


Abstract
The quantification of transcriptomic features is the basis of the analysis of RNA-seq data. We present an integrated alignment workflow and a simple counting-based approach to derive estimates for gene, exon and exon–exon junction expression. In contrast to previous counting-based approaches, EQP takes into account only reads whose alignment pattern agrees with the splicing pattern of the features of interest. This leads to improved gene expression estimates as well as to the generation of exon counts that allow disambiguating reads between overlapping exons. Unlike other methods that quantify skipped introns, EQP offers a novel way to compute junction counts based on the agreement of the read alignments with the exons on both sides of the junction, thus providing a uniformly derived set of counts. We evaluated the performance of EQP on both simulated and real Illumina RNA-seq data and compared it with other quantification tools. Our results suggest that EQP provides superior gene expression estimates and we illustrate the advantages of EQP's exon and junction counts. The provision of uniformly derived high-quality counts makes EQP an ideal quantification tool for differential expression and differential splicing studies. EQP is freely available for download at https://github.com/Novartis/EQP-cluster.
INTRODUCTION
In recent years, RNA-seq has become a widely used approach for expression profiling studies. Usually, the aim of such studies is to determine the set of differentially expressed genes between two or more conditions or, in rarer cases, to analyze the changes in the expressed splicing patterns of genes. In order to address such questions, the reads delivered by the sequencing machines need to be converted to expression estimates that can then be used as the basis for statistical modeling and analysis.
The different steps of a RNA-seq workflow are shown in Figure 1. In order to generate expression estimates, the reads are usually aligned to a reference sequence, e.g. the genome or the transcripts. The information about the overlap between the read alignments and the genomic features is then used to derive the expression values. Two approaches can be distinguished here. In the first approach, feature expression estimates are computed by simply counting the number of reads that overlap with the feature reference sequences (1–4). Genes, exons and junctions can be quantified in this way; however, it is not possible to obtain meaningful transcript expression estimates by counting since reads that map to the same gene are often shared between different transcripts preventing the determination of their transcript of origin. This problem is addressed in the second approach that is based on the use of statistical models and optimization algorithms to distribute the reads between the transcripts resulting in transcript expression estimates. The expression estimate of a gene can then be obtained as the sum of the expression estimates of its transcript isoforms.
Figure 1.
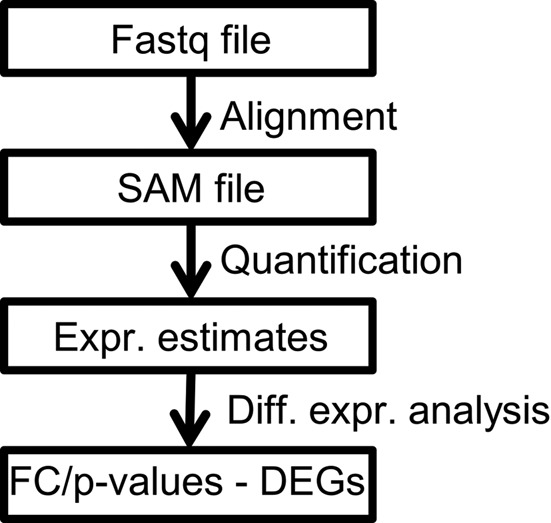
The major steps of a standard RNA-seq workflow. First, reads in Fastq format are aligned against reference sequences. Expression estimates are derived from the alignments. The estimates are then used to identify differentially expressed (or spliced) genes, usually by computing fold changes and P-values.
If the goal of an expression profiling experiment is to identify differentially expressed genes, then gene expression estimates obviously suffice. For counting-based gene quantification the script htseq-count of the HTSeq framework is widely used (3); its approach can be considered as a standard and has been reimplemented in other tools such as featureCounts (1) and the R package GenomicRanges (4). An alternative is represented by the recently released Bioconductor package QuasR which provides a complete RNA-seq workflow in R including read alignment and generation of gene, exon and junction counts (5). The computation of gene expression estimates via transcript abundances is provided by the popular tool Cufflinks (6,7) and numerous other approaches (8–16).
If the goal of an expression profiling experiment is to identify differentially spliced genes, then either the direct use of transcript abundance estimates (7,17–19) or of exon or junction counts is required (20–25). In contrast to transcript abundance estimates, the simpler problem of the generation of exon and junction counts has received relatively little attention–the main options for exon counts being the DEXSeq helper script dexseq-count (20) and, more recently, QuasR. Nevertheless, as we will show, the different approaches to exon and junction quantification can lead to significant differences in the reported counts.
Here, we introduce the exon quantification pipeline (EQP), a new counting-based quantification approach designed to estimate the expression level of genes, exons and exon–exon junctions in RNA-seq experiments. EQP performs the steps of alignment and quantification to generate these three types of counts. For the analysis, EQP requires the sequencing reads (either in Fastq format or pre-aligned in SAM/BAM format to the reference genome), the genome of the species in Fasta format, the transcript sequences in Fasta format, and the corresponding gene, transcript and exon annotation in a GTF file. We assessed the performance of EQP on experimental and simulated RNA-seq data sets by comparing it with the results of different quantification methods. We show that EQP delivers more accurate gene expression estimates as well as exon and junction counts which are well suited to quantify overlapping exons. Altogether, our results showcase the potential of EQP to serve as a high quality quantification tool for differential expression and differential splicing studies.
MATERIALS AND METHODS
The exon quantification pipeline (EQP)
EQP consists of two distinct parts: the alignment module and the quantification module. The workflow of EQP is designed to provide a high degree of granularity allowing for the use of distributed computational resources as given, for instance, by a cluster with a job scheduling system or a cloud-based infrastructure. It provides the option to split the Fastq input files into blocks of a user-definable read number which can then be processed independently; at the same time, it also filters out reads with too few non-A or non-T bases to filter out polyA tail reads. Generally, all processing operations of EQP are based on a read by read ordering – rather than a genome-coordinate based ordering after the alignment step.
The EQP alignment module
Before EQP can be run, it is necessary to create a number of auxiliary files that are needed in the alignment module and for the generation of the counts. As input for the setup EQP requires a genome Fasta file, a transcript Fasta file and a GTF file. The main output of the setup step is a file containing the mapping of the genomic exons to the transcripts in BED format (26) and an exon–exon junction Fasta file. The latter file contains all possible junctions of exons that belong to the same gene. More precisely, only the parts of the exons up to the read length on each side of the junction are used in the creation of the Fasta file. If an exon is shorter than the read length, EQP pads junction entries with all possible upstream or downstream exons (depending on whether the exon is upstream or downstream of the skipped intron) in order to avoid imbalanced junction entries. This procedure can lead to a very high number of combinations for genes with many small exons as several padding steps may be required on each side until the sum of the exon lengths exceeds the read length. In EQP such genes are identified based on a threshold on the number of junction entries and for these genes short exons are only padded by their direct upstream and downstream flanking exons.
The EQP alignment module aligns the Fastq files containing the single or paired-end reads against the externally provided transcript and genome Fasta file as well as the custom created junction Fasta file using Bowtie2 (27); the three alignment computations can be run independently, thus providing an additional opportunity for parallelization. For each read and alignment file, the type of alignment (single-read or paired-end), the number of alignments and the sum of the number of mismatches of the alignments (taken from the optional SAM NM field (28)) are computed. Then the best set of alignments (either the transcript, junction or genome alignments) is selected based on three criteria (similar to (29): the alignment type (paired-end is preferred over single-read), the mean number of mismatches and the number of alignments; a slight preference is given to transcript alignments by adding a small penalty per read to genome alignments. If no paired-end alignment exists, then the best alignments are chosen separately for each read. The result of the selection process is a combined SAM file consisting of SAM entries that originate from the three different original alignment files. As an optional step, EQP can generate a genome alignment file containing spliced genome alignments for all mapped reads which can then be used to visualize the alignments in a genome browser such as IGV (30).
The EQP quantification module
The counts computed by EQP are based on weighted reads where the weight w of a read that aligns n times to the genome is given as w = 1 / n similar to the treatment of multi-reads by Cufflinks (6). EQP allows setting an upper bound B on the number of allowed genomic alignments of a read. The default is 100 but, for instance, setting B = 1 results in counting only uniquely aligning reads and setting B = 10 in counting quasi-unique alignments (31).
For the computation of the read weights EQP takes the combined SAM file as input, converts it into a BED file, and intersects the converted BED file with a BED file containing the transcript, the genome and the junction coordinates of the genomic exons using BEDTools (26) – see Supplementary Information for an example. The resulting intersection file contains the relevant information to calculate the reads’ spliced genomic alignments.
In EQP a read is counted for a feature, e.g. a gene or exon, if and only if the induced exon boundaries of its alignment completely agree (are compatible) with the exon boundaries of the feature. This is illustrated in Figure 2; a more precise definition is given in the Supplementary Information. In particular, compatibility implies that no intron spanned by the read overlaps with the feature and vice versa.
Figure 2.

The alignment of read R is compatible with the exons E1, E2 and E3 but not with the exons E4, E5 and E6. It is also compatible with gene G since it is completely contained in the compatible exons of G. Of the 12 possible exon–exon junctions between exons of G only the junctions between E1 and E2 and E2 and E3 are compatible with the alignment of R.
The compatibility requirement ensures that the counted reads represent evidence for the fully matured expression of the feature and excludes reads originating from the sequencing of pre-mRNA transcripts. This is especially important for the computation of exon and junction counts as these are mainly used in the analysis of differential splicing and, thus, should only capture the mRNA signal.
Based on the intersection BED file and a file containing the mapping of the genomic exons to genes, exons or junctions, the respective counts are then computed. Note that the generation of junction counts (as well as gene and exon counts) is independent of aligning the reads against the junction Fasta file. All three types of counts can be generated from transcript or genome alignments alone. In particular, EQP allows computing the gene, exon and junction counts from an externally provided genome alignment file generated by a splice-aware aligner such as, for instance, Tophat2 (32) or STAR (33) if a GTF file with the genomic exon coordinates is provided.
Tools and data sets used for benchmarking
Experimental and simulated RNA-seq data sets were used to evaluate the performance of EQP by comparing it with the results of different quantification pipelines. For the alignment module, we considered SpliceMap (34), the aligner used in QuasR, the two popular aligners Tophat2 (32) and STAR (33) as well as the genome alignments generated by EQP; for the quantification step we used the software packages htseq-count (3), featureCounts (1), Cufflinks2 (7) and QuasR (5) for the generation of gene counts, dexseq-count (20) and QuasR for exon counts, and QuasR for junction counts as shown in Figure 3.
Figure 3.

An overview of the data sets (white), the alignment methods (light grey) and the quantification methods (dark grey) used for comparison with EQP.
The experimental data set consists of RNA-seq data of the Universal Human Reference RNA (UHRR or SEQC-A) sample and the Human Brain Reference (HBR or SEQC-B) sample that were generated in the context of the RNA Sequencing Quality Control (SEQC) initiative (35). There are a number of reasons for choosing an RNA-seq benchmarking data set based on the UHRR and HBR samples: (i) these two samples have been extensively profiled and analyzed in the MicroArray and the RNA SEQC initiatives (36); (ii) RNA-seq data for these samples were previously used for comparison in the publication of different RNA-seq quantification tools (14) as well as in an independent comparison study of RNA-seq quantification pipelines (37); (iii) Taqman qRT-PCR data exist for both samples for 1000 genes.
The RNA-seq data set is part of GEO series GSE47774 and consists of paired-end reads of length 100 bp for each of the two samples with four technical replicates; each of the replicates was sequenced on eight (multiplexed) lanes of a HiSeq 2000, yielding ∼10 M reads per lane resulting in 64 Fastq file pairs with more than 80 M reads per replicate or more than 320 M reads per sample. The Supplementary Information contains a list of the accession numbers of the data used. The comparison is based on the processing of each of the 64 Fastq input file pairs separately. The Taqman qRT-PCR data were downloaded from Gene Expression Omnibus under accession number GSE5350.
The second RNA-seq data set is taken from an independent evaluation of RNA-seq quantification pipelines (38). It was simulated using Flux simulator (39) on reference data from Ensembl v66 (40). All transcripts of protein coding genes were simulated to have approximately the same expression level. A number of different settings for the read type (single or paired-end), read length and sequencing depth were used in the simulation. Here, we use the paired-end data for a read length of 100 bp and sequencing depths ranging from ∼3 to ∼36 M reads; for each sequencing depth a data set consists of two samples with four libraries each.
RESULTS
Comparison of the alignment module
We start out with a comparison of the EQP alignment module with the aligners Tophat2, STAR and SpliceMap on the SEQC data set. It should be noted that these aligners solve a more general problem than EQP, as EQP's alignment module is based only on un-spliced alignments against various reference data sets and, therefore, does not detect any spliced reads for novel, unannotated exons. For EQP, STAR and Tophat2 an effort was made to obtain similar alignment results (allowing up to 100 genomic mapping locations for one read) and to provide as much auxiliary input from the reference data sets as possible. For SpliceMap which was run via the QuasR R interface we used the default alignment parameters as provided by QuasR which, in particular, implies that at most one genomic mapping location is reported. The exact versions and calls for each aligner are listed in the Supplementary Information.
We used human reference data of Ensembl 76 for the alignment which is the first Ensembl version based on the GRCh38 genome build; we excluded the alternative scaffolds as these would have resulted in a combined genome Fasta file of more than 35 GB consisting mostly of Ns which is a challenge for aligner index construction.
Although our main goal is to assess the usefulness and influence of the different aligners in the generation of expression estimates, it is still informative to compare some basic alignment statistics as shown in Figure 4.
Figure 4.

Comparison of the EQP alignment module to other aligners. (A) The percentage of reads of the SEQC samples aligned to the reference genome (left) and the percentage of reads overlapping exons (right) for the four aligners EQP (Bowtie2), STAR, SpliceMap and Tophat2. The error bars are the standard deviation across all 64 input Fastq files. (B) The mean number of mismatches and the mean number of soft masked bases per (paired-end) read.
EQP and STAR align a very similar number of reads to the reference (93.9% and 93.6%) whereas Tophat2 and SpliceMap yield fewer alignments (83.4% and 74.6%). In terms of reads aligned to exons, the aligners EQP and Tophat2 are essentially indistinguishable (82.8% and 83.1%) suggesting an excellent selectivity of Tophat2. STAR gave a slightly lower number of reads (80.7%) whereas SpliceMap also maps considerably fewer reads to the exons (67%). The number of reads aligned to the exons is an important parameter for the RNA-seq analysis because exonic reads contribute to the generation of gene, exon and junction expression counts.
The percentage of aligned reads in itself is not a sufficient measure of alignment quality since high alignment percentages may contain many false positive alignments. Therefore, it is also important to assess the accuracy of the alignments produced by the different alignment algorithms. We consider the mean number of mismatches between the reference and the reads as a coarse measure for the overall alignment quality (as reported in the optional SAM NM field). As shown in the left half of Figure 4B, Tophat2 and SpliceMap have the least number of mean mismatches (0.46 and 0.44), with EQP having slightly more (0.61) and STAR being a clear outlier with about twice as many mismatches as EQP (1.22). Local aligners such as STAR and SpliceMap soft mask mismatches at the beginning and end of the alignments and count only mismatches in the aligned middle part. For this reason, we also investigated the number of soft masked bases. As it can be seen from the right half of Figure 4B, Tophat2 is a global aligner that does not make use of soft masking, EQP masks ∼0.1 bp per read pair whereas STAR and SpliceMap mask ∼1% of all aligned bases (1.8 bp and 2.6 bp, resp., of 2 × 100 bp per read pair).
The ability of an aligner to find the location of spliced reads in the reference is an important feature for the analysis of differential splicing. In Figure 5 the number of introns that are covered by the different aligners is shown. Tophat2 clearly has the highest sensitivity with over 0.5 M spanned introns, more than twice the number of introns discovered by EQP which is the second most sensitive spliced aligner with 0.23 M introns. However, Tophat2 discovers many introns which are likely to be spurious since over 75% of the introns are spanned by less than five reads. For the other aligners only about 50% of the introns are spanned by less than five reads indicating a higher accuracy than Tophat2 in agreement with the results reported in (41).
Figure 5.

Mean number of introns spanned by at least one, three or five reads per sample. The error bars are the standard deviation across all 64 input SEQC Fastq files.
Comparison of computational resources used by the aligners
As a last point we also want to report on the runtime and memory performance of the different aligners. We ran EQP, STAR and Tophat2 on six cores in our Linux cluster environment with a Sun Grid Engine job scheduling system. In general, we observed about an order of magnitude speed-up of STAR over EQP which in turn took about half the time of Tophat2 (for ∼10 M reads ∼45 min for STAR, ∼5 h for EQP and ∼9 h for Tophat2). Here, the time of EQP is measured as the sum of the three independent alignment calls; however, in practice, the alignment calls are often executed in parallel leading to significantly reduced computation times. SpliceMap was run on a different but comparable hardware with eight cores as it could not be integrated into the job scheduling system of the cluster and needed about 20–25% more time than Tophat2 – see Supplementary Information for more details. Finally, since EQP, Tophat2 and SpliceMap are all Bowtie-based, their memory requirements are 2–4 GB whereas STAR requires more than 25 GB.
In summary, our results indicate that the EQP alignment module has the highest sensitivity compared to the other tested aligners (considering both the percentages of reads aligned to the reference and to the exons), a high accuracy (few mismatches on average and no soft masked bases), a high sensitivity for introns and low requirements on computational resources.
Comparison of gene counts
As mentioned before, we compared the gene counts generated by EQP to the gene counts of htseq-count, featureCounts, QuasR and Cufflinks. In the following, we briefly present the different tools.
htseq-count is a widely used software and one of the first tools developed for the count-based quantification of RNA-seq data. It uses three natural, set-theoretic criteria (called modes) to assign a read to a feature: (i) if the read has a non-empty overlap with the feature (called mode union), (ii) if the read is contained in the feature (called mode intersection-strict) and (iii) a relaxed containment criterion (called mode intersection-nonempty). EQP's evidence-based counting criterion can be seen as more stringent version of mode intersection-strict. In all three modes, htseq-count excludes ambiguous reads (i.e. reads that map to multiple genes) based on the insightful observation that the fold-change can be more accurately estimated if only unambiguous reads are counted.
The three counting modes of htseq-count can be considered as a de facto standard which has been reimplemented in the BioConductor package GenomicRanges (4) as well as the subRead package tool featureCounts (1,42). featureCounts implements only the mode union, however, with highly superior speed and more flexible options; for instance, it is possible to specify whether or not to count ambiguous reads or multi-mappers. Interestingly, featureCounts does not exactly reproduce the results of htseq-count with mode union due to a difference in the interpretation of the coordinates in the GTF file containing the genome annotation (1).
QuasR is a recently released, elegant and versatile BioConductor package which provides a simple R interface to the complete RNA-seq workflow; this includes QC, read alignment using either Bowtie or SpliceMap, and quantification. Besides RNA-seq it can also be used in a number of different count-based applications such as ChIP-seq, small-RNA-seq and bisulfite sequencing. With respect to the functionality of RNA-seq it is the most similar to EQP in that it also provides gene, exon and junction counts. In fact, many of the features of EQP were inspired by the Perl-based predecessor of QuasR as, for instance, the weighting of reads (which is, however, not included in the published version of QuasR). QuasR's counting criterion is based on a single read position that is the first base by default but can be chosen freely. A read is counted for a feature if and only if the designated aligned read position falls into the feature; this leads to the elegant property that a gene count in QuasR equals the sum of its exon counts (if there are no overlapping exons). QuasR is the only tool which provides read counts (as opposed to fragment counts) even if paired end data are supplied.
In stark contrast to the simple counting based approaches discussed above, Cufflinks2 provides transcript and gene abundance estimates by employing a highly sophisticated statistical inference engine that allows disambiguating reads that map to several transcripts; in addition, it can take biases in library preparation protocols into account. In this respect Cufflinks2 solves a considerably more complicated problem than just providing gene counts. In this comparison, however, we only make use of Cufflinks’ gene abundance estimates and ignore the transcript abundance estimates. Cufflinks2 is also able to assemble and quantify a set of de-novo transcripts; this mode can be used if no genome annotation is available or to integrate and extend an existing genome annotation.
Comparison of gene counts using the SEQC RNA-seq data
We start the comparison of the different tools by considering the root-mean-square deviation (RMSD) of the log-fold changes between different lanes and the mean of log-fold change of the Taqman qPCR data which we consider as a gold standard for our purposes. The use of fold changes is motivated by the fact that fold changes are often an important parameter in the analysis of RNA-seq data which aim to determine differentially expressed genes. Usually, this is combined with a measure of the significance of the fold change in order to take the biological variability into account, e.g. a P-value. However, we do not include this second measure in our assessment since the SEQC RNA-seq data consist only of technical replicates with no biological variability.
To obtain the fold change values the gene counts are first library-size normalized using DESeq (43) except for Cufflinks2's FPKM gene abundance estimates which are taken as is; note that length normalization as used in FPKM is not necessary for the computation of fold changes. In the following we use the term CPM (count per million) to denote the library-size normalized counts (which are scaled to sum to one million on average). To be able to also assign a fold change value to sample pairs in which the count(s) in one (or both) sample(s) are zero we add a pseudo count of 0.1 to the RNA-seq derived CPM counts; see the Supplementary Information for more details about the computation of the fold change correlations.
In Figure 6, we illustrate the agreement of the Taqman data with the results of three RNA-seq quantification methods. The scatter plots display on the log-fold changes between SEQC-A and SEQC-B samples for 995 of 1000 Taqman genes which could be mapped to Ensembl gene ids. The plots for the different methods are very similar; some outliers are present in all three plots suggesting systematic differences between Taqman and RNA-seq expression measurements. To be able to numerically assess the agreement of the RNA-seq with the Taqman measurements we use the RMSD of the Taqman and RNA-seq log-fold changes between SEQC-A and SEQC-B.
Figure 6.

Scatter plots of Taqman mean gene expression fold changes versus the RNA-seq gene count fold changes of the first lanes for the samples SEQC-A and SEQC-B using the three quantification methods EQP/Bowtie2, htSeq intersection-strict/STAR and Cufflinks2/Tophat2.
The results of the comparison are summarized in Figure 7. Overall the results of all approaches are quite similar with the mean RMSD values ranging between 1.2 and 1.34. If we disregard the results for SpliceMap as outliers, then the first observation is that the choice of the quantification method influences the results more than the choice of the aligner (ANOVA P-value < 2−16 for the quantification and 0.63 for the alignment methods). This is consistent with the results reported in (38). The poor performance of SpliceMap can be partially explained by the fact that we used the QuasR default option of allowing at most one alignment per read for SpliceMap whereas we used the EQP default option of allowing up to 100 genomic alignments for the other aligners. For the best quantification methods using STAR leads to lower RMSD values than using Tophat2 and, in particular, when considering EQP the results are indistinguishable from results of EQP's own alignment method.
Figure 7.

The distribution of RMSD values for different quantification methods and aligners. Each box plot reflects 32 values computed between the Taqman mean gene expression fold changes and the gene count fold changes of sample SEQC-A versus SEQC-B for different lanes of the RNA-seq data. For EQP we consider four parameter settings: counting reads with up to 100 or up to 10 genomic alignments (EQP and EQP-quasi-unique), with unique genomic alignments (EQP-unique), and with unambiguous genomic alignments (EQP-unambiguous).
The first four columns on the left of Figure 7 are based on different options for the usage of EQP. With respect to the comparison of the Taqman fold changes it seems that the exclusion of multi-mappers or ambiguous reads confers a slight advantage. Concerning the three modes of htseq-count, the RMSD values clearly decrease with increasing the stringency of the read assignment criterion (in the order of union, intersection-non-empty and intersection-strict). This is consistent with the fact that EQP yields the lowest RMSD values since, as mentioned before, the read assignment criterion used by EQP can be viewed as an even more stringent version of intersection-strict. In general, EQP, QuasR and htseq-count with mode intersection-strict behave very similar across all aligners. Cufflink2 shows the lowest agreement with the Taqman fold changes even if Tophat2 is used as an aligner, again consistent with previous results (37,38). All together, these results indicate that EQP provides slightly superior or on par gene expression estimates as compared to the other methods tested – independent of the aligner.
A very similar overall picture can be seen if we consider the coefficient of determination instead of the RMSD values; see the Supplementary Information.
Comparison of gene counts using simulated RNA-seq data
The simulated RNA-seq data were originally generated by Fonseca et al. (38) to assess the correlation of the data generated by the different quantification methods on the count level. Here, we use the same data set to compare the quantification methods on the count-level as well. It should be noted that a count-level comparison puts htseq-count at a disadvantage since its exclusion of ambiguous reads leads to inferior count correlations.
We performed the comparisons on a reduced set of methods; in particular, we excluded the SpliceMap alignments due to the considerable time investment of generating the alignments without the support of a cluster. We also excluded the alignments produced by EQP from this comparison as they behave very similar to STAR and Tophat2.
The RNA-seq data were simulated for a range of sequencing depth consisting of 3 M, 9 M, 18 M and 36 M reads. For each read depth, there are two samples with four libraries. In order to use expression values that reflect the relative expression of each gene as accurately as possible we also length normalize the CPM counts by dividing by the gene length (which is computed as the length of the ‘genomic footprint’ of a gene, that is, the number of all genomic bases covered by some exon of the gene) – again except for Cufflinks’ FPKM values. As in (38), we use the Spearman correlation to measure the agreement between the true counts and the expression estimates.
In Figure 8, we show the results for the library size 9 M reads which is a number close to the ∼10 M reads for the SEQC samples considered above. The plots for the other library sizes (3 M, 18 M and 36 M reads) show very similar results and can be found in the Supplementary Information. With respect to the count correlation Cufflinks2 shows the highest correlation values.
Figure 8.

Spearman correlation between gene expression estimates and true gene counts based on the comparison of the eight different libraries of the simulated data.
Quantifier execution times
We also assessed the run times of the quantifiers on the SEQC data (with the exception of QuasR). Though the total processing time is usually dominated by the alignment step, it is still interesting to compare the time spent on the quantification step. If the genome alignment file is provided, then EQP and featureCounts have a comparable execution times of about 20–25 min on average, followed by htSeq-count with 35–40 min (independent of the counting mode). Cufflinks2 needs considerably more time with a median of 3–3.5 h (see Supplementary Information for more details).
Assessment of exon counts
As mentioned above, we compare EQP's exon counts with the counts generated by the dexseq-count helper script included in the DEXSeq package (20) and QuasR. One difficulty is that there is no agreement about what an exon count should represent, leading to different types of counts which cannot be compared.
The exon counts generated by dexseq-count are based on a modified exon model in which the entries in the GTF file are ‘flattened’, i.e. overlapping exons are subdivided into disjoint intervals called exonic parts which can belong to more than one exon and are quantified using htseq's union count mode. This elegantly reduces the multitude of possible splicing events to mostly exonic part skipping events but can obscure differential exon usage for overlapping exons and makes the counts more difficult to interpret. QuasR and EQP on the other hand use whole exons as their basis for quantification. As a consequence, the counts generated by dexseq-count and EQP or QuasR are only comparable for exons that do not overlap with another exon.
As is shown in Table 1, overlapping exons represent a significant fraction (up to over 60%) of the overall number of exons for different organisms. Annotation sources and differences in the treatment of these features can lead to considerably different results. However, if we restrict ourselves to non-overlapping exons, the exon counts of dexseq-count and EQP agree to a high degree as shown in Figure 9A.
Table 1. The number and percent of exons which overlap with another exon and the number and percent of exons which share the left or right boundary with another exon for different annotation sources and organisms.
| Annotation source | Organism | Num exons | Num ov. exons | Perc. ov. exons | Num sh. bd. exons | Perc. sh. bd. exons |
|---|---|---|---|---|---|---|
| Ensembl 76 | human | 597 495 | 372 779 | 62.39 | 347 209 | 58.11 |
| Ensembl 76 | mouse | 362 381 | 167 129 | 46.11 | 155 522 | 42.91 |
| Ensembl 76 | rat | 216 258 | 5945 | 2.74 | 4699 | 2.17 |
| Refseq NCBI | human | 318 929 | 78 185 | 24.51 | 73 295 | 22.98 |
| Refseq NCBI | mouse | 343 914 | 104 542 | 30.39 | 98 154 | 28.54 |
| Refseq NCBI | rat | 282 271 | 56 008 | 19.84 | 52 396 | 18.56 |
Figure 9.

Logarithmic exon count scatter plots for the Tophat2 alignments of the first lane of sample SEQC-A with x- and y-axis labels in untransformed counts. (A) Correlation between EQP and dexseq-count on exons that do not overlap other exons. (B) Correlation between EQP and QuasR on all exons.
Differences still exist due to the stricter read assignment rules of EQP and the exclusion of ambiguous reads for dexseq-count. Since both QuasR and EQP use exons, their counts can be compared directly as shown in Figure 9B, with the different count criteria leading to significant differences between the counts.
The exon expression values of EQP and dexseq-count depend on both the exon and the read length as a read is counted for all the exons or exonic parts it overlaps; since in QuasR the assignment of a read to an exon is based on a single position, the exon expression values depend only on the exon length. In fact, for QuasR the number of potential positions for the last exon of a gene is actually the exon length minus the read length; however, since 3′-exons are usually considerably longer than the currently used read lengths this does not pose a serious problem. For short exons this means that the signal generated by EQP and dexseq-count is amplified as there are more read alignment positions that potentially lead to an assignment; however, this effect is offset by the fact that QuasR reports read counts as opposed to fragment counts.
Although a direct comparison between the different counts is problematic, it is still possible to investigate the sensitivity of the quantification methods on the fold change level. In Table 2 we show the number of exons or exonic parts which have an absolute fold change value of at least two between the samples SEQC-A and SEQC-B averaged over the 2 × 32 input data sets. Across all aligners used, EQP reports ∼5–15% more exons to have a fold change of at least two than either dexseq-count or QuasR – underlining the importance of the count criteria.
Table 2. The mean number of exons or exonic parts that have an absolute fold change value of at least two for different aligners and different quantification methods.
| Bowtie2-EQP | STAR | Tophat2 | SpliceMap | |
|---|---|---|---|---|
| EQP | 65 316.19 | 72 247.31 | 72 361.78 | 67 752.12 |
| dexseq-count | 62 496.12 | 62 982.22 | 60 927.31 | 65 038.12 |
| QuasR | 55 141.88 | 57 653.25 | 61 340.56 | 56 502.50 |
To further illustrate the consequences of the different exon count criteria, in particular for overlapping mutually exclusive exons, we use an example based on the STAR alignments of the first lane of sample SEQC-A in the two exons E1 and E2 of the gene RAN (Ras-related nuclear protein; ENSG00000132341) as displayed in Figure 10. A closer examination of the reads covering these exons shows that only a handful of reads are consistent with exon E1 (see the green box in the inset) whereas many reads are compatible with exon E2. This would suggest a higher expression of exon E2 and a very low expression of exon E1.
Figure 10.

An IGV screenshot of the genomic region of the 5′-prime end of gene RAN (Ras-related nuclear protein; ENSG00000132341). RAN is a Ras family GTPase involved in multiple cellular functions, including regulation of DNA replication, cell cycle progression, nuclear structure formation, RNA processing-exportation and nuclear protein importation. It contains the two overlapping mutually exclusive exons 12:130872584-130872617:+ (E1) and 12:130872584-130872629:+ (E2) shown in the third track. The first two tracks show the base coverage and the read alignments generated by STAR of the first lane of sample SEQC-A. The third track shows the gene annotation with the exons E1 and E2, and the fourth contains the exonic parts (e.g. ep1 and ep2) used by dexseq-count. The inset shows the first alignments against exon E2 in more detail; the green rectangle in the inset encloses the four alignments that are also compatible with E1.
As can be seen from Table 3, neither the exon counts of QuasR nor the counts for the exonic parts of dexseq-count are consistent with the mutually exclusive use of exon E2 over E1 whereas the count distribution of EQP clearly reflects the exclusion of E1.
Table 3. The exon counts of EQP and QuasR and the counts for the exonic parts of dexseq-count for the features shown in Figure 10.
| Exon | EQP | QuasR | Exonic part | dexseq-count |
|---|---|---|---|---|
| E1 | 4 | 155 | ep1 | 482 |
| E2 | 517.5 | 230 | ep2 | 553 |
Finally, EQP's count criterion also has the advantage that it allows for a proper definition and detection of ambiguous reads for exons, that is, reads that are compatible with exactly one exon without losing the vast majority of the reads. For the SEQC samples more than half of the reads (54.7–59.4%) can be unambiguously assigned to a single exon.
Junction counts
Junction counts depend to a high degree on the aligner used since number of spliced reads corresponds directly to the coverage of junctions. If the junction counts are based on genomic alignments, then they are usually given as the number of reads spanning introns between two exons as reported by QuasR (5,15). However, as shown in Table 1, for many reference data sets a significant fraction of the exons shares one (or both) of its boundaries with another exon leading to ambiguities if only intron-spanning counts are reported. In EQP all possible combinations of exons that border an intron are quantified separately which leads to a higher resolution for junction expression estimates and allows associating junction counts directly with pairs of exons. It should be noted that the junction reads are already included in the exon counts generated by EQP.
The difference between intron-spanning and junction counts can also be illustrated using Figure 10. The exons E1 and E2 share the left boundary but only very few of the reads spanning the intron to the left of these exons are consistent with E2 whereas most of them are consistent with E1 which is reflected in the reported counts (see Supplementary Information).
A different approach to generate junction counts is to use a junction data base similar to the one used in the alignment module of EQP (21,44). The reads are aligned against the junction data base and the junction counts are given by how many reads align against each junction. One drawback of this simple and effective approach is that the alignments used for quantifying the junctions are different from the ones used for the gene and exon counts; this can lead to additional biases and inconsistencies solely due to the alignment differences. Note that, in particular, paired-end read information is poorly utilized in this approach as the junction entries typically consist of relatively short sequences, extending at most the read length to either side of the junction. EQP, on the other hand, generates junction counts from genomic alignments which are consistent with gene and exon counts.
DISCUSSION
The generation of expression estimates for genes, exons and junctions is usually the basis for the analysis of differentially expressed and differentially spliced genes. In this paper we present EQP as a new tool to derive gene, exon and junction counts from the output of a sequencer. EQP covers both the alignment and the quantification steps of a RNA-seq workflow. We assess the performance of each of these steps on RNA-seq experimental data from the SEQC project (35) and simulated data generated for the comparison of gene quantification methods reported by Fonsenca et al. (38).
Overall, EQP's alignment and quantification modules provide an good combination of speed, accuracy, sensitivity and the ability to quantify superior expression estimates as compared to other methods.
EQP's alignment module has low requirements on the computational resources and is particularly suited for well-annotated model species as it strongly depends on high quality reference data. This module provides highly sensitive and accurate alignments on experimental data generated from human reference RNA samples. When compared to other aligners (Tophat2, STAR, SpliceMap), EQP shows a higher percentage of reads aligned to the reference and to the exons, lower number of mismatches and no soft masking. The ability to accurately locate reads on the exons is one of the most important features for a RNA-seq aligner, as exonic reads are subsequently used to infer gene expression levels in the quantification step. The high sensitivity of EQP's alignment module is also confirmed by its ability to find the locations of spliced reads in the reference. This is supported by the high number of introns spanned by the reads shown in Figure 5. For this metric EQP is second only to Tophat2, which, however, discovers many spurious introns supported only by one read.
As input EQP's quantification module can use the alignments generated by either its own alignment module or by a different genomic aligner of choice. It uses a novel count criterion based on the agreement of the splicing pattern of a read alignment with a feature. It offers a variety of ways to quantify genes (using weighted or unweighted reads, limiting the number of alignments of a read, allowing or excluding ambiguous reads) and delivers unified expression estimates for genes, exons and junctions. In combination with EQP alignments, EQP's quantification method provides fold change values with a higher similarity to Taqman-based fold changes when compared to results of combinations of different aligners (Tophat2, STAR, Splicemap, EQP) and quantification methods (htseq-count, featureCount, Cufflinks2, QuasR, EQP). QuasR and htseq-count behave very similar to EQP's quantification, while Cufflink2 has the lowest agreement with the Taqman RT-PCR results on the SEQC samples. However, the performance of Cufflinks2 improves considerably on the simulated data set probably due to the fact that the simulations do not exhibit the same biases of a real RNA-seq data set thus facilitating the estimation of transcript abundance estimates.
The differences between the methods are most pronounced for exon counts for which even the unit to be measured differs between the tools. EQP and QuasR directly measure the exons as defined in the GTF file whereas dexseq-count introduces exonic parts as the measuring unit. In addition, the three methods use three distinct counting criteria leading to different properties of the counts. EQP's evidence-based counting criterion is well-suited to deal with overlapping, mutually exclusive exons whereas QuasR and dexseq-count are less sensitive. This is also illustrated by the fact that EQP reports considerably more exons with a fold change of at least two. Moreover, with increasing read length the (length normalized) exon expression of EQP converges to the sum of the (length normalized) expression of those transcripts in which the exon is contained; in contrast, in QuasR more and more reads will be assigned to the first exons of the transcripts. This difference becomes even more important if we consider that recent technology advances are moving toward sequencing of full-length transcript isoforms (45) though not yet in high-throughput mode. In many ways the problem of computing exon counts is similar to the problem of inferring transcript abundances due to the many overlapping exons so that similar approaches could be applied here as well. Since the problem is much more localized and the overlap patterns are simpler for exons than for transcripts such abundance estimates are likely to be more robust. Exon abundance estimates derived in this way could then even be used as an additional input for the estimation of transcript or gene abundances (46).
Supplementary Material
Acknowledgments
The authors thank M. Beibel, M. Borowski, F. Nigsch and S. Bergling for helpful discussions and critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Novartis Pharma AG.
Conflict of interest statement. Both authors are employees and own shares of Novartis Pharma AG.
REFERENCES
- 1.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 2.Schmid M.W., Grossniklaus U. Rcount: simple and flexible RNA-Seq read counting. Bioinformatics. 2015;31:436–437. doi: 10.1093/bioinformatics/btu680. [DOI] [PubMed] [Google Scholar]
- 3.Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaidatzis D., Lerch A., Hahne F., Stadler M.B. QuasR: Quantify and Annotate Short Reads in R. Bioinformatics. 2015;31:1130–1132. doi: 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotech. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang H., Wong W.H. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W., Jiang T. Transcriptome assembly and isoform expression level estimation from biased RNA-Seq reads. Bioinformatics. 2012;28:2914–2921. doi: 10.1093/bioinformatics/bts559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nariai N., Hirose O., Kojima K., Nagasaki M. TIGAR: transcript isoform abundance estimation method with gapped alignment of RNA-Seq data by variational Bayesian inference. Bioinformatics. 2013;29:2292–2299. doi: 10.1093/bioinformatics/btt381. [DOI] [PubMed] [Google Scholar]
- 11.Nariai N., Kojima K., Mimori T., Sato Y., Kawai Y., Yamaguchi-Kabata Y., Nagasaki M. TIGAR2: sensitive and accurate estimation of transcript isoform expression with longer RNA-Seq reads. BMC Genomics. 2014;15:S5. doi: 10.1186/1471-2164-15-S10-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suo C., Calza S., Salim A., Pawitan Y. Joint estimation of isoform expression and isoform-specific read distribution using multisample RNA-Seq data. Bioinformatics. 2014;30:506–513. doi: 10.1093/bioinformatics/btt704. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z., Wang X., Zhang X. Using non-uniform read distribution models to improve isoform expression inference in RNA-Seq. Bioinformatics. 2011;27:502–508. doi: 10.1093/bioinformatics/btq696. [DOI] [PubMed] [Google Scholar]
- 14.Hu Y., Liu Y., Mao X., Jia C., Ferguson J.F., Xue C., Reilly M.P., Li H., Li M. PennSeq: accurate isoform-specific gene expression quantification in RNA-Seq by modeling non-uniform read distribution. Nucleic Acids Res. 2014;42:e20. doi: 10.1093/nar/gkt1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turro E., Su S.-Y., Goncalves A., Coin L., Richardson S., Lewin A. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome Biol. 2011;12:R13. doi: 10.1186/gb-2011-12-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y., Jiang H. rSeqDiff: Detecting differential isoform expression from RNA-Seq data using hierarchical likelihood ratio test. PLoS One. 2013;8:e79448. doi: 10.1371/journal.pone.0079448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vitting-Seerup K., Porse B., Sandelin A., Waage J. spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics. 2014;15:81. doi: 10.1186/1471-2105-15-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turro E., Astle W.J., Tavaré S. Flexible analysis of RNA-seq data using mixed effects models. Bioinformatics. 2014;30:180–188. doi: 10.1093/bioinformatics/btt624. [DOI] [PubMed] [Google Scholar]
- 20.Anders S., Reyes A., Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasche A., Lienhard M., Yaspo M.-L., Lehrach H., Herwig R. ARH-seq: identification of differential splicing in RNA-seq data. Nucleic Acids Res. 2014;42:e110. doi: 10.1093/nar/gku495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katz Y., Wang E.T., Airoldi E.M., Burge C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffith M., Griffith O.L., Mwenifumbo J., Goya R., Morrissy A.S., Morin R.D., Corbett R., Tang M.J., Hou Y.-C., Pugh T.J., et al. Alternative expression analysis by RNA sequencing. Nat. Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 24.Shen S., Park J.W., Huang J., Dittmar K.A., Lu Z.-x., Zhou Q., Carstens R.P., Xing Y. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen S., Park J.W., Lu Z.-X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Subgroup G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grant G.R., Farkas M.H., Pizarro A., Lahens N., Schug J., Brunk B., Stoeckert C.J., Hogenesch J.B., Pierce E.A. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq Unified Mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C., Gong B., Bushel P.R., Thierry-Mieg J., Thierry-Mieg D., Xu J., Fang H., Hong H., Shen J., Su Z., et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotech. 2014;32:926–932. doi: 10.1038/nbt.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Au K.F., Jiang H., Lin L., Xing Y., Wong W.H. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium S.M.-I. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014;32:903–914. doi: 10.1038/nbt.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandramohan R., Po-Yen W., Phan J.H., Wang M.D. Benchmarking RNA-Seq quantification tools. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2013;2013:647–650. doi: 10.1109/EMBC.2013.6609583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fonseca N.A., Marioni J., Brazma A. RNA-Seq gene profiling - A systematic empirical comparison. PLoS One. 2014;9:e107026. doi: 10.1371/journal.pone.0107026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griebel T., Zacher B., Ribeca P., Raineri E., Lacroix V., Guigó R., Sammeth M. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 2012;40:10073–10083. doi: 10.1093/nar/gks666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gatto A., Torroja-Fungairiño C., Mazzarotto F., Cook S.A., Barton P.J.R., Sánchez-Cabo F., Lara-Pezzi E. FineSplice, enhanced splice junction detection and quantification: a novel pipeline based on the assessment of diverse RNA-Seq alignment solutions. Nucleic Acids Res. 2014;42:e71. doi: 10.1093/nar/gku166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao Y., Smyth G.K., Shi W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nix D., Courdy S., Boucher K. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tilgner H., Grubert F., Sharon D., Snyder M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. U.S.A. 2014;111:9869–9874. doi: 10.1073/pnas.1400447111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laiho A., Elo L.L. A note on an exon-based strategy to identify differentially expressed genes in RNA-Seq experiments. PLoS One. 2014;9:e115964. doi: 10.1371/journal.pone.0115964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.