

Abstract
AIM
To determine if manipulation of dietary advanced glycation end product (AGE), intake affects non-alcoholic fatty liver disease (NAFLD) progression and whether these effects are mediated via RAGE.
METHODS
Male C57Bl6 mice were fed a high fat, high fructose, high cholesterol (HFHC) diet for 33 wk and compared with animals on normal chow. A third group were given a HFHC diet that was high in AGEs. Another group was given a HFHC diet that was marinated in vinegar to prevent the formation of AGEs. In a second experiment, RAGE KO animals were fed a HFHC diet or a high AGE HFHC diet and compared with wildtype controls. Hepatic biochemistry, histology, picrosirius red morphometry and hepatic mRNA were determined.
RESULTS
Long-term consumption of the HFHC diet generated significant steatohepatitis and fibrosis after 33 wk. In this model, hepatic 4-hydroxynonenal content (a marker of chronic oxidative stress), hepatocyte ballooning, picrosirius red staining, α-smooth muscle actin and collagen type 1A gene expression were all significantly increased. Increasing the AGE content of the HFHC diet by baking further increased these markers of liver damage, but this was abrogated by pre-marination in acetic acid. In response to the HFHC diet, RAGE-/- animals developed NASH of similar severity to RAGE+/+ animals but were protected from the additional harmful effects of the high AGE containing diet. Studies in isolated Kupffer cells showed that AGEs increase cell proliferation and oxidative stress, providing a likely mechanism through which these compounds contribute to liver injury.
CONCLUSION
In the HFHC model of NAFLD, manipulation of dietary AGEs modulates liver injury, inflammation, and liver fibrosis via a RAGE dependent pathway. This suggests that pharmacological and dietary strategies targeting the AGE/RAGE pathway could slow the progression of NAFLD.
Keywords: Advanced glycation end-products, Fructose, Steatohepatitis, Non-alcoholic fatty liver disease, Hepatic fibrosis, Oxidative stress
Core tip: A novel high fructose, high cholesterol diet produces hepatic non-alcoholic steatohepatitis (NASH) with fibrosis in 33 wk and increasing the Advanced glycation end products (AGEs), content of this diet via baking increases hepatic fibrosis whilst vinegar marination decreases dietary AGE levels, abrogating the harmful effects of AGEs. RAGE-/- animals appeared to be protected from the additional harmful effects of a high AGE containing diet suggesting the central role of RAGE in progression of NASH. Increased cell proliferation and oxidative stress in isolated primary Kupffer cells with the addition of AGEs suggests they are an important mechanism in which AGEs contribute to liver injury.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease in the world and is strongly linked to the burgeoning rates of diabetes and obesity[1]. Although most patients with NAFLD are asymptomatic and do not develop clinically significant liver injury, many progress to non-alcoholic steatohepatitis (NASH), cirrhosis and liver cancer. However, the host and/or environmental risk factors that determine whether patients with simple hepatic steatosis go on to develop NASH and its complications remain unclear[2].
Advanced glycation end products (AGEs), also known as glycotoxins, are a complex group of compounds that are formed when active sugar moieties become bound to proteins causing browning and other irreversible modifications[3]. Foods that are highly processed or dry heated at high temperatures, such as broiled foods, have particularly high AGE content[4]. However, they are also formed endogenously and this occurs at an increased rate in diabetes, most likely because of hyperglycaemia. There is now considerable evidence that accumulation of AGEs are implicated in the pathogenesis of diabetic renal, neurological, retinal and vascular complications[5].
AGEs act on several receptors but the receptor for advanced glycation end products (RAGE), a member of the immunoglobulin superfamily of cell-surface molecules, is the best characterised of these receptors[6]. Engagement with RAGE, which in turn increases inflammation and oxidative stress is thought to be the main way in which AGEs impart these pathogenic effects[7]. This receptor is expressed in a number of cell types, including endothelial cells, vascular smooth muscle cells, peripheral blood mononuclear cells, macrophages (including Kupffer cells) and HSCs[8].
A number of lines of evidence to suggest that, in NAFLD, AGEs may be a “second hit” that contributes to the progression from simple steatosis to NASH and liver fibrosis[9]. Several studies have shown that RAGE plays a role in acute liver injury and that blockade of RAGE can ameliorate toxic, ischaemic and cholestatic liver damage[10-12]. In chronic liver injury, hepatic expression of RAGE is significantly increased[13], and in NAFLD AGE levels correlate with the severity of fibrosis, leading to speculation that AGEs play a primary role in disease pathogenesis[14]. At a cellular level, AGEs induces ROS via oxidative stress[15] and this is a significant mechanism underlying the pathogenesis of NASH[16]. Furthermore, diabetes, which increases AGE formation RAGE expression and oxidative stress, worsens the progression of fibrosis in a number of human liver diseases, including NAFLD[17] and hepatitis C[18].
In a previous study, we showed that in a short term methionine choline deficient (MCD) model of NAFLD, a high AGE diet increased hepatic AGE content and exacerbated liver injury, oxidative stress and liver fibrosis and that AGEs produced RAGE dependent profibrotic effects in activated hepatic stellate cells (HSCs)[19]. However, the relevance of the MCD model to human disease is limited since the metabolic profile of MCD fed animals is generally the converse of human NAFLD[20]. In contrast, long-term murine models based on a high-calorie diet high in fat, fructose and cholesterol [high fructose, high cholesterol (HFHC) diet], yield NASH associated with many of the metabolic features of human NASH[21]. The aim of the current study was to explore whether increasing dietary AGE consumption could precipitate the development of NASH in a novel HFHC model of NAFLD. We also performed complementary studies in RAGE KO animals to determine the role of RAGE signalling in our liver disease model.
MATERIALS AND METHODS
Experimental design
Experiments were approved by the Austin Health Animal Ethics Committee and performed according to the National Health and Medical Research Council (NHMRC) of Australia Guidelines for animal experimentation.
Male C57Bl6 mice (n = 10/group) were fed a high fat, high fructose, high cholesterol (HFHC) diet [Specialty Feeds SF11-109 (21% saturated fat, 10% fructose, 2% cholesterol)] for 33 wk and compared with animals on normal chow (Specialty Feeds AIN93G). This is a novel dietary model combining amounts of saturated fat (especially animal fats such as ghee) and fructose to mimic unhealthy Western diets[22]. This model is different to other high fat diets in that we gave amounts of fat actually consumed by patients for a longer period to induce fibrosis[23]. Two percent cholesterol was incorporated as this has been shown to be a critical driver for fibrosis in murine dietary models of NASH[22] and 10% fructose was added, both due to its prevalence in Western diet as well as its potent ability to induce AGE formation[24] (See Supplementary materials 2). In two further groups, the effects of further increasing dietary AGE content by baking (described below), were studied in animals fed normal chow and in animals fed the HFHC diet. A final group was fed a baked HFHC diet that was marinated in acetic acid (vinegar 4% w/v) prior to heating to prevent the formation of AGEs, as is seen in Mediterranean style diets[25]. In a second experiment, RAGE KO animals were fed a HFHC diet or a high AGE HFHC diet for 33 wk and compared with WT controls. At the completion of each experiment, insulin resistance was measured via HOMA-IR and oral glucose tolerance testing (OGTT) performed as previously described[26]. Livers were harvested for assessment of liver injury and serum alanine aminotransaminase (ALT) levels were measured by auto-analyser (Beckman Instruments, Fullerton, CA).
Production of AGEs
Dietary AGE content was increased by baking at 160 °C for 1 h which we have previously shown produces an approximately 4 fold increase in AGE levels[12]. CML is the predominant AGE in food and the extent of advanced glycation in the diet was assessed by measuring CML content using high performance liquid chromatography with fluorescence detection against authentic standards of CML[27].
For cell studies, AGEs were prepared in vitro by incubating bovine serum albumin (BSA, 50 mg/mL) with 0.5 mol/L glucose in 100 mmol/L sodium phosphate buffer, pH 7.4. This was incubated at 37 °C for 6 wk, followed by dialysis against phosphate buffered saline (PBS) for 48 h at 4 °C to remove any free glucose. AGE-BSA and BSA were then passed through an endotoxin column (Detoxigel, Pierce, Rockford, IL, United States) to remove any possible endotoxin contaminants. The extent of advanced glycation was assessed by CML levels with ELISA and by isotope dilution, selected ion monitoring gas chromatography-mass spectrometry[12,27].
Liver histology
Paraffin embedded, paraformaldehyde fixed sections of liver (4 μm) were stained with haematoxylin and eosin and picrosirius red (Polysciences Inc. Warrington, PA) for assessment of liver fibrosis and steatohepatitis by an independent pathologist. The NAFLD Activity Score (NAS) system was used to quantify steatohepatitis using ten × 200 light microscopic fields in a blinded fashion as previously described[28]. Each field was scored using the following criteria: For hepatic steatosis: grade 0, no fat; grade 1, steatosis occupying less than 33% of the hepatic parenchyma; grade 2, 34%-66% of the hepatic parenchyma; grade 3, more than 66% of the hepatic parenchyma; for inflammatory cell infiltration: grade 0:none; grade 1, 1-2 foci/field; grade 2, 3-4 foci/field; grade 3, more than 4 foci/field (steatosis 0-3, lobular inflammation 0-2, hepatocellular ballooning 0-3 and fibrosis 0-4)[28]. Collagen content of the liver was quantified histologically using computerized quantification of picrosirius red staining, as described previously[12]. This was assessed at × 100 magnification in a total of ten fields per section (per animal), using computerized quantification and results were expressed as proportion of picrosirius red staining per field.
Assessment of hepatic oxidative stress
Superoxide anion and ROS are key mediators of liver injury and cellular dysfunction associated with the progression of fatty liver disease. Immunohistochemistry for HNE, a marker of lipid peroxidation, was performed on 4 μm sections of paraffin embedded liver as previously described[12]. The primary HNE antibody (1:500 Alpha Diagnostic International, San Antonio, Texas, United States) in 0.1% normal goat serum with PBS was applied and then incubated with biotinylated secondary goat anti-mouse antibody (1/100) followed by incubation with avidin-biotin horseradish peroxidase. Peroxidase conjugates were subsequently localized using diaminobenzidine tetrahydrochloride chromogen (Sigma-Aldrich, Sydney, Australia). The relative positive staining in each group was determined by computerized quantification (MCID; Imaging Research, Ontario, Canada) at × 100 magnification (10 fields per animal).
Quantitative real time PCR
Total RNA was extracted using TRI reagent (Sigma-Aldrich) and reverse transcribed to cDNA using a protocol previously described[29]. Gene expression was normalized to the expression of the endogenous control, ribosomal 18S. Each sample was run and analysed in duplicate. The normalized values from normal chow livers were used as the calibrator with a given value of 1 and the other groups compared with this calibrator. The gene probe, forward and reverse primer sequences are detailed in Table 1.
Table 1.
Primer and probe sequences used for real time qPCR
| Gene name | Probe/primer | Sequence |
| IL-6 | Probe | 5-FAM ATTGCCATTGCACAACT-3 |
| Forward | 5-GGGAAATCGTGGAAATGAGAAA-3 | |
| Reverse | 5-AAGTGCATCATCGTTGTTCATACA-3 | |
| MCP1 | Probe | 5-FAM-AATGGGTCCAGACATAC-3 |
| Forward | 5-GTCTGTGCTGACCCCAAGAAG-3 | |
| Reverse | 5-TGGTTCCGATCCAGGTTTTTA-3 | |
| TNFα | Probe | 5-FAM-TCACCCACACCGTCAG-3 |
| Forward | 5-GGCTGCCCCGACTACGT-3 | |
| Reverse | 5-TTTCTCCTGGTATGAGATAGCAAATC-3 | |
| RAGE | Probe | 5-FAM-CACAGCCCGGATTG-3 |
| Forward | 5-GCTGTAGCTGGTGGTCAGAACA-3 | |
| Reverse | 5-CCCCTTACAGCTTAGCACAAGTG-3 | |
| alphaSMA | Probe | 5-FAM-TGCCAGATCTTTTCC-3 |
| Forward | 5-GACGCTGAAGTATCCGATAGAACA-3 | |
| Reverse | 5-GGCCACACGAAGCTCGTTAT-3 | |
| COL1A1 | Probe | 5-FAM-ATCGACCCTAACCAAG-3 |
| Forward | 5-GACTGGAAGAGCGGAGAGTACTG-3 | |
| Reverse | 5-CCTTGATGGCGTCCAGGTT-3 | |
| CTGF | Probe | 5-FAM-C ACTGCCTGGTCCAGAC-3 |
| Forward | 5-GCTGCCTACCGACTGGAAGA-3 | |
| Reverse | 5-CTTAGAACAGGCGCTCCACTCT-3 |
Kupffer cell experiments
Kupffer cells (KCs) were isolated from rat livers as described previously[30,31]. Further details can be found in Supplementary materials 1. KCs were cultured in M199 medium (Invitrogen), supplemented with 10% (v/v) FCS (foetal calf serum) containing 100 units/mL penicillin and 100 μg/mL streptomycin (Invitrogen), at 37 °C in an oxygenated, humidified cabinet containing 5% CO2.
Twenty thousand KCs per well were plated in black 96 well plates at 37 °C. The cells were incubated with 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) (10 μmol/L, Sigma-Aldrich, St Louis, MO, United States) for 30 min, and washed twice with PBS. The cells were then treated with either AGEs or vehicle BSA at physiological concentrations of 100 μg/mL. Measurement of intracellular ROS generation was performed using 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) as described previously[32]. Briefly, fluorescence measurements were taken with excitation and emission wavelengths of 485 nm and 520 nm respectively on an Optima Microplate Reader (BMG Labtech, Mornington, Australia).
KCs were assayed for their proliferative response to AGEs using a BrdU cell proliferation assay (Roche Applied Science, IN, United States) as per manufacturer’s instructions. KCs were cultured in 96 well plates, with 10000 cells per well and treated with BSA and AGEs at 100 μg/mL.
Statistical analysis
Results are expressed as mean ± SE and analysed by analysis of variance (ANOVA) and Student’s two-tail, unpaired t-test where appropriate with Prism 5 software (GraphPad Software, San Diego, United States). Data that were not normally distributed with equal variance were log transformed prior to analysis. P < 0.05 was considered significant.
RESULTS
Novel 33 week high fat, HFHC dietary model of NAFLD produced both steatohepatitis and significant fibrosis
The HFHC diet was well tolerated and the initial body weights of the experimental groups were similar. All the HFHC groups (HFHC, HFHC baked and HFHC baked + acetic acid) gained more weight, as expected, over the 33 wk compared with normal chow mice, and there was no final difference in weight gain, liver weight or liver to body weight ratio among the HFHC groups (Table 2). The daily weight of chow consumed each day was the same in all the HFHC groups
Table 2.
Animal characteristics and metabolic findings
| Body weight (g) | Epididymal fat (g) | Liver weight (g) | Blood glucose (mmol/L) | OGTT (AUC) | |
| Normal chow | 35.4 ± 1.5a | 1.4 ± 0.2a | 1.3 ± 0.1a | 8.0 ± 0.7 | 654.1 ± 94.4 |
| HFHC | 43.4 ± 1.7 | 2.7 ± 0.2 | 3.7 ± 0.4 | 8.2 ± 0.6 | 528.7 ± 65.2 |
| HFHC baked | 45.5 ± 1.2 | 2.8 ± 0.2 | 3.4 ± 0.5 | 7.8 ± 0.3 | 506.2 ± 70.2 |
| HFHC baked and acetic acid | 44.6 ± 0.9 | 2.6 ± 0.1 | 3.6 ± 0.2 |
Data are mean ± SEM at 33 wk of normal chow or high fat high cholesterol feeding.
P < 0.01 vs HFHC groups. AUC: Area under the curve; OGTT: Oral glucose tolerance test.
The diet produced features similar to human NASH with ballooning of hepatocytes and oxidative stress as assessed by HNE accumulation and steatosis grade > 3 (Figure 1A-C), with significant increases in ALT and lobular inflammation (Figure 1D and E). The diet also induced hepatic fibrosis with the typical “river delta” tendrils of lobular fibrosis seen in fibrotic human NASH (Figure 2A). This was associated with an elevation in profibrotic and proinflammatory cytokine expression in the liver (Figure 2B and C, Supplementary materials 3). Thus using a combination of physiological amounts of fat, fructose and a prolonged duration of feeding, a reliable model of NASH with fibrosis was generated. Importantly this diet did not result in insulin resistance in any of the groups as measured by blood glucose OGTT (Table 2).
Figure 1.
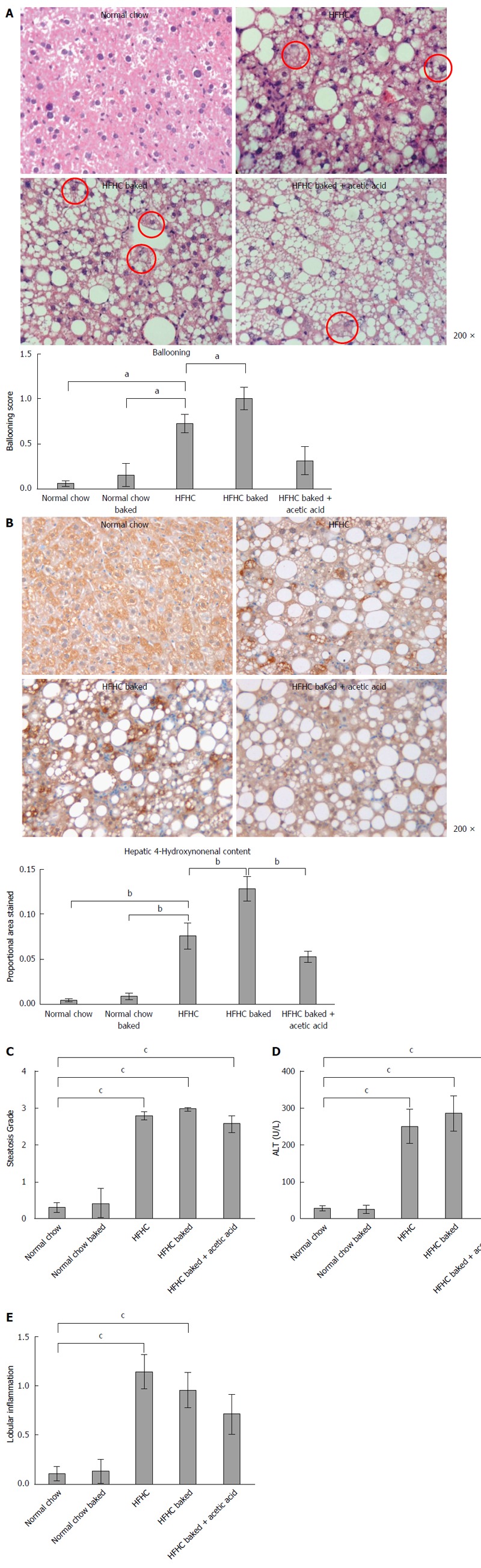
Novel 33 wk high fructose, high cholesterol dietary model induced changes of non-alcoholic steatohepatitis. A: Haematoxylin and eosin stained sections showing ballooning after high fructose, high cholesterol (HFHC) diets (examples of ballooning circled). At right, ballooning scores determined morphometrically; B: Oxidative stress revealed by the immunohistochemical localisation of 4-hydroxynonenal; C: Steatosis grade (grade 0, no fat; grade 1, steatosis occupying less than 33% of the hepatic parenchyma; grade 2, 34%-66% of the hepatic parenchyma; grade 3, more than 66% of the hepatic parenchyma); D: Plasma levels of alanine transaminase (ALT) in the different treatment groups; and E: Lobular inflammation (grade 0:none; grade 1, 1-2 foci/field; grade 2, 3-4 foci/field; grade 3, more than 4 foci/field). Ballooning and oxidative stress were significantly increased by raising the AGE content of the HFHC diet. These changes were attenuated when dietary AGE content was reduced by acetic acid premarination. aP < 0.05, bP < 0.01, cP < 0.001.
Figure 2.

Elevation in profibrotic and proinflammatory cytokine expression in the liver. A: Picrosirius red staining of collagen showing centrilobular sinusoidal fibrosis induced by the 33 wk high fructose, high cholesterol (HFHC) dietary model (c). In animals receiving the HFHC diet, fibrosis was further increased by raising the advanced glycation end products (AGEs) content of the diet (d) and attenuated when dietary AGE content was reduced by acetic acid premarination prior to baking (e). However increasing the AGE content of normal chow did not cause increased fibrosis (compare a, b). Hepatic gene expression of COL1A (B) and αSMA (C) were also significantly increased by raising the AGE content of the diet. Expression of RAGE, a key receptor for AGEs, was elevated in the HFHC model but this was not affected by varying the AGE content of the diet, aP < 0.05, bP < 0.01 (D).
Diet high in AGEs did not cause steatosis or steatohepatitis in the normal liver (normal chow animals)
Both normal chow and normal chow baked groups had similar food intake and there was no difference in weight gain, liver weight or liver to body weight ratio between the two groups. In line with previous studies[19], baking the normal chow diet increased AGE content by over 6 fold (Figure 3) but feeding with baked chow did not change liver biochemistry, produce steatosis, oxidative stress or fibrosis (Figures 1 and 2).
Figure 3.
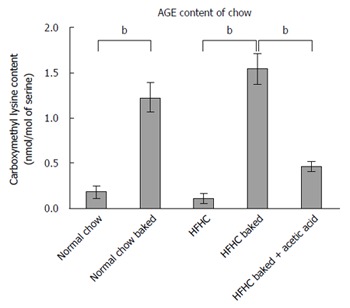
Levels of carboxymethyl lysine in the different diets. Baking the high fructose, high cholesterol (HFHC) diet and normal chow increased CML content of feed by > 6 fold. Levels of CML were markedly reduced by premarinating with vinegar prior to baking, bP < 0.01.
Changes in dietary AGE content modulate oxidative stress and hepatocyte ballooning in HFHC induced steatohepatitis
Baking the HFHC diet markedly increased CML content (Figure 3). Other studies have shown that pre-marination of food in vinegar decreases AGE levels to much lower levels comparable to raw food or boiled food. Importantly, the pre-marination of the HFHC baked diet with acetic acid reduced CML levels in the baked diet to non-baked levels (Figure 3). These AGE levels in the HFHC baked diet are similar to the CML levels found in a moderate to high AGE typical Western diet[33].
Steatosis was significantly increased in all HFHC groups but not further increased by baking to increase its AGE content (Figure 1C). However, the ballooning produced by HFHC feeding was further increased by the high AGE/HFHC diet and this effect was inhibited by the reduction in dietary AGE content achieved by vinegar pre-marination (Figure 1A). Oxidative stress is strongly implicated in liver injury and ballooning degeneration in NASH. Higher levels of HNE were detected in the livers of the HFHC high AGE group compared to those fed the HFHC diet alone. Furthermore, hepatic HNE content was reduced to the levels observed in the HFHC group when AGE content of the diet was reduced by acetic acid (Figure 1B). These findings implicate increased generation of oxidative stress in the pathogenesis of AGE mediated injury in this model.
We previously showed that AGEs increase activation and proliferation of hepatic stellate cells and lead to the production of ROS by these cells[19]. However KCs are responsible for hepatic AGE uptake and play a central role in NASH pathogenesis. We therefore isolated primary KCs to determine the effects of AGEs on these key drivers of inflammation and injury in NAFLD. As shown in Figure 4A, the generation of ROS by isolated Kupffer cells, as measured by 2,3-DCFDA, was significantly increased by the addition of AGEs to the medium compared to vehicle. Moreover, Kupffer cell proliferation as assessed by BrDU incorporation was also significantly increased in Kupffer cells exposed to AGEs compared to BSA vehicle alone (Figure 4B).
Figure 4.

Effects of advanced glycation end products on reactive oxygen species and proliferation in isolated Kupffer cells exposed to physiological amounts of dietary advanced glycation end products compared with bovine serum albumin vehicle. A: Advanced glycation end products (AGEs) increased ROS generation as measured by 2’,7’-dichlorodihydrofluorescein diacetate. B: AGEs increased cell proliferation as assessed by BrDU incorporation, aP ≤ 0.05.
Lobular inflammation, ALT levels and proinflammatory cytokine expression were all significantly increased in the HFHC model (Figure 1D and E, supplement 3) but were not affected by the dietary AGE content.
Modulation of dietary AGE content increased markers of liver fibrosis in the HFHC model
As outlined above, in this novel HFHC model, fibrosis was largely confined to zone 3 and produced a sinusoidal pericellular fine “river delta” pattern. This is in keeping with human NASH where fibrosis tends to follow a centrizonal pattern[34]. The proportional area stained was further increased when the HFHC diet had been baked to increase AGE content (Figure 2A). In addition to more pronounced zone 3 fibrosis, periportal fibrosis was also observed in HFHC high AGE fed animals following the pattern of fibrosis progression in human NAFLD[34]. However, the portal pattern of inflammation was not affected by changing dietary AGE content.
In keeping with the results of picrosirius red staining, COL1A gene expression was significantly increased in both HFHC groups compared with normal chow controls and levels were higher in the mice with a high oral intake of AGEs compared with HFHC alone (Figure 2B). Activation of myofibroblasts, as assessed by hepatic α-SMA gene expression was also significantly increased in the HFHC model and further increased (P < 0.05) with high oral intake of AGEs (Figure 2C).
The role of AGEs in mediating increased liver fibrosis was further supported by the finding that picrosirius staining, COL 1A gene expression and hepatic α-SMA gene expression were reduced to those observed in animals fed the HFHC diet alone, with vinegar pre-marination to reduce AGE content during baking (Figure 2B and C).
Pathological effects of dietary AGEs are RAGE dependent
In diabetes, many of the harmful effects of AGEs are thought to be mediated via AGE/RAGE signalling[35]. RAGE expression, as measured by immunohistochemistry[19], was minimal in healthy liver from animals fed normal chow however RAGE expression was increased in all HFHC groups compared to animals fed standard chow (Figure 2D).
Given the putative role of RAGE in mediating the harmful effects of AGEs and the finding that its expression was increased in our dietary model, we performed a further study in which we examined whether RAGE deletion abrogated liver injury and the harmful effect of AGEs in the HFHC model. As in the initial study, in this experiment, HFHC diet induced liver fibrosis and 4HNE staining were increased by raising dietary AGE content and the area of fibrosis was expanded to include the periportal region (Figure 5A-C).
Figure 5.
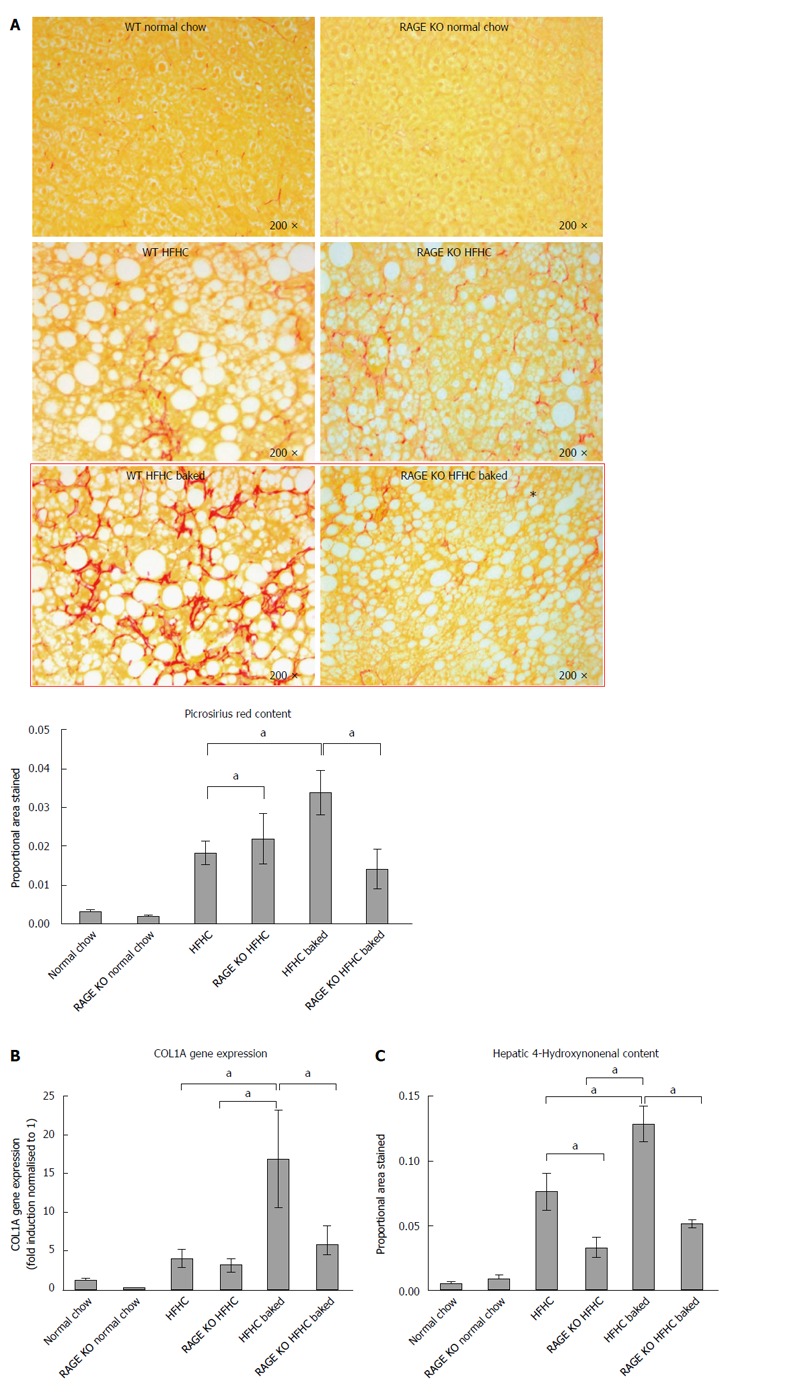
Effects of the high fructose, high cholesterol diet and dietary advanced glycation end products manipulation on fibrosis in the receptor for advanced glycation end products KO animals. A: The absence of receptor for advanced glycation end (RAGE) attenuated the effects of increasing the advanced glycation end products (AGEs) content of the high fructose, high cholesterol (HFHC) diet as evidenced by a reduction in picrosirius red staining to levels observed in animals on the HFHC diet alone; B: There was a commensurate reduction in COL1A gene expression; C: A reduction in hepatic 4-hydroxynonenal content (aP < 0.05).
RAGE KO animals fed the HFHC diet had similar levels of fibrosis to wild type animals (5A). However RAGE KO animals appeared to be protected from the increased liver fibrosis induced by AGEs. Compared to wild type animals fed the high AGE/HFHC diet, there was less picrosirius staining and COL 1A gene expression in the RAGE deleted animals (Figure 5A-B). The increased oxidative stress in animals fed a HFHC baked diet was also abrogated significantly in corresponding RAGE KO animals (Figure 5C). However, HNE levels did not return back to normal chow levels. These findings suggest that AGEs act via RAGE to increase liver fibrosis in this model of experimental NASH.
DISCUSSION
Our experiments show that AGEs may be important modifiable dietary cofactors which contribute to the development of liver fibrosis in NAFLD. The current findings are in keeping with previous studies which showed that AGEs administered intraperitoneally or in the diet exacerbate liver injury and fibrosis[12,19]. However these previous experiments were conducted in short term models of liver disease of limited relevance to human NAFLD[36]. For the present studies, therefore, a novel model of NAFLD was developed using physiological amounts of fat, cholesterol and fructose that occur in human diets and it reliably produced slowly progressive steatosis, ballooning, oxidative stress and predominantly pericentral sinusoidal fibrosis with a tempo of disease consistent with the long progressive natural history of human NASH. In this study, all of these key features of high fat diet induced NASH were exacerbated by increasing dietary AGE content
The two-hit hypothesis of NASH pathogenesis suggests that a second injury or cofactor is required for progression from simple benign steatosis to harmful steatohepatitis or fibrosis, cirrhosis and hepatocellular carcinoma[37]. There has been considerable interest in factors which could serve as this “second hit”. In line with our previous study in MCD animals[19], we found that a high AGE containing diet has no effect on liver biochemistry or histology in animals without hepatic steatosis. However, we showed they act as a co-factor to increase injury in diseased livers.
AGEs have been shown to exert their effects through several receptors, the best studied of which is RAGE. Activation of RAGE stimulates multiple signal transduction pathways[38], ultimately leading to the generation of ROS[39]. This culminates in the activation of NF-κB, a redox sensitive transcription factor, which in turn translocates into the nucleus[40]. The promoter region of the RAGE gene contains an NF-κB binding site and therefore, one of the important consequences of NF-κB activation and translocation is upregulation of RAGE itself. This sets up a positive feedback loop and ensures maintenance of RAGE signalling. Furthermore, the generation of ROS triggered by RAGE activation causes increased AGE formation and contributes to a vicious cycle of AGE formation, generation of oxidative stress and further RAGE activation[7]. Our study showed that RAGE expression was minimal in healthy livers but upregulated in a non-diabetic model of fatty liver disease. These differences in RAGE expression may explain why the high AGE diet had no harmful effects in healthy controls but exacerbated liver injury in animals with NAFLD. However, since the dietary AGE content of normal mouse chow is so high it is also feasible that the dietary AGE content of normal mouse chow is so high that mice become preconditioned against the effects of further increases. In keeping with these findings, although RAGE KO animals developed NASH in response to the HFHC diet they were protected from the additional harmful effects of the high AGE diet.
Oxidative stress and the accumulation of superoxide is a key mediator of ballooning, cellular dysfunction and fibrosis in non-alcoholic steatohepatitis[7,41]. HNE, an end-product of peroxidation of membrane N-6-polyunsaturated fatty acids, is a particularly good marker of lipid oxidation during liver injury and is related to the intensity of necroinflammation[42]. Given the long- term nature of our model, assessment of oxidative stress in the liver was therefore performed by measuring HNE adducts which reflect accumulation of oxidative stress over time. This showed that HNE content was significantly elevated in all the HFHC groups (Figure 1C). Consistent with the known effects of AGEs, we found that a diet high in AGEs significantly increased oxidative stress in HFHC induced NASH. This was associated with increased ballooning, stellate cell activation as assessed by aSMA expression and increased fibrosis. These findings are consistent with our previous work which showed that in activated primary murine hepatic stellate cells which express RAGE, ROS production, cell activation and proliferation were markedly increased in the presence of AGEs. These effects were inhibited by RAGE blockade or NADPH oxidase inhibition. However, Kupffer cells (KCS) express RAGE[43] and uptake of AGEs in the liver occurs primarily via these cells[11]. It is known that KCs play a key role in promoting hepatic steatosis, steatohepatitis and ballooning in NASH[44]; and generation of ROS in KCs rather than by HSCs has been associated with NAFLD disease progression[45]. Given the important role of KCs in NAFLD progression and their key role in scavenging AGEs[46], we explored the effect of AGEs on primary KCs and showed that AGEs increase cell proliferation and the generation of ROS by these cells.
As expected, in our HFHC model there was marked upregulation of a number of key proinflammatory and fibrogenic cytokines. It is unclear why, despite showing that AGEs increase oxidative stress, hepatocyte ballooning and fibrosis, we did not find they increased inflammatory infiltration of the liver or in inflammatory cytokines expression. In keeping with this finding, in a very similar dietary model of NASH, Lo et al[47] found that although diabetes worsened steatosis induced liver fibrosis this was not associated with measurable increases in liver inflammatory infiltration or proinflammatory cytokine levels. These finding suggest that profibrotic effects of AGEs and diabetes may not be mediated primarily through increasing hepatic inflammation.
The formation of AGEs in foods involves the condensation of an amino group with the carbonyl group of a reducing carbohydrate (glucose, fructose, maltose, lactose or ribulose) to form intermediate Amadori products. Oxidation of Amadori products leads to a more stable compound, CML, used in studies as an indicator of the AGE content of foods[4]. A common method of high dietary AGE formation is via heating at high temperatures in non-aqueous environments (e.g., baking or broiling)[4]. High levels of AGEs are thus found in many common foods such as baked breads and biscuits/cookies, toasted breakfast cereals, grilled steak, brewed beer, and roasted coffee beans. For example, toasting white bread increases its AGE content by more than 3 times[48]. Interestingly, acidifying such foods prior to cooking by marinating them in acetic acid (vinegar) or lemon juice, reduces their CML content substantially without compromising palatability[49]. In this study, we have found reducing AGEs production by vinegar marination prior to baking can abrogate the harmful effects of a high AGE diet in an animal model of NAFLD. This may be one mechanism by which the Mediterranean diet which includes the use of vinegar and lemon juice marination is beneficial for metabolic health and in NAFLD[50].
It has been shown that patients with NASH have higher levels of circulating AGEs than those with simple steatosis[46]. However whether this reflects an increased dietary exposure to AGEs or greater endogenous AGE production in patients who have both fatty liver and glucose intolerance is unclear. Studies examining the relationship between dietary exposure to AGEs and the histological severity of liver injury in non-diabetic patients will help clarify this issue.
In conclusion, we show that high dietary AGE exposure worsens liver pathology in experimental NASH, implicating AGE/RAGE signalling in fatty liver disease progression. Decreasing dietary AGEs by altering food selection and cooking methods may thus offset the possible harmful effects afforded by a high AGE diet. If confirmed in human studies, our findings have broad implications for the way we process foods and the dietary advice given to patients with NAFLD. They also suggest a possible role for therapies targeting the AGE/RAGE pathway in the treatment and prevention of NASH.
ACKNOWLEDGMENTS
Many thanks to A/Prof Trishe Leong from the Department of Anatomical Pathology, Austin Health, for her expert histopathological assistance.
COMMENTS
Background
There is considerable interest in identifying the factors which drive progression of uncomplicated non-alcoholic fatty liver disease (NAFLD) to non-alcoholic steatohepatitis (NASH), cirrhosis and liver cancer. Advanced glycation end products (AGEs) are a complex group of compounds formed in foods that are highly processed or dry heated at high temperatures which have been implicated in the pathogenesis of diabetic complications. Our previous work in a methionine choline deficient model of NASH has shown that AGEs may be an important environmental risk factor that serves as a second hit driving simple steatosis to steatohepatitis and cirrhosis. The aims of the current study were to determine whether AGEs drive NAFLD progression in a long term high fat, high cholesterol, high fructose dietary model of NASH that mimics many of the features of human NASH. We also aimed to examine the role of the receptor for AGEs (RAGE) and Kupffer cells in this process.
Research frontiers
There is major interest in identifying so-called second hits which drive liver inflammation and fibrosis in NAFLD. AGEs have been implicated in progression of a number of disease processes, including diabetic nephropathy, retinopathy, vasculopathy and neuropathy. This study investigates whether AGEs could represent an important potential dietary and pharmacological target in the management of the burgeoning problem of fatty liver disease.
Innovations and breakthroughs
This is the first study which examines the impact of altering dietary AGE content in a long term physiological model of NASH that utilises proportions of fats and fructose found typically in Western diets. We have found that changes in AGE content influence liver inflammation and fibrosis. This is also the first study to show that in RAGE knock out animals, these deleterious effects of AGEs are abrogated, suggesting a RAGE dependent pathway. Studies by Leung et al in isolated primary Kupffer cells also show AGEs increase cell proliferation and oxidative stress, suggesting a likely mechanism by which these compounds contribute to liver injury.
Applications
Food sourcing and preparation methods that reduce AGE content could influence the progression of NAFLD. This study also suggests that pharmacological therapies which target the AGE/RAGE pathway may have a role in treatment of NAFLD.
Terminology
AGEs, also known as glycotoxins, are a complex group of compounds that are formed when active sugar moieties become bound to proteins causing browning and other irreversible modifications. Foods that are highly processed or dry heated at high temperatures, such as broiled foods, have particularly high AGE content. However, they are also formed endogenously and this occurs at an increased rate in diabetes, most likely because of hyperglycaemia. There is now considerable evidence that accumulation of AGEs are implicated in the pathogenesis of diabetic renal, neurological, retinal and vascular complications. AGEs act on several receptors but the receptor for advanced glycation end products (RAGE), a member of the immunoglobulin superfamily of cell-surface molecules, is the best characterised of these receptors. Engagement with RAGE, which in turn increases inflammation and oxidative stress, is thought to be the main way in which AGEs impart these pathogenic effects. This receptor is expressed in a number of cell types, including endothelial cells, vascular smooth muscle cells, peripheral blood mononuclear cells, macrophages (including Kupffer cells) and HSCs.
Peer-review
A well designed and organized study.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Australia
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Austin Health Animal Ethics Committee (Project number A2011/04342).
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Austin Health Animal Ethics Committee (Project number A2011/04342).
Conflict-of-interest statement: To the best of our knowledge, no conflict of interest exists.
Data sharing statement: Data set available from the corresponding author at chris.leung@y7mail.com.
Peer-review started: May 6, 2016
First decision: June 20, 2016
Article in press: August 10, 2016
P- Reviewer: Busuttil RW, Mendez-Sanchez N, Tanoglu A S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 2.Arteel GE. Beyond reasonable doubt: who is the culprit in lipotoxicity in NAFLD/NASH? Hepatology. 2012;55:2030–2032. doi: 10.1002/hep.25721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henle T. Protein-bound advanced glycation endproducts (AGEs) as bioactive amino acid derivatives in foods. Amino Acids. 2005;29:313–322. doi: 10.1007/s00726-005-0200-2. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg T, Cai W, Peppa M, Dardaine V, Baliga BS, Uribarri J, Vlassara H. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104:1287–1291. doi: 10.1016/j.jada.2004.05.214. [DOI] [PubMed] [Google Scholar]
- 5.Aso Y, Inukai T, Tayama K, Takemura Y. Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol. 2000;37:87–92. doi: 10.1007/s005920070025. [DOI] [PubMed] [Google Scholar]
- 6.Stitt AW, Bucala R, Vlassara H. Atherogenesis and advanced glycation: promotion, progression, and prevention. Ann N Y Acad Sci. 1997;811:115–127; discussion 127-129. doi: 10.1111/j.1749-6632.1997.tb51994.x. [DOI] [PubMed] [Google Scholar]
- 7.Miyata T, Hori O, Zhang J, Yan SD, Ferran L, Iida Y, Schmidt AM. The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway. Implications for the pathogenesis of dialysis-related amyloidosis. J Clin Invest. 1996;98:1088–1094. doi: 10.1172/JCI118889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 9.Patel R, Baker SS, Liu W, Desai S, Alkhouri R, Kozielski R, Mastrandrea L, Sarfraz A, Cai W, Vlassara H, et al. Effect of dietary advanced glycation end products on mouse liver. PLoS One. 2012;7:e35143. doi: 10.1371/journal.pone.0035143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekong U, Zeng S, Dun H, Feirt N, Guo J, Ippagunta N, Guarrera JV, Lu Y, Weinberg A, Qu W, et al. Blockade of the receptor for advanced glycation end products attenuates acetaminophen-induced hepatotoxicity in mice. J Gastroenterol Hepatol. 2006;21:682–688. doi: 10.1111/j.1440-1746.2006.04225.x. [DOI] [PubMed] [Google Scholar]
- 11.Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, Dun H, Lu Y, Qu W, Schmidt AM, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 2004;39:422–432. doi: 10.1002/hep.20045. [DOI] [PubMed] [Google Scholar]
- 12.Goodwin M, Herath C, Jia Z, Leung C, Coughlan MT, Forbes J, Angus P. Advanced glycation end products augment experimental hepatic fibrosis. J Gastroenterol Hepatol. 2013;28:369–376. doi: 10.1111/jgh.12042. [DOI] [PubMed] [Google Scholar]
- 13.Lohwasser C, Neureiter D, Popov Y, Bauer M, Schuppan D. Role of the receptor for advanced glycation end products in hepatic fibrosis. World J Gastroenterol. 2009;15:5789–5798. doi: 10.3748/wjg.15.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyogo H, Yamagishi S, Iwamoto K, Arihiro K, Takeuchi M, Sato T, Ochi H, Nonaka M, Nabeshima Y, Inoue M, et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:1112–1119. doi: 10.1111/j.1440-1746.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 15.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–592. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Santos JC, Valentim IB, de Araújo OR, Ataide Tda R, Goulart MO. Development of nonalcoholic hepatopathy: contributions of oxidative stress and advanced glycation end products. Int J Mol Sci. 2013;14:19846–19866. doi: 10.3390/ijms141019846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Ratziu V, Munteanu M, Charlotte F, Bonyhay L, Poynard T. Fibrogenic impact of high serum glucose in chronic hepatitis C. J Hepatol. 2003;39:1049–1055. doi: 10.1016/s0168-8278(03)00456-2. [DOI] [PubMed] [Google Scholar]
- 19.Leung C, Herath CB, Jia Z, Goodwin M, Mak KY, Watt MJ, Forbes JM, Angus PW. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J Hepatol. 2014;60:832–838. doi: 10.1016/j.jhep.2013.11.033. [DOI] [PubMed] [Google Scholar]
- 20.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 21.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology. 2010;52:934–944. doi: 10.1002/hep.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, Teoh NC, Farrell GC. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol. 2013;59:144–152. doi: 10.1016/j.jhep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig Dis Sci. 2010;55:931–940. doi: 10.1007/s10620-009-0815-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takagi Y, Kashiwagi A, Tanaka Y, Asahina T, Kikkawa R, Shigeta Y. Significance of fructose-induced protein oxidation and formation of advanced glycation end product. J Diabetes Complications. 1995;9:87–91. doi: 10.1016/1056-8727(94)00022-g. [DOI] [PubMed] [Google Scholar]
- 25.David E. A Book of Mediterranean Food (New York Review Books Classics) New York City: New York Review Books; 2002. [Google Scholar]
- 26.Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 27.Nobécourt E, Tabet F, Lambert G, Puranik R, Bao S, Yan L, Davies MJ, Brown BE, Jenkins AJ, Dusting GJ, et al. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2010;30:766–772. doi: 10.1161/ATVBAHA.109.201715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 29.Herath CB, Warner FJ, Lubel JS, Dean RG, Jia Z, Lew RA, Smith AI, Burrell LM, Angus PW. Upregulation of hepatic angiotensin-converting enzyme 2 (ACE2) and angiotensin-(1-7) levels in experimental biliary fibrosis. J Hepatol. 2007;47:387–395. doi: 10.1016/j.jhep.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patella S, Phillips DJ, Tchongue J, de Kretser DM, Sievert W. Follistatin attenuates early liver fibrosis: effects on hepatic stellate cell activation and hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol. 2006;290:G137–G144. doi: 10.1152/ajpgi.00080.2005. [DOI] [PubMed] [Google Scholar]
- 31.Kitani H, Takenouchi T, Sato M, Yoshioka M, Yamanaka N. A simple and efficient method to isolate macrophages from mixed primary cultures of adult liver cells. J Vis Exp. 2011;(51):pii 2757. doi: 10.3791/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guimarães EL, Empsen C, Geerts A, van Grunsven LA. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52:389–397. doi: 10.1016/j.jhep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Stirban A, Tschoepe D. Comment on “Advanced glycation endproducts in food and their effects on health” by Poulsen et al. (2013) Food and Chemical Toxicology 60, 10-37. Food Chem Toxicol. 2014;64:411. doi: 10.1016/j.fct.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53:810–820. doi: 10.1002/hep.24127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goh SY, Cooper ME. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–1152. doi: 10.1210/jc.2007-1817. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P, Dooley S. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304:G449–G468. doi: 10.1152/ajpgi.00199.2012. [DOI] [PubMed] [Google Scholar]
- 37.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 38.Huang JS, Guh JY, Chen HC, Hung WC, Lai YH, Chuang LY. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J Cell Biochem. 2001;81:102–113. doi: 10.1002/1097-4644(20010401)81:1<102::aid-jcb1027>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 39.Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, Tazuma S, Chayama K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J Gastroenterol. 2008;43:298–304. doi: 10.1007/s00535-007-2152-7. [DOI] [PubMed] [Google Scholar]
- 40.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, Torok NJ. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 41.Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 42.Poli G, Biasi F, Leonarduzzi G. 4-Hydroxynonenal-protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol Aspects Med. 2008;29:67–71. doi: 10.1016/j.mam.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 43.Smedsrød B, Melkko J, Araki N, Sano H, Horiuchi S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem J. 1997;322(Pt 2):567–573. doi: 10.1042/bj3220567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B, Kersten S, Müller M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511–522. doi: 10.1002/hep.23337. [DOI] [PubMed] [Google Scholar]
- 45.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, Penfold SA, Bach LA, Cooper ME, Forbes JM. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes. 2008;57:460–469. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 46.Hyogo H, Yamagishi S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr Pharm Des. 2008;14:969–972. doi: 10.2174/138161208784139701. [DOI] [PubMed] [Google Scholar]
- 47.Lo L, McLennan SV, Williams PF, Bonner J, Chowdhury S, McCaughan GW, Gorrell MD, Yue DK, Twigg SM. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non-alcoholic steatohepatitis. J Hepatol. 2011;55:435–444. doi: 10.1016/j.jhep.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 48.Vlassara H, Striker G. Glycotoxins in the diet promote diabetes and diabetic complications. Curr Diab Rep. 2007;7:235–241. doi: 10.1007/s11892-007-0037-z. [DOI] [PubMed] [Google Scholar]
- 49.Uribarri J, Woodruff S, Goodman S, Cai W, Chen X, Pyzik R, Yong A, Striker GE, Vlassara H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110:911–16.e12. doi: 10.1016/j.jada.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoffman R, Gerber M. Evaluating and adapting the Mediterranean diet for non-Mediterranean populations: a critical appraisal. Nutr Rev. 2013;71:573–584. doi: 10.1111/nure.12040. [DOI] [PubMed] [Google Scholar]