

Abstract
Introduction
The cause of persistent injury-associated anemia is multifactorial and includes acute blood loss, an altered erythropoietin (EPO) response, dysregulation of iron homeostasis, and impaired erythropoiesis in the setting of chronic inflammation/stress. Hepcidin plays a key role in iron homeostasis and is regulated by anemia as well as inflammation. EPO is a main regulator of erythropoiesis induced by hypoxia. A unique rodent model of combined lung injury (LC)/hemorrhagic shock (HS)/chronic restraint stress (CS) was utilized to produce persistent injury-associated anemia to further investigate the roles of EPO, hepcidin, iron, ferritin, and the expression of EPO receptors (EPOr).
Methods
Male Sprague-Dawley rats were randomly assigned into one of the four groups of rodent models: naïve, CS alone, combined LCHS, or LCHS/CS. Plasma was used to evaluate levels of EPO, hepcidin, iron, and ferritin. RNA was isolated from bone marrow and lung tissue to evaluate expression of EPOr. Comparisons between models were performed by t-tests followed by one-way analysis of variance.
Results
After seven days, only LCHS/CS was associated with persistent anemia despite significant elevation of plasma EPO. LCHS and LCHS/CS led to a persistent decrease in EPOr expression in bone marrow on day seven. LCHS/CS significantly decreased plasma hepcidin levels by 75% on day one and 84% on day seven as compared to LCHS alone. Hepcidin plasma levels are inversely proportional to EPO plasma levels (Pearson R=-0.362, p<0.05).
Conclusion
Tissue injury, hemorrhagic shock, and stress stimulate and maintain high levels of plasma EPO while hepcidin levels are decreased. In addition, bone marrow EPOr and plasma iron availability are significantly reduced following LCHS/CS. The combined deficit of reduced iron availability and reduced bone marrow EPOr expression may play a key role in the ineffective EPO response associated with persistent injury-associated anemia.
Keywords: trauma, chronic stress, erythropoiesis, iron, erythropoietin receptor
Introduction
Severely injured trauma patients often develop persistent injury-associated anemia. 1, 2 While acute anemia is present in majority of trauma patients admitted to the intensive care unit (ICU), it persists throughout the duration of critical illness and results in a large number of blood transfusions and the anemia can last for weeks to months. 2, 3 The persistence of injury-associated anemia is multifactorial. The acute component is related to blood loss, traumatic injuries, and initial surgical management. However, the persistence of injury-associated anemia is combined with a prolonged hypercatecholamine state, ongoing inflammation, bone marrow dysfunction with abnormal erythropoiesis, an altered erythropoietin (EPO) response, as well as dysregulation of iron homeostasis. 1, 4–10
In severely injured trauma patients, the roles of EPO, hepcidin, and iron in persistent injury-associated anemia have not been fully elucidated. EPO is produced by the kidney and is the main regulator of erythropoiesis. 11, 12 In effective erythropoiesis, the binding of EPO to its EPO receptor (EPOr) is crucial for differentiation of erythroid progenitor cells and the production of mature red blood cells (RBCs). 13–18 Although EPOr is located on multiple cell types, it is mainly found within bone marrow on erythroid progenitor cells such as burst-forming unit erythroid (BFU-Es) and colony-forming unit erythroid cells (CFU-Es).15 Specifically, the binding between EPOr and EPO is required for the proliferation and survival of CFU-Es to produce mature erythrocytes.16–18 Previous work has shown that serum EPO levels are significantly elevated following burns and trauma.5, 19, 20 However, other groups found inappropriately low and insufficient serum EPO levels for the degree of anemia in critically ill adults. 11, 21–25 It is postulated that critically ill patients have an insufficient EPO response relative to the degree of anemia.
Hepcidin, a major regulator of iron homeostasis, creates a link between inflammation, iron metabolism, and anemia. Hepcidin is released mainly from the liver in response to inflammation and infection.26 However, in the setting of anemia, tissue hypoxia, EPO, and iron utilization also regulate the expression of hepcidin.26 The regulation of hepcidin in persistent injury-associated anemia remains uncertain. Severely injured patients develop a prolonged inflammatory state concomitant with their multiple traumatic injuries and acute blood loss. With acute blood loss, increased EPO production is expected to suppress hepcidin expression to make iron available for erythropoiesis but alternatively continued inflammation can increase hepcidin expression in order to reduce iron bioavailability for pathogens. 26–28
Iron bioavailability is another component important in restoring hemoglobin (Hb) levels among critically ill patients with persistent anemia.10, 11 Patients often present with low iron levels and significantly high ferritin levels. 10, 11, 19, 26 Ferritin levels are a reflection of iron storage but following severe trauma, ferritin levels are also markers of inflammation.10, 11, 19, 26 Adequate iron levels are necessary for EPO in order function properly and initiate erythropoiesis.26 A limited supply of iron can lead to an ineffective EPO response, as Hb synthesis requires an adequate iron supply. However, in large multicenter prospective randomized trial, intravenous iron supplementation did not significantly affect Hb concentration or transfusion rates.10
The mechanisms involved in persistent-injury associated anemia are complex. In this study our goal is to investigate the regulation of hepcidin and EPO a clinically relevant model of lung contusion, hemorrhagic shock and chronic stress (LCHS/CS) that reproduces persistent injury-associated anemia.
Materials and Methods
Animals
All rodent models were performed in male Sprague-Dawley rats (Charles River, Wilmington, MA), weighing 250 to 300g and were housed in pairs in the animal facility at the University of Florida in Gainesville, Florida. The animals were provided free access to water and standard laboratory food. The Institutional Animal Care and Use Committee at the University of Florida approved all rodent models.
Experimental Groups
Animals were randomly assigned into one of the four models: naïve (N=5), chronic restraint stress (CS) alone (N=5), combined lung contusion followed by hemorrhagic shock (LCHS) (N=9), or LCHS followed by daily chronic restraint stress (LCHS/CS) (N=9). LCHS/CS has previously been shown to create a persistent injury-associated anemia seven days after the initial injury. 20 Naïve rodents underwent daily handling. Animals in each group were sacrificed 24 hours following CS alone, LCHS, or LCHS/CS. Additional animals were sacrificed after seven days of CS, LCHS, and LCHS/CS. The following were collected post sacrificing: lung tissue, bone marrow, peripheral blood, and urine. Specimens were immediately frozen and stored at −80°C.
Lung Contusion and Hemorrhagic Shock
After intra-peritoneal pentobarbital, a unilateral lung contusion was made by using a blast wave of a percussive nail gun (Sears Brand, Chicago, IL) applied directly to a 12mm metal plate that was placed in the right axilla of the rat. Ten minutes after the animal has undergone the lung contusion, the right femoral artery and right internal jugular vein were cannulated. Both tubes contained heparinized saline (10units/ml). The femoral artery tubing was then connected to a continuous blood pressure monitoring device (BP-2 Digital Blood Pressure Monitor; Columbus Instruments, Columbus, Ohio) for measurement of mean arterial pressure and heart rate. Blood was then withdrawn from the internal jugular to maintain a mean arterial pressure of 30–35 mmHg for 45 minutes. After this time, shed blood was re-infused at 1mL/min.
Restraint Stress
Daily restraint stress was utilized to recreate CS. This was performed in the morning daily to account for circadian rhythms. Animals were placed in nose cone rodent holders (Kent Scientific Corporation, Torrington, CT, USA) for two hours daily for six days. During the two hours, rats were repositioned in the cone every 30 minutes during which an alarm sounded for two minutes. This was performed to prevent habituation during the two-hour period. Animals in the LCHS/CS model were given 24 hours to recover from the injury prior to starting the CS model. Those animals in LCHS alone group underwent daily handling and were restricted from food and water during this time each morning.
Hematological Analysis
Heparinized blood samples were used for the analysis of complete blood count. Specifically, peripheral white (WBC) and red blood cells (RBC) counts, Hb, hematocrit (Hct), mean corpuscular volume (MCV), mean corpuscular hemoglobin concentration (MCHC), and red blood cell distribution width (RDW) were measured in all animals on days one and seven after injury (VetScan HM5, Abaxis, CA, USA).
EPO, Hepcidin, Ferritin, and Iron measurements
Plasma EPO was evaluated using the standard sandwich ELISA (R&D Systems, Minneapolis, MN, USA). Plasma and urine hepcidin was measured using the standard competitive ELISA kit (MyBioSource.com, San Diego, CA, USA). Plasma iron and ferritin were evaluated using the standard competitive ELISA kit (Abcam, Cambridge, MA, USA). All samples were run in duplicate following the manufacturer’s protocol.
EPOr mRNA
RNA was isolated from bone marrow and lung tissue using RNeasy Mini Kit (Qiagen, Germantown, MD, USA) and then measured by an Eon plate reader (Biotek, Winooski, VT, USA). Complementary DNA (cDNA) was prepared from the RNA using high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Polymerase chain reaction (PCR) was performed for EPO receptor (EPOr) expression and β-actin using the following primers: EPOr forward primer 5-GCTCCTATGACCACCCACAT-3 and reverse 5-GGTGGTGAAGAGACCCTCAA-3 and β-actin forward 5-AGCCATGTACGTAGCCATCC-3 and reverse 5-CTCTCAGCTGTGGTGGTGAA-3. REDTaq Ready Mix PCR Reaction Mix (Sigma-Aldrich, St. Louis, MO, USA) was used to perform PCR analysis with the following cycles: first cycle at 94°C for 3 minutes, followed by 40 cycles of 94°C for 30 seconds, 56°C for 30 seconds, and 72°C for 30 seconds. The amplified products were run on a 1.5% Agarose Gel containing Ethidium Bromide and visualized by ChemiDoc MP imaging system (Bio-Rad, Hercules, CA, USA). Densitometry of the amplified products was performed using Image Processing and Analysis in Java (ImageJ-NIH, Bethesda, MD, USA).
Statistical Analysis
All statistical analysis was performed using Graph Pad Prism Version 6 (GraphPad Software, Inc, San Diego, CA). The data was presented as mean±SD and comparisons between multiple rodent models were performed using one-way analysis of variance (ANOVA) followed by Tukey-Kramer’s multiple comparisons or Student’s t-test. Data homoscedasticity was evaluated using the Brown-Forsythe test. When data heteroscedasticity was identified (three data points), it was corrected by logarithmic transformation of the data and analyzed using ANOVA followed by Tukey-Kramer’s multiple comparison. Statistical significance remained unchanged and data was presented as mean±SD. In addition, a Pearson correlation test was computed to assess the relationship between EPO and hepcidin. *p < 0.05 vs. naïve and **p < 0.05 vs. LCHS were considered significant.
Results
Analysis of Hematologic Data
CS, LCHS, and LCHS/CS had significant decrease in Hb and Hct on day one (Table SDC 1). Only the LCHS/CS model had a persistent decrease in Hb with a moderate anemia on day seven as compared to naïve (10.6±0.8* vs. 14.3±0.3 g/dL, *p<0.0001) (Table SDC 1). In addition, the addition of CS to LCHS created as persistent injury associated anemia on day seven as compared to LCHS alone (10.6±0.8** vs. 12.3±0.7 g/dL, **p=0.0163). CS, LCHS, and LCHS/CS all led to a significant decrease in RBCs along with a 30, 72, and 60% increase in WBCs on day seven when compared to naïve (Table SDC 1). Table SDC 1 also shows that only LCHS/CS resulted in a significant increase in both MCV and RDW and a significant decrease in MCHC on day seven when compared to naïve.
The impact of severe trauma and stress on EPO and EPOr expression
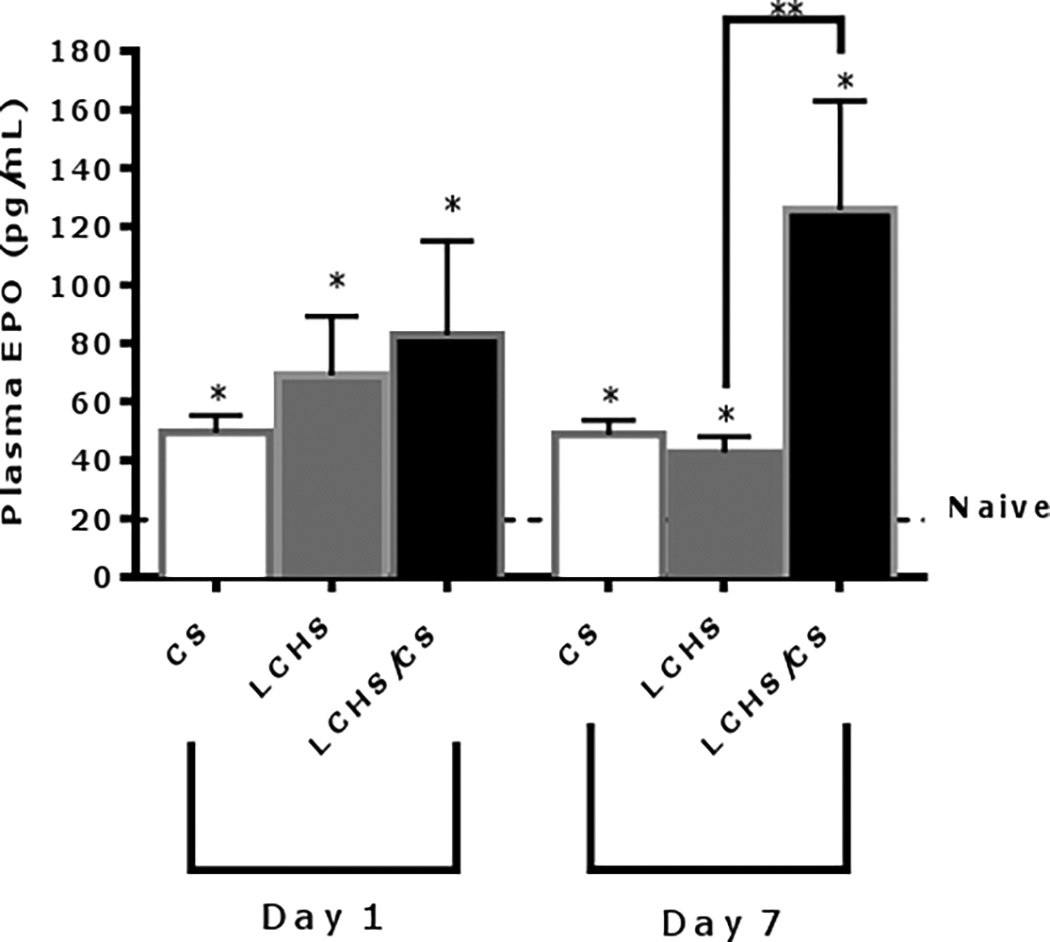
Within 24 hours CS, LCHS, and LCHS/CS, all resulted in significant increase in plasma EPO levels compared to naïve (Figure 1). On day seven, plasma EPO levels remained significantly elevated as compared to naïve in all three groups. However, plasma EPO levels in the CS alone and LCHS groups decreased on day seven as compared to day one. The addition of CS to LCHS led to persistent significant elevation of plasma EPO as compared to LCHS alone (Figure 1). This correlates with the finding of persistent anemia in the LCHS/CS group.
Figure 1.
Plasma EPO levels 1 and 7 days after injury. CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation. ANOVA experiment-wise p<0.0001; *p <0.05 vs. naïve (Day 1: CS, LCHS, and LCHS: p<0.0001; Day 7: CS: p<0.0001, LCHS: p=0.0004, LCHS/CS: p<0.0001); **p <0.05 vs. LCHS (Day 7: LCHS/CS: p<0.0001)
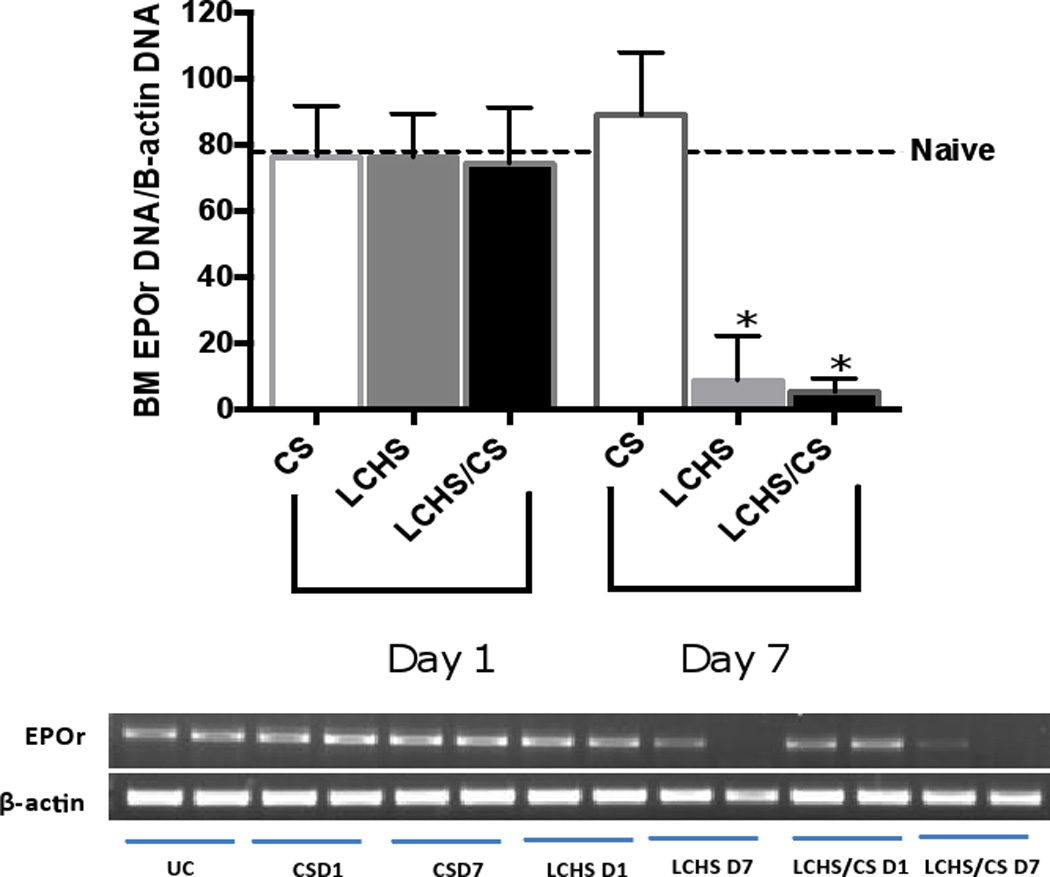
Successful erythropoiesis requires EPO to activate the EPOr on erythroid progenitor cells to allow for proliferation and differentiation. We therefore studied EPOr expression both in bone marrow as well as in injured lung tissue. One day following CS, LCHS, and LCHS/CS there was no difference in EPOr expression in bone marrow by PCR (Figure 2). On day seven, both LCHS and LCHS/CS significantly suppressed bone marrow EPOr expression by 83% and 91% compared to naïve (Figure 2).
Figure 2.
Following injury, day 1 and 7 quantitative PCR analysis of bone marrow EPO expression. CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation. ANOVA experiment-wise p<0.0001; *p <0.05 vs. naïve (Day 7 LCHS: p<0.0001, LCHS/CS: p: 0.0002)
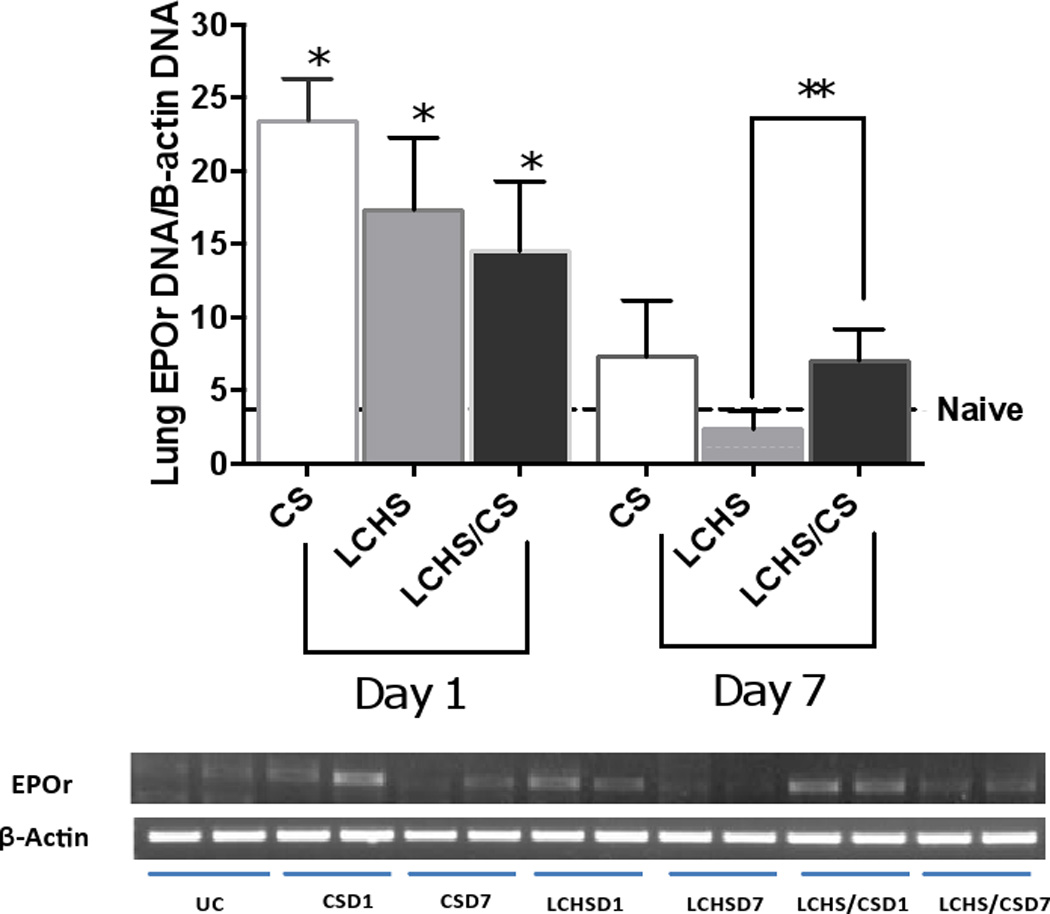
At the site of lung injury, lung EPOr levels were significantly increased on day one as compared to naïve animals (Figure 3). On day seven, CS and LCHS lung EPOr expression returned to that of naïve animals (Figure 3). Yet LCHS/CS maintained a persistently elevated lung EPOr expression on day seven as compared to LCHS (7.0±2.1** vs. 2.3±1.3, p=0.0085).
Figure 3.
Following injury, day 1 and 7 quantitative PCR analysis of lung EPO expression. CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation. ANOVA experiment-wise p<0.0001; *p <0.05 vs. naïve (Day 1: CS: p<0.0001, LCHS: p=0.0010, LCHS/CS: p=0.0191); **p <0.05 vs. LCHS (Day 7: LCHS/CS: p=0.0085)
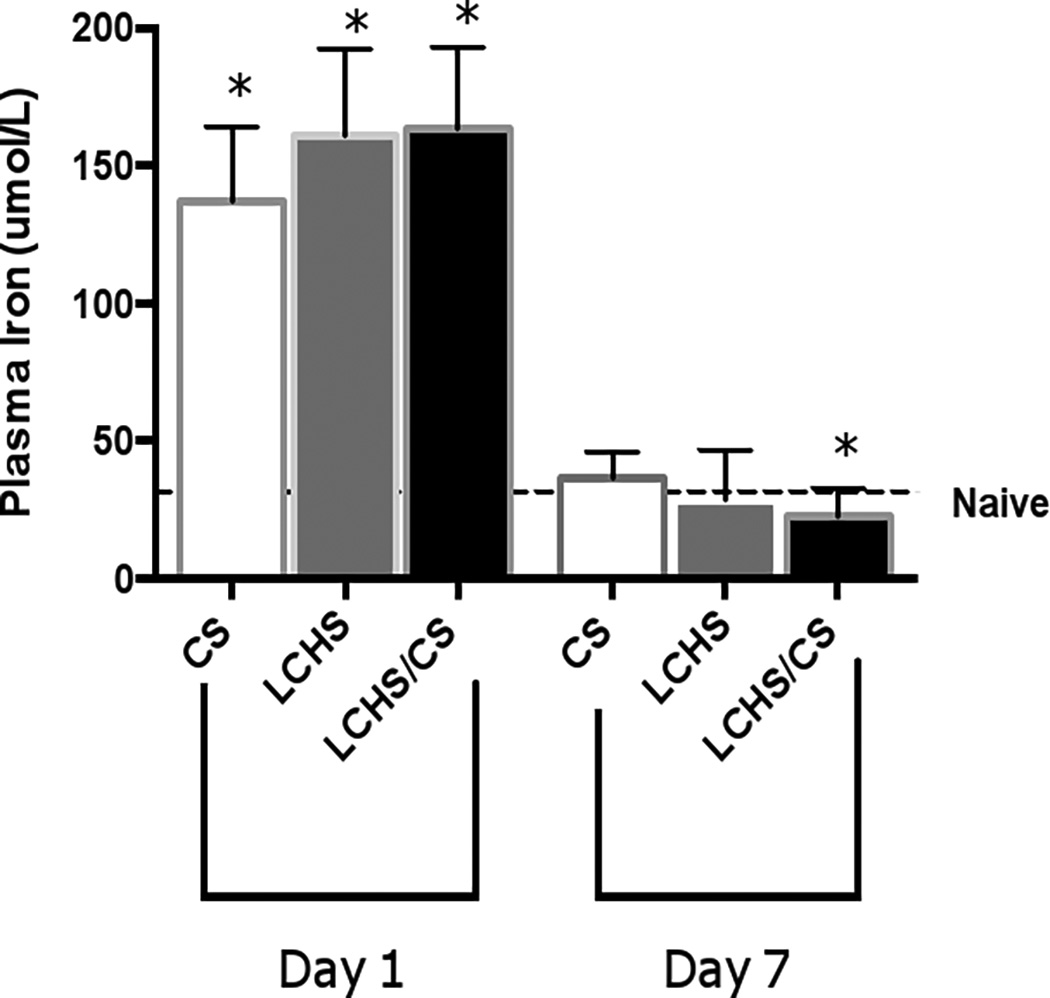
The role of iron following severe trauma and stress
After 24 hours of CS, LCHS, and LCHS/CS, plasma iron levels significantly increased when compared with naïve (Figure 4). On day seven, both CS and LCHS iron levels returned to baseline naïve levels (Figure 4). The addition of CS to LCHS led to a significant decrease in plasma iron levels on day seven compared to naïve (22.4±10* vs. 34.3±6.5 umol/L, p=0.003). Iron depletion following LCHS/CS may play a critical role in the ineffective EPO response that results in persistent injury-associated anemia.
Figure 4.
Plasma iron levels 1 and 7 days after injury. CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation. ANOVA experiment-wise p<0.0001; *p <0.05 vs. naïve (Day 1: CS, LCHS, and LCHS/CS: p<0.0001; Day 7: LCHS/CS: p=0.003)
There was no change in bone marrow transferrin on day seven following CS and LCHS as compared to naïve animals as measured by PCR (data not shown). There was a decrease in bone marrow transferrin expression following LCHS/CS on day seven but it was not significant (data not shown).
Ferritin is a marker of severe trauma and stress
After 24 hours, plasma ferritin levels were significantly elevated following CS, LCHS, and LCHS/CS (Figure 5). After seven days, CS and LCHS plasma ferritin levels decreased but still remained significantly elevated as compared to naïve (142±42* and 130±12* vs. 83.4±14 ng/mL, p=0.0177 and 0.0007). However, after seven days, the addition of CS to LCHS led to persistent increase of plasma ferritin as compared LCHS alone (266±52** vs. 130±12 ng/mL, p=0.0012). Following LCHS/CS, plasma ferritin appears to be a marker for continuous inflammation rather than iron storage.
Figure 5.
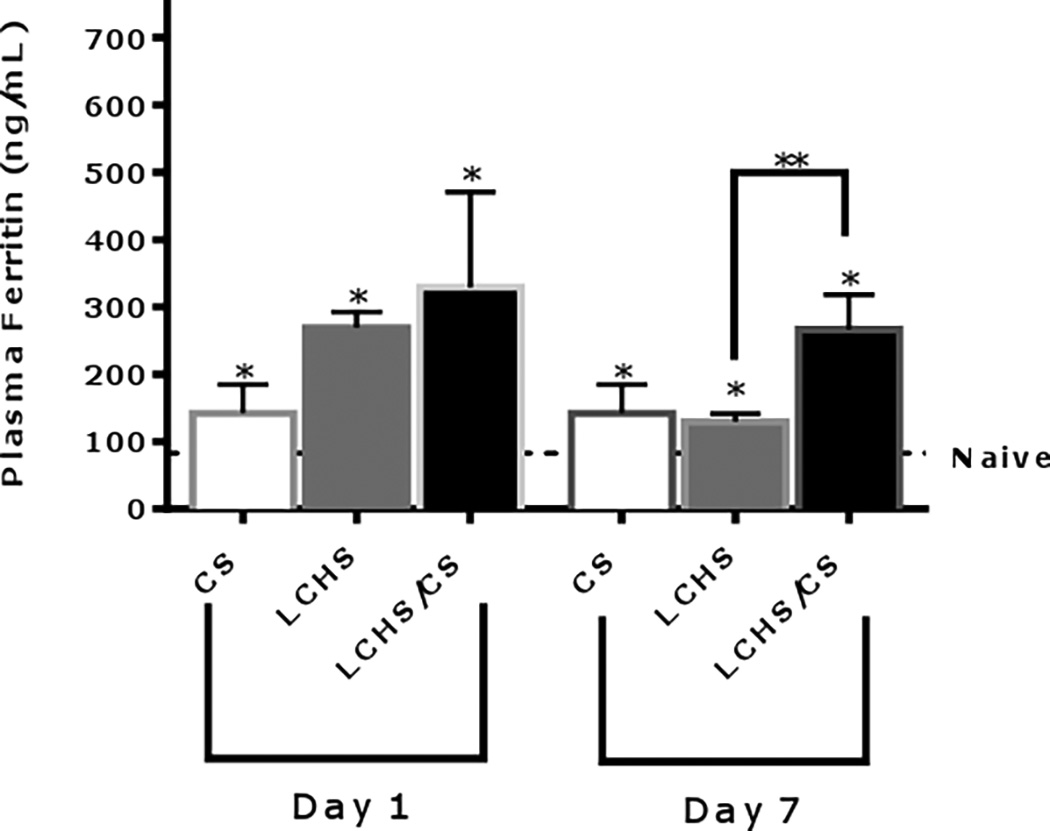
Plasma ferritin levels 1 and 7 days after injury. CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation. ANOVA experiment-wise p<0.0001; *p <0.05 vs. naïve (Day 1: CS: p=0.0177, LCHS and LCHS/CS: p<0.0001; Day 7: CS: p=0.0177, LCHS: p=0.0007; LCHS/CS: p<0.0001); **p <0.05 vs. LCHS (Day 7: LCHS/CS: p=0.0012)
The influence of chronic stress following severe trauma on hepcidin
CS alone significantly increased plasma hepcidin on day one but by day seven plasma hepcidin levels have returned to naïve levels (Figure 6). Similarly, LCHS alone significantly increased plasma hepcidin on day one and the level returns to that of naïve on day seven (Figure 6). However, the addition of CS to LCHS significantly decreased plasma hepcidin levels by 75% on day one compared to LCHS. By day seven after LCHS/CS there is a persistent decrease in plasma hepcidin levels by 49% as compared to LCHS alone (Figure 6). Urine hepcidin levels were measured in all groups and there was no significant change from naïve on either day one or day seven in any group (data not shown).
Figure 6.
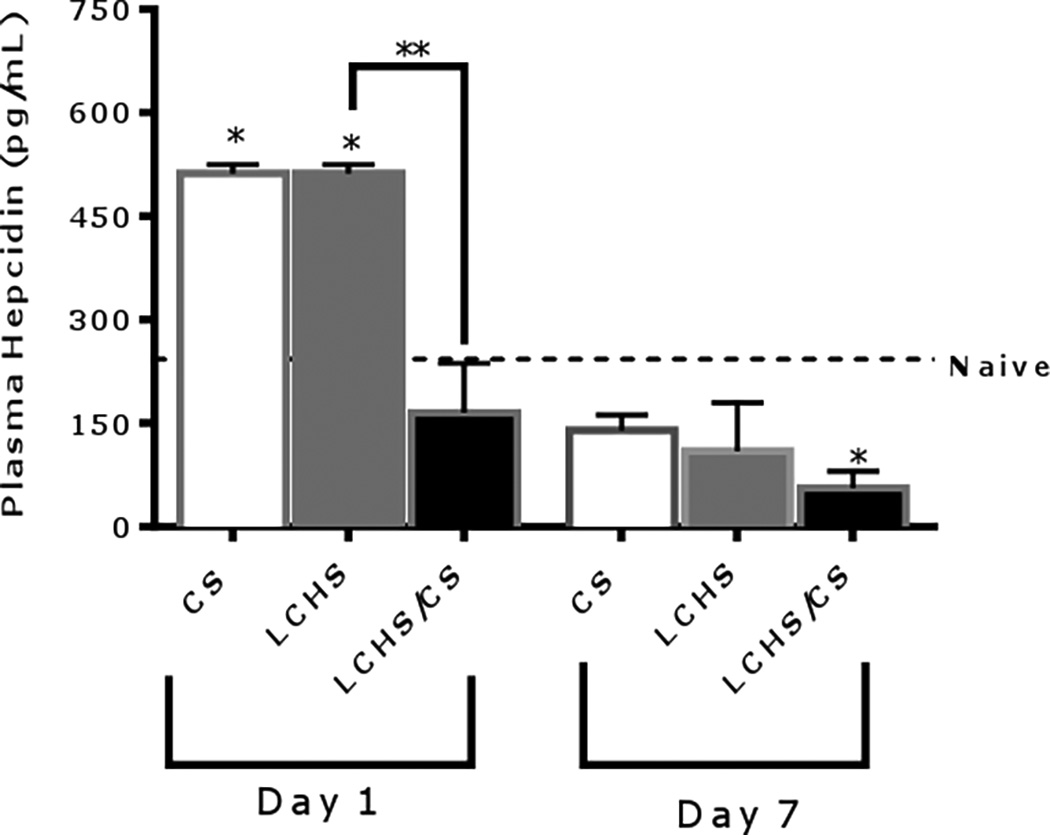
Plasma hepcidin levels 1 and 7 days after injury. CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation. ANOVA experiment-wise p<0.0001; *p <0.05 vs. naïve (Day 1: CS and LCHS: p<0.0001; Day7: LCHS/CS: p=0.0286); **p <0.05 vs. LCHS (Day 1: LCHS/CS: p=0.0034)
Relationship between EPO and hepcidin following severe trauma and stress
On day seven, we assessed the relationship of plasma EPO and hepcidin. In all models, there was an inverse relationship between plasma EPO and hepcidin (Figure, SDC 2A, 2B). LCHS/CS had the highest degree of elevation of plasma EPO and the most significant decrease in plasma hepcidin (Figure, SDC 2A). There is a significant inverse correlation between plasma hepcidin levels and plasma EPO levels (Pearson R=-0.362, p=.049) (Figure, SDC 2B). A marked elevation of plasma EPO appears to be the strongest correlation for the persistent suppression of plasma hepcidin.
Discussion
Among severely injured trauma patients, persistent-injury associated anemia develops and lasts for weeks to months following injury.29 This is the first study to demonstrate links between EPO, hepcidin, iron, EPOr expression and persistent injury-associated anemia in a clinically relevant model of lung injury, hemorrhagic shock, and chronic stress. LCHS/CS stimulated and maintained high levels of plasma EPO but these high EPO levels were not sufficient for resolution of persistent injury-associated anemia. This is the first study to demonstrate reduced bone marrow expression of EPOr following LCHS/CS. Iron availability is also significantly reduced following LCHS/CS and this iron deficit combined with reduced bone marrow EPOr expression may play a key role in the ineffective EPO response associated with persistent injury-associated anemia. In addition, we demonstrated that plasma ferritin is a useful marker of severe trauma and stress and that plasma hepcidin levels are significantly decreased with the addition of chronic stress to LCHS.
Both trauma patients and rodent models have been shown to result in persistently high levels of catecholamines, that are associated with bone marrow dysfunction and clinically manifests as a persistent injury-associated anemia 1, 4–6, 20, 30. In this study, the addition of CS to the LCHS model resulted in a moderate persistent injury-associated anemia on day seven, which is associated with a decrease RBC number. These findings correlated with a significant decline in the MCHC following LCHS/CS. Overall, these results are consistent with our previously published data and correlate with the persistent injury-associated anemia that is seen in the ICU.20
Previous studies have evaluated the role of EPO in the development of anemia after severe trauma. 5, 19 In a study among twenty-four burn patients with persistent anemia, EPO levels were increased 2–6 fold at days three, seven, and fourteen compared to controls regardless of the level of Hb (Hb: 8.9–9.5 g/dL). 19 Similarly, Livingston et al. 5 found that plasma EPO among severely injured trauma patients were significantly elevated compared to controls despite a mean Hb of 8.3±1.0 g/dL. The lack of restoration in Hb levels may be due to an inadequate production of EPO relative to that required for effective erythropoiesis. 5 In the current study, plasma EPO levels were significantly elevated from day one to seven despite the presence of anemia. The addition of CS following LCHS maintained a higher degree of EPO elevation on day seven. Our findings are supported by Robinson et al. 31, who demonstrated that in mouse models as early as 24 hours after HS, serum EPO levels increased 20-fold and remained elevated after 72 hours, but decreased in comparison to day one. This suggests that in addition to injury and hemorrhagic shock, a persistent insult, such as chronic stress is likely involved in persistent elevation of EPO.
If insufficient levels of EPO were solely responsible for persistent injury associated anemia then adjunctive erythropoiesis-stimulating agents (ESAs) such as recombinant human erythropoietin (rhEpo) should be effective. Unfortunately, clinical studies have not demonstrated this effectiveness. Corwin et al. 8, 23, 32, 33 studied the use of rhEpo in critically ill trauma and surgical patients in multiple trials. Compared to placebo, rhEpo reduced number of blood transfusions in one of the three studies but overall had no significant effect on improving Hb concentration, mortality, and length of stay in ICU or total hospital. 8, 23, 32, 33 Use of rhEpo in was associated with an increase in thrombotic events in those patients who did not receive venous thromboembolism prophylaxis.23 Therefore, other factors in addition to EPO must a play a role in erythropoiesis after injury.
In addition to adequate levels of EPO in the blood, sufficient expression of EPOr in the bone marrow is crucial for the production of mature RBCs. 13–18, 33, 34 Nalbant et al. 15 demonstrated the relationship between EPOr expression and anemia. They demonstrated a 12-fold increase in bone marrow EPOr levels nine days post phlebotomy (Hb: 3.7–4.2 g/dL) when compared to pre-phlebotomy (Hb: 9.7±1.1 g/dL). By 28 days, there was only a 1–2 fold increase in bone marrow EPOr level with Hb levels that were restored to baseline (Hb: 9.5±1.5 g/dL). 15 Similarly, Pontikoglou et al. 35 revealed that if bone marrow EPOr expression was 50% less, anemia remained unresolved (Hb: 11.5±0.4 g/dL). This is the first study to demonstrate decreased bone marrow EPOr expression after trauma. One day after injury and stress, bone marrow EPOr expression decreased and remained low seven days following LCHS/CS, which correlated with the persistence of anemia. The discrepancy between plasma EPO levels and bone marrow EPOr expression is likely multifactorial. The mechanisms involved in EPOr regulation have not been fully elucidated. In order for differentiation and maturation of erythroid progenitor cells to occur, the binding between EPO and EPOr is key. EPOr expression has been shown to be linked to the cell cycle and we previously demonstrated that the LCHS/CS model led to significantly reduced growth of bone marrow CFU-E, BFU-E, and colony forming units-granulocyte, erythrocyte, monocyte, megakaryocytes (CFU-GEMM).20 Others have shown little or no growth of CFU-Es when progenitor EPOr deficient cells were cultured. 17, 18 Moreover, cells that did grow CFU-Es were smaller and with less Hb than those in wild-type colonies. 17 Insufficient levels of bone marrow EPOr expression likely play a role in effective EPO binding, growth of bone marrow erythroid progenitors, and signaling for maturation of RBCs.
When examining EPOr expression at the site of injury in the lungs, we found a significant elevation in expression on day one following CS, LCHS and LCHS/CS and on day seven there was persistent elevation of EPOr expression following LCHS/CS. These findings correlated with previous work done showing the bone marrow hematopoietic progenitor cells mobilize out of the bone marrow to sites of injury and participate in healing of injured tissues. 36, 37 Hematopoietic progenitor cell mobilization to the sites of injury correlated with the persistent expression of lung EPOr on day seven seen following LCHS/CS in this study. Thus, the loss of hematopoietic progenitor cells from the bone marrow likely contributes to ongoing bone marrow dysfunction after trauma.
Iron availability is essential for effective erythropoiesis. 10, 11, 19, 22 Previous studies have shown that serum iron levels are low among severely injured patients with persistent anemia. 10, 11, 19 Rodriguez et al. 11 showed decreased serum iron levels on day three in critically ill patients with anemia. The current study demonstrated despite an initial increase in plasma iron, LCHS/CS led to a persistent anemia and lower plasma iron levels on day seven. This similar to what was shown in critically ill burn patients who had elevated iron levels on the day of injury that decreased significantly on day three.19 The lack of availability of plasma iron for erythropoiesis is a potential mechanism for persistent injury-associated anemia. However, in a multicenter randomized controlled trial of intravenous iron supplementation in critically ill trauma patients there was no improvement in iron deficiency, anemia, or transfusion requirement.10 Similar to the clinical use of rhEPO, other factors in addition to intravenous iron must a play a role in erythropoiesis after injury. As limitation of this study, reticulocyte Hb content, hypochromic erythrocytes, and transferrin saturation were not evaluated. So an assessment of bioavailable iron for bone marrow deposition has not been fully elucidated.
This study demonstrates that ferritin levels reflect acute, inflammatory changes following injury and stress. While all groups had increased ferritin levels at 24 hours, only the LCHS/CS group had persistently high ferritin levels throughout the seven days. Our findings are supported by previous work in which critically ill patients with persistent anemia developed significantly elevated ferritin levels. 10, 25 Therefore, following severe traumatic injury, ferritin serves as a reliable sensitive inflammatory marker.
Anemia secondary to acute blood loss is the main stimulator in the cascade that leads to suppression of hepcidin. 28 Yet, Sihler et al.9 demonstrated that urine hepcidin levels in mildly anemic trauma patients (Hb: 12.4 g/dL) were significantly elevated 9. The increase in urinary hepcidin was attributed to blood transfusions, as these patients received an average 2.5 units of blood.9 In trauma patients, inflammation rather than anemia appears play a significant role in the increase in hepcidin. In our rodent model of LCHS/CS, we found that plasma hepcidin was elevated on day one following CS and LCHS alone but significantly suppressed following LCHS/CS. The addition of daily CS to LCHS led to a 75% decrease in hepcidin on day one and 49% on day seven as compared to LCHS alone. These results parallel that by Lasocki et al 26, who also confirmed that phlebotomy-induced anemia in mice decreased hepcidin mRNA. We hypothesize that anemia and erythropoietic stimulation have a greater impact on hepcidin suppression in our rodent model of LCHS/CS.
The relationship between hepcidin and EPO has been studied in other models. 12, 26, 38–43 Fein et al. 39 showed that hepatoma and rat pancreatic cells stimulated in vitro with EPO inhibited the expression of hepcidin by 50% when compared to control cells. Previous work by Sasaki et al. 12 showed that rhEPO treatments on mice significantly decreased serum hepcidin levels at 24 hours when compared to controls. Both in vitro or in vivo models, hepcidin production is suppressed in the presence of anemia directly or indirectly through EPO.26, 40–43 Our study correlated with these findings in other models. Throughout the seven days, a strong inverse correlative relationship was noted between plasma EPO and hepcidin levels. By day seven, LCHS/CS maintained a significant elevation of plasma EPO with an equally significant suppression of plasma hepcidin. Although EPO is a negative regulator of hepcidin, inflammation has also been shown to stimulate hepcidin.40–43 In our model, the presence of persistent injury-associated anemia prevented the stimulatory effects of chronic stress and inflammation to produce hepcidin. Similarly, in phlebotomy-induced anemia mice models, the effects of zymosan injection used to mediate an increase in hepcidin, were completely ineffective and hepcidin levels remained suppressed.26 Further work is required to elucidate the complex mechanisms of hepcidin regulation by iron, erythroid activity, and inflammation.
In conclusion, this study outlines the complex relationship between EPO, hepcidin, iron, and the development of persistent injury-associated anemia. LCHS/CS led to significant plasma EPO elevation and hepcidin suppression. Despite elevated plasma EPO levels following LCHS/CS, reduced bone marrow EPOr expression and plasma iron levels were inadequate to correct the anemia. Anemia following trauma is not one entity but is comprised of both acute blood loss and persistent injury-associated anemia. Further investigation of the mechanisms involved in regulation of bone marrow EPOr expression and potential combined treatment modalities in these critically injured patients are warranted.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health. AMM was supported by NIH NIGMS grant R01 GM105893-01A1. PAE was supported by P30 AG028740 from the National Institute on Aging and by the NIH NIGMS grant R01 GM113945-01. Finally, AMM and PAE were all supported by P50 GM111152-01 (NIGMS).
Footnotes
Presented in part at the 10th Annual Academic Surgical Congress in Las Vegas, NV February 3–5, 2015
This submission has not been published elsewhere and the authors have nothing to disclose.
Authors’ contributions
IGA contributed to the formation of the experimental rodent model design, performed all rodent survival surgeries, assisted with tissue collection/processing, hematology and protein analysis, did significant writing and statistical analysis of the manuscript.
KBK assisted with the formation of the experimental rodent model design, hematology, and protein analysis, performed tissue collection/processing, and protein isolation.
MAS performed rodent survival surgeries, sample preparations, protein and statistical analysis of the data.
PAE provided assistance with the research question, interpretation of the data, statistical analysis, and writing of the manuscript.
AMM conceived the concept for the experimental rodent model design, research question, interpretation of data, and contributed significantly to the writing of the manuscript and final edits. All authors have read and approved the final manuscript.
Table SDC 1. Peripheral Blood Hematologic Parameters after Injury
Figure SDC 2A, 2B. Relationship between plasma EPO and hepcidin levels 7 days after injury. Plasma hepcidin levels were inversely proportional to plasma EPO levels (Pearson R=-0.362, p<0.05). CS= chronic stress; LCHS= LC followed by HS; LCHS= LCHS followed by daily CS after resuscitation.
Contributor Information
Ines G. Alamo, Email: Ines.Alamo@surgery.ufl.edu.
Kolenkode B. Kannan, Email: Kolenkode.Kannan@surgery.ufl.edu.
Michael A. Smith, Email: mikesmith@ufl.edu.
Philip A. Efron, Email: Philip.Efron@surgery.ufl.edu.
Alicia M. Mohr, Email: alicia.mohr@surgery.ufl.edu.
References
- 1.Bible LE, Pasupuleti LV, Alzate WD, Gore AV, Song KJ, Sifri ZC, Livingston DH, Mohr AM. Early propranolol administration to severely injured patients can improve bone marrow dysfunction. J Ttrauma and Acute Care Surg. 2014;77(1):54–60. doi: 10.1097/TA.0000000000000264. discussion 59-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Napolitano LM. Scope of the problem: epidemiology of anemia and use of blood transfusions in critical care. Crit Care. 2004;8(Suppl 2):S1–S8. doi: 10.1186/cc2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Abraham E, MacIntyre NR, Shabot MM, Duh MS, Shapiro MJ. The CRIT Study: Anemia and blood transfusion in the critically ill--current clinical practice in the United States. Crit Care Med. 2004;32(1):39–52. doi: 10.1097/01.CCM.0000104112.34142.79. [DOI] [PubMed] [Google Scholar]
- 4.Mohr AM, ElHassan IO, Hannoush EJ, Sifri ZC, Offin MD, Alzate WD, Rameshwar P, Livingston DH. Does beta blockade postinjury prevent bone marrow suppression? J Tauma. 2011;70(5):1043–1049. doi: 10.1097/TA.0b013e3182169326. discussion 9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Livingston DH, Anjaria D, Wu J, Hauser CJ, Chang V, Deitch EA, Rameshwar P. Bone marrow failure following severe injury in humans. Ann Surg. 2003;238(5):748–753. doi: 10.1097/01.sla.0000094441.38807.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badami CD, Livingston DH, Sifri ZC, Caputo FJ, Bonilla L, Mohr AM, Deitch EA. Hematopoietic progenitor cells mobilize to the site of injury after trauma and hemorrhagic shock in rats. J Trauma. 2007;63(3):596–600. doi: 10.1097/TA.0b013e318142d231. discussion -2. [DOI] [PubMed] [Google Scholar]
- 7.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 8.Corwin HL, Gettinger A, Rodriguez RM, Pearl RG, Gubler KD, Enny C, Colton T, Corwin MJ. Efficacy of recombinant human erythropoietin in the critically ill patient: a randomized, double-blind, placebo-controlled trial. Crit Care Med. 1999;27(11):2346–2350. doi: 10.1097/00003246-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Sihler KC, Raghavendran K, Westerman M, Ye W, Napolitano LM. Hepcidin in trauma: linking injury, inflammation, and anemia. J Trauma. 2010;69(4):831–837. doi: 10.1097/TA.0b013e3181f066d5. [DOI] [PubMed] [Google Scholar]
- 10.Pieracci FM, Stovall RT, Jaouen B, Rodil M, Cappa A, Burlew CC, Holena DN, Maier R, Berry S, Jurkovich J, et al. A multicenter, randomized clinical trial of IV iron supplementation for anemia of traumatic critical illness*. Crit Care Med. 2014;42(9):2048–2057. doi: 10.1097/CCM.0000000000000408. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez RM, Corwin HL, Gettinger A, Corwin MJ, Gubler D, Pearl RG. Nutritional deficiencies and blunted erythropoietin response as causes of the Amemia of critical illness. J Crit Care. 2001;16(1):36–41. doi: 10.1053/jcrc.2001.21795. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki Y, Noguchi-Sasaki M, Yasuno H, Yorozu K, Shimonaka Y. Erythropoietin stimulation decreases hepcidin expression through hematopoietic activity on bone marrow cells in mice. Int J Hematol. 2012;96(6):692–700. doi: 10.1007/s12185-012-1217-4. [DOI] [PubMed] [Google Scholar]
- 13.Constantinescu SN, Keren T, Russ WP, Ubarretxena-Belandia I, Malka Y, Kubatzky KF, Engelman DM, Lodish HF, Henis YI. The erythropoietin receptor transmembrane domain mediates complex formation with viral anemic and polycythemic gp55 proteins. J Biol Chem. 2003;278(44):43755–43763. doi: 10.1074/jbc.M302974200. [DOI] [PubMed] [Google Scholar]
- 14.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 15.Nalbant D, Saleh M, Goldman FD, Widness JA, Veng-Pedersen P. Evidence of receptor-mediated elimination of erythropoietin by analysis of erythropoietin receptor mRNA expression in bone marrow and erythropoietin clearance during anemia. J Pharm Exp Ther. 2010;333(2):528–532. doi: 10.1124/jpet.109.163568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 17.Lin CS, Lim SK, D'Agati V, Costantini F. Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes & Dev. 1996;10(2):154–164. doi: 10.1101/gad.10.2.154. [DOI] [PubMed] [Google Scholar]
- 18.Kieran MW, Perkins AC, Orkin SH, Zon LI. Thrombopoietin rescues in vitro erythroid colony formation from mouse embryos lacking the erythropoietin receptor. Proc Nat Acad Sci USA. 1996;93(17):9126–9131. doi: 10.1073/pnas.93.17.9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deitch EA, Sittig KM. A serial study of the erythropoietic response to thermal injury. Ann Surg. 1993;217(3):293–299. doi: 10.1097/00000658-199303000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bible LE, Pasupuleti LV, Gore AV, Sifri ZC, Kannan KB, Mohr AM. Chronic restraint stress after injury and shock is associated with persistent anemia despite prolonged elevation in erythropoietin levels. J Trauma Acute Care Surg. 2015;79(1):91–96. doi: 10.1097/TA.0000000000000686. discussion 6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hobisch-Hagen P, Wiedermann F, Mayr A, Fries D, Jelkmann W, Fuchs D, Hasibeder W, Mutz N, Klingler A, Schobersberger W. Blunted erythropoietic response to anemia in multiply traumatized patients. Crit Care Med. 2001;29(4):743–747. doi: 10.1097/00003246-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 22.von Ahsen N, Muller C, Serke S, Frei U, Eckardt KU. Important role of nondiagnostic blood loss and blunted erythropoietic response in the anemia of medical intensive care patients. Crit Care Med. 1999;27(12):2630–2639. doi: 10.1097/00003246-199912000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Jelkmann I, Jelkmann W. Impact of erythropoietin on intensive care unit patients. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie. 2013;40(5):310–318. doi: 10.1159/000354128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogiers P, Zhang H, Leeman M, Nagler J, Neels H, Melot C, Vincent JL. Erythropoietin response is blunted in critically ill patients. Intensive Care Med. 1997;23(2):159–162. doi: 10.1007/s001340050310. [DOI] [PubMed] [Google Scholar]
- 25.van Iperen CE, Gaillard CA, Kraaijenhagen RJ, Braam BG, Marx JJ, van de Wiel A. Response of erythropoiesis and iron metabolism to recombinant human erythropoietin in intensive care unit patients. Crit Care Med. 2000;28(8):2773–2778. doi: 10.1097/00003246-200008000-00015. [DOI] [PubMed] [Google Scholar]
- 26.Lasocki S, Millot S, Andrieu V, Letteron P, Pilard N, Muzeau F, Thibaudeau O, Montravers P, Beaumont C. Phlebotomies or erythropoietin injections allow mobilization of iron stores in a mouse model mimicking intensive care anemia. Crit Care Med. 2008;36(8):2388–2394. doi: 10.1097/CCM.0b013e31818103b9. [DOI] [PubMed] [Google Scholar]
- 27.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Inv. 2002;110(7):1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108(12):3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bateman AP, McArdle F, Walsh TS. Time course of anemia during six months follow up following intensive care discharge and factors associated with impaired recovery of erythropoiesis. Crit Care Med. 2009;37(6):1906–1912. doi: 10.1097/CCM.0b013e3181a000cf. [DOI] [PubMed] [Google Scholar]
- 30.Molina PE. Neurobiology of the stress response: contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock. 2005;24(1):3–10. doi: 10.1097/01.shk.0000167112.18871.5c. [DOI] [PubMed] [Google Scholar]
- 31.Robinson Y, Matenov A, Tschoke SK, Weimann A, Oberholzer A, Ertel W, Hostmann A. Impaired erythropoiesis after haemorrhagic shock in mice is associated with erythroid progenitor apoptosis in vivo. Acta Anaesth Scand. 2008;52(5):605–613. doi: 10.1111/j.1399-6576.2008.01656.x. [DOI] [PubMed] [Google Scholar]
- 32.Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Shapiro MJ, Corwin MJ, Colton T Group EPOCCT. Efficacy of recombinant human erythropoietin in critically ill patients: a randomized controlled trial. JAMA. 2002;288(22):2827–2835. doi: 10.1001/jama.288.22.2827. [DOI] [PubMed] [Google Scholar]
- 33.Corwin HL, Gettinger A, Fabian TC, May A, Pearl RG, Heard S, An R, Bowers PJ, Burton P, Klausner MA, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Eng J Med. 2007;357(10):965–976. doi: 10.1056/NEJMoa071533. [DOI] [PubMed] [Google Scholar]
- 34.Elliott S, Pham E, Macdougall IC. Erythropoietins: a common mechanism of action. Exp Hemat. 2008;36(12):1573–1584. doi: 10.1016/j.exphem.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Pontikoglou C, Liapakis G, Pyrovolaki K, Papadakis M, Bux J, Eliopoulos GD, Papadaki HA. Evidence for downregulation of erythropoietin receptor in bone marrow erythroid cells of patients with chronic idiopathic neutropenia. Exp Hemat. 2006;34(10):1312–1322. doi: 10.1016/j.exphem.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Pasupuleti LV, Cook KM, Sifri ZC, Alzate WD, Livingston DH, Mohr AM. Do all beta-blockers attenuate the excess hematopoietic progenitor cell mobilization from the bone marrow following trauma/hemorrhagic shock? J Trauma Acute Care Surg. 2014;76(4):970–975. doi: 10.1097/TA.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baranski GM, Pasupuleti LV, Sifri ZC, Cook KM, Alzate WD, Rameshwar P, Livingston DH, Mohr AM. Beta Blockade Protection of Bone Marrow Following Injury: A Critical Link between Heart Rate and Immunomodulation. J Bone Marrow Res. 2013;1 doi: 10.4172/2329-8820.1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pirie K, Myles P, Wood E. Anemia and iron-restricted erythropoiesis in traumatic critical illness. J Trauma Acute Care Surgery. 2016;80(3):538–545. doi: 10.1097/TA.0000000000000939. [DOI] [PubMed] [Google Scholar]
- 39.Fein E, Merle U, Ehehalt R, Herrmann T, Kulaksiz H. Regulation of hepcidin in HepG2 and RINm5F cells. Peptides. 2007;28(5):951–957. doi: 10.1016/j.peptides.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Nicolas G, Viatte L, Bennoun M, Beaumont C, Kahn A, Vaulont S. Hepcidin, A New Iron Regulatory Peptide. Blood Cells Molecules Dis. 2002;29(3):327–335. doi: 10.1006/bcmd.2002.0573. [DOI] [PubMed] [Google Scholar]
- 41.Shike H, Lauth X, Westerman ME, Ostland VE, Carlberg JM, Van Olst JC, Shimizu C, Bulet P, Burns JC. Bass hepcidin is a novel antimicrobial peptide induced by bacterial challenge. Eur J Biochem / FEBS. 2002;269(8):2232–2237. doi: 10.1046/j.1432-1033.2002.02881.x. [DOI] [PubMed] [Google Scholar]
- 42.Kelley-Loughnane N, Sabla GE, Ley-Ebert C, Aronow BJ, Bezerra JA. Independent and overlapping transcriptional activation during liver development and regeneration in mice. Hepatology. 2002;35(3):525–534. doi: 10.1053/jhep.2002.31351. [DOI] [PubMed] [Google Scholar]
- 43.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–4433. doi: 10.1182/blood-2011-01-258467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.