reporter for meiotic prophase arrest. (A) Schematic representation of a centromeric plasmid (pSS51) carrying the PCH2 promoter and the coding sequence for the first N-terminal 90 amino acids fused in frame with the bacterial lacZ gene. (B) Dityrosine fluorescence and β-galactosidase assays of the indicated strains transformed with the pSS51 plasmid after 48 h on sporulation plates. (C) Kinetics of meiotic divisions and β-galactosidase activity in meiotic time courses of wild-type and zip1 strains containing the pSS51 plasmid. (D) Schematic representation of the substitution of the PCH2 gene for the pch2-lacZ construct at the genomic locus. (E) Dityrosine fluorescence and β-galactosidase assays of the indicated strains after 48 h on sporulation plates. (F) Scheme of the genetic screen. (G) Representative β-galactosidase assay of a plate from zip1 pch2Δ-lacZ transformed with the genomic high-copy library. Black triangles indicate positive controls deliberately placed at known positions on every plate of the screen. The red arrow points to a positive ‘blue’ candidate. (H) Dityrosine fluorescence and β-galactosidase assays of the zip1 pch2Δ-lacZ strain transformed with the indicated plasmids. Strains for (B) and (C) are: BR2495 (wild type), MY63 (zip1), DP174 (zip1 dot1) and S3483 (ndt80). Strains for (E), (F), (G) and (H) are: DP221 (zip1 pch2Δ-lacZ) and DP228 (zip1 pch2Δ-lacZ/PCH2). DP221 was transformed with empty vector or the indicated high-copy plasmids.
We next generated diploid strains in which both copies of the PCH2 gene were replaced by the pch2Δ-lacZ construct at the genomic locus (Figure 1D). As expected, pch2Δ-lacZ behaved like a null mutant; it bypassed checkpoint arrest as manifested by the presence of dityrosine fluorescence in zip1 pch2Δ-lacZ, but no β-galactosidase was detected (Figure 1E). Notably, introduction of a single-copy or a high-copy PCH2 restored the meiotic block in zip1 pch2Δ-lacZ and resulted in conspicuous β-galactosidase accumulation (Figure 1E). Thus, we reasoned that we could use this assay to screen for high-copy suppressors of the unscheduled sporulation resulting from the defective checkpoint in zip1 pch2Δ-lacZ. After transformation with a 2μ-based high-copy genomic library and replica-plate to sporulation medium, we scored for the appearance of blue color as an indicator of restored meiotic prophase arrest (Figure 1F and G). The plasmids contained in the colonies displaying β-galactosidase activity were recovered, analyzed by restriction digestion and DNA sequencing, and re-transformed into the original zip1 pch2Δ-lacZ strain to confirm that the suppression phenotype was linked to the plasmid. In addition to clones containing PCH2, we isolated two different high-copy suppressor genes (PTS, for Pch Two-Suppressors). PTS10 encoded a C-terminal truncated version of the Mot1 transcriptional regulator (50). Since 2μPTS10 only conferred a transient and partial restoration of the meiotic arrest (Figure 1H) it was not further analyzed. In contrast, the effect of 2μPTS11 was comparable to that of the 2μPCH2 control (Figure 1H). Sequencing analysis revealed that the HOP1 gene was present in the 2μPTS11 plasmids isolated from the library. To confirm that high levels of Hop1 were indeed responsible for the phenotype we analyzed a previously characterized 2μHOP1 plasmid (8,45) and found a similar effect on suppression of zip1 pch2Δ-lacZ sporulation (Figure 1H). Thus, HOP1 overexpression restores meiotic arrest in the checkpoint-deficient zip1 pch2Δ-lacZ mutant.
Overexpression of HOP1 suppresses the checkpoint defect of pch2, sir2 and dot1, but not that of rad17, ddc1 and rad24 mutants
To determine whether the effect of Hop1 overproduction was exclusively exerted when the checkpoint was inactivated by the absence of Pch2, we analyzed mutants in other checkpoint genes also required for the zip1 meiotic block. The Rad17, Ddc1 and Rad24 proteins are members of the 9-1-1 and RFC complexes acting upstream in the checkpoint pathway and contributing to the activation of the Mec1-Ddc2 sensor kinase (6,51,52). On the other hand, the H3K79 methyltransferase Dot1 is required for proper phosphorylation and localization of the Hop1 adaptor and the Mek1 checkpoint effector kinase (8). Dot1 regulates the chromosomal distribution of the nucleolar-associated Pch2 and Sir2 proteins (8,19). As shown in Figure 2, Hop1 overproduction significantly reduced sporulation efficiency in zip1 pch2, zip1 sir2 and zip1 dot1, but had no effect on zip1 rad17, zip1 ddc1 and zip1 rad24 mutants. Thus, HOP1 overexpression specifically supports meiotic checkpoint function when the Dot1-Pch2 module is not operative.
Figure 2.
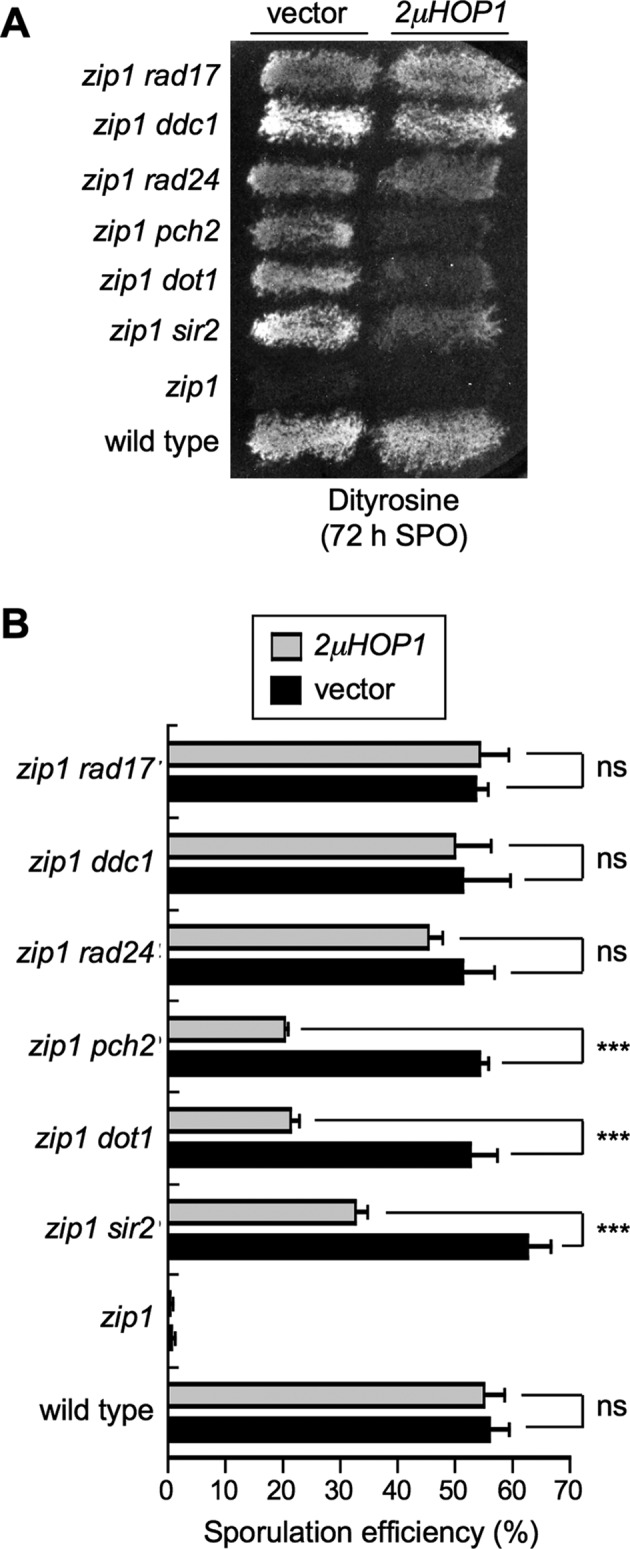
High-copy HOP1 specifically suppresses the checkpoint defect of zip1 pch2, zip1 sir2 and zip1 dot1. (A) Dityrosine fluorescence assay of the indicated strains transformed with empty vector (YEp352) of high-copy HOP1 (R1692) after 3 days on sporulation plates. (B) Microscopic quantification of the sporulation efficiency of the strains analyzed in (A). Three independent counts were performed. Means and standard deviations are shown. (ns), no significant difference; (***), P < 0.001. Strains are: BR2495 (wild type), MY63 (zip1), DP174 (zip1 dot1), DP223 (zip1 pch2), DP267 (zip1 sir2), S4295 (zip1 rad24), S4278 (zip1 ddc1) and S4286 (zip1 rad17).
HOP1 overexpression restores meiotic checkpoint activity in zip1 pch2
To further explore the effect of HOP1 overexpression, we next analyzed meiotic kinetics and molecular markers of checkpoint activation, such as Mek1 phosphorylation (in phostag gels) and Cdc5 inhibition, during meiotic time courses of wild-type, zip1 and zip1 pch2 strains with or without Hop1 overproduction (Figure 3). In the wild type, the Mek1 kinase was only weakly and transiently activated (Figure 3A) (8); HOP1 overexpression slightly prolonged Mek1 activation resulting in a subtle meiotic delay (Figure 3A and B). As expected, the synapsis-deficient zip1 mutant showed extensive Mek1 phosphorylation, delayed Cdc5 production and a significant meiotic delay; Hop1 overproduction slightly enhanced those effects (Figure 3C and D). Consistent with the defective checkpoint, Mek1 hyperactivation was not observed in the zip1 pch2 mutant; moreover, Cdc5 production and meiotic divisions displayed roughly wild-type kinetics (Figure 3E and F). However, high doses of Hop1 markedly restored Mek1 phosphorylation, delayed Cdc5 production and provoked a significant meiotic delay in zip1 pch2 (Figure 3E and F).
Figure 3.

HOP1 overexpression largely restores meiotic arrest, Mek1 activation, Mek1 localization and delayed Cdc5 production in zip1 pch2. Western blot analysis of the indicated proteins and meiotic kinetics of (A and B) wild type, (C and D) zip1 and (E and F) zip1 pch2 transformed with empty vector (pRS426) or high-copy HOP1 (R1692). Strains are: DP421 (wild type), DP422 (zip1) and DP1029 (zip1 pch2). (G) Analysis of Mek1 phosphorylated forms in ndt80-arrested strains. The black arrowhead marks the Mec1/Tel1-dependent band and the white arrowheads point to the forms resulting from Mek1 autophosphorylation (8). Strains are: DP428 (zip1) and DP881 (zip1 pch2), transformed with pRS426 (empty vector) or R1692 (OE-HOP1). (H) Analysis of Mek1 localization by immunofluorescence of spread meiotic chromosomes using anti-GFP antibodies. Representative nuclei are shown. Strains are DP582 (zip1) and DP1111 (zip1 pch2).
To elude the possible effect of the different kinetics of meiotic progression in the strains analyzed in these assays, we assessed Mek1 activation and localization in pachytene-arrested ndt80 cells. To achieve full activation, Mek1 undergoes Mec1/Tel1-dependent phosphorylation followed by in trans autophosphorylation (6,9). The different forms resulting from these two phosphorylation events can be resolved in phostag gels (8) (Figure 3G, black and white arrows). We found that Mec1/Tel1-dependent phosphorylation occurred in the absence of Pch2 (Figure 3G, black arrow). However, the bands corresponding to Mek1 autophosphorylation were absent in zip1 pch2 transformed with empty vector (Figure 3G, white arrows) indicating that, like Dot1, Pch2 is specifically required for Mek1 autophosphorylation. Strikingly, complete Mek1 activation was reestablished in zip1 pch2 cells overproducing Hop1 (Figure 3G).
The zip1-induced meiotic checkpoint also promotes the formation of chromosome-associated Mek1 foci (8,53) (Figure 3H). Like Mek1 phosphorylation, formation of Mek1 foci was also impaired in zip1 pch2, which displayed fewer and dimmer foci. In contrast, numerous bright Mek1 foci were observed in the zip1 pch2 mutant overexpressing HOP1 (Figure 3H).
Taken together, these results indicate that Pch2 is required for Mek1 autophosphorylation and localization when the synapsis checkpoint is activated by the lack of Zip1. In the absence of Pch2, artificially-induced high levels of Hop1 can support checkpoint activity suggesting that Hop1 function is somehow compromised in the zip1 pch2 mutant resulting in impaired Mek1 activation.
Pch2 is required for Hop1 phosphorylation induced by the synapsis checkpoint
Several lines of evidence indicate that, in unperturbed meiosis, Pch2 promotes the turnover of Hop1 from chromosomes; the pch2 single mutant displays a more abundant and continuous localization of Hop1 along synapsed chromosomes (18,22). Thus, in principle, it was rather surprising that high doses of Hop1 could compensate for the absence of Pch2 supporting Mek1 activation and checkpoint function in the zip1 pch2 mutant. To investigate this apparent contradiction, we examined Hop1 production, localization and phosphorylation in wild type, pch2, zip1 and zip1 pch2 prophase-arrested ndt80 cells. Pachytene checkpoint activation leads to Mec1/Tel1-dependent phosphorylation of Hop1 at defined S/T-Q sites; in particular, phosphorylation of the T318 residue is critical for Hop1 checkpoint function promoting Mek1 activation (6,7). As expected, both on chromosome spreads and in whole-cell lysates, we found higher levels of Hop1 in the pch2 single mutant, compared with the wild type (Figure 4Aa,b; B and D). Paralleling the accumulation of total Hop1, an increased number of phospho-Hop1T318 foci (Figure 4Ae,f; C) and higher levels of the phosphorylated protein (Figure 4D) were also observed in pch2. Nevertheless, the ratio of phospho-Hop1T318 relative to total Hop1 was only moderately increased in pch2 compared with the wild type (Figure 4E). In fact, the Mek1 kinase is minimally activated in pch2 as manifested by the almost complete absence of autophosphorylation (Figure 4D, white arrow), the low level of histone H3T11 phosphorylation, which is a target of Mek1 (54) and, therefore, a useful reporter of its kinase activity (Figure 4D), and the rather normal kinetics of meiotic progression of pch2, which only shows a minor delay (20) (see also Figure 8D below).
Figure 4.

Pch2 promotes Hop1T318 phosphorylation in zip1. (A) Immunofluorescence of spread meiotic chromosomes stained with DAPI (blue) and anti-Hop1 (a-d panels) or anti-phospho-Hop1T318 (e–h panels) antibodies (red). Representative nuclei are shown. (B and C) Quantification of total Hop1 and phospho-Hop1T318 fluorescence signal, respectively, on the spreads analyzed in (A). Each spot in the plot represents the intensity of a nucleus scored. (D) Western blot analysis of the indicated proteins and phosphorylation events. (E) Quantification of relative Hop1T318 phosphorylation analyzed as in (D). The ratio of phospho-Hop1T318 versus total Hop1 chemiluminiscence signal is represented. Means and standard deviations from three independent experiments are shown. Strains are: DP424 (wild type), DP1058 (pch2), DP428 (zip1), DP881 (zip1 pch2) and DP700 (hop1). Spreads and lysates were prepared 24 h after meiotic induction of ndt80 cells. For DP700 the sample was taken at 17 h.
Figure 8.

The ATPase activity of Pch2 is required for its checkpoint function. (A) Schematic representation of the Pch2 protein indicating the AAA+ domain, the conserved Walker A and Walker B motifs, and the mutations introduced at both sites. (B) Western blot analysis of Pch2, Pch2-K320A or Pch2-E399Q production (detected with anti-HA antibodies) during meiosis. (C) Dityrosine fluorescence assay. (D) Time course of meiotic nuclear divisions; the percentage of cells containing two or more nuclei is represented. (E) Western blot analysis of Hop1T318 phosphorylation and Mek1 activation. PGK was used as a loading control. Strains are: DP1151 (wild type), DP1164 (pch2Δ), DP1163 (pch2-K320A), DP1287 (pch2-E399Q), DP1152 (zip1), DP1161 (zip1 pch2Δ), DP1162 (zip1 pch2-K320A) and DP1288 (zip1 pch2-E399Q).
On the other hand, the zip1 mutant displayed continuous Hop1 signal along unsynapsed chromosome axes (55) (Figure 4Ac and B), numerous and strong phospho-Hop1T318 foci (Figure 4Ag and C), and high levels of phospho-Hop1T318 protein (Figure 4D). Importantly, the phospho-Hop1T318/total Hop1 ratio was significantly raised in zip1 (Figure 4E), leading to full activation of Mek1 and robust H3T11 phosphorylation (Figure 4D). Deletion of PCH2 in zip1 resulted in increased global levels of the Hop1 protein detected by Western blot, as compared with the zip1 single mutant (Figure 4D); however, it was not massively incorporated on the axes; Hop1 chromosomal distribution in the zip1 pch2 double mutant was less continuous than in zip1 (Figure 4Ad and B). Importantly, zip1-induced Hop1T318 phosphorylation was dramatically reduced in the absence of Pch2 (Figure 4Ah, C, D and E), thus explaining the impaired Mek1 activation and defective checkpoint response in zip1 pch2 (Figures 3 and 4D).
These observations reveal that the critical role of Pch2 in the zip1-induced checkpoint is to promote Hop1 phosphorylation at T318. Confirming this notion, overexpression of wild-type HOP1, but not that of a phosphorylation-deficient hop1-T318A mutant (6), restored Hop1T318 phosphorylation and checkpoint function (i.e. full Mek1 activation) in zip1 pch2 (Figure 5).
Figure 5.
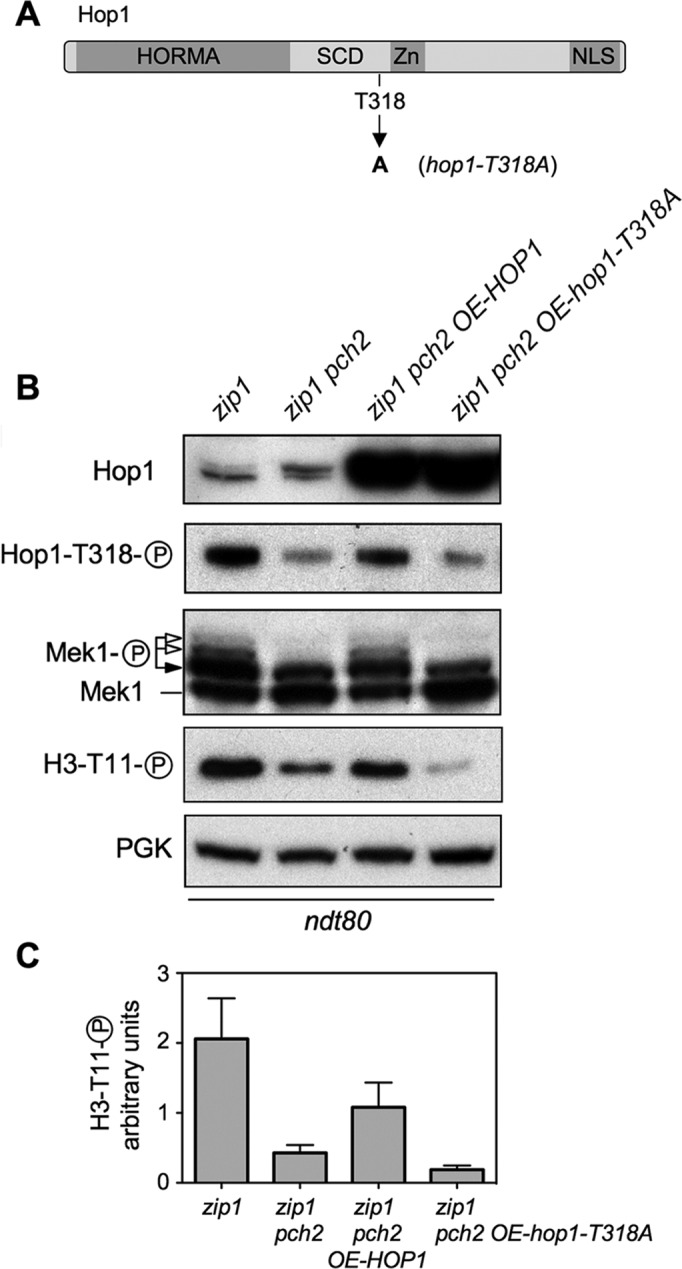
Hop1T318 phosphorylation is the critical checkpoint event impaired in zip1 pch2. (A) Schematic representation of the Hop1 protein domains and the position of the mutated T318 phosphosite (6) (B) Overexpression of wild-type HOP1, but not the hop1-T318A mutant, restores Mek1 activation in zip1 pch2. Western blot analysis of Hop1, phospho-Hop1T318, Mek1 and phospho-H3T11 in ndt80-arrested cells. (C) Quantification of the phospho-H3T11 signal from three experiments. Note that Mek1 activity markedly increases, but is not fully restored in zip1 pch2 OE-HOP1 cells due to plasmid-loss events in the meiotic cultures (43); those cells that lose the plasmid do not overproduce Hop1 and do not contribute to Mek1 phosphorylation. Strains are DP428 (zip1) and DP881 (zip1 pch2) transformed with pRS426 (empty vector), pSS316 (OE-HOP1) or pSS317 (OE-hop1-T318A).
The absence of PP4 restores checkpoint activity in zip1 pch2 to a small extent
It has been reported that the protein phosphatase 4 (PP4) counteracts Mec1/Tel1-dependent Hop1T318 phosphorylation (7); therefore, to further explore the role of Pch2 in controlling Hop1T318 phosphorylation we examined the effect of deleting the PPH3 gene, which encodes the catalytic subunit of PP4. We analyzed the kinetics of meiotic divisions (Figure 6A) and the activation of molecular markers of the checkpoint, including Hop1T318, Mek1 and histone H3T11 phosphorylation (Figure 6B). Consistent with a role for PP4 in shutting off pachytene checkpoint signaling, the pph3 mutant showed a transient Hop1-Mek1 activation (Figure 6B) manifested as a short meiotic delay (Figure 6A); moreover, the zip1 pph3 double mutant displayed persistent checkpoint activity and strong meiotic arrest (Figure 6A and B). Although Hop1T318 phosphorylation is severely impaired in zip1 pch2 (Figures 4D, E and 6B), the lack of PP4 resulted in higher levels of Hop1T318 phosphorylation and the ensuing Mek1 kinase activity (Figure 6B) leading to delayed meiotic divisions in zip1 pch2 pph3 compared to zip1 pch2 (Figure 6A). However, checkpoint function (i.e. Hop1T318 phosphorylation) was not completely restored in the zip1 pch2 pph3 triple mutant, which showed faster meiotic progression than that of zip1 and a weaker and shorter Mek1 activation (Figure 6A and B).
Figure 6.

Impact of the PP4 phosphatase on Pch2-dependent meiotic checkpoint. (A) Time course of meiotic nuclear divisions; the percentage of cells containing two or more nuclei is represented. (B) Western blot analysis of Hop1T318 phosphorylation and Mek1 activity at the indicated time points in meiosis. PGK was used as a loading control. Strains are: DP421 (wild type), DP1247 (pph3), DP422 (zip1), DP1249 (zip1 pph3), DP1029 (zip1 pch2) and DP1245 (zip1 pch2 pph3).
All together, these observations confirm that Pch2 specifically supports Hop1T318 phosphorylation when the synapsis checkpoint is triggered by the absence of Zip1 and reveal that the control of Hop1T318 phosphorylation by Pch2 is not, at least exclusively, exerted by modulating PP4 action.
To determine whether the defective Hop1T318 phosphorylation in zip1 pch2 stems from an impaired general activation of the Mec1-Ddc2 complex, we performed immunofluorescence of spread meiotic chromosome using an antibody that recognizes the phosphorylated S/T-Q motifs. As shown in Supplementary Figure S1A, phospho-S/T-Q foci similarly decorated both zip1 and zip1 pch2 chromosomes. We also examined the localization of the Mec1-Ddc2 complex monitoring the induction of Ddc2-GFP foci when the meiotic checkpoint is triggered in zip1 cells (43). We found that the formation of Ddc2 foci is not altered by the absence of Pch2 in ndt80-arrested cells (Supplementary Figure S1B). These observations suggest that Pch2 does not impact widespread Mec1 signaling.
Unsynapsed chromosomes and unrepaired DSBs remain in zip1 pch2 ndt80
Although the precise defect(s) triggering the checkpoint in zip1 mutants (defective synapsis and/or unrepaired DSBs) remains unclear, since pch2 affects meiotic DSB metabolism, it was formally possible that the reduced Hop1T318 phosphorylation observed in zip1 pch2 resulted from the absence or the disappearance of the checkpoint-activating signal. However, the fact that Ddc2 foci formation and S/T-Q phosphorylation are conspicuous in zip1 pch2 (Supplementary Figure S1) indicates that the zip1 defects triggering the checkpoint are still present in the absence of Pch2. We also monitored the presence of Rad51 foci as a marker for unrepaired DSBs (56). To avoid again the effect of meiotic progression on DSB repair outcome we performed the analysis in ndt80 cells. No or few Rad51 foci were observed in most wild-type (1.8 ± 0.3 SEM foci per nucleus; n = 40) and pch2 (2.0 ± 0.5 SEM foci per nucleus; n = 35) nuclei, but the number was markedly increased in the zip1 mutant (5.5 ± 0.5 SEM foci per nucleus; n = 35). Importantly, although the fraction of unrepaired DSBs was reduced in zip1 pch2 (3.5 ± 0.5 SEM foci per nucleus; n = 45), likely due to intersister (IS) repair, the double mutant still showed prominent Rad51 foci (Figure 7A). In addition, chromosome axes are unsynapsed in zip1 pch2; i.e., the synapsis defect continues to exist (Figure 4Ac,d). Thus, these observations indicate that the inability of zip1 pch2 to phosphorylate Hop1 at T318 does not stem from the absence of defects triggering the checkpoint and point to a more direct role for Pch2 in controlling Hop1-Mek1 activation.
Figure 7.

Effect of Pch2 on cell cycle progression and resolution of zip1-induced recombination intermediates. (A) Localization and quantification of Rad51 foci as markers for unrepaired DSBs on spread meiotic nuclei of ndt80 cells after 24 h of meiotic induction. Strains are: DP424 (wild type), DP1058 (pch2), DP428 (zip1) and DP881 (zip1 pch2). (B) Time course of meiotic nuclear divisions. The percentage of cells with two or more nuclear masses is represented. (C) Quantification of Ddc2-GFP foci throughout meiosis. The percentage of cells containing a single non-meiotic Ddc2 focus (green bars) or multiple meiotic Ddc2 foci (red bars) from three different counts is represented. Note that rad51 cells accumulate spontaneous non-meiotic Ddc2 foci at time zero. Between 150 and 800 cells were scored for each strain at every time point. (D) Western blot analysis of phospho-H3T11 (as a reporter for Mek1 activity), Cdc5 (as a marker for meiosis I entry) and PGK (as a loading control) from the same cultures analyzed in (B) and (C). Strains for (B), (C) and (D) are: DP448 (wild type), DP449 (zip1), DP1379 (zip1 pch2) DP1381 (zip1 rad51) and DP1382 (zip1 pch2 rad51).
The Pch2-Hop1-Mek1 signaling module impacts meiotic cell cycle progression
Since the analyses described above were performed in ndt80-arrested cells, we also explored the possibility that the mutation of PCH2 suppresses zip1 meiotic arrest exclusively by allowing the repair of DSBs by recombination between sister chromatids as a result of the impaired Mek1 function in zip1 pch2 (28). If that were the case; that is, Mek1 only controls recombination partner choice and not cell cycle events, compromising repair by mutation of recombination factors would restore meiotic cell cycle arrest in zip1 pch2 cells; therefore, we deleted RAD51 to interfere with DNA repair. In parallel with monitoring meiotic divisions, we used the presence of multiple Ddc2-GFP foci as a reporter for unrepaired Spo11-dependent meiotic recombination intermediates (43), H3T11 phosphorylation as a reporter for Mek1 activity and Cdc5 production as a molecular indicator of prophase I exit and entry into meiotic divisions (Figure 7B, C and D). In the wild type, only a transient peak of Ddc2-containing cells was detected because recombination is normally completed. In the zip1 mutant, cells containing multiple Ddc2 foci accumulate, leading to Mek1 activation and delayed Cdc5 production. As expected, the zip1 pch2 double mutant showed impaired Mek1 activation allowing normal meiotic progression and the disappearance of recombination intermediates (Figure 7B, C and D). Notably, whereas at late time points a fraction of zip1 cells underwent meiotic divisions after a delay, the zip1 rad51 double mutant displayed a much tighter arrest (Figure 7B) and markedly reduced and delayed Cdc5 production (Figure 7D). Likewise, Ddc2 multiple foci and Mek1 activation persisted for longer in zip1 rad51 (Figure 7C and D; Supplementary Figure S2). These observations indicate that compromising recombination by the rad51 mutation prevents the resolution of recombination intermediates in zip1 leading to a stronger meiotic block. Importantly, deletion of PCH2 reduced Mek1 activation and restored meiotic progression and earlier Cdc5 production in zip1 rad51 despite the persistence of multiple Ddc2 foci; that is, unrepaired recombination intermediates (Figure 7B,C,D; Supplementary Figure S2).
We also analyzed the effect of Rad54, which is a Rad51 accessory factor, and a direct Mek1 target (12). In addition to the action of Hed1, phosphorylation of Rad54 by Mek1 reduces its affinity for Rad51 binding; therefore, mutation of RAD54 can be used also as a tool to interfere with meiotic recombinational repair (57–59). We observed that, like in zip1 pch2, the sporulation block of zip1 was still relieved in the zip1 pch2 rad54 triple mutant (Supplementary Figure S3A). Consistent with impaired repair in the absence of Rad54, spore viability was further reduced in zip1 pch2 rad54 (29.1%, n = 144) compared with zip1 pch2 (47.2%; n = 144). Moreover, we used chromosome spreads to monitor the presence of Ddc2 foci in combination with spindle staining as a sensitive cytological assay for checkpoint function (19,43,51). Whereas zip1 rad54 cells arrested in prophase with unseparated SPBs and numerous Ddc2 foci (35.4 ± 2.5 SEM foci per nucleus; n = 13), zip1 rad54 pch2 nuclei displaying elongated meiosis I spindle coexisting with persistent recombination intermediates (14.0 ± 3.0 SEM Ddc2 foci per nucleus; n = 10) could be detected (Supplementary Figure S3B). In wild-type nuclei, recombination intermediates never coexist with metaphase spindles (19,43,51). These findings indicate that, in the absence of Pch2, a late cell cycle event has been initiated before an earlier one has been completed; that is, checkpoint function is impaired.
In summary, these observations argue that Pch2 controls a general checkpoint response via Hop1-Mek1 regulation that includes the cell-cycle arrest outcome and not only the regulation of recombination.
To determine whether the effect of Pch2 was exerted exclusively in response to the meiotic defects resulting from the lack of Zip1, we examined other mutants affecting SC dynamics, such as zip3 and ecm11. Zip3 is a SUMO ligase constituting the so-called synapsis initiating complex (60,61) and Ecm11 is a component of the central element of the SC (39,62). Similar to zip1, although to different extents, both ecm11 and zip3 single mutants displayed delayed meiotic progression (Supplementary Figure S4A), increased Hop1-T318 phosphorylation and induced Mek1 activity (Supplementary Figure S4B). Deletion of PCH2 resulted in faster meiotic progression and impaired Hop1 and Mek1 activation (Supplementary Figure S4). Therefore, Pch2 impact on Hop1-Mek1 signaling is also required to restrain meiotic divisions in other mutants altering SC proper development.
The ATPase activity of Pch2 is required for its checkpoint function
Pch2 is a hexameric ring AAA+ ATPase (63). In order to investigate whether Pch2 ATPase activity is required for its meiotic checkpoint function, conserved critical residues in the AAA+ domain of Pch2 were mutated to abolish its catalytic activity (64). In particular, the lysine 320 in the Walker A motif required for ATP binding was changed to alanine, and the glutamic acid at position 399 in the Walker B motif, required for ATP hydrolysis, was substituted for glutamine to generate the pch2-K320A and pch2-E399Q mutants, respectively (Figure 8A). We used the delitto perfetto technique, which leaves no additional modifications, to introduce these mutations in the genomic loci of strains carrying functional N-terminal HA- or MYC-tagged versions of PCH2 (18). Both Pch2-K320A and Pch2-E399Q mutant proteins were produced at normal levels and with normal kinetics (Figure 8B). Like pch2Δ, the pch2-K320A and pch2-E399Q single mutants showed no prominent phenotype in unperturbed meiosis; they were able to complete meiosis and sporulation with kinetics and efficiency similar to the wild type, producing high levels of viable spores (Figure 8C and D; Table 1). Interestingly, when the meiotic checkpoint was triggered by the lack of Zip1, we found that, like pch2Δ, the pch2-K320A and pch2-E399Q mutations completely suppressed the sporulation defect of zip1 (Figure 8C); the zip1 pch2-K320A and zip1 pch2-E399Q double mutants displayed fairly normal kinetics of meiotic divisions (Figure 8D), but generate largely inviable meiotic products (Table 1). These observations imply that the ATPase activity of Pch2 is absolutely required to restrain meiotic progression when the checkpoint is induced by the absence of Zip1. To further confirm this interpretation we assessed the effect of the pch2-K320A and pch2-E399Q mutations on checkpoint function by monitoring molecular markers of checkpoint activation impacted by Pch2 (see above), such as Hop1T318 phosphorylation, Mek1 hyperphosphorylation and histone H3T11 phosphorylation (Figure 8E). Consistent with the alleviation of meiotic arrest (Figure 8C and D), the high levels of phospho-Hop1T318 and Mek1 hyperactivation observed in zip1 were drastically reduced in both zip1 pch2-K320A and zip1 pch2-E399Q double mutants (Figure 8E). Like in zip1 pch2Δ, HOP1 overexpression restored Hop1T318 phosphorylation and Mek1 activation in both ATPase mutants (Supplementary Figure S5). Thus, both ATP binding to the Walker A motif and ATP hydrolysis by the Walker B module are critical for Pch2's meiotic checkpoint function.
Table 1. Sporulation and spore viability.
| Genotype | Sporulation frequency (%) | Spore Viability (%) |
|---|---|---|
| Wild type | 66.8 | 95.3 |
| pch2Δ | 73.9 | 90.7 |
| pch2-K320A | 64.6 | 96.5 |
| pch2-E399Q | 80.3 | 93.0 |
| zip1 | 2.0 | nd |
| zip1 pch2Δ | 83.3 | 35.2 |
| zip1 pch2-K320A | 77.3 | 46.5 |
| zip1 pch2-E399Q | 80.3 | 31.2 |
| Wild type + OE-HOP1 | 65.3 | 92.0 |
| pch2Δ + OE-HOP1 | 20,3 | 94.4 |
Sporulation frequency and spore viability was determined as explained in Materials and Methods. nd, not determined.
OE-: overexpression.
ATP binding to Pch2 is required for its localization and complex stability
We also analyzed the localization of wild-type Pch2 and the ATPase-dead versions on pachytene chromosome spreads. As previously described, the wild-type Pch2 protein showed a conspicuous accumulation in a particular chromosomal region lacking Hop1 that corresponds to the rDNA array (18) (Figure 9Aa, Ad). In addition, faint foci outside the rDNA can be found on wild-type chromosomes displaying an exclusive localization with Hop1 (18,22) (Supplementary Figure S6, white arrows). Surprisingly, despite being present at normal levels in whole cell extracts (Figure 8B), the Walker A-deficient Pch2-K320A protein was not detectable at any location on meiotic chromosomes of either wild-type or zip1 spread nuclei (Figure 9Ab and Ae). In contrast, the Pch2-E399Q protein showed normal rDNA localization (Figure 9Ac and Af) even though it is catalytically inactive (63) and lacks checkpoint function (Figure 8). Remarkably, like in pch2Δ (Figure 4Ab), the Hop1 protein was also more abundant on chromosomes and displayed a linear instead of a dotty pattern in both pch2-K320A and pch2-E399Q single mutants (Figure 9Ab and Ac). On the contrary, but also similar to zip1 pch2Δ (Figure 4Ad), Hop1 localization was disrupted and discontinuous in zip1 pch2-K320A and zip1 pch2-E399Q double mutants (Figure 9Ae and Af). In addition, the characteristic exclusion of Hop1 from the rDNA region (18,55) was lost in the ATPase-deficient mutants (Figure 9A; arrows); in fact, the Pch2-E399Q protein extensively colocalized with Hop1 at the rDNA (Figure 9Ac and Af). Moreover, in contrast to the wild type, chromosomal foci of Pch2-E399Q also colocalized with Hop1 (Supplementary Figure S6, yellow arrows).
Figure 9.

Localization of ATPase-deficient versions of Pch2. (A) Immunofluorescence of meiotic chromosomes stained with anti-HA or anti-MYC antibodies to detect Pch2, Pch2-K320A or Pch2-E399Q (red), anti-Hop1 antibodies (green) and DAPI (blue). Strains are: DP1243 (wild type), DP1193 (pch2-K320A), DP1262 (pch2-E399Q), DP1244 (zip1), DP1192 (zip1 pch2-K320A) and DP1263 (zip1 pch2-E399Q). (B) Immunofluorescence of whole meiotic cells stained with anti-HA antibodies (to detect Pch2 or Pch2-K320A; red), anti-Hop1 antibodies (green) and DAPI (blue). The contour of the cells is outlined in the rightmost panels. Representative cells 24 h after meiotic induction in zip1 ndt80 background are shown. The arrows point to the rDNA region, which is distinguishable by the accumulation of Pch2 and the absence of Hop1. This region is not recognizable in the pch2-K320A mutant due to the mislocalization of Pch2-K320A and Hop1. Strains are: DP1190 (wild type) and DP1192 (pch2-K320A).
Since the Pch2-K320A version was not associated to the meiotic chromatin, we next studied the subcellular localization of Pch2 and Pch2-K320A by immunofluorescence in whole meiotic zip1 cells. The wild-type Pch2 was prominently detected in a discrete lateral nuclear region devoid of Hop1, presumably the nucleolus (Figure 9B, arrow). On the other hand, Pch2-K320A did not accumulate in the nucleus and was present rather evenly dispersed throughout the cell (Figure 9B). Interestingly, total cellular levels of Hop1 were higher in the zip1 pch2-K320A mutant (Figure 9B), although the protein was not massively incorporated onto the chromosome axes (Figure 9Ae).
Mutation of this conserved lysine in the Walker A motif of AAA+ family members commonly abolishes ATP binding and therefore ATPase activity in vitro, as has been demonstrated for Pch2 (63) but, in addition, it often leads to dissociation of the hexameric complex into monomers, as has been described for the HslU and SV40 LTag AAA+ ATPases (65). Therefore, we analyzed whether the pch2-K320A mutation compromises the formation of the Pch2 hexameric complex in vivo. We generated wild-type and pch2-K320A heterozygous diploid strains in which one copy of the PCH2 (or pch2-K320A) gene was tagged with the HA epitope and the other copy with the Myc epitope (Figure 10). In the wild type (PCH2-HA/PCH2-Myc), the anti-HA antibody immunoprecipitated both Pch2-HA and Pch2-Myc, consistent with the formation of a stable complex. In contrast, the anti-HA antibody failed to immunoprecipitate the Myc-tagged subunits in the pch2-K320A-HA/pch2-K320A-Myc strain (Figure 10A), suggesting that ATP binding is required for the integrity or stability of the Pch2 hexameric ring in vivo (Figure 10B).
Figure 10.

Mutation of the Pch2 ATP-binding site impairs the stability of the hexameric complex. (A) Whole cell extracts (WCE) prepared after 16 h in meiosis were immunoprecipitated with anti-HA antibodies. WCE and immunoprecipitates (IP) were analyzed by Western blot with both anti-HA and anti-Myc antibodies, as indicated. Strains are: DP1325 (PCH2-HA/PCH2-Myc; lane 1), DP1329 (PCH2/PCH2-Myc; lane 2) and DP1337 (pch2-K320A-HA/pch2-K320A-Myc; lane 3). To discard a problem with detection levels, in lane 2 (HA-untagged control) and lane 3, three more times of IP compared to lane 1 were loaded. (B) Schematic interpretation of the result described in (A). K and A represent a lysine and an alanine, respectively, at position 320 of Pch2.
In summary, the ATPase activity of Pch2 is absolutely required for the checkpoint response to zip1 meiotic defects and orchestrates proper Hop1 subcellular distribution, chromosomal localization and phosphorylation. ATP binding to the Walker A motif of Pch2, but not ATP hydrolysis, is essential for its nuclear accumulation and association to the meiotic chromosomes.
DISCUSSION
Suppression of pch2 checkpoint defect by Hop1 overproduction
Previous studies have reported that the Pch2 protein is an important player in the meiotic checkpoint response triggered by the absence of Zip1, a major structural component of the central region of the synaptonemal complex. This work provides new insights into the role of Pch2 in this process; we show that the critical function of Pch2 in the zip1-induced checkpoint is to promote Mec1/Tel1-dependent Hop1 phosphorylation at T318 and, therefore, the ensuing Mek1 full activation leading to the meiotic cell cycle block. We also show here that the ATPase activity of Pch2 is essential for this checkpoint function.
We report the isolation of HOP1 in a genetic screen for high-copy suppressors of the pch2 checkpoint defect. Monitoring various cytological and molecular markers, we demonstrate that high levels of Hop1 restore checkpoint function in a zip1 pch2 mutant restraining meiotic cell cycle progression. This finding was unanticipated, since it has been well established that Pch2 negatively regulates Hop1 abundance (at least on synapsed chromosomes) (18,21,22). Since Hop1 chromosomal levels are higher in the pch2 mutant it was unexpected that additional amounts of Hop1 provided by an overexpression plasmid could suppress the phenotype resulting from the lack of Pch2. Nevertheless, the Hop1 remodeling function reported for Pch2 has been only analyzed in the context of synapsed chromosomes, where Pch2 determines an alternating pattern of Hop1 abundance (21) (Figure 11). We show here in the context of unsynapsed chromosomes that, although global cellular levels of Hop1 are higher in zip1 pch2 compared with zip1, the extensive incorporation of Hop1 onto the lateral elements of zip1 chromosomes is not further increased when PCH2 is deleted. However, the rDNA region, normally devoid of Hop1, is decorated by the Hop1 protein in both pch2 and zip1 pch2 mutants, consistent with the nucleolar localization of Pch2 in both wild type and zip1 nuclei. Notably, although Pch2 and Hop1 display largely exclusive localization patterns on wild-type chromosomes, we show that the catalytically-inactive Pch2-E399Q protein colocalizes with Hop1 both at chromosomal foci and the rDNA confirming that the ATPase activity of Pch2 is required for displacing Hop1 from the meiotic chromatin in vivo like it does in vitro (63).
Figure 11.

A model for Pch2 checkpoint function. (A) In unperturbed wild-type meiosis, nucleolar Pch2 excludes Hop1 from the rDNA preventing recombination at this chromosome XII array. In turn, chromosomal Pch2 dictates Hop1 discontinuous axis distribution sustaining proper synapsis and recombination. In the pch2 single mutant, Hop1 localizes to the nucleolar region and unwanted rDNA recombination occurs. Hop1 is also more abundant on chromosome axes disturbing normal recombination events that slightly increase Hop1-T318 phosphorylation. Nevertheless, most recombination defects of the pch2 mutant are only evident when DSBs are limiting (28,56). (B) In the synapsis-deficient zip1 mutant, Pch2 is absent from the chromosomes and concentrated in the rDNA. This configuration supports continuous distribution of Hop1 along unsynapsed axes and high levels of Hop1-T318 phosphorylation relaying the checkpoint signal to Mek1 activation. When PCH2 is deleted (or the ATPase inactivated), Hop1 loading and phosphorylation is impaired leading to inoperative checkpoint. (C) Despite the weakened Hop1/Mek1 activation, HOP1 overexpression in zip1 pch2 provides enough protein to restore high levels of global Hop1 phosphorylation and reinstate checkpoint function (see text for additional details).
Regulation of Hop1 phosphorylation
Phosphorylation of Hop1 at T318 by the Mec1/Tel1 checkpoint kinases is a requisite for Mek1 autophosphorylation and, therefore, for checkpoint activity. We show here that zip1-induced Hop1-T318 phosphorylation is drastically reduced in the absence of Pch2 or in ATPase-dead pch2 mutants. This reduction is manifested both globally using western blot analysis of whole cell extracts and locally on chromosome spreads, indicating a general requirement for Pch2 to maintain high levels of Hop1-T318 phosphorylation when synapsis defects occur. The fact that overexpression of wild-type HOP1, but not that of a hop1-T318A mutant, restores the checkpoint in zip1 pch2 reveals Hop1-T318 phosphorylation as the relevant target of Pch2's checkpoint function. Notably, although HOP1 overexpression in the wild type has only minimal effects, it reduces sporulation efficiency in the pch2 single mutant (Table 1), suggesting that Pch2 activity is required to maintain the proper balance of Hop1 abundance and phosphorylation also in the presence of synapsed chromosomes. We also note that Zip1 may contribute to the accumulation of phospho-Hop1T318 in pch2 mutant chromosomes.
We show that meiotic defects persist in zip1 pch2; thus, in principle, two possibilities can be envisaged to explain the regulation of Hop1-T318 phosphorylation by the Pch2 ATPase: Pch2 might favor the action of the Mec1/Tel1 kinases on Hop1-T318 or, alternatively, may inhibit phospho-Hop1-T318 phosphatase(s). In line with the first option, Pch2 (together with Xrs2) promotes Tel1-dependent Hop1 phosphorylation in response to unresected DSBs in a rad50S mutant (29). However, Tel1 is not required for the zip1-induced synapsis checkpoint (29) pointing to a Tel1-independent function for Pch2 in this particular scenario. We also show here that the absence of Pch2 does not alter the localization of the Mec1-Ddc2 complex suggesting that Pch2 is not required for global Mec1 activity. Nevertheless, a local requirement for Pch2 in facilitating the access of Mec1 to the Hop1-T318 substrate on chromosome axes cannot be ruled out. In this scenario, Pch2's ATPase activity could be required to induce some conformational change in the vicinity of Hop1 enabling its phosphorylation at T318 by Mec1. The interaction of Red1 with SUMO chains promotes Hop1-T318 phosphorylation (66). It is possible that Pch2 differentially modulates this interaction in response to SC defects. In line with this possibility, a role for Pch2 in orchestrating the interdependence between Hop1 and Red1 for Hop1 phosphorylation has been proposed (67). Nevertheless, these studies have not been performed under checkpoint-inducing conditions (i.e. zip1 mutant) where Pch2 may have different and/or additional functions (see below).
On the other hand, if Pch2 negatively regulates the phosphatase(s) involved in removing the phosphorylation of Hop1-T318, the lack of this phosphatase should reestablish the meiotic block in a zip1 pch2 mutant. It has been proposed that PP4 is the main phosphatase reversing Hop1-T318 phosphorylation (7); therefore, we investigated the impact of PP4 in the response to zip1 defects. In the BR1919 genetic background used in this study, the zip1 mutant shows a checkpoint-induced meiotic block in prophase, but at later time points at least a fraction of the cells resume cell cycle progression and complete the meiotic divisions. At the molecular level this meiotic resumption is manifested by an eventual decrease in Hop1-T318 phosphorylation and reduced Mek1 signaling (Figure 6). We found that the zip1 pph3 double mutant displays more persistent Mek1 activation and a tighter meiotic block arguing that, indeed, PP4 is crucial for deactivation and/or adaptation of the zip1-induced checkpoint. However, we have observed that the absence of PP4 function only confers a partial restoration of Hop1-T318 phosphorylation in zip1 pch2, indicating that Pch2 does not primarily act on this phosphatase or that PP4 may not be the only phosphatase capable of dephosphorylating Hop1-T318. The simplest interpretation of this result is that the low levels of Hop1-T318 phosphorylation in zip1 pch2, perhaps resulting from crippled Mec1/Tel1 function, are increased to some extent when the inhibitory action of PP4 is removed. Regardless of the identity of the direct target of Pch2, these observations confirm that the crucial role of Pch2 in the zip1 checkpoint is the fine regulation of Hop1-T318 phosphorylation status. In fact, these experiments reveal that manipulation of Hop1 phosphorylation in zip1 pch2 by other means, such as deleting PPH3, but without altering HOP1 expression levels also has an impact on the checkpoint.
Relevance of the ATPase activity for Pch2 checkpoint function
Recent in vitro analyses of the S. cerevisiae Pch2 protein have elegantly demonstrated that Pch2 is indeed an AAA+ ATPase that assembles into hexameric rings in the presence of nucleotides (63). That study also assessed the in vivo meiotic impact of mutations in the ATPase domain of Pch2 by determining the decrease in spore viability resulting from combining a csm4 mutant with the loss of Pch2 function. However, since Csm4 is involved in chromosome movement during prophase driven by telomere attachment to the nuclear envelope (68–70) and there are multiple meiotic processes influenced by Pch2, the physiological basis of this synthetic spore viability defect with csm4 is not obvious. Therefore, we analyzed the contribution of the ATPase activity to the well-established Pch2 role in the zip1-induced checkpoint. In contrast to the intermediate phenotype reported for the pch2-K320A mutant based on its combination with csm4 (63), we show here that mutants defective in either ATP binding site (pch2-K320A) or ATP hydrolysis (pch2-E399Q) phenocopy the pch2 deletion mutant for the checkpoint defect indicating that both activities are absolutely required for Pch2 checkpoint function. However, we observe a striking difference between both catalytically-inactive proteins in terms of localization on meiotic chromosome spreads. Whereas Pch2-E399Q displays the normal accumulation at the rDNA region on chromosome XII and some fainter interstitial chromosomal foci, the Pch2-K320A version completely fails to bind to the chromosomes. We found that the Pch2-K320A mutation preventing ATP binding compromises the integrity of the hexameric ring; this observation could explain the defect in localization, which may require an intact complex for its correct targeting. On the other hand, Pch2-E399Q does localize like the wild-type Pch2 protein; however, in contrast to Pch2, which does not overlap with Hop1 staining, the Pch2-E399Q version displays extensive colocalization with Hop1 both at chromosomal foci (in otherwise wild-type strains) and the rDNA, where Hop1 fails to be excluded in this mutant. These observations are consistent with the notion that Pch2/Trip13 utilizes the forces generated from ATP hydrolysis to displace or reorganize HORMA domain proteins, such as Hop1/HORMAD1 in meiosis or MAD2 in the SAC response (71). The effect of mammalian Trip13 on the conformational change of MAD2 is exerted with the help of the p31 adaptor (35). To determine whether yeast Pch2 requires additional cofactors or adaptors to disassemble Hop1 from meiotic chromosomes awaits further investigation. Nevertheless, in vitro assays show that purified Pch2 can directly alter Hop1 DNA binding properties (63).
A dual role for Pch2 on Hop1 regulation?
The Pch2 protein is predominantly found in the unsynapsed region of chromosome XII, which lacks Zip1 and Hop1, preventing recombination within the rDNA array (18,26). In addition, Pch2 is also present in SC-associated chromosomal foci promoting an alternating pattern of Zip1 and Hop1 during SC development (21). Curiously, whereas in yeast SK1 strains Pch2 chromosomal foci are conspicuous (22), in the BR strains used in this work, most Pch2 is detected in the nucleolar area with SC-associated foci showing a much weaker staining ((18); Figure 9 and Supplementary Figure S6). Importantly, in the synapsis-deficient zip1 mutant, the chromosomal Pch2 protein is no longer detectable and it is exclusively observed in the rDNA ((18); Figure 9). These observations suggest that two populations of Pch2 with different requirements for chromosome binding may exist: the nucleolar Pch2 pool, which does not require Zip1 for being targeted to the unsynapsed rDNA array and the interstitial chromosomal Pch2 protein, which is likely dependent on Zip1 assembly for its localization. Both pools of Pch2 possess the ability to displace Hop1 from the chromosome axes because Hop1 is more abundant along the SC and accumulates in the rDNA region in pch2 mutants lacking ATPase activity. However, since Pch2 function is critically required for the zip1-induced meiotic arrest, the fact that in the zip1 mutant Pch2 is only present at the rDNA location implies that the nucleolar Pch2 protein is responsible for the checkpoint function by sustaining high levels of Hop1-T318 phosphorylation in response to synapsis defects (Figure 11).
How can Pch2 control Hop1 phosphorylation from the nucleolus where, precisely, Hop1 is excluded? A role for nucleolar proteins in the control of cell cycle events is a well-documented phenomenon (72). Perhaps, the most paradigmatic example is the regulation of mitotic exit in budding yeast by the FEAR/MEN pathways orchestrating the release of the Cdc14 phosphatase from the nucleolus during anaphase (73–75). In a similar scenario it is possible that, when synapsis is defective, Pch2 traps in the nucleolus a crucial regulator required for exit from pachytene. Upon synapsis completion this factor would be released promoting entry into meiosis I. The PP4 phosphatase was a good candidate to meet these requirements because it promotes Hop1-T318 dephosphorylation and, hence, checkpoint inactivation, but our results indicate that Pch2 does not primarily acts via PP4 regulation (see above). In addition, during meiotic prophase PP4 appears to be localized at centromeres, not the nucleolus (76). We have also tested another phosphatase, PP1, which displays nucleolar localization at least during some stages of the cell cycle (77). In contrast to a previous study reporting a role for Glc7 -the catalytic subunit of PP1- in reversing Mek1 activation (38), we have found a minimal, if any, effect of either a glc7-T152K allele or a meiotic-depletion glc7-md mutant in Hop1 or Mek1 phosphorylation, and no evidence of functional relationship between Glc7 and Pch2 (AL and PSS, unpublished results). Alternatively, nucleolar Pch2 could control the localization of a Mec1 activator in response to synapsis defects. Since the N-terminal domain of Xrs2 physically interacts with Pch2 (29), it will be interesting to further investigate how this interaction impinges on Hop1 regulation in response to SC defects.
In sum, our results are consistent with a differential regulation of Hop1 by chromosomal and nucleolar Pch2 in a wild-type (checkpoint OFF) versus a zip1 (checkpoint ON) situation (Figure 11). The identification of Pch2 co-factors that may modulate its action on Hop1 depending on the specific chromosomal context or synapsis status will help to elucidate the basis of this differential effect.
Supplementary Material
Acknowledgments
The authors are grateful to Rodrigo Bermejo for a pph3Δ strain, Félix Prado for delitto perfetto reagents, Jesús Carballo for the phospho-Hop1-T318 antibody, Olga Calvo for the anti-GFP antibody, and Amy MacQueen and Shirleen Roeder for plasmids and strains. We also thank Vanessa Silva Dutra de Carvalho for help with some experiments, Isabel Acosta for technical assistance and Jesús Carballo and Andrés Clemente for helpful discussions and ideas.
Footnotes
Present addresses:
David Ontoso, Molecular Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Santiago Cavero, Department of Experimental and Health Sciences, Pompeu Fabra University, 08003 Barcelona, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Economy and Competitiveness (MINECO) of Spain [BFU2012-35748 and BFU2015-65417-R]; Predoctoral contract from the University of Salamanca (Spain) (to E.H.); Predoctoral fellowship (JAE-predoc) (to D.O.) and a postdoctoral fellowship (JAE-doc) (to S.C.) from the CSIC (Spain). Funding for open access charge: Ministry of Economy and Competitiveness (MINECO) of Spain [BFU2012-35748 and BFU2015-65417-R].
Conflict of interest statement. None declared.
REFERENCES
- 1.MacQueen A.J., Hochwagen A. Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends Cell. Biol. 2011;21:393–400. doi: 10.1016/j.tcb.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Roeder G.S., Bailis J.M. The pachytene checkpoint. Trends Genet. 2000;16:395–403. doi: 10.1016/s0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- 3.Subramanian V.V., Hochwagen A. The meiotic checkpoint network: step-by-step through Meiotic Prophase. Cold Spring Harb. Perspect. Biol. 2014;6:e1002369. doi: 10.1101/cshperspect.a016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwacha A., Kleckner N. Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell. 1997;90:1123–1135. doi: 10.1016/s0092-8674(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 5.Sym M., Engebrecht J.A., Roeder G.S. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell. 1993;72:365–378. doi: 10.1016/0092-8674(93)90114-6. [DOI] [PubMed] [Google Scholar]
- 6.Carballo J.A., Johnson A.L., Sedgwick S.G., Cha R.S. Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell. 2008;132:758–770. doi: 10.1016/j.cell.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 7.Chuang C.N., Cheng Y.H., Wang T.F. Mek1 stabilizes Hop1-Thr318 phosphorylation to promote interhomolog recombination and checkpoint responses during yeast meiosis. Nucleic Acids Res. 2012;40:11416–11427. doi: 10.1093/nar/gks920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ontoso D., Acosta I., van Leeuwen F., Freire R., San-Segundo P.A. Dot1-dependent histone H3K79 methylation promotes activation of the Mek1 meiotic checkpoint effector kinase by regulating the Hop1 adaptor. PLoS Genet. 2013;9:e1003262. doi: 10.1371/journal.pgen.1003262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niu H., Li X., Job E., Park C., Moazed D., Gygi S.P., Hollingsworth N.M. Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol. Cell. Biol. 2007;27:5456–5467. doi: 10.1128/MCB.00416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niu H., Wan L., Baumgartner B., Schaefer D., Loidl J., Hollingsworth N.M. Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol. Biol. Cell. 2005;16:5804–5818. doi: 10.1091/mbc.E05-05-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terentyev Y., Johnson R., Neale M.J., Khisroon M., Bishop-Bailey A., Goldman A.S. Evidence that MEK1 positively promotes interhomologue double-strand break repair. Nucleic Acids Res. 2010;38:4349–4360. doi: 10.1093/nar/gkq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niu H., Wan L., Busygina V., Kwon Y., Allen J.A., Li X., Kunz R.C., Kubota K., Wang B., Sung P., et al. Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol. Cell. 2009;36:393–404. doi: 10.1016/j.molcel.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acosta I., Ontoso D., San-Segundo P.A. The budding yeast polo-like kinase Cdc5 regulates the Ndt80 branch of the meiotic recombination checkpoint pathway. Mol. Biol. Cell. 2011;22:3478–3490. doi: 10.1091/mbc.E11-06-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leu J.Y., Roeder G.S. The pachytene checkpoint in S. cerevisiae depends on Swe1-mediated phosphorylation of the cyclin-dependent kinase Cdc28. Mol. Cell. 1999;4:805–814. doi: 10.1016/s1097-2765(00)80390-1. [DOI] [PubMed] [Google Scholar]
- 15.Tung K.S., Hong E.J., Roeder G.S. The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis-specific transcription factor Ndt80. Proc. Natl. Acad. Sci. U.S.A. 2000;97:12187–12192. doi: 10.1073/pnas.220464597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Chang C.Y., Wu J.F., Tung K.S. Nuclear localization of the meiosis-specific transcription factor Ndt80 is regulated by the pachytene checkpoint. Mol. Biol. Cell. 2011;22:1878–1886. doi: 10.1091/mbc.E10-12-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eichinger C.S., Jentsch S. Synaptonemal complex formation and meiotic checkpoint signaling are linked to the lateral element protein Red1. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11370–11375. doi: 10.1073/pnas.1004248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.San-Segundo P.A., Roeder G.S. Pch2 links chromatin silencing to meiotic checkpoint control. Cell. 1999;97:313–324. doi: 10.1016/s0092-8674(00)80741-2. [DOI] [PubMed] [Google Scholar]
- 19.San-Segundo P.A., Roeder G.S. Role for the silencing protein Dot1 in meiotic checkpoint control. Mol. Biol. Cell. 2000;11:3601–3615. doi: 10.1091/mbc.11.10.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu H.Y., Burgess S.M. Two distinct surveillance mechanisms monitor meiotic chromosome metabolism in budding yeast. Curr. Biol. 2006;16:2473–2479. doi: 10.1016/j.cub.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borner G.V., Barot A., Kleckner N. Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3327–3332. doi: 10.1073/pnas.0711864105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi N., Barot A., Jamison C., Borner G.V. Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet. 2009;5:e1000557. doi: 10.1371/journal.pgen.1000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wojtasz L., Daniel K., Roig I., Bolcun-Filas E., Xu H., Boonsanay V., Eckmann C.R., Cooke H.J., Jasin M., Keeney S., et al. Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 2009;5:e1000702. doi: 10.1371/journal.pgen.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farmer S., Hong E.J., Leung W.K., Argunhan B., Terentyev Y., Humphryes N., Toyoizumi H., Tsubouchi H. Budding yeast Pch2, a widely conserved meiotic protein, is involved in the initiation of meiotic recombination. PLoS One. 2012;7:e39724. doi: 10.1371/journal.pone.0039724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roig I., Dowdle J.A., Toth A., de Rooij D.G., Jasin M., Keeney S. Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS Genet. 2010;6:e1001062. doi: 10.1371/journal.pgen.1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vader G., Blitzblau H.G., Tame M.A., Falk J.E., Curtin L., Hochwagen A. Protection of repetitive DNA borders from self-induced meiotic instability. Nature. 2011;477:115–119. doi: 10.1038/nature10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanders S., Alani E. The pch2Δ; mutation in baker's yeast alters meiotic crossover levels and confers a defect in crossover interference. PLoS Genet. 2009;5:e1000571. doi: 10.1371/journal.pgen.1000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zanders S., Sonntag Brown M., Chen C., Alani E. Pch2 modulates chromatid partner choice during meiotic double-strand break repair in Saccharomyces cerevisiae. Genetics. 2011;188:511–521. doi: 10.1534/genetics.111.129031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho H.C., Burgess S.M. Pch2 acts through Xrs2 and Tel1/ATM to modulate interhomolog bias and checkpoint function during meiosis. PLoS Genet. 2011;7:e1002351. doi: 10.1371/journal.pgen.1002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eytan E., Wang K., Miniowitz-Shemtov S., Sitry-Shevah D., Kaisari S., Yen T.J., Liu S.T., Hershko A. Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet) Proc. Natl. Acad. Sci. U.S.A. 2014;111:12019–12024. doi: 10.1073/pnas.1412901111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson C.R., Hwang T., Chen P.H., Bhalla N. TRIP13PCH-2 promotes Mad2 localization to unattached kinetochores in the spindle checkpoint response. J. Cell Biol. 2015;211:503–516. doi: 10.1083/jcb.201505114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K., Sturt-Gillespie B., Hittle J.C., Macdonald D., Chan G.K., Yen T.J., Liu S.T. Thyroid hormone receptor interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic checkpoint-silencing protein. J. Biol. Chem. 2014;289:23928–23937. doi: 10.1074/jbc.M114.585315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banerjee R., Russo N., Liu M., Basrur V., Bellile E., Palanisamy N., Scanlon C.S., van Tubergen E., Inglehart R.C., Metwally T., et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat. Commun. 2014;5:4527. doi: 10.1038/ncomms5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin K.J., Patrick D.R., Bissell M.J., Fournier M.V. Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS One. 2008;3:e2994. doi: 10.1371/journal.pone.0002994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye Q., Rosenberg S.C., Moeller A., Speir J.A., Su T.Y., Corbett K.D. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife. 2015;4:e07367. doi: 10.7554/eLife.07367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rockmill B., Roeder G.S. Meiosis in asynaptic yeast. Genetics. 1990;126:563–574. doi: 10.1093/genetics/126.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal S., Roeder G.S. Zip3 provides a link between recombination enzymes and synaptonemal complex proteins. Cell. 2000;102:245–255. doi: 10.1016/s0092-8674(00)00029-5. [DOI] [PubMed] [Google Scholar]
- 38.Bailis J.M., Roeder G.S. Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell. 2000;101:211–221. doi: 10.1016/S0092-8674(00)80831-4. [DOI] [PubMed] [Google Scholar]
- 39.Humphryes N., Leung W.K., Argunhan B., Terentyev Y., Dvorackova M., Tsubouchi H. The Ecm11-Gmc2 complex promotes synaptonemal complex formation through assembly of transverse filaments in budding yeast. PLoS Genet. 2013;9:e1003194. doi: 10.1371/journal.pgen.1003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstein A.L., McCusker J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 41.Voth W.P., Jiang Y.W., Stillman D.J. New ‘marker swap’ plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast. 2003;20:985–993. doi: 10.1002/yea.1018. [DOI] [PubMed] [Google Scholar]
- 42.Schneider B.L., Seufert W., Steiner B., Yang Q.H., Futcher A.B. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- 43.Refolio E., Cavero S., Marcon E., Freire R., San-Segundo P.A. The Ddc2/ATRIP checkpoint protein monitors meiotic recombination intermediates. J. Cell Sci. 2011;124:2488–2500. doi: 10.1242/jcs.081711. [DOI] [PubMed] [Google Scholar]
- 44.Stuckey S., Mukherjee K., Storici F. In vivo site-specific mutagenesis and gene collage using the delitto perfetto system in yeast Saccharomyces cerevisiae. Methods Mol. Biol. 2011;745:173–191. doi: 10.1007/978-1-61779-129-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hollingsworth N.M., Ponte L. Genetic interactions between HOP1, RED1 and MEK1 suggest that MEK1 regulates assembly of axial element components during meiosis in the yeast Saccharomyces cerevisiae. Genetics. 1997;147:33–42. doi: 10.1093/genetics/147.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlson M., Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell. 1982;28:145–154. doi: 10.1016/0092-8674(82)90384-1. [DOI] [PubMed] [Google Scholar]
- 47.Rockmill B. Chromosome spreading and immunofluorescence methods in Saccharomyes cerevisiae. Methods Mol. Biol. 2009;558:3–13. doi: 10.1007/978-1-60761-103-5_1. [DOI] [PubMed] [Google Scholar]
- 48.Santos B., Duran A., Valdivieso M.H. CHS5, a gene involved in chitin synthesis and mating in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997;17:2485–2496. doi: 10.1128/mcb.17.5.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu L., Ajimura M., Padmore R., Klein C., Kleckner N. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995;15:6572–6581. doi: 10.1128/mcb.15.12.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collart M.A. The NOT, SPT3, and MOT1 genes functionally interact to regulate transcription at core promoters. Mol. Cell. Biol. 1996;16:6668–6676. doi: 10.1128/mcb.16.12.6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lydall D., Nikolsky Y., Bishop D.K., Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- 52.Majka J., Burgers P.M. Clamping the Mec1/ATR checkpoint kinase into action. Cell Cycle. 2007;6:1157–1160. doi: 10.4161/cc.6.10.4221. [DOI] [PubMed] [Google Scholar]
- 53.Hong E.J., Roeder G.S. A role for Ddc1 in signaling meiotic double-strand breaks at the pachytene checkpoint. Genes Dev. 2002;16:363–376. doi: 10.1101/gad.938102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Govin J., Dorsey J., Gaucher J., Rousseaux S., Khochbin S., Berger S.L. Systematic screen reveals new functional dynamics of histones H3 and H4 during gametogenesis. Genes Dev. 2010;24:1772–1786. doi: 10.1101/gad.1954910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith A.V., Roeder G.S. The yeast Red1 protein localizes to the cores of meiotic chromosomes. J. Cell Biol. 1997;136:957–967. doi: 10.1083/jcb.136.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joshi N., Brown M.S., Bishop D.K., Borner G.V. Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol. Cell. 2015;57:797–811. doi: 10.1016/j.molcel.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arbel A., Zenvirth D., Simchen G. Sister chromatid-based DNA repair is mediated by RAD54, not by DMC1 or TID1. EMBO J. 1999;18:2648–2658. doi: 10.1093/emboj/18.9.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y., Gaines W.A., Callender T., Busygina V., Oke A., Sung P., Fung J.C., Hollingsworth N.M. Down-regulation of Rad51 activity during meiosis in yeast prevents competition with Dmc1 for repair of double-strand breaks. PLoS Genet. 2014;10:e1004005. doi: 10.1371/journal.pgen.1004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raschle M., Van Komen S., Chi P., Ellenberger T., Sung P. Multiple interactions with the Rad51 recombinase govern the homologous recombination function of Rad54. J. Biol. Chem. 2004;279:51973–51980. doi: 10.1074/jbc.M410101200. [DOI] [PubMed] [Google Scholar]
- 60.Cheng C.H., Lo Y.H., Liang S.S., Ti S.C., Lin F.M., Yeh C.H., Huang H.Y., Wang T.F. SUMO modifications control assembly of synaptonemal complex and polycomplex in meiosis of Saccharomyces cerevisiae. Genes Dev. 2006;20:2067–2081. doi: 10.1101/gad.1430406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lynn A., Soucek R., Borner G.V. ZMM proteins during meiosis: crossover artists at work. Chromosome Res. 2007;15:591–605. doi: 10.1007/s10577-007-1150-1. [DOI] [PubMed] [Google Scholar]
- 62.Voelkel-Meiman K., Taylor L.F., Mukherjee P., Humphryes N., Tsubouchi H., Macqueen A.J. SUMO localizes to the central element of synaptonemal complex and is required for the full synapsis of meiotic chromosomes in budding yeast. PLoS Genet. 2013;9:e1003837. doi: 10.1371/journal.pgen.1003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen C., Jomaa A., Ortega J., Alani E.E. Pch2 is a hexameric ring ATPase that remodels the chromosome axis protein Hop1. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E44–E53. doi: 10.1073/pnas.1310755111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanson P.I., Whiteheart S.W. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell. Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 65.Wendler P., Ciniawsky S., Kock M., Kube S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta. 2012;1823:2–14. doi: 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 66.Lin F.M., Lai Y.J., Shen H.J., Cheng Y.H., Wang T.F. Yeast axial-element protein, Red1, binds SUMO chains to promote meiotic interhomologue recombination and chromosome synapsis. EMBO J. 2010;29:586–596. doi: 10.1038/emboj.2009.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lo Y.H., Chuang C.N., Wang T.F. Pch2 prevents Mec1/Tel1-mediated Hop1 phosphorylation occurring independently of Red1 in budding yeast meiosis. PLoS One. 2014;9:e85687. doi: 10.1371/journal.pone.0085687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conrad M.N., Lee C.Y., Chao G., Shinohara M., Kosaka H., Shinohara A., Conchello J.A., Dresser M.E. Rapid telomere movement in meiotic prophase is promoted by NDJ1, MPS3, and CSM4 and is modulated by recombination. Cell. 2008;133:1175–1187. doi: 10.1016/j.cell.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 69.Kosaka H., Shinohara M., Shinohara A. Csm4-dependent telomere movement on nuclear envelope promotes meiotic recombination. PLoS Genet. 2008;4:e1000196. doi: 10.1371/journal.pgen.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wanat J.J., Kim K.P., Koszul R., Zanders S., Weiner B., Kleckner N., Alani E. Csm4, in collaboration with Ndj1, mediates telomere-led chromosome dynamics and recombination during yeast meiosis. PLoS Genet. 2008;4:e1000188. doi: 10.1371/journal.pgen.1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vader G. Pch2(TRIP13): controlling cell division through regulation of HORMA domains. Chromosoma. 2015;124:333–339. doi: 10.1007/s00412-015-0516-y. [DOI] [PubMed] [Google Scholar]
- 72.Pederson T. The nucleolus. Cold Spring Harb. Perspect. Biol. 2011;3:a000638. doi: 10.1101/cshperspect.a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shou W., Seol J.H., Shevchenko A., Baskerville C., Moazed D., Chen Z.W., Jang J., Charbonneau H., Deshaies R.J. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97:233–244. doi: 10.1016/s0092-8674(00)80733-3. [DOI] [PubMed] [Google Scholar]
- 74.Stegmeier F., Visintin R., Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108:207–220. doi: 10.1016/s0092-8674(02)00618-9. [DOI] [PubMed] [Google Scholar]
- 75.Visintin R., Hwang E.S., Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- 76.Falk J.E., Chan A.C., Hoffmann E., Hochwagen A. A Mec1- and PP4-dependent checkpoint couples centromere pairing to meiotic recombination. Dev. Cell. 2010;19:599–611. doi: 10.1016/j.devcel.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 77.Bloecher A., Tatchell K. Dynamic localization of protein phosphatase type 1 in the mitotic cell cycle of Saccharomyces cerevisiae. J. Cell Biol. 2000;149:125–140. doi: 10.1083/jcb.149.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.