

Abstract
Background
BMPER, an orthologue of Drosophila melanogaster Crossveinless-2, is a secreted factor that regulates bone morphogenetic protein activity in endothelial cell precursors and during early cardiomyocyte differentiation. Although previously described in the heart, the role of BMPER in cardiac development and function remain unknown.
Methods
BMPER-deficient hearts were phenotyped histologically and functionally using echocardiography and Doppler analysis. Since BMPER −/− mice die perinatally, adult BMPER +/− mice were challenged to pressure-overload-induced cardiac hypertrophy and hindlimb ischemia to determine changes in angiogenesis and regulation of cardiomyocyte size.
Results
We identify for the first time the cardiac phenotype associated with BMPER haploinsufficiency. BMPER messenger RNA and protein are present in the heart during cardiac development through at least E14.5 but is lost by E18.5. BMPER +/− ventricles are thinner and less compact than sibling wild-type hearts. In the adult, BMPER +/− hearts present with decreased anterior and posterior wall thickness, decreased cardiomyocyte size and an increase in cardiac vessel density. Despite these changes, BMPER +/− mice respond to pressure-overload-induced cardiac hypertrophy challenge largely to the same extent as wild-type mice.
Conclusion
BMPER appears to play a role in regulating both vessel density and cardiac development in vivo; however, BMPER haploinsufficiency does not result in marked effects on cardiac function or adaptation to pressure overload hypertrophy.
Keywords: BMPER, BMP, Heart, Development, Cell size, Angiogenesis
1. Introduction
Bone morphogenetic protein (BMP)-binding endothelial cell precursor-derived regulator (BMPER) is a secreted factor that is an extracellular modulator of BMP activity and has been described [at the messenger RNA (mRNA) level] in the adult heart, lung, skin and brain as well as primary chondrocytes [1,2]. BMPER’s role in regulating the development of the vasculature has recently been described in a zebrafish model [3]. BMPER is expressed at sites of high BMP activity, including vascular precursor cells located in the aortic arches and the intermediate cell mass during embryo development [3]. Decreasing BMPER expression compromises the generation of the caudal vein and disturbs the pattern of intersomatic vessel distribution, demonstrating a requirement for BMPER in the early steps of vascular formation [3]. In addition to having a role in vascular development, BMPER also plays a role in oxygen-induced angiogenesis, as recently described in a mouse model of oxygen-induced retinopathy [4]. Compared to wild-type mice, retinas from BMPER haploinsufficient mice have an increased rate of retinal revascularization when challenged with a hypoxic insult [4]. BMPER +/− retinas also have more branching points and angiogenic sprouts at the leading edge of the newly formed vasculature [4]. Taken together, these studies suggest a role for BMPER both in the formation of the developing vasculature and in angiogenesis in the mature retina.
In the present study, we report for the first time the cardiac phenotype associated with BMPER haploinsufficiency. Although BMPER protein is present in the developing heart, we found that BMPER protein is not present in the adult heart. BMPER deficiency during development leads to a distinct cardiac phenotype whereby BMPER haploinsufficient mice have a reduction in cardiomyocyte size at baseline and an increase in cardiac vessel density that does not progress with age. Despite these defects, BMPER +/− hearts respond to pressure-overload-induced cardiac hypertrophy and hindlimb ischemia largely to the same extent as wild-type mice controls. These studies indicate that BMPER haplosufficiency during development leads to mild vascular anomalies but does not lead to any physiological impairment in the ability to respond to cardiovascular challenges.
2. Materials and methods
2.1. Animals
BMPER-deficient mice on a mixed C57BL/6/129S background were created in our laboratory as previously described [5]. Briefly, exons 1 and 2 were deleted and replaced with a green fluorescent protein expression cassette. The BMPER LacZ mice (B6SJL-BMPERtm1Emdr/J mice) were purchased from The Jackson Laboratory (Stock 7554) from stocks submitted by E. M. de Robertis [6,7]. A targeting vector was used to place an in-frame LacZ reporter gene in the first exon of the gene at the initiator methionine and a neomycin resistance casette was inserted downstream of LacZ. The targeting vector was introduced to LW1 129S4/SvJae-derived embryonic stem cells. They were then back-crossed to B6SJLF1/J for 17 generations before submission. All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of North Carolina and conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.2. Experimental design
A series of experiments were performed on BMPER −/−, BMPER +/− and sibling wild-type controls. Using timed breeding, mouse embryos were harvested at E18.5, euthanized and fixed for further histological analysis (4–5 hearts per group, outlined in Supplemental Table 1). Baseline echocardiography was performed at 12 weeks of age (9–17 mice, outlined in Table 1). Microarray analyses were performed on 4 wild-type and 4 BMPER +/− hearts at 12 weeks of age. Trans-aortic constriction (TAC) was performed on 6 wild-type and 14 BMPER +/− mice; 2 wild-type mice were lost to unrelated medical issues and 3 wild-type and 3 BMPER +/− mice included in the baseline did not undergo TAC and were used for other experiments (Table 1 and Fig. 5). Western blot analysis was performed on 3 wild-type and 4 BMPER +/− hearts from 12-week-old mice. Vessel density analyses were performed on 5 wild-type and 8 BMPER +/− mice at baseline (8–12 weeks of age) and 6 wild-type and 5 BMPER +/− after TAC. Hindlimb ischemia was performed on 7 wild types and 7 BMPER +/− (4 experimental and 3 sham per group) at 8–12 weeks of age. Real-time polymerase chain reaction (PCR) analysis of E18.5 hearts was performed on 4 wild-type, 3 BMPER +/− and 3 BMPER −/− mice. Real-time PCR analysis of adult hearts was performed on 3 wild-type and 3 BMPER +/− hearts.
Table 1.
Transthoracic echocardiography on conscious BMPER +/− and sibling wild-type mouse hearts at baseline and after TAC to induce pressure overload and cardiac hypertrophy (±S.E.)
| Baseline wild type (N=9) |
Baseline BMPER +/− (N=17) |
1 week TAC wild type (N=6) |
1 week TAC BMPER +/− (N=14) |
2 weeks TAC wild type (N=5) |
2 weeks TAC BMPER +/− (N=12) |
3 weeks TAC wild type (N=4) |
3 weeks TAC BMPER +/− (N=12) |
4 weeks TAC wild type (N=4) |
4 weeks TAC BMPER +/− (N=14) |
|
|---|---|---|---|---|---|---|---|---|---|---|
| LV anterior wall; diastole | 1.08±0.02* | 0.87±0.01* | 1.38±0.04 | 1.17±0.02** | 1.45±0.02**,& | 1.38±0.04**,& | 1.38±0.04& | 1.40±0.04& | 1.43±0.06& | 1.44±0.02& |
| LV anterior wall; systole | 1.68±0.03 | 1.53±0.03* | 1.85±0.03 | 1.83±0.03 | 2.04 ±0.07†,** | 2.05±0.04†,** | 2.08±0.06†,** | 2.05±0.05†,** | 2.05±0.06†,** | 2.06±0.04†,** |
| LV posterior wall; diastole | 1.03±0.03 | 0.90±0.03* | 1.12±0.02 | 1.10±0.03 | 1.32±0.06†,‡ | 1.27±0.04†,‡ | 1.27±0.07† | 1.28±0.05†,‡ | 1.30±0.07† | 1.31±0.04†,‡ |
| LV posterior wall; systole | 1.60±0.3 | 1.50±0.03* | 1.65±0.02 | 1.54±0.05 | 1.81±0.09 | 1.87±0.05**,& | 1.81±0.13 | 1.81±0.04**,& | 1.90±0.13**,& | 1.91±0.04**,& |
| Body Weight (g) | 24.4±1.4 | 24.6±0.4 | 22.6±1.4 | 24.2±0.9 | 23.0±1.5 | 25.2±1.0 | 24.3±1.6 | 24.7±1.0 | 24.6±1.8 | 25.9±1.0 |
| HR (bpm) | 674±13 | 652±15 | 682±18 | 670±15 | 680±15 | 677±10 | 680±11 | 687±10 | 660±17 | 689±13 |
| LVEDD (mm) | 3.10±0.14 | 3.30±0.09 | 2.78±0.13• | 3.15±0.08** | 2.54±0.09•,& | 2.83±0.10•,& | 2.60±0.16•,& | 2.87±0.08•,& | 2.80±0.19• | 2.91±0.07•,‡ |
| LVESD (mm) | 1.52±0.10 | 1.61±0.07 | 1.30±0.09•,& | 1.60±0.10• | 1.16±0.06•,&,** | 1.28±0.07•,&,** | 1.19±0.06•,&,** | 1.28±0.06•,&,** | 1.29±0.13•,& | 1.33±0.04•,& |
| LV mass (corrected) | 107.7±5.2 | 89.2±6.3* | 108.3±7.3 | 118.0±5.8 | 125.8±12.2 | 132.5±6.6*** | 158.4±23.0*** | 166.8±7.0***,** | 136.9±17.0*** | 146.4±5.0***,** |
| LV mass/BW | 4.52±0.34 | 3.62±0.18* | 4.8±0.25 | 4.90±0.18 | 5.34±0.42 | 5.11±0.26 | 6.52±0.73***,**,‡,‡‡ | 6.80±0.18***,‡ | 5.70±0.63***,•• | 5.73±0.24***,**,•• |
| EF% | 83.5±1.2 | 83.5±0.8 | 85.2±1.2 | 81.6±2.3 | 86.5±0.5 | 85.7±0.9 | 86.3±0.9 | 86.0±1.0 | 85.4±1.6 | 85.4±0.5 |
| FS% | 51.3±1.3 | 51.5±0.9 | 52.9±1.4 | 50.0±1.9 | 54.0±0.6 | 54.7±1.0 | 54.0±1.2 | 54.0±1.2 | 53.2±1.9 | 53.2±0.6 |
| LV vol; d (ml) | 42.45±4.55** | 48.46±3.44 | 31.07±3.50•,& | 41.50±3.10 | 25.2±2.7•,&,*** | 33.5±2.7• | 27.8±4.7•,&,*** | 33.4±2.5• | 32.19±5.60• | 35.2±1.6• |
| LV vol; s (ml) | 7.35±1.22 | 8.26±0.87 | 4.69±0.81• | 7.96±1.42 | 3.4±0.4•,*** | 4.9±0.7• | 3.7±0.6• | 4.8±0.6• | 4.87±1.24 | 5.18±0.34• |
| RWT; d | 0.34±0.02‡ | 0.28±0.01 | 0.42±0.02•,‡ | 0.37±0.02• | 0.54±0.03•,& | 0.46±0.02•,&,***,‡ | 0.49±0.01•,&,*** | 0.46±0.03•,&,***,‡ | 0.49±0.05•,&,*** | 0.46±0.02•,&,***,‡ |
| RWT; s | 1.07±0.08 | 0.95±0.04 | 1.27±0.08• | 1.09±0.07 | 1.59±0.14•,**,*** | 1.47±0.07•,*** | 1.51±0.07•,*** | 1.44±0.09•,*** | 1.51±0.13•,*** | 1.41±0.05•,*** |
Serial echocardiography was performed in mice undergoing TAC. TAC was performed on 6 wild-type and 14 BMPER +/− mice; 2 wild-type mice were lost to unrelated medical issues and 3 wild-type and 3 BMPER +/− mice included in the baseline did not undergo TAC and were used for other experiments. A one-way ANOVA was performed to determine statistical significance, followed by a Holm-Sidak test for pairwise comparisons of all groups.
Abbreviations: LV mass index [(ExLVD3d−LVED3d)×1.055]; ExLVD, external left ventricular diameter; bpm, heart beats per minute; LVEDD, left ventricular end diastolic dimension; LVESD, left ventricular end systolic dimension; FS, fractional shortening, calculated as (LVEDD−LVESD)/LVEDD×100; EF%, ejection fraction calculated as (end Simpson’s diastolic volume−end Simpson’s systolic volume)/end Simpson’s diastolic volume×100; LV vol, LV volume, calculated as (8/3π)×(endocardial area2/endocardial long-axis length); RWT, relative wall thickness calculated as (IVSd+LVPWd)/LVEDD.
P<.05 vs. all other groups.
P<.05 vs. 1 week TAC wild type.
P<.05 vs. baseline wild type.
P<.03 vs. 1 week TAC BMPER +/−.
vs. baseline wild type and 1 week TAC BMPER.
P<.05 vs. 2 weeks TAC wild type.
P<.05 vs. 2 weeks TAC BMPER +/−.
P<.05 vs. BMPER +/− baseline.
P<.05 vs. 3 weeks TAC BMPER +/−.
Fig. 5.
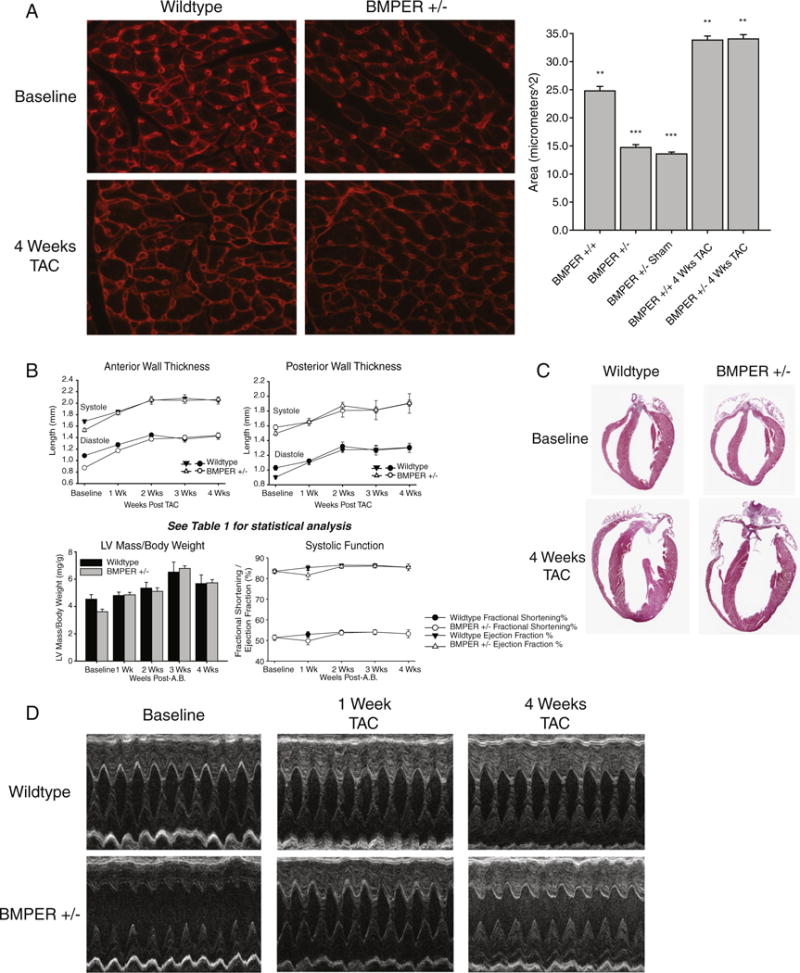
Despite baseline defects in cardiomyocyte size, BMPER +/− hearts hypertrophy in response to pressure overload to the same extent as wild-type hearts. (A) Analysis of the cardiomyocyte cross-sectional areas reveals a baseline deficit in cardiomyocyte size. After 4 weeks of TAC, cardiomyocyte cross-sectional areas significantly increase in BMPER +/− to the same extent as sibling wild-type controls, indicating no affect on cardiac hypertrophy. (B) Echocardiographic analysis of BMPER +/− at baseline and after TAC. See Table 1 for detailed statistical analysis. (C and D) Differences in baseline LV thickness can be seen both histologically and by echocardiography in BMPER +/− at baseline; after 4 weeks of TAC, these reveal that differences in wall size are no longer present after the induction of cardiac hypertrophy. A one-way ANOVA was used to compare BMPER +/− to wild-type mice at all the time points investigated. *P<.05 vs. BMPER +/−; **P<.05 vs. BMPER +/− sham; ***P<.05 vs. BMPER +/+, BMPER +/+ 4 weeks TAC, BMPER +/− 4 weeks TAC. (A and C) Cross-sectional area analysis: N=2 hearts per group; 100 measurements from 4 independent heart sections were analyzed. (B and D) Echocardiographic analysis: N=4–17 per group/time point (detailed in Table 1). TAC was performed on 6 wild-type and 14 BMPER +/− mice; 2 wild-type mice were lost to unrelated medical issues and 3 wild-type and 3 BMPER +/− mice included in the baseline did not undergo TAC and were used for other experiments.
2.3. Blood pressure determination
Blood pressure was determined by tail cuff using the CODA 8 noninvasive blood pressure system (Kent Scientific Corporation, Torrington, CT). Mice were acclimated for 5 cycles, followed by 25 cycles of measurement. Their blood pressure was measured daily for 4 consecutive days; measurements included in the analysis were taken on days 3 and 4. The maximum occlusion pressure was set at 250, the deflation time was 15 and the minimal volume accepted for inclusion was 15. The platform was continuously heated at 37°C.
2.4. Histology and immunochemistry
Hearts were perfused with 4% paraformaldehyde/phosphate-buffered saline (PBS) and processed for histology using standard methods then stained with hematoxylin and eosin, Masson’s trichrome, Movat’s pentachrome or TRITC-conjugated Triticum vulgaris lectin as previously described [8,9]. Cardiomyocyte cross-sectional area was determined on lectin-stained sections using NIH Image J (Version 1.38X). Three hundred measurements per group from at least 10 sections from 3 mice per group were included in the calculations.
Embryonic mouse hearts were excised, fixed in 4% paraformaldehyde and processed for cryosectioning. Cryosectioning was performed by the UNC Histology Research Facility. Sections were labeled with a goat anti-Crossveinless-2 antibody (=BMPER, R&D Systems) and detected as described in Zakin et al. [6].
Fresh frozen hearts were cryosectioned and stained for β-galactosidase activity as previously described [10]. Briefly, hearts were perfused with cold PBS and fresh fix solution (0.2% glutaraldehyde, 5 mM ethyleneglycotetraacetic acid, pH 7.3, PBS) and fixed at 4°C for 2 h. The fixed hearts were embedded in Tissue-Tek OCT compound and frozen (stored at −80°C). They were then cryosectioned (8-μm sections), placed on slides and allowed to dry. Prior to staining, sections were refixed in cold PBS containing 0.2% glutaraldehyde for 10 min. Sections were washed three times for 5 min in LacZ wash buffer (2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% Nonidet-P40 in PBS) and then stained in Xgal staining solution at 37°C overnight. Sections were rinsed in PBS, counterstained, moved through a graded ethanol series for dehydration and then mounted.
2.5. Histological analysis of cardiac vessel density and tricuspid/mitral valve cell counts
Vessel density was determined on paraffin-sectioned adult hearts; 3–4 sections stained with TRITC-lectin (10 images total) were analyzed per heart. In each image, the number of lectin-positive vessels surrounding the 10 roundest cardiomyocytes was counted. These values were then averaged per heart to yield the average vessel density per cardiomyocyte. Manual valve cell counts included both the mesenchymal cells and the endothelial cell layer and included 3–5 frontal sections per value when focused centrally.
2.6. Conscious echocardiography and Doppler analysis of the heart
Echocardiographic analysis was performed (blinded to mouse genotype and treatment) on conscious mice at indicated time points using a VisualSonics Vevo 770 ultrasound biomicroscopy system (VisualSonics, Inc., Toronto, Ontario, Canada), using the model 707B scan head (30 MHz) as previously described [8,9]. Two-dimensional guided M-mode echocardiography was performed in the parasternal long-axis view at the level of the papillary muscle. Wall thickness was then determined by measurements of epicardial to endocardial leading edges. Mitral valve flow velocity was determined by Doppler analysis using the VisualSonics Vevo 770 ultrasound biomicroscopy system (VisualSonics, Inc.) on lightly anesthetized mice [2% (vol/vol) isoflurane/100% oxygen] as previously described [11,12]. Mitral valve flow Doppler was acquired by positioning the transducer angled cranially in a supine mouse at 45° in an epigastric position to achieve an apical four-chamber view. The heights of Peak E and A were determined on mitral valve sequential waveforms in at least 5 waveforms. Aortic valve flow Doppler was acquired by positioning the transducer caudally at ~45°. M-mode and Doppler measurement data represent 3–6 averaged cardiac cycles from at least 2 scans per mouse. The mean performance index (MPI) was determined by measuring isovolumetric relaxation time and isovolumetric contraction time/ET from both mitral and aortic Doppler waveforms [13,14].
2.7. In vivo hindlimb ischemia
Hindlimb ischemia was performed on mice as previously described [15].
2.8. RNA isolation and real-time PCR analysis
Total RNA was isolated from ~30 mg of frozen mouse heart using the RNeasy system (Qiagen, Inc., Valencia, CA). Tissue was homogenized and total RNA was purified according to the manufacturer’s protocols for animal tissues. mRNA expression analysis was determined using a two-step reaction. Complementary DNA (cDNA) was made using High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). One microliter of cDNA product was then amplified on an ABI Prism 7500 Sequence Detection System in 20 μl final volume using the TaqMan® Universal PCR Master Mix. The PCR reaction including 1 μl of mouse specific TaqMan probes for BMPER (Mm01175807_m1), BMP2 (Mm01340178_m1), BMP4 (Rn00563202_m1), BMP6 (Mm00432095_m1), BMP7 (Mm00432102_m1), BMP10 (Mm01183889_m1), SMURF1 (Mm00547102_m1), VEGF-A (Mm00437304_m1), ANG1 (Mm00456496_m1), ANG2 (Mm00545822_m1), ID1 (Mm00775963_g1) and SMAD6 (Mm00484738_m1) were run in triplicate (Life Technologies Corporation, Carlsbad, CA). The relative expression of mRNA was determined using 18S (Hs99999901_s1), GUSB (Mm01197698) or GAPDH (Mm99999915_g1) as an internal sample loading control (Life Technologies Corporation). For the GUSB PCR, the Universal Probe Library Probe #42 (Cat#04688015001) was used together with the following primers: GUSB-F: 5′-CTCTGGTGGCCTTACCTGAT-3′ and GUSB-R: 5′-TCAGTTGTTGTCACCTTCACCT-3′.
2.9. In situ hybridization
Embryonic mouse hearts were excised, fixed in diethylpyrocarbonate-treated 4% paraformaldehyde and processed for cryosectioning. Cryosectioning was performed by the UNC Histology Research Facility. The BMPER in situ hybridization probe was prepared as described by Moser et al. [1], and in situ hybridizations were carried out by the UNC In Situ Hybridization Core Facility.
2.10. Western blot analysis of BMP signaling
Western blot analysis was performed on ventricular lysates prepared by homogenizing ventricular tissue in lysis buffer (Cell Signaling Technology, Danvers, MA; Cat#9308) in the presence of phosphatase and protease inhibitors as previously described [16]. Anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (ERK1/2) Thr202/Try204 (Cell Signaling Technology, Cat#4376) and anti-total p44/42 MAPK (ERK1/2) (Cell Signaling Technology, Cat#9201) were used according to the manufacturer’s protocols. Similarly, phospho-SMAD1 (Ser463/465)/SMAD5 (Ser463/465), SMAD8 (Ser426/428) (Cell Signaling Technology, Cat#9511) and anti-total SMAD (Santa Cruz Biotechnology, Santa Cruz, CA; Cat#sc7153) were used according to the manufacturer’s protocols.
2.11. RNA extraction and microarray analysis
Total RNA was isolated from heart ventricles from 12-week-old wild-type and BMPER +/− mice using the RNeasy Mini Kit (Qiagen, Inc.). Each genotype was represented by four biological replicates. RNA integrity (RIN score>7) was verified by assay on an Agilent BioAnalyzer 2100 and subsequently used for microarray analysis using the Agilent 4×44K two-color mouse microarray (Agilent G4122F, Cat#014868) co-hybridized with a common mouse reference RNA. Loess normalization and probe quality control was applied using Feature Extraction (v9.5.1.1, Agilent). Probes were median centered across all arrays and probes that were identified as high quality (“detected”) in at least five of the eight samples were selected for differential gene expression analysis (17,886 probes). Missing data points were imputed using the k nearest-neighbor algorithm (k=5). The complete data set is available online through the Gene Expression Omnibus (series record GSE40983). Principal component analysis and hierarchical clustering was performed using Partek Genomics Suite (Saint Louis, MO), and significance analysis of microarrays (SAM) was used as previously described [17].
2.12. Statistical analysis
A one-way analysis of variance (ANOVA) or Student’s t test was performed using Sigma Stat 3.5 (Systat Software, Inc., San Jose, CA). If significance was determined by the one-way ANOVA, an all pairwise multiple comparison procedure (Holm-Sidak method) was performed between all the groups. Basic statistics were performed using Microsoft Excel 2007 (Microsoft, Seattle, WA). Results are expressed as averages±standard error (S.E.), with statistical significance defined as P<.05 (or as otherwise indicated in the figure legends).
3. Results
3.1. BMPER expression during development regulates adult cardiac phenotype in vivo
BMPER +/− mice were bred to obtain BMPER −/−, BMPER +/− and sibling wild-type mice, as previously described [5]. Since BMPER −/− mice die perinatally [5,6,18], we characterized each genotype histologically at day E18.5, the day prior to birth. Three distinct findings were evident in the BMPER +/− hearts. First, the walls of both the right and left ventricles appear thinner in the BMPER +/− hearts compared to sibling wild-type and BMPER −/− hearts (Fig. 1A). Objectively, BMPER +/− hearts are statistically thinner in the apex upon measuring the base, midway and apex points (Supplemental Table 1). Evidence for a delay in the ventricular compaction process was also identified in both ventricles (Fig. 1B–D). Subtle changes in the mitral valve were also detected in BMPER +/− and BMPER −/− hearts compared to wild-type hearts (Fig. 1E). These changes consisted of the mitral valve being consistently elongated compared to wild-type hearts. The BMPER +/− hearts showed a slight elongation (2 of 4 mice) but not severe enough to cause the prolapsed/wrinkled appearance of the null mice. No defects in the tricuspid or aortic valve were detected based on standard published definitions [19,20]. No differences in mitral valve cell number were identified among the wild-type, BMPER +/− and BMPER −/− mitral valves (598.6±42.1, 329.4±155.6, 506.3±50.3; N=3, 2, 5, respectively analyzed by one-way ANOVA). Similarly, no changes in the tricuspid valve cell number were seen between the genotypes (449.8±66.0, 376.4±46.3, 389.6±56.7; N=3, 2, 5, respectively analyzed by one-way ANOVA). Because of the more severe phenotype of the BMPER −/− mitral valves, we further analyzed them for detailed abnormalities. Using Movat’s staining, we identified that BMPER −/− mitral valve changes (Fig. 1F), ranging from mostly normal (middle panel) to more broadly thickened compared to wild-type mitral valves (left panel). Differences in the extracellular matrix (ECM) of the mitral valve was noted; BMPER −/− valves contained less glycosaminoglycans (blue) compared to wild-type mitral valves, while having increased elastin content (yellow) in some BMPER −/− mitral valves (Fig. 1F, middle). These changes may suggest that there is a different distribution of endothelial and mesenchymal cells resulting in differences in ECM deposition.
Fig. 1.
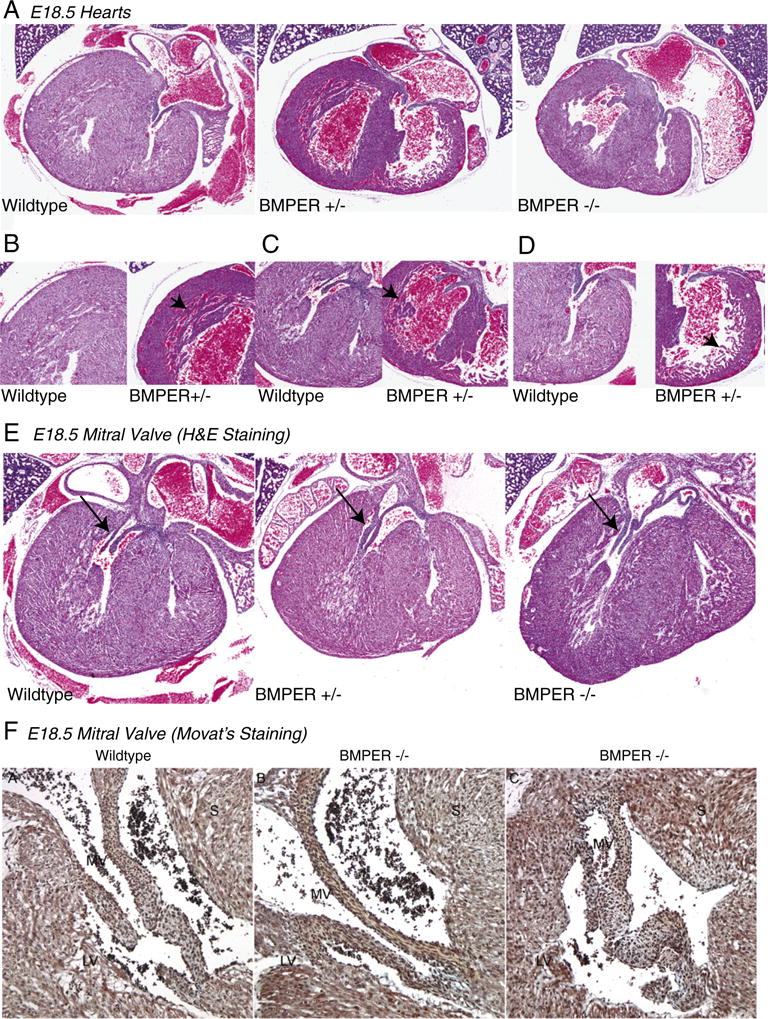
BMPER hypomorphs (+/−) demonstrate thinner right and LV walls, mild changes in mitral valves and occasional VSDs. (A) Compared to wild-type hearts, BMPER +/− hearts have thinner ventricular walls than the wild-type and BMPER −/− hearts. BMPER +/− hearts have decreased compaction of the left (B and C) and right ventricle (D), indicated by arrowheads, compared to sibling E18.5 wild-type hearts. (E) BMPER +/− mice also had consistently demonstrated subtle differences in the shape and length of their mitral valves compared to wild-type valves (arrows). (F) Movat’s staining of a wild-type heart (left) with rich blue (glycosaminoglycans) in the mitral valve connecting to the heart and an example of the diversity of BMPER −/− mitral valves seen, including a mostly normal mitral valve (MV) (middle) and thicker, septated MV (right). Elastin (yellow) is prevalent in the BMPER −/− valve mesenchyme and glycosaminoglycans are also highly prevalent in the BMPER −/− MVs (particularly the abnormal heart on the right). Bright red is fibrin, red is muscle and nuclei/elastic fibers are black. S, septum. (A–E) N=4–5 mice per group (outlined in Supplemental Table 1). (B) N=2 per group.
3.2. BMPER is localized to the mitral and tricuspid valves in the E14.5 heart but is absent in the adult heart
We next investigated the localization of BMPER in the developing heart, focusing on the mitral valve based on the above observation and recent studies indicating a link between BMP/TGFβ signaling and mitral valve prolapse [21]. By in situ hybridization, we detected faint amounts of BMPER mRNA localized to the mitral and tricuspid valves of wild-type mice at E14.5 (Fig. 2A and B). Similarly, BMPER protein was detected by immunohistochemistry localized to the mitral and tricuspid valves at E14.5 (Fig. 2C). Specifically, BMPER mRNA was localized to the endothelial cells in the mitral and tricuspid valves by in situ hybridization (Fig. 2A and B), whereas BMPER protein was distinctly absent from the endothelial cells and localized to the mesenchymal cell of the valve below (Fig. 2C), suggesting that endothelial cells secrete BMPER to the surrounding mesenchymal cells during cardiac development. Staining for BMPER mRNA and protein using these methods in the hearts of older E18.5 embryos did not reveal the presence of BMPER (data not shown). Since BMPER’s activity has been shown to antagonize BMP2, BMP4 and BMP6 [1] in addition to enhancing BMP4 activity depending on dose [5], we hypothesized that decreased levels of BMPER expression in BMPER +/− and BMPER −/−hearts might affect BMP-regulated genes. To test this hypothesis, we performed quantitative PCR (qPCR) analysis on wild-type, BMPER +/− and BMPER −/− hearts. We identified increases in BMP10 in the BMPER +/− hearts compared to wild-type hearts (Fig. 2D), but not BMP2, SMAD1, BMP6 or BMP7 compared to littermate wild-type controls. We next determined the localization of BMPER in the adult heart. Immunohistochemical and in situ hybridization analysis using established techniques failed to detect BMPER expression in the adult heart. We also examined LacZ-BMPER mice, in which BMPER expression in the adult heart has been previously described [6]. In hearts from 4- to 6-week-old BMPER +/− mice, we did not identify any LacZ activity anywhere in the heart, including the ventricles (Fig. 2E–G), consistent with the lack of BMPER expression seen by E18.5. This suggested that BMPER expression is present up through at least E14.5 during development, with minimal effects on BMP-regulated gene expression, but is absent in the fetal and adult heart, contrasting previous studies, which identified BMPER mRNA by Southern blot [1].
Fig. 2.
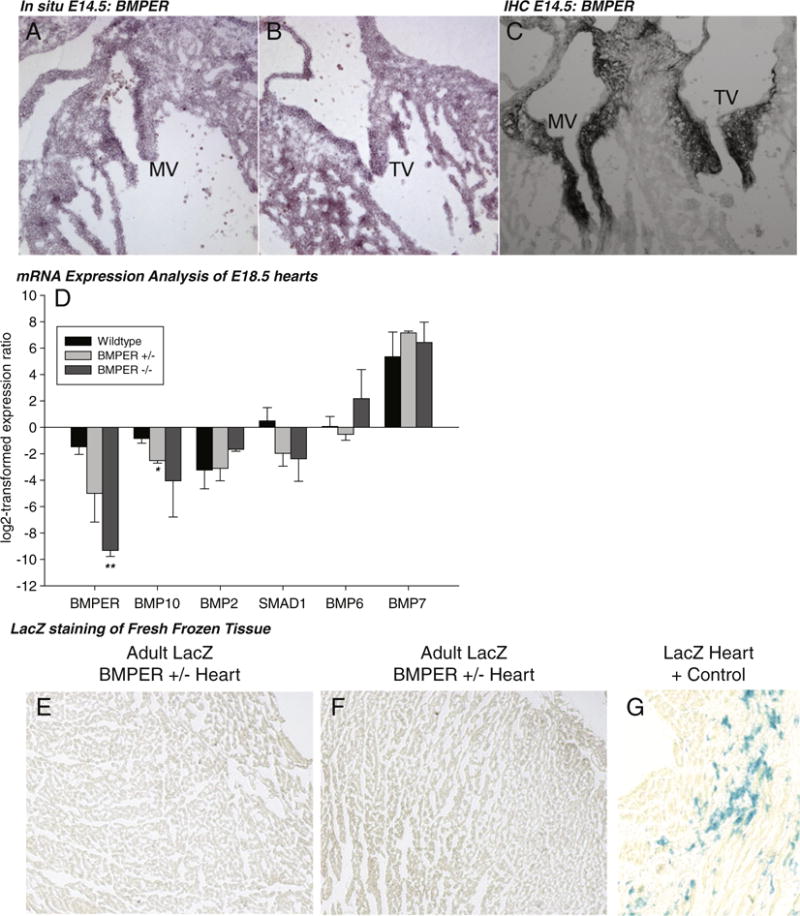
BMPER is not expressed in the late embryo stage and adult hearts. (A and B) BMPER is present by in situ hybridization and (C) immunohistochemistry in wild-type hearts at E14.5, localized to the mitral and tricuspid valves. However, by day E18.5, BMPER is not detected by these two methods (data not shown). (D) Real-time qPCR analysis of BMPER-regulated gene expression in E18.5 hearts. N=4 wild type; N=3 BMPER +/−; N=3 BMPER −/−. *P=.01 vs. wild type; **P=.001 vs. wild type. (D and E) In the adult heart of the LacZ-BMPER mouse (BMPER +/−), no LacZ activity is detected throughout the hearts (N=2). (F) Parallel stained positive control LacZ heart section.
3.3. BMPER +/− adult hearts exhibit thin left ventricular walls
Echocardiographic analysis of the hearts of 12-week-old adult BMPER +/− and wild-type mice revealed both anterior and posterior wall thinning in the 12-week-old adult BMPER +/− hearts compared to sibling matched wild-type controls. Despite having thinner anterior and posterior ventricular walls, no systolic dysfunction, as measured by fractional shortening and ejection fraction, were noted in the BMPER +/− mice (Fig. 3A–C). This phenotype of anterior and posterior wall thinning in the BMPER +/− mice persisted but did not increase in severity, for up to 1 year of age when the experiment was completed (Supplemental Fig. 1, Supplemental Table 2). Since BMPER was initially identified as a regulator of endothelial progenitor cells (EPCs), we next determine if the vasculature resistance was affected in BMPER +/− mice by analyzing systemic blood pressure using tail cuff measurements. We did not detect any differences in systolic, diastolic or mean arterial pressures in BMPER +/− vs. wild-type 12-week-old mice (Fig. 3D). These findings demonstrate a static left ventricular (LV) wall thinning in BMPER +/− mice in the absence of any changes in systolic function or systemic blood pressure, a phenotype not previously reported in genetic mouse models.
Fig. 3.
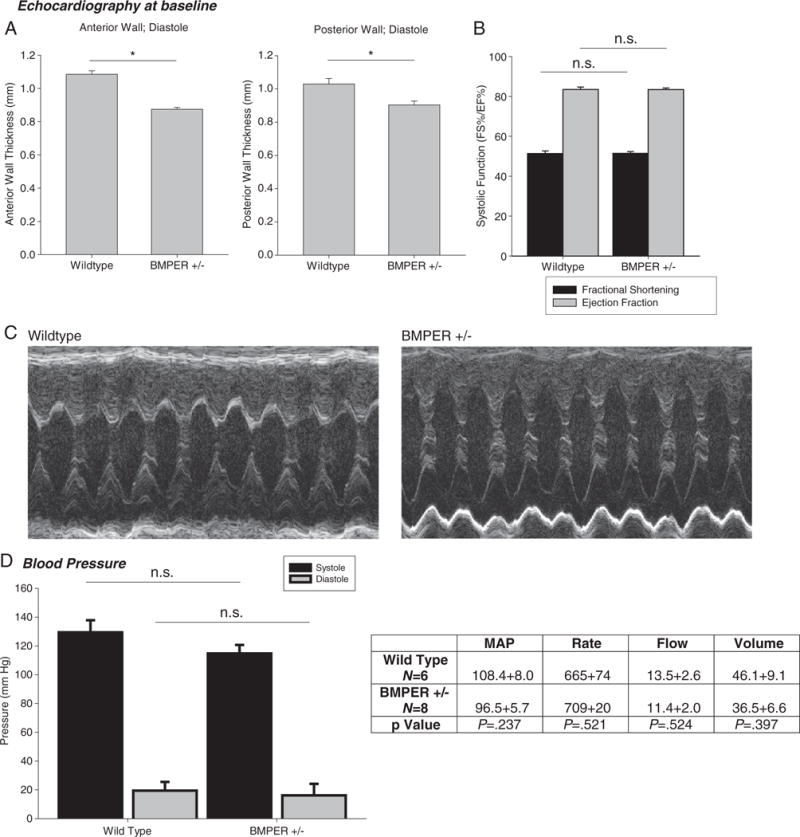
Echocardiographic and Doppler analysis of BMPER hypomorphs reveals decreases in LV wall thickness but no systolic dysfunction. (A) Both anterior and posterior LV wall thicknesses are significantly decreased in 12-week-old BMPER +/− mice. (B and C) However, systolic function measured by fractional shortening or ejection fraction is not significantly different in BMPER +/− hearts. (D) Determination of blood pressure in BMPER +/− mice did not reveal any differences in systolic, diastolic or mean arterial pressures. (A and B) N=9 (wild type); N=17 (BMPER +/−). (D) N=6 (wild type); N=8 (BMPER +/−). A Student’s t test was used to compare the BMPER +/− to their wild-type sibling control groups. *P<.01. n.s., not significant.
3.4. Adult BMPER +/− hearts have phenotypically normal mitral and aortic valves and demonstrate no diastolic or systolic dysfunction
Because we had noted phenotypic changes in the LV wall thickness and mitral valves of BMPER +/− mice (2 of 4 were slightly elongated in histological analysis, but neither with the folded over phenotype of the BMPER −/− hearts), we next analyzed the functional integrity of the aortic and mitral valves in BMPER +/− mice using standard measures of diastolic and systolic function by Doppler analysis. BMPER +/− aortic valve waveform and peak velocity indicated no defect in cardiac function or aortic valve integrity, respectively (Fig. 4A and B). Similarly, BMPER +/− mitral valve waveforms were unaffected compared to wild-type controls (Fig. 4C). To determine diastolic function, the E/A ratio, calculated by the height of the E and A waveforms in the mitral valve, is used as a measure of diastolic function [22]. Using this measure, we determined that the diastolic function did not differ between BMPER +/− and wild-type mice (Fig. 4D). Utilization of the MPI, calculated from both aortic and mitral valve Doppler measurements, as a means to detect either systolic or diastolic dysfunction in the heart, was also determined for the BMPER +/− hearts. No differences in diastolic/systolic function were identified (Fig. 4E). Consistent with these findings, we did not detect any histological differences in the adult MV, TV or AV (Fig. 4F).
Fig. 4.
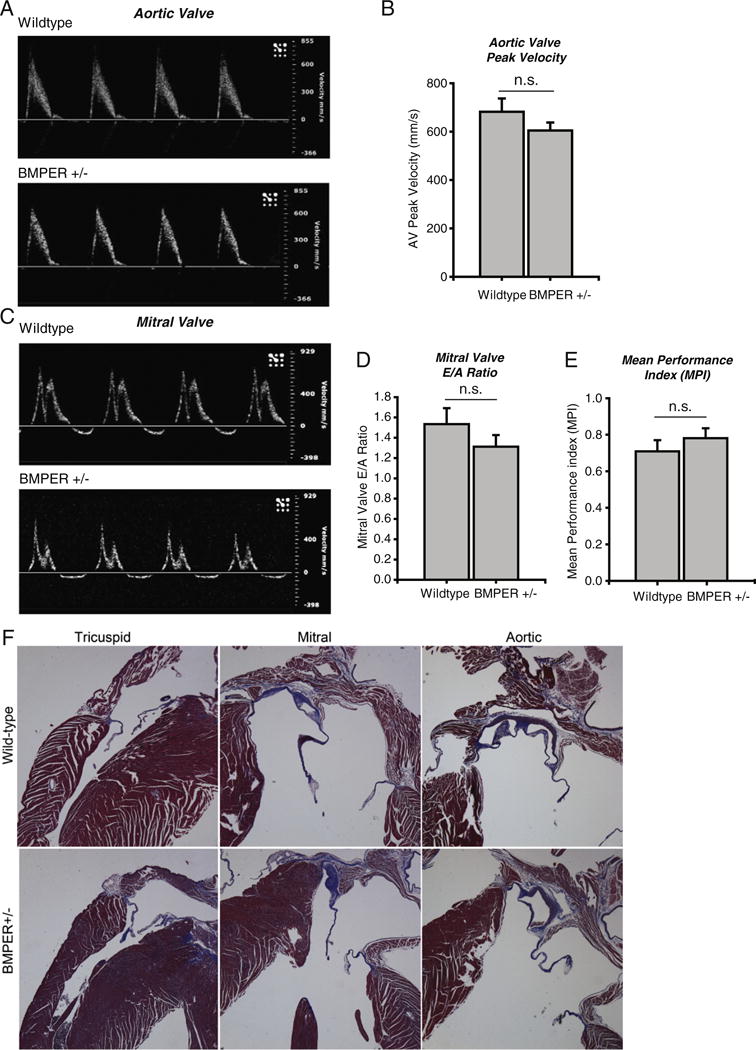
Doppler analysis of aortic and mitral valves of adult BMPER +/− mice reveal no functional defects in valves or cardiac function. A. Doppler waveform analysis of a representative aortic valve demonstrates that BMPER +/− hearts do not differ from wild-type controls. B. Aortic valve peak velocity is not affected in BMPER +/− hearts. C. Representative BMPER +/− waveforms of the mitral valve indicate no defects in function. D. The mitral valve E/A ratio, a measure of diastolic function and the E. Mean Performance Index, a measure of both systolic and diastolic function demonstrate that BMPER +/− hearts do not differ from wild-type controls. N=7 BMPER +/−; N=6 wild-type. A Student’s t-test was used to compare the BMPER +/− to their wild-type sibling control groups. n.s.=not significant. F. Morphological analysis of adult wild-type and BMPER +/− tricuspid, mitral, and aortic valves, representative of N=2/group.
3.5. Despite a baseline decrease in cardiomyocyte size and increased cardiac vessel density, BMPER +/− adult hearts respond to pressure-overload-induced cardiac hypertrophy similar to wild-type mice
At baseline, BMPER +/− hearts exhibited a relative baseline decrease in cardiomyocyte size identified by cross-sectional area analysis of cardiomyocytes in BMPER +/− compared to wild-type hearts (Fig. 5A). Since BMPER haploinsufficiency resulted in a decrease in cardiomyocyte size at baseline, we next sought to determine if BMPER +/− cardiomyocytes retained the ability to respond to stimuli that regulate cardiomyocyte size. We challenged BMPER +/− mice to pressure-overload-induced cardiac hypertrophy using the TAC model. Despite decreased anterior and posterior wall thicknesses prior to TAC (Fig. 5B–D), BMPER +/− hearts hypertrophied to the same extent as wild-type mice in as little as 2 weeks, without any differences in systolic function (Table 1). After 4 weeks of TAC, these deficits in cardiomyocyte size were completely compensated for in the hearts from BMPER +/− mice (Fig. 5A), indicating that the developmental deficiency in cardiac BMPER did not affect the pathologic hypertrophy response in adult cardiomyocytes, despite a relative decrease in cardiomyocyte size at baseline. Histological analysis of the BMPER +/− hearts at baseline and 4 weeks after TAC did not reveal any morphological differences from wild-type hearts (Supplemental Fig. 2), indicating that no differences in fibrosis and/or cellular infiltrates occurred between the different genotypes.
BMPER has been reported to both antagonize and enhance BMP activities [1,2,18,23–25]. Since BMPER does not appear to be present in the adult heart, we hypothesized that there would be no difference in BMP signaling between BMPER +/− and wild-type hearts. Since BMP signaling can activate ERK1/2 and SMAD pathways [26–28], we first performed a Western blot analysis of phospho-ERK1/2 and phospho-SMAD. As anticipated, no differences between BMPER +/− and wild-type mice were identified in either phospho-ERK1/2 or phospho-SMAD levels (Supplemental Fig. 3). We next analyzed BMPER +/− hearts for BMP-regulated gene expression by real-time qPCR analysis. Of the genes we investigated, we found that BMPER +/− hearts displayed a significant increase in BMP2 expression (Fig. 6A). Since BMP2 has been implicated in promoting angiogenesis in the heart [29], we next investigated the density of cardiac capillaries in the ventricles. Surprisingly, we identified that adult BMPER +/− hearts exhibited a significant increase in cardiac vessel density compared to wild-type controls (Fig. 6B). Four weeks after challenge with TAC, we found that the cardiac vessel density increased in the wild-type mice to the level of BMPER +/− baseline hearts. The vessel density in BMPER +/− hearts, however, did not change after TAC (Fig. 6B). Previous studies have demonstrated that TAC induces a relatively mild global cardiac ischemia [30,31], which is believed to act as a potent stimulus for the induction of angiogenesis driving the associated increased cardiac vessel density seen in the resulting cardiac hypertrophy [32]. The baseline increase in cardiac vessel density seen in the BMPER +/− hearts may be one potential mechanism by which BMPER +/− hearts are able to adapt or compensate to the same extent as wild-type hearts in the face of pressure-overload-induced cardiac hypertrophy; i.e., the increase in vessel density would allow greater perfusion to the ischemic tissue thereby allowing a greater degree of growth in response to TAC.
Fig. 6.
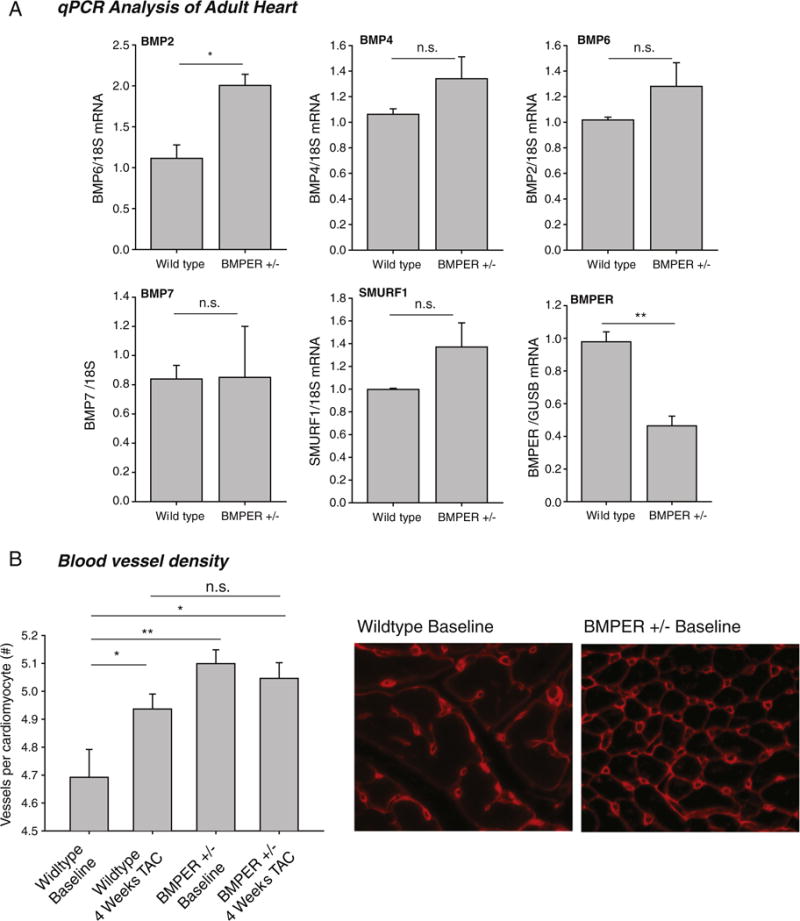
BMPER +/− hearts have an increased vessel density compared to sibling controls; this difference is lost after pressure-overload-induced hypertrophy. (A) Analysis of BMPER-related genes in adult hearts revealed increases in BMP2 message, but not BMP4, BMP6, BMP7 or SMURF1. A Student’s t test was performed to determine significance between groups. *P<.05; **P<.01. N=3 per group. n.s., not significant. (B) Blood vessel density analysis of BMPER +/− hearts reveals significantly more vessels per cardiomyocyte than controls at baseline. Four weeks after pressure overload induced by TAC, these differences between BMPER +/− and wild-type hearts are not seen. A one-way ANOVA was used to compare BMPER +/− to wild-type mice at all the time points investigated. If significance was determined, an all pairwise multiple comparison procedure (Holm-Sidak method) was then performed to determine significance between the groups. *P<.05; **P<.01. N=5 wild type; N=6 wild type TAC; N=10 BMPER +/−; N=5 BMPER +/− TAC. n.s., not significant.
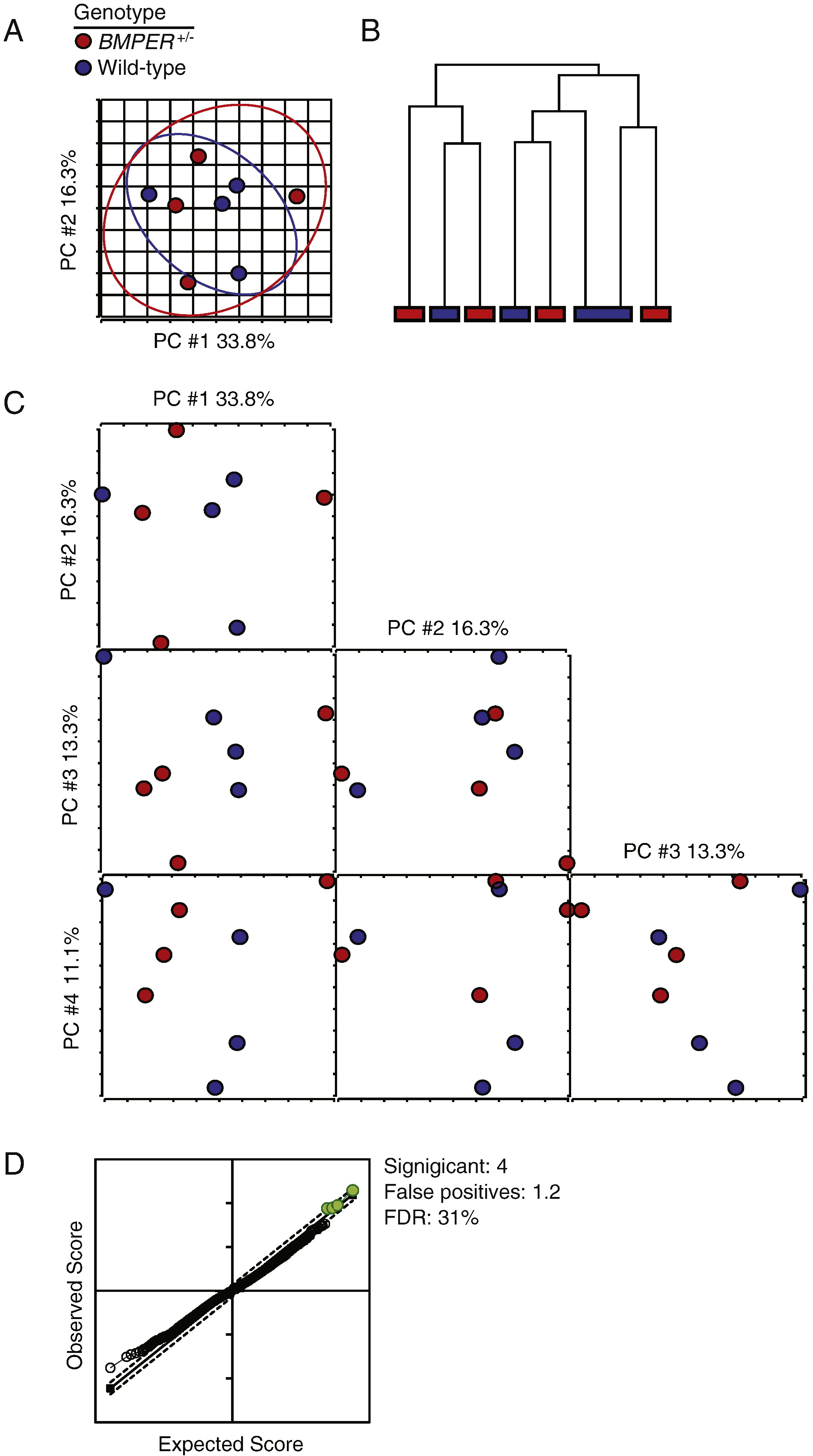
To begin to understand the underlying mechanisms involved in the BMPER+/− phenotype of decreased cardiomyocyte size and increased vessel density, we performed microarray analyses on the cardiac ventricles (Supplemental Fig. 4). Surprisingly, using multiple unsupervised algorithms including principal component analysis (Supplemental Fig. 4A) and unsupervised hierarchical clustering using Pearson’s dissimilarity metric (Supplemental Fig. 4B), we did not detect any global differences in gene expression between the BMPER+/− and wild-type hearts. To attempt to identify any small discrete differences in gene expression, we extended the principal component analysis out to the fourth principal component yet failed to see any segregation in variance that was attributable to the genotype of the samples (Supplemental Fig. 4C). Similarly, we did not see a significant difference in any probe using either an unpaired t test (corrected P<0.05) or SAM (FDR<10%). Only at false discovery rates (FDRs) greater than 31% did we find any significant difference in gene expression comparing genotypes (Supplemental Fig. 4D). These data convincingly demonstrate that there is no measurable change in gene expression between adult wild-type and BMPER+/− hearts.
3.6. BMPER +/− mice respond to hindlimb ischemic injury to the same extent as controls
The hindlimb ischemia challenge is a common experimental design to test the ability of angiogenic growth factors to expedite and/or augment collateral artery development. The remodeling that occurs in the hindlimb skeletal muscle vasculature is not restricted to angiogenesis but includes postnatal vasculogenesis as well [33]. Several groups have established the role of bone-marrow-derived EPCs in the angiogenic response to ischemia; EPCs are present in the circulation and respond to certain cytokines and/or tissue ischemia as well as home into sites of neovascularization [33]. BMPER was originally described as being highly expressed in EPCs [1]. Therefore, in order to determine whether BMPER plays a role in vasculogenesis, we next determined if BMPER +/− mice were able to compensate for other vascular related injuries that induced vasculogenesis, namely hindlimb ischemia. We challenged BMPER +/− mice to hindlimb ischemia and then measured the amount of perfusion immediately after ligation (to ensure that the ligation was complete) and then at 7, 14 and 21 days after ligation on both the affected and opposite control hindlimb. The only significant differences in hindlimb perfusion after ligation between BMPER hypomorphs and sibling wild-type mice were found on day 14 (Supplemental Fig. 5). This result indicates a minor role for BMPER in peripheral vasculature adaptations that occur in response to ischemia.
4. Discussion
In these studies, we identify a role for BMPER in the developing heart and in regulating adult cardiomyocyte size and vessel density, despite its absence in the heart by E18.5. The decreased wall thickness and normal function were identified in mice up to 1 year of age. Despite these changes, BMPER +/− mice largely respond to cardiac hypertrophy and ischemic insults to the same extent as wild-type mice. These studies indicate for the first time a role for BMPER in the developing heart that affects the adult phenotype, despite the fact that BMPER is absent in the adult heart.
A surprising finding in the present study is the phenotypic changes seen in the BMPER +/− hearts at E18.5, whereas the wild-type and BMPER −/− hearts are relatively unaffected with regard to size and LV compaction and apex thickness (Fig. 1). BMPER is known to regulate the activity of BMP in a tissue- and stage-dependent manner. BMPER can also regulate BMP activity in a biphasic manner. Our laboratory recently identified that BMPER regulates the signaling of different BMP proteins in a ratiometric fashion [5]. Specifically, when the concentration of BMPER is less than the concentrations of BMP4, BMPER enhanced BMP4 activity [5]. As molar concentrations of BMPER exceed BMP4, BMPER dynamically switches from an activator to an inhibitor of BMP4 signaling through a novel endocytic trap-and-sink mechanism [5]. Ambrosio et al. have also shown that BMPER interacts with Chordin to coordinate BMP diffusion; when Chordin levels are low, BMPER cooperates with Chordin to sequester BMPER from its receptors [34]. When Chordin increases, BMPER switches from a BMP antagonist to a Chordin antagonist, resulting in the enhancement of BMP signaling [34]. In the context of the present study, it is possible that the differences in the observed phenotype between the BMPER −/− and BMPER +/− mice may be due to differences in the local ratio of BMPER to BMPs, resulting in either pro-or anti-BMP activity during the development of the heart, although we did not test this hypothesis directly in the current study.
Very little has been published on how BMPs might regulate cell size. However, germane to the finding that BMPER +/− mice have a decrease in cardiomyocyte size is a recent study demonstrating BMP2’s regulation of osteoclast size. It has been reported that BMP2 directly enhances RANKL-mediated osteoclast differentiation by increasing the size and number of osteoclasts [35]. Conversely, increasing the expression of the BMP antagonist TWSG1 in primary osteoclasts decreases the size and number of osteoclasts but does not affect proliferation or apoptosis [36]. The addition of BMP2 to the TWSG1-treated osteoclasts reversed the effects of TWSG1 by increasing cell size, illustrating the interplay between BMP2 and TWSG1 in the regulation of cell size [36]. In the context of the BMPER +/− mice with decreased cardiomyocyte size, our findings may suggest that BMPER haploinsufficiency results in inhibition of BMP signaling that would normally enhance cardiomyocyte cell growth during development. However, given the complexity of BMPER signaling pathways and its regulation of BMPs and BMP antagonists such as Chordin, the specific mechanism during development where this would happen is currently not known.
BMPER regulates BMPs during the development of the vasculature and has a key role in organogenesis during development [1,3]. BMPER was first discovered in a screen for differentially expressed transcripts in developing flk-1-positive embryoid bodies (endothelial cell precursors) involved in vasculogenesis [1]. In these initial studies, BMPER was found to interact with BMP2, BMP4 and BMP6 and to antagonize BMP4-dependent SMAD activation [1]. When BMPER is knocked out experimentally in mice, defects in organogenesis and perinatal death occurred within 24 h of birth [18]. Specifically, vertebral column and eye defects have been identified in BMPER −/− mice, which are substantially enhanced when further deleting one copy of the BMP4 genes, suggesting a pro-BMP role in the development of these organs [18]. BMPER −/− mice also exhibit defects in nephron generation of the kidney and exhibit a kidney hypoplasia upon perinatal demise, which is further enhanced by the additional deletion of the pro-BMP protein Kcp (Crim2) [18]. The role of BMPER in the heart and in the development of vessels has not previously been described.
Recent studies have implicated BMP signaling in the development of the heart and in the process of cardiomyocyte differentiation in vitro [37–40]. The concept that BMPER has a role in cardiac development is not surprising given that BMP2, BMP4 and BMP7, which BMPER regulates, have been implicated in cardiac induction, specification and development, as well as congenital heart diseases, as recently reviewed [41,42]. BMPs interact with BMP receptors (BMP receptors type I and II) and have inhibitors, such as Noggin, which have been shown to finely regulate BMP activity to regulate cardiac differentiation. Interruption of this fine-tuning can lead to defects in cardiac development. For example, mutations in BMP receptor type I (ALK2) have been found in patients with atrial septal defects (ASDs) [43] and mice with compound heterozygosity for BMP2 and BMP4 demonstrate ventricular septal defects (VSDs), among other abnormalities [44]. Studies investigating early cardiomyocyte differentiation using P19 cells have found that BMPER expression is bimodal during differentiation [42]. Moreover, BMPER is expressed prior to the induction of the transcription factors Nkx2.5 and Tbx5, both critical to cardiac differentiation, and acts to suppress BMP signaling [42]. Specifically, RNAi knockdown of BMPER results in increased SMAD1/5/8 activation and impaired cardiomyocyte differentiation [42]. At the level of the cardiomyocyte, decreasing BMPER during differentiation appears to impair differentiation in these studies. In the current study, we identified a delay in the compaction of the ventricles of BMPER +/− mice at E18.5, which may be due to impaired differentiation that would parallel these findings in P19 cells with RNAi knockdown of BMPER. The size of the P19 cells was not addressed, but BMPER +/− mice in the current study had significant decreases in cross-sectional areas, suggestive of decreased cardiomyocyte size.
The model described here is a germ line haploinsufficiency model that should be acknowledged as a possible limitation of the present studies. While the present study focused on determining the localization and presence of BMPER in the heart (direct effects), the observed phenotype may be due to indirect effects due to defects in other systems. This is the primary reason we carefully measured blood pressure in the BMPER +/− and sibling wild-type controls to ensure that differences in heart size were not due to changes in blood pressure. While the perinatal BMPER −/− mice have been shown to have defects in the vertebral column, eyes and in nephron generation in the kidney [6,7,18], none of these defects were identified in the BMPER +/− mice [6,7,18]. The perinatal death of BMPER −/− mice has been identified by multiple groups, but the specific cause of death has not been identified despite the possibility that the mild lung defects identified may play a role [5,6,18]. Indeed, in the present study, BMPER −/− hearts exhibited previously unreported changes that might contribute to the perinatal lethality including ASDs and VSDs. When BMPER +/− mice on an APOE −/− background are challenged with a high-fat diet, the observed atherosclerotic lesions are enhanced due to BMPER’s inhibition of vascular inflammatory adhesion molecules [45]. Additionally, when BMPER +/− mice are challenged with retinal revascularization, they display increased numbers of branching points and angiogenic sprouts demonstrating a role in inhibiting pathological angiogenesis [4]. Beyond these studies, there are no clear physiological reasons to explain the observed BMPER +/− cardiac phenotype by indirect effects on other organ systems.
The relationship among angiogenesis, vessel density and cardiac mass has only recently been reported. Tirziu et al. reported a transgenic mouse with a regulatable expression of the secreted angiogenic growth factor PR39 in cardiomyocytes. They identified that the endothelial cell mass in this model increased 3 weeks after induction of PR39 expression, resulting in cardiac hypertrophy in the absence of other stimuli [46]. Similarly, treating mice with VEGF-B resulted in the stimulation of vascular growth in normal adult mouse hearts, which also lead to an increase in cardiac mass [46]. When constitutively expressed cardiac-specific VEGF-B transgenic mice were created, the hearts showed a concentric cardiac hypertrophy without significant defects in cardiac function [47]. The increase in cardiomyocyte size was paralleled with increased capillary size of cardiac blood vessels, but the number of blood vessels per cell remained unchanged [47]. Cardiomyocyte lipid metabolism was also affected, with increased ceramide and decreased triglyceride levels found in the transgenic hearts, linking VEGF-B with lipid metabolism [47]. These findings differ from our current study whereby increases in vessel density, not size, were seen, paralleling decreases in cardiomyocyte size, which may or may not be related.
In response to pressure-overload-induced cardiac hypertrophy caused by TAC, previous studies have identified that EPCs defined as stem cell antigen 1+/vascular endothelial growth factor receptor 2+ cells increase up to 200% in the blood and bone marrow [48]. This increase in EPCs results in increased extracardiac neoangiogenesis of approximately 50% [48]. These studies demonstrate that the heart has an angiogenic response to TAC, which may be evidenced in the current study by increases in cardiac vessel density 4 weeks after TAC. This response appeared to be “pre-set” in the BMPER +/− mice with enhanced vessel density at baseline to the level that wild-type mice achieved in response to TAC. This may be one mechanism by which BMPER +/− hearts compensate in response to TAC despite baseline defects.
Despite the lack of evidence of BMPER being present in the adult heart, including cardiomyocytes and the vasculature, the current study indicates a key role of BMPER in regulating cardiomyocyte size and vessel density in the heart during development. Surprisingly, these apparent changes in BMPER +/− mice do not appear to leave the heart at any greater risk when challenged with pressure overload or leave the vasculature at any more or less risk when challenged with hindlimb ischemia. Nor do the gene expression profiles significantly differ between BMPER +/− and their wild-type siblings. These studies indicate that BMPER present in the heart during development is key in regulating multiple aspects of BMP-mediated cardiac development.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by NHLBI R01HL061656 (to CP), the Children’s Cardiomyopathy Foundation (to MW), the American Heart Association Scientist Development Grant (to MW), and the Leducq Foundation (to CP and MW). The authors wish to thank Janice Weaver in the UNC Animal Histopathology Laboratory for assistance in preparing histological specimens, Jonathan Lowery for his insightful discussion of the data and Jinzhu Duan for his guidance with the Xgal staining.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.carpath.2012.10.005.
Disclosures: None of the authors (M.W., L.D., R.R., P.L., I.M., J.S. and C.P.) have any conflict of interest to declare.
References
- 1.Moser M, Binder O, Wu Y, Aitsebaomo J, Ren R, Bode C, et al. BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol Cell Biol. 2003;23:5664. doi: 10.1128/MCB.23.16.5664-5679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binnerts ME, Wen X, Cante-Barrett K, Bright J, Chen HT, Asundi V, et al. Human crossveinless-2 is a novel inhibitor of bone morphogenetic proteins. Biochem Biophys Res Commun. 2004;315:272. doi: 10.1016/j.bbrc.2004.01.048. [DOI] [PubMed] [Google Scholar]
- 3.Moser M, Yu Q, Bode C, Xiong JW, Patterson C. BMPER is a conserved regulator of hematopoietic and vascular development in zebrafish. J Mol Cell Cardiol. 2007;43:243. doi: 10.1016/j.yjmcc.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreno-Miralles I, Ren R, Moser M, Hartnett ME, Patterson C. Bone morphogenetic protein endothelial cell precursor-derived regulator regulates retinal angiogenesis in vivo in a mouse model of oxygen-induced retinopathy. Arterioscler Thromb Vasc Biol. 2011 doi: 10.1161/ATVBAHA.111.230235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley R, Ren R, Pi X, Wu Y, Moreno I, Willis M, et al. A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J Cell Biol. 2009;184:597. doi: 10.1083/jcb.200808064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zakin L, Metzinger CA, Chang EY, Coffinier C, De Robertis EM. Development of the vertebral morphogenetic field in the mouse: interactions between Crossveinless-2 and Twisted Gastrulation. Dev Biol. 2008;323:6. doi: 10.1016/j.ydbio.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zakin L, Chang EY, Plouhinec JL, De Robertis EM. Crossveinless-2 is required for the relocalization of Chordin protein within the vertebral field in mouse embryos. Dev Biol. 2010;347:204. doi: 10.1016/j.ydbio.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2011;31:429. doi: 10.1038/emboj.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jearawiriyapaisarn N, Moulton HM, Sazani P, Kole R, Willis MS. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res. 2010;85:444. doi: 10.1093/cvr/cvp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou YQ, Foster FS, Parkes R, Adamson SL. Developmental changes in left and right ventricular diastolic filling patterns in mice. Am J Physiol Heart Circ Physiol. 2003;285:H1563. doi: 10.1152/ajpheart.00384.2003. [DOI] [PubMed] [Google Scholar]
- 13.Stypmann J, Engelen MA, Troatz C, Rothenburger M, Eckardt L, Tiemann K. Echocardiographic assessment of global left ventricular function in mice. Lab Anim. 2009;43:127. doi: 10.1258/la.2007.06001e. [DOI] [PubMed] [Google Scholar]
- 14.Tei C, Ling LH, Hodge DO, Bailey KR, Oh JK, Rodeheffer RJ, et al. New index of combined systolic and diastolic myocardial performance: a simple and reproducible measure of cardiac function—a study in normals and dilated cardiomyopathy. J Cardiol. 1995;26:357. [PubMed] [Google Scholar]
- 15.Gherghe CM, Duan J, Gong J, Rojas M, Klauber-Demore N, Majesky M, et al. Wnt1 is a proangiogenic molecule, enhances human endothelial progenitor function, and increases blood flow to ischemic limbs in a HGF-dependent manner. FASEB J. 2011;25:1836. doi: 10.1096/fj.10-172981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li HH, Du J, Fan YN, Zhang ML, Liu DP, Li L, et al. The ubiquitin ligase MuRF1 protects against cardiac ischemia/reperfusion injury by its proteasome-dependent degradation of phospho-c-Jun. Am J Pathol. 2011;178:1043. doi: 10.1016/j.ajpath.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikeya M, Kawada M, Kiyonari H, Sasai N, Nakao K, Furuta Y, et al. Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development. 2006;133:4463. doi: 10.1242/dev.02647. [DOI] [PubMed] [Google Scholar]
- 19.Mitruka SN, Lamberti JJ. Congenital Heart Surgery Nomenclature and Database Project: mitral valve disease. Ann Thorac Surg. 2000;69:S132. doi: 10.1016/s0003-4975(99)01360-0. [DOI] [PubMed] [Google Scholar]
- 20.Dearani JA, Danielson GK. Congenital Heart Surgery Nomenclature and Database Project: Ebstein’s anomaly and tricuspid valve disease. Ann Thorac Surg. 2000;69:S106. doi: 10.1016/s0003-4975(99)01265-5. [DOI] [PubMed] [Google Scholar]
- 21.Lincoln J, Yutzey KE. Molecular and developmental mechanisms of congenital heart valve disease. Birth Defects Res A Clin Mol Teratol. 2011;91:526. doi: 10.1002/bdra.20799. [DOI] [PubMed] [Google Scholar]
- 22.Galderisi M. Diastolic dysfunction and diastolic heart failure: diagnostic, prognostic and therapeutic aspects. Cardiovasc Ultrasound. 2005;3:9. doi: 10.1186/1476-7120-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coles E, Christiansen J, Economou A, Bronner-Fraser M, Wilkinson DG. A vertebrate crossveinless 2 homologue modulates BMP activity and neural crest cell migration. Development. 2004;131:5309. doi: 10.1242/dev.01419. [DOI] [PubMed] [Google Scholar]
- 24.Kamimura M, Matsumoto K, Koshiba-Takeuchi K, Ogura T. Vertebrate crossveinless 2 is secreted and acts as an extracellular modulator of the BMP signaling cascade. Dev Dyn. 2004;230:434. doi: 10.1002/dvdy.20069. [DOI] [PubMed] [Google Scholar]
- 25.Serpe M, Umulis D, Ralston A, Chen J, Olson DJ, Avanesov A, et al. The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Dev Cell. 2008;14:940. doi: 10.1016/j.devcel.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Chen YG. Fine-tune of intrinsic ERK activity by extrinsic BMP signaling in mouse embryonic stem cells. Protein Cell. 2012 doi: 10.1007/s13238-012-2925-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maegdefrau U, Bosserhoff AK. BMP activated Smad signaling strongly promotes migration and invasion of hepatocellular carcinoma cells. Exp Mol Pathol. 2012;92:74. doi: 10.1016/j.yexmp.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Xu XL, Dai KR, Tang TT. The role of Smads and related transcription factors in the signal transduction of bone morphogenetic protein inducing bone formation (Zhongguo xiu fu chong jian wai ke za zhi) Chin J Reparative Reconstr Surg. 2003;17:359. [PubMed] [Google Scholar]
- 29.Song X, Liu S, Qu X, Hu Y, Zhang X, Wang T, et al. BMP2 and VEGF promote angiogenesis but retard terminal differentiation of osteoblasts in bone regeneration by up-regulating Id1. Acta Biochim Biophys Sin. 2011;43:796. doi: 10.1093/abbs/gmr074. [DOI] [PubMed] [Google Scholar]
- 30.Bache RJ, Arentzen CE, Simon AB, Vrobel TR. Abnormalities in myocardial perfusion during tachycardia in dogs with left ventricular hypertrophy: metabolic evidence for myocardial ischemia. Circulation. 1984;69:409. doi: 10.1161/01.cir.69.2.409. [DOI] [PubMed] [Google Scholar]
- 31.Bache RJ, Dai XZ. Myocardial oxygen consumption during exercise in the presence of left ventricular hypertrophy secondary to supravalvular aortic stenosis. J Am Coll Cardiol. 1990;15:1157. doi: 10.1016/0735-1097(90)90258-q. [DOI] [PubMed] [Google Scholar]
- 32.Breisch EA, Houser SR, Carey RA, Spann JF, Bove AA. Myocardial blood flow and capillary density in chronic pressure overload of the feline left ventricle. Cardiovasc Res. 1980;14:469. doi: 10.1093/cvr/14.8.469. [DOI] [PubMed] [Google Scholar]
- 33.Murasawa S, Asahara T. Endothelial progenitor cells for vasculogenesis. Physiology (Bethesda) 2005;20:36. doi: 10.1152/physiol.00033.2004. [DOI] [PubMed] [Google Scholar]
- 34.Ambrosio AL, Taelman VF, Lee HX, Metzinger CA, Coffinier C, De Robertis EM. Crossveinless-2 is a BMP feedback inhibitor that binds Chordin/BMP to regulate Xenopus embryonic patterning. Dev Cell. 2008;15:248. doi: 10.1016/j.devcel.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sotillo Rodriguez JE, Mansky KC, Jensen ED, Carlson AE, Schwarz T, Pham L, et al. Enhanced osteoclastogenesis causes osteopenia in twisted gastrulation-deficient mice through increased BMP signaling. J Bone Miner Res. 2009;24:1917. doi: 10.1359/JBMR.090507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pham L, Beyer K, Jensen ED, Rodriguez JS, Davydova J, Yamamoto M, et al. Bone morphogenetic protein 2 signaling in osteoclasts is negatively regulated by the BMP antagonist, twisted gastrulation. J Cell Biochem. 2011;112:793. doi: 10.1002/jcb.23003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattacharya S, Macdonald ST, Farthing CR. Molecular mechanisms controlling the coupled development of myocardium and coronary vasculature. Clin Sci (Lond) 2006;111:35. doi: 10.1042/CS20060003. [DOI] [PubMed] [Google Scholar]
- 38.Dyer LA, Kirby ML. The role of secondary heart field in cardiac development. Dev Biol. 2009;336:137. doi: 10.1016/j.ydbio.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 40.Yamagishi T, Ando K, Nakamura H. Roles of TGFβ and BMP during valvulo-septal endocardial cushion formation. Anat Sci Int. 2009;84:77. doi: 10.1007/s12565-009-0027-0. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Greene SB, Martin JF. BMP signaling in congenital heart disease: new developments and future directions. Birth Defects Res A Clin Mol Teratol. 2011;91:441. doi: 10.1002/bdra.20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harada K, Ogai A, Takahashi T, Kitakaze M, Matsubara H, Oh H. Crossveinless-2 controls bone morphogenetic protein signaling during early cardiomyocyte differentiation in P19 cells. J Biol Chem. 2008;283:26705. doi: 10.1074/jbc.M801485200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joziasse IC, Smith KA, Chocron S, van Dinther M, Guryev V, van de Smagt JJ, et al. ALK2 mutation in a patient with Down’s syndrome and a congenital heart defect. Eur J Hum Genet : EJHG. 2011;19:389. doi: 10.1038/ejhg.2010.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchimura T, Komatsu Y, Tanaka M, McCann KL, Mishina Y. Bmp2 and Bmp4 genetically interact to support multiple aspects of mouse development including functional heart development. Genesis. 2009;47:374. doi: 10.1002/dvg.20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pi X, Lockyer P, Dyer LA, Schisler JC, Russell B, Carey S, et al. Bmper inhibits endothelial expression of inflammatory adhesion molecules and protects against atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2214. doi: 10.1161/ATVBAHA.112.252015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest. 2007;117:3188. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karpanen T, Bry M, Ollila HM, Seppanen-Laakso T, Liimatta E, Leskinen H, et al. Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ Res. 2008;103:1018. doi: 10.1161/CIRCRESAHA.108.178459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller P, Kazakov A, Semenov A, Bohm M, Laufs U. Pressure-induced cardiac overload induces upregulation of endothelial and myocardial progenitor cells. Cardiovasc Res. 2008;77:151. doi: 10.1093/cvr/cvm037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.