

Abstract
Myelodysplastic syndromes (MDS) are heterogeneous myeloid disorders with prevalent mutations in several splicing factors, but the splicing programs linked to specific mutations or MDS in general remain to be systematically defined. We applied RASL-seq, a sensitive and cost-effective platform, to interrogate 5502 annotated splicing events in 169 samples from MDS patients or healthy individuals. We found that splicing signatures associated with normal hematopoietic lineages are largely related to cell signaling and differentiation programs, whereas MDS-linked signatures are primarily involved in cell cycle control and DNA damage responses. Despite the shared roles of affected splicing factors in the 3′ splice site definition, mutations in U2AF1, SRSF2, and SF3B1 affect divergent splicing programs, and interestingly, the affected genes fall into converging cancer-related pathways. A risk score derived from 11 splicing events appears to be independently associated with an MDS prognosis and AML transformation, suggesting potential clinical relevance of altered splicing patterns in MDS.
Keywords: pre-mRNA splicing, myelodysplastic syndromes (MDS), RASL-seq, splicing factor mutations, diagnostic and prognostic splicing signatures
INTRODUCTION
Myelodysplastic syndromes (MDS) are a heterogeneous group of chronic hematological malignancies defined by clonal hematopoiesis, impaired differentiation, peripheral blood cytopenias, and a risk of progression to acute myeloid leukemia (AML) (Swerdlow et al. 2008). In clinical practice, the disease-related criteria used to evaluate patients with MDS include those described in the Revised International Prognostic Scoring System (IPSS-R) (Greenberg et al. 2012), the WHO classification-based Prognostic Scoring System (WPSS) (Malcovati et al. 2007), and M.D. Anderson Cancer Center (MDACC) risk stratification (Garcia-Manero et al. 2008). They are all largely reliant on morphological features that require visual examination of bone marrow aspirate or biopsy, which is known to have significant inter-observer variability even among expert hematopathologists (Font et al. 2013). Molecularly based diagnostic and prognostic criteria might provide better biomarkers than dysplasia since they likely reflect the underlying biology of the disease. To that end, several groups have examined the utility of somatic mutations as relevant clinical criteria, which appear to work well for myeloproliferative neoplasms with highly recurrent mutations and a high degree of specificity in appropriate clinical contexts. This is, however, not the case for myelodysplastic syndromes, which are nearly as heterogeneous at the genetic level as they are clinically (Bejar et al. 2011; Papaemmanuil et al. 2013; Haferlach et al. 2014). Alternative biomarkers that capture disease-related biological features may therefore improve our ability to diagnose MDS and predict outcomes in these disorders.
Gene expression profiling characterizes molecular processes downstream from somatic mutations. As such, it may integrate the effects of diverse somatic and epigenetic lesions into common phenotypic patterns. Gene expression profiling has been successfully used to identify potential biomarkers for diagnosis and prognosis of various cancers, including acute myeloid leukemia (AML) (Payton et al. 2009), MDS (Pellagatti et al. 2013), and breast cancer (van't Veer et al. 2002). However, results from profiling the same diseases, like breast cancer, by different groups are sometimes inconsistent with one another (Koscielny 2010). Furthermore, gene expression profiling alone is insufficient to capture additional complexities of regulated gene expression under disease conditions, such as alternative isoform utilization (Feero et al. 2010).
Alternative splicing (AS) of pre-mRNA is known to play key roles in generating genomic and proteomic diversity and complexity, as >90% of multi-exon pre-mRNAs undergo AS (Pan et al. 2008; Wang et al. 2008), with many alternatively spliced gene products exhibiting distinct or even opposing biological functions (Tress et al. 2007). Recent analysis of a large splicing array data set suggests that splicing signatures may be more effective than gene expression profiles for the characterization of cancers (Zhang et al. 2013). Several unique mRNA isoforms have been linked to specific cancer types, including breast (Eswaran et al. 2013), ovarian (Venables et al. 2009), lung (Misquitta-Ali et al. 2011), pancreatic (Omenn et al. 2010), head and neck (Li et al. 2014), digestive tract (Miura et al. 2011), renal (Malouf et al. 2014), gastric malignancies (Liu et al. 2014), neuroblastoma (Chen et al. 2015), and AML (Adamia et al. 2014). These alternatively spliced transcripts, reflecting an independent layer and critical component of regulated gene expression, may thus serve as a new class of biomarkers.
Biological differences in gene expression and alternative splicing are particularly relevant to MDS, given the high frequency of somatic splicing factor mutations in these disorders. About two-thirds of patients with MDS carry a mutation in a splicing regulator, such as U2AF1, SRSF2, SF3B1, and ZRSR2 (Papaemmanuil et al. 2011; Graubert et al. 2012; Makishima et al. 2012; Thol et al. 2012). Nearly all of the splicing factors mutated in MDS characterized to date are associated with the U2 small nuclear ribonucleoprotein particle (snRNP) of the spliceosome, which defines functional 3′ splice sites in mammalian genomes (Sharp and Burge 1997). The observation that splicing factor mutations in MDS are largely mutually exclusive suggests that these mutant splicing factors may induce a shared set of mRNA isoforms that may contribute to the development and progression of MDS. Several studies utilizing deep sequencing examined the splicing patterns associated with these mutations (Przychodzen et al. 2013; Dolatshad et al. 2015). However, it has been technically challenging to obtain quantitative data from the large number of patient samples to deduce potential disease mechanisms imposed by specific genetic lesions.
Here we address this challenge by selectively interrogating a large cohort (n = 5502) of annotated alternative splicing events in hematopoietic cells. We profiled 115 MDS and 54 healthy blood and bone marrow samples using RNA-mediated oligonucleotide annealing, selection, and ligation coupled with next-generation sequencing (RASL-seq) (Li et al. 2012). Compared to transcriptome analysis by standard RNA-seq, the RASL-seq platform is designed to measure specific and quantitative information on potential isoform switches in biological samples with high sensitivity and cost-effectiveness. While this technology does not permit de novo discovery of novel RNA processing events, it generates robust data for global comparison and characterization of splicing programs in different cell types or in response to specific perturbations (Zhou et al. 2012b; Sun et al. 2015). With this approach, we examined unique splicing signatures associated with normal hematopoietic cell lineages as well as with MDS; established splicing patterns defined by different splicing factors, and explored how specific sets of splicing events might serve as biomarkers for MDS diagnosis and prognosis.
RESULTS
Lineage commitment and disease status defined by alternative splicing
We previously determined the mutation status of SRSF2, U2AF1, SF3B1, and ZRSR1 in a large cohort of MDS patients (Thol et al. 2012). Since this initial study, we have collected and characterized additional samples, and extracted total RNA from a total of 115 samples from 112 MDS patients and 54 samples from 39 healthy individuals (Fig. 1A). The MDS group contains samples from bone marrow (BM, n = 93) or peripheral blood (PB, n = 22), whereas the healthy group comprises samples from BM, PB, and sorted cells, including CD34+ hematopoietic progenitor cells from bone marrow, common myeloid progenitor cells (CMP), granulocytes, monocytes, B lymphocytes, and T lymphocytes (Fig. 1A; Supplemental Table S1). The median age of patients was 67 yr (range: 26–92); 71 patients (63%) were males; 59 (53%) had IPSS low or intermediate-1 risk scores; 74 (66%) were transfusion dependent; 31 (28%) progressed to AML; and 18 (14%) received allogeneic stem cell transplantation (Supplemental Table S2).
FIGURE 1.
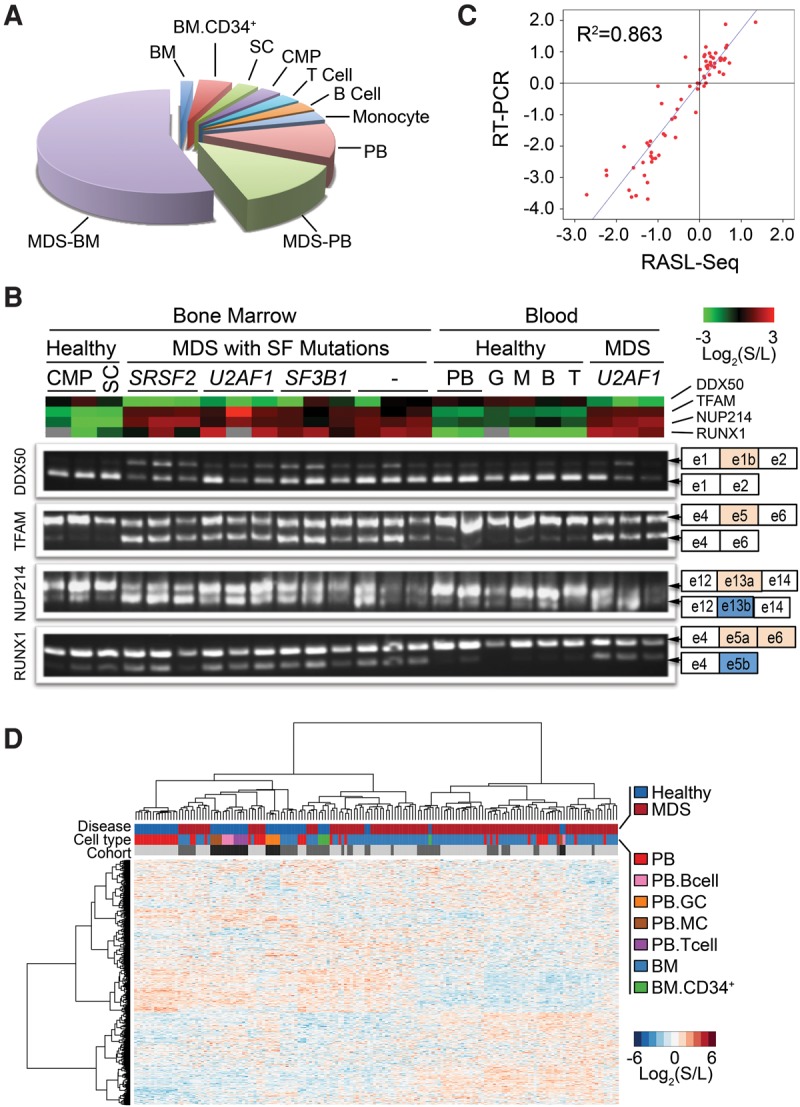
Characterization of lineage commitment and disease status by alternative splicing. RASL-seq was applied to 115 MDS samples and 54 samples from healthy volunteers to assess global pre-mRNA splicing. (A) Pie chart showing type and origin of investigated samples. Bone marrow (BM); peripheral blood (PB); common myeloid progenitor cells (CMP); stem cell (SC). (B) RT-PCR validation of four events across multiple samples with different sample origins, different splicing factor mutations from MDS patients, and healthy volunteers. (Top) Heatmap view of RASL-seq data. (Bottom) Corresponding RT-PCR products for validation. (C) Scatter plot of RASL-seq versus RT-PCR validated data. (S) Short isoform; (L) long isoform. (D) Global view of RASL-seq data [normalized Log2 (short isoform/long isoform)] using unsupervised hierarchical clustering. Monocyte (MC); granulocyte (GC); bone marrow (BM). Light gray, gray, and dark gray colored bars represent three separate sample cohorts.
To characterize the splicing profile in our cohort, we chose RASL-seq for a rapid, quantitative, and cost-effective survey of 5502 curated alternative splicing events in the human genome, including those conserved between mice and humans (Sugnet et al. 2004; Yeo et al. 2005) and those we manually annotated by searching the literature. While the current RASL oligonucleotide pool was designed for splicing profiling in diverse biological systems, numerous annotated alternative splicing events are related to cancer, and like those detected by RNA-seq, the predominant form of alternative splicing events is cassette exon (Supplemental Table S3). We obtained a total of 480 million mappable sequencing reads and identified 1956 alternative splicing events with sufficient counts of both isoforms. The mean and median counts of the sum of short and long isoforms per event and per sample were consistently distributed (Supplemental Fig. S1). To validate the performance of RASL-seq on human samples from different tissue origins, we designed PCR primers (Supplemental Table S4) for four specific splicing events from genes known to associate with hematological malignancy, including two myeloid cancer-related genes, RUNX1 (alternative terminal exon) and NUP214 (alternative 3′ exon), the mitochondrial transcription factor TFAM (cassette exon), and the multifunction factor DDX50 (cassette exon). RASL-seq results were aligned with the corresponding RT-PCR data (Fig. 1B), showing a high overall concordance between ratios derived from RASL-seq and RT-PCR (R2= 0.86, Fig. 1C). The high-quality RASL-seq data permitted us to compare mRNA isoform signatures associated with different hematopoietic cell lineages, and with those from different MDS patients characterized by distinct mutations and clinical features.
Initial analysis of the RASL-seq generated data set by using unsupervised hierarchical clustering largely segregated healthy samples from those with MDS (see the color key in the first row on top of Fig. 1D). More than half of the MDS samples (those in the right side, Fig. 1D) showed an overall pattern largely distinct from that of the healthy samples (left cluster), while the rest of MDS samples (middle clusters) resembled healthy controls, which likely reflect early disease states of those patients. Normal peripheral blood samples and normal hematopoietic progenitor cells (BM-CD34+) formed distinct clusters (clusters indicated in the second row of colored key, Fig. 1D), although some of these normal samples were also mixed with MDS samples. As we calculated the log ratio of short isoform versus long isoform from each alternative splicing event and then normalized the ratio based on the averaged log ratio across all samples, this treatment eliminated potential batch-specific clustering (the three cohorts of samples separately analyzed were indicated by different gray bars in the third row, Fig. 1D). Together, these data suggest that splicing signatures may be developed to segregate healthy versus disease whole blood samples as well as different lineages of normal hematopoietic cells.
Identification of the hematopoietic lineage-specific splicing signature
The challenge in studying blood disorders is the presence of heterogeneous cell populations and depletion and/or expansion of various cell types in those populations (Walter et al. 2012; Woll et al. 2014). We reasoned that we might address this problem by first identifying cell lineage-associated splicing events in healthy controls, and then focusing on those relatively cell type-independent splicing events for characterizing disease samples. We therefore first characterized different normal hematopoietic cell lineages by performing principal component analysis (PCA) and a supervised multiple logistic regression analysis with five defined cell lineages (lin-CD34+ progenitor cells, granulocytes, monocytes, B- and T-lymphocytes, and unsorted mononuclear cells from PB and BM) (see Supplemental Table S1). After 10-fold cross validation, we identified a collection of 200 events (here termed Hemo-SP for Hematopoiesis-specific Splicing Program), which efficiently differentiated five sorted lineage-specific cells as well as PB and BM mononuclear cells (Fig. 2A; Supplemental Table S5A).
FIGURE 2.
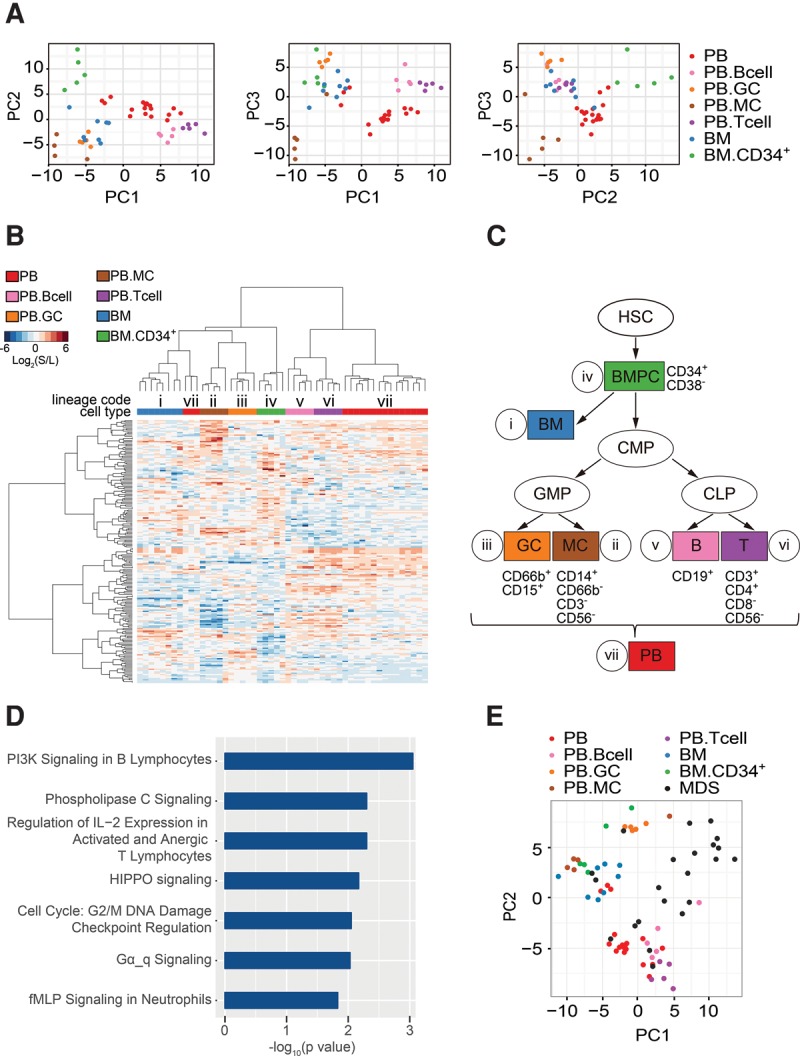
Characterization of hematopoietic lineage-defining splicing programs. (A) Principal component analysis (PCA) represents the results of regression analysis that identified a splicing program that differentiates between normal hematopoietic cell lineages (Hemo-SP panel). (B) Unsupervised hierarchical clustering of normal hematopoietic cell samples using the 200-event cell lineage-specific panel (Hemo-SP). A code was assigned to each lineage. Unsupervised hierarchical clustering of MDS blood samples with fixed 200-event panel as used. (C) A demonstration tree of hematopoietic lineages. Hematopoietic stem cell (HSC); bone marrow progenitor cells (BMPC); bone marrow (BM); common myeloid progenitor cell (CMP); granulocyte (GC); monocyte (MC); B-lymphocyte (B); T-lymphocyte (T); peripheral blood (PB). As CMP and SC were very well correlated across all events, and each group had limited sample numbers, we combined these two groups together as CMP. (D) Canonical pathways identified by Ingenuity IPA analysis of the hematopoietic lineage-specific panel (Hemo-SP). (E) Segregation of MDS samples from different cell types from healthy controls based on Hemo-SP by PCA.
Using this Hemo-SP program, we displayed the data among the seven hematological cell types with unsupervised hierarchical clustering (Fig. 2B). Even though some cell types share various commonalities in certain events, the overall patterns are quite distinct among individual cell types, demonstrating the power of our approach in extracting unique, cell type-specific splicing signatures. Notably, the lymphoid lineages largely resemble peripheral blood (v, vi, and vii, Fig. 2B,C), which is clearly distinct from cells in the myeloid lineages and bone marrow (i to iv and vii, Fig. 2B,C). Ingenuity IPA analysis revealed many enriched alternatively spliced genes involved in various canonical pathways, including those in PI3K signaling in B lymphocytes and the regulation of IL-2 expression in naïve and activated T lymphocytes, in the HIPPO pathway known to play a critical role in hematopoietic stem cells (Jansson and Larsson 2012), and in Gα signaling, which is fundamental to hematopoietic cell differentiation and function (Fig. 2D; Supplemental Table S5B; Wilkie et al. 1991). These observations suggest that regulated splicing of these genes contributes to hematopoietic lineage commitment and proliferation. Importantly, PCA analysis based on Hemo-SP showed that MDS PB samples are largely segregated from healthy PB and BM, indicating unique cell populations in MDS patients (Fig. 2E).
Characterization of MDS-linked splicing programs
We next wished to identify MDS-specific, but relatively hematopoietic cell lineage-independent splicing signatures. For this purpose, we removed the splicing events in the Hemo-SP signature and then applied the same multiple logistic regression model to analyze the rest of the RASL-seq data by using 115 MDS samples in comparison with 26 healthy individuals. Training of the regression model identified a panel of 204 splicing events capable of robustly differentiating MDS from healthy samples (here termed MDS-Dx for MDS diagnostic panel, Fig. 3A; Supplemental Table S6A). This is also evident from unsupervised hierarchical clustering of MDS and healthy samples (indicated by colored bars on top of Fig. 3B). Notably, even though BM and PB (blue or red in the second bar) were clustered from one another in the healthy sample group, MDS samples clustered independently of cell sources. This suggests that splicing is dysregulated in MDS and that the MDS-specific splicing pattern is preserved in cells from either PB or BM. A close examination of the cluster tree suggests that ∼40% MDS samples, regardless of their PB or BM origins, were still segregated with healthy BM and CD34+ cells (Fig. 3B), suggesting that this group may be relatively early in disease development compared to those that were largely segregated from healthy samples. Further analysis provides support to this notion, indicating that the MDS samples closely clustered to healthy samples were more linked to the low-risk prognostic signature (Supplemental Fig. S9, see below).
FIGURE 3.
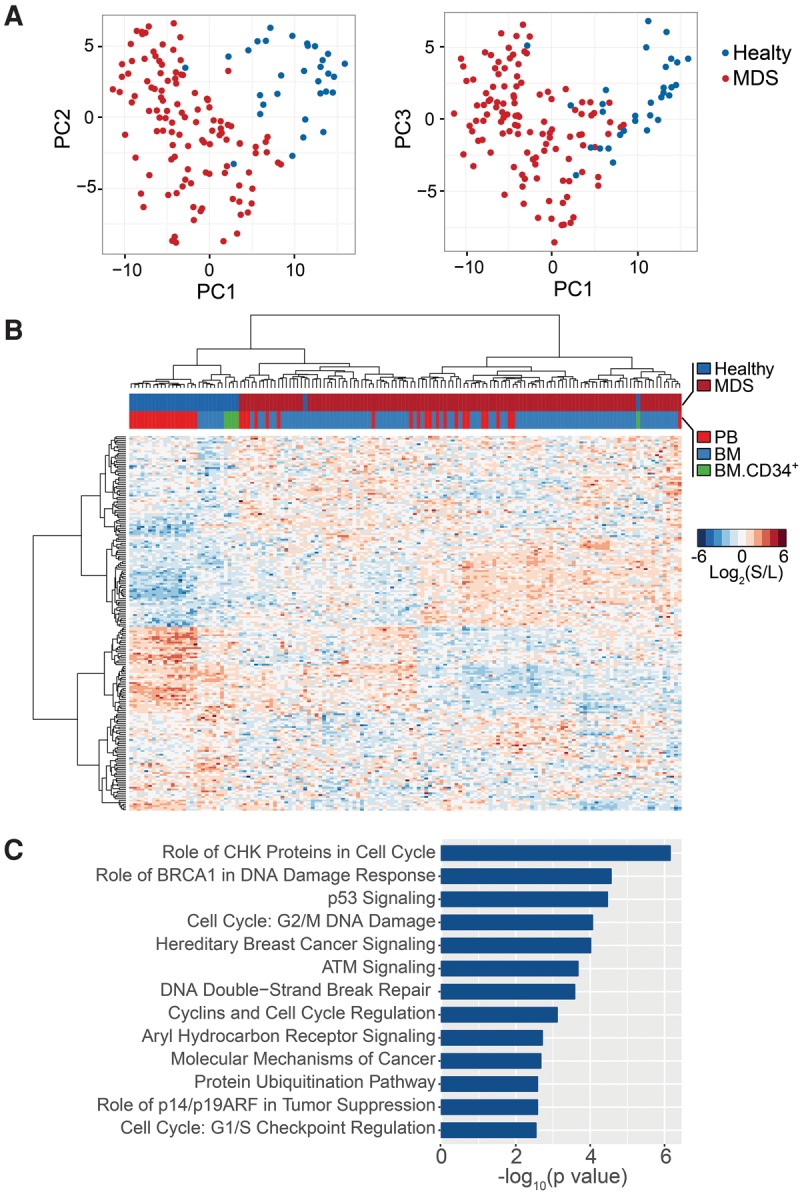
Characterization of MDS-defining splicing programs. (A) PCA identified a 204-event panel (MDS-Dx) that differentiates MDS from healthy samples (both bone marrow and peripheral blood were included, while healthy lineage sorted samples were excluded). (B) Unsupervised hierarchical clustering of MDS and healthy samples with the MDS-Dx panel. (C) Pathway analysis of the MDS-Dx panel using Ingenuity IPA.
To gain functional insights into the genes in this MDS-Dx panel, we used Ingenuity IPA to identify top canonical pathways linked to MDS. We found specific enrichment of alternatively spliced genes involved in cell cycle regulation, DNA damage response/repair, self-renewal, and cancer progression (Fig. 3C; Supplemental Table S6B). Therefore, by setting aside cell lineage-associated splicing events, we were able to identify critical splicing events that may directly contribute to the etiology and/or progression of MDS. This approach may be generally applicable to characterizing gene signatures associated with other blood disorders.
Functional insights into MDS-linked splicing events
We next performed String network analysis to gain further insights into the genes in the MDS-Dx panel, observing two distinct sub-networks (Fig. 4A). The first sub-network contains a large number of genes (individually listed in Fig. 4B) involved in cell cycle control and DNA damage response (i.e., RB1, E2F6), protein ubiquitination (i.e., DNAJC3, DNAJC8), hematological physiology (i.e., LMO2, TRAF3), regulation of apoptosis (i.e., BAX, BNIP2), and epigenetic control of gene expression, and the other sub-network consists of a group of RNA-binding splicing regulators, including multiple SR protein family members (i.e., SRSF2, SRSF3, SRSF5, SRSF7, SRSF10, and SRSF11), specific hnRNP proteins (i.e., HNRNPD and HNRNPH1), various other well-characterized splicing regulators (i.e., RALY, SNRNP70, TRA2A, and U2SURP), and those involved in the nonsense-mediated mRNA decay (NMD) pathway (i.e., UPF3A and SMG7). This observation suggests that many altered splicing events in MDS may result from induced splicing of various splicing regulators.
FIGURE 4.

Network analysis of key altered splicing events in MDS. (A) Protein network analysis of MDS-Dx by String9.1 and Cytoscape. Line connections represent the evidence supported association. (B) Genes in the two concentrated networks are involved in splicing regulation or in cell cycle control and DNA damage repair. (C) Analysis of the effect of MDS-Dx alternative splicing (cassette exon) on the reading frame of gene transcripts. Fisher's exact test was used to compare in-/out-of-frame events of MDS-Dx, and its disease pathway-related events with overall and detectable events ([*] P < 0.05, [**] P < 0.01). For in-frame events in enriched pathways in MDS-Dx, the in/out of the protein domain was further analyzed.
Inclusion or skipping of an exonic region as a result of alternative splicing may or may not disrupt the reading frame of a given mRNA transcript, and out-of-frame changes are more likely to generate functionally distinct or loss-of-function gene products. Among all annotated genes for the current study and those with detectable isoform expression, ∼45% would alter the reading frame between the mRNA isoforms from the same gene (Fig. 4C). Interestingly, among the MDS-Dx panel of 204 events, more genes (>60%) are associated with out-of-frame changes, as exemplified by the alternative exon in BAX, MADD, MLH1, and E2F6 (Supplemental Fig. S2A). The frame-shift may convert a transcript to become NMD-sensitive, which happened to many splicing regulators (Ni et al. 2007). The frame-shift becomes more evident within the events of 39 genes in various enriched pathways identified by IPA, >75% of which are out of frame. Even for the remaining in-frame events in this subgroup, about half of such in-frame events are located within a functional domain of individual proteins (Fig. 4C), as exemplified by the alternative exons in PRKDC (a key kinase in phosphatidylinositol signaling) and MDM2 (a p53 E3 ligase) (Supplemental Fig. S2B). These findings suggest that MDS-specific alternative splicing events may directly contribute to MDS pathogenesis by creating functionally distinct or defective proteins.
Altered splicing programs by splicing factor mutations in MDS
The identification of prevalent mutations in some key components of the spliceosome machinery (i.e., SF3B1, SRSF2, U2AF1, and ZRSR2) suggests that those mutations may be key drivers of MDS. Puzzling, however, is the observation that mutations in SRSF2 and U2AF1 appear to associate with poor prognosis of the disease while mutations in SF3B1 seem to predict good prognosis (Papaemmanuil et al. 2011). ZRSR2 was reported as an essential component of the minor spliceosome (U12 dependent) assembly (Madan et al. 2015), which is the least frequent compared to the mutation frequencies of the other three splicing factor genes. As the functions of these splicing factors converge on the definition of 3′ splice sites, a popular hypothesis is that mutations in these genes may affect a common set of splicing events that may directly contribute to MDS. Because RNA-seq experiments carried out so far have not yielded a sufficient number of altered splicing events for testing this hypothesis, we took advantage of our RASL-seq data set by segregating MDS samples with or without specific splicing factor mutations to determine whether individual splicing factor mutations have convergent or divergent consequences on alternative splicing.
We took our regression model to compare 115 MDS samples with or without mutations in specific splicing factor genes. This analysis led to the identification of 197 mutation-associated events for SRSF2 (Fig. 5A; Supplemental Table S7), 206 events for U2AF1 (Fig. 5B; Supplemental Table S8), and 191 events for SF3B1 mutated patients (Fig. 5C; Supplemental Table S9). Because of insufficient sample size, we had to exclude ZRSR2 from this analysis. It is also worth pointing out that, while mutations in SRSF2 occurred in a single location in the gene, multiple mutations occurred in two separate locations in U2AF1. In our cohort, for example, among 12 patients that carried U2AF1 mutations, six contained the Q157P mutation; one had the Q157R mutation; four carried the S34F mutation, and one contained a noncanonical C163 frame-shift mutation. Because there are insufficient samples in different mutation classes, we had to characterize them as a cohort, rather than individually analyzed.
FIGURE 5.
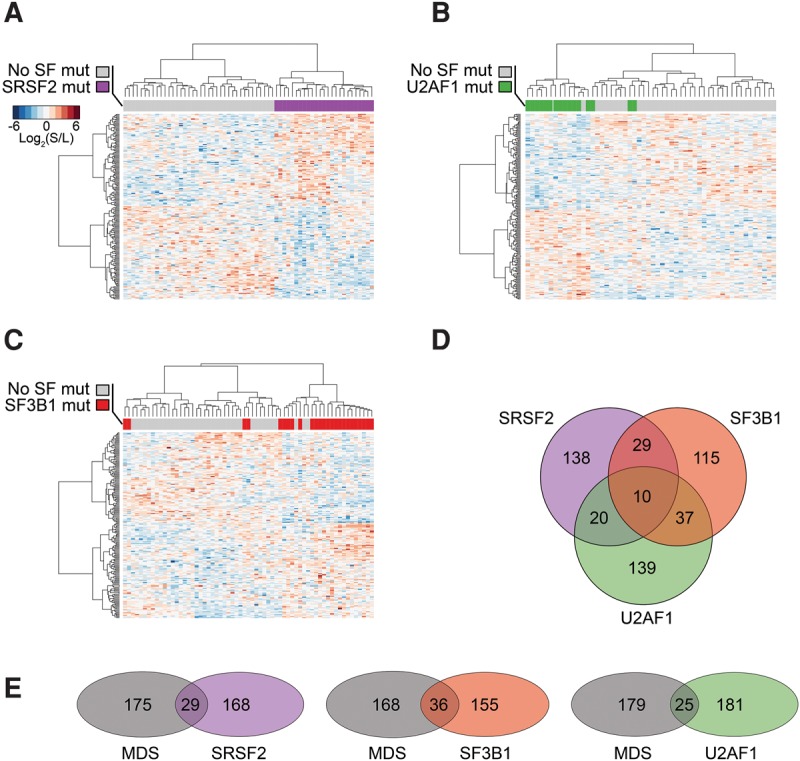
Splicing factor mutations in MDS affect distinct splicing programs, but the affected genes converge in similar dysregulated pathways. (A) Unsupervised hierarchical clustering using the SRSF2 mutation-related splicing program. (B) Unsupervised hierarchical clustering using the U2AF1 mutation-related splicing program. (C) Unsupervised hierarchical clustering using the SF3B1 mutation-related splicing program. (D) Venn-diagram of regression model-identified splicing programs associated with SRSF2, U2AF1, and SF3B1 mutations, and their relationships. (E) The overlap of SRSF2, U2AF1, and SF3B1 mutation-related programs with the MDS-Dx panel. No SF mut: Samples without SRSF2, U2AF1, SF3B1, or ZRSR2 mutations.
Strikingly, the identified splicing events were able to efficiently differentiate MDS samples without SF mutations from those that carry specific mutations in SRSF2, U2AF1, and SF3B1 (Fig. 5A–C). Surprisingly, however, the three splicing programs showed little overlap (Fig. 5D; Supplemental Table S10), indicating that mutations in individual splicing factors are unlikely to cause a common set of alternative splicing events to induce MDS. Consistent with this possibility, the collection of significantly altered splicing events associated with each splicing factor mutation contributes a small subset to the splicing program that distinguishes MDS from healthy samples (MDS-Dx) (Fig. 5E). Interestingly, however, Ingenuity IPA analysis suggested that mutations in the three splicing factors widely affected genes involved in DNA damage response pathways, even though different genes were affected by different splicing factor mutations in these pathways (Supplemental Fig. S3A–C; Supplemental Tables S11–S13). This observation suggests that, instead of causing a common set of alternative splicing events, mutations in each splicing factor may modulate critical genes in some common pathways to cause the disease.
We further analyzed the splicing events overlapping with MDS-Dx splicing factor mutations that fall in enriched pathways related to disease progression. The SRSF2 mutation-affected splicing program has eight events overlapping with MDS-Dx and all are changed in the same direction (Supplemental Fig. S4A). These genes, including CDK7, CCNL1, CARD16, SNCA, and PYCARD, function as regulators of cell cycle progression, cancer, and apoptosis. Similar to the SRSF2 mutation-affected splicing program, a small subset of the U2AF1 mutation-affected splicing program overlapping with MDS-Dx and also showed the changes in the same directions (Supplemental Fig. S4B). These overlapped genes include RB1, CARD16, ZDHHC16, MYB, CD300LF, DDB2, and TET2, which have functions related to cancer, apoptosis, and hematopoiesis. In contrast, the genes shared between the SF3B1 mutation-affected splicing program and MDS-Dx, inducing MDM2, RAD17, SNRNP 70, and SRSF10, largely showed changes in opposite directions (Supplemental Fig. S4C). This might bear some functional relevance to the reports that SF3B1 mutations are associated with better survival, while SRSF2 and U2AF1 mutations correlated with worse overall survival in MDS patients (Papaemmanuil et al. 2011; Makishima et al. 2012).
Splicing factor mutations induce genes with a unique 3′ splice site consensus
U2AF1, SF3B1, and SRSF2 are functionally connected to the U2 snRNP complex critical for 3′ splice site selection. The observation that the splicing signatures of these mutated genes showed little overlap motivated us to further analyze the number of alternative 5′ or 3′ splicing events in different mutation-induced programs relative to unaltered events. We found that, while unaltered events covered roughly equal numbers of alternative 5′ and 3′ splice sites; SF3B1 and U2AF1 mutation-affected programs were more enriched with alternative 3′ splice sites; and the SRSF2 mutation-affected program was associated with a high frequency of alternative 5′ events (Supplemental Fig. S5). This is consistent with the roles of SF3B1 and U2AF1 in 3′ splice site selection, while SRSF2 is a more general regulator of splice site selection by multiple mechanisms as we reported earlier (Pandit et al. 2013).
We further analyzed consensus sequences associated with cassette exons in specific splicing factor mutation-induced programs. Besides the motif GTAAGT at 5′ donor sites and the canonical 3′ acceptor sites present in all groups, we observed the changed consensus at the +1 position of the 3′ acceptor site in U2AF1 mutation-induced events (red box in Supplemental Fig. S6), a position likely regulated by one of the U2AF1 zinc finger motifs as noted earlier (Ilagan et al. 2014; Okeyo-Owuor et al. 2014). In our cohort, six out of 12 MDS samples carried the U2AF1 mutation at Q157P, which is also the most frequent mutation identified among AML patients (Ilagan et al. 2014), thus counting for similarly altered 3′ splice sites.
Recent studies revealed that mutant SRSF2 showed increased binding to the CCNG motif (N = any nucleotide) and decreased binding to the GGNG motif, leading to enriched CCNG among enhanced splicing events and enriched GGNG among repressed splicing events (Kim et al. 2015; Zhang et al. 2015). To determine whether this trend was also represented in our MDS patients, we identified 140 elevated inclusion and 123 increased skipping events linked to SRSF2 mutations (P < 0.01, |Fold Change| ≥ 2), and then calculated 4-mer enrichment as previously described (Zhang et al. 2015). While the difference is not obvious with CCNG, we detected a dramatic enrichment of GGNG motifs among increased exon skipping events (Supplemental Fig. S7). This observation is consistent with compromised SRSF2 binding to the GGNG motif to cause exon skipping. We suspect that many induced exon inclusion events may result from various indirect effects, therefore masking the anticipated enrichment of the CCNG motif among enhanced exons, as our analysis was based on complex human samples, rather than engineered cell or animal models. Together, these motif analyses further reinforced distinct splicing programs induced by different splicing factor mutations in MDS patients.
A critical splicing signature linked to MDS prognosis
To determine if differential splicing events detected by RASL-seq in MDS patient samples have any prognostic value, we applied a lasso penalized Cox regression model (Coxnet) (Pellagatti et al. 2013) to identify isoform ratios associated with overall survival. This analysis was restricted to 96 MDS patients with available survival information. After subjecting candidate events to 10-fold internal cross validation, we identified a panel of 11 events with prognostic significance (Supplemental Table S14, here referred to as MDS-PGx for the MDS-prognostic signature). Patients were assigned a risk score based on the weighted expression of these 11 events, and then split into equally sized tertiles, as MDS-PGx good risk, intermediate risk, and poor risk. With 3.85 yr median follow-up time for this cohort, the results demonstrated significant differences in overall survival (Fig. 6A).
FIGURE 6.
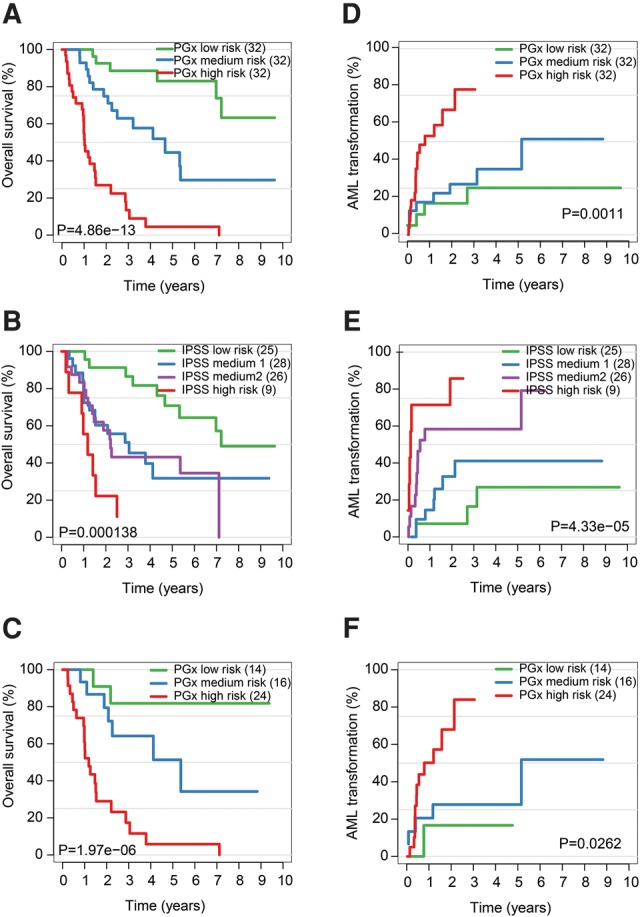
A critical splicing signature linked to MDS prognosis. (A) Kaplan–Meier curve for overall survival demonstrating how application of the MDS-PGx score efficiently stratifies MDS patients into three distinct risk groups. (B) Overall survival curves for the same patients stratified by the IPSS show substantial overlap in the intermediate risk groups. (C) IPSS Intermediate-1 and Intermediate-2 risk patients (from B) were further stratified by the MDS-PGx score. (D) Time to AML transformation is shown for MDS patients stratified by the MDS-PGx score. (E) Time to AML transformation is shown for MDS patients stratified by their IPSS risk group. (F) Time to AML transformation of IPSS Intermediate-1 and Intermediate-2 risk patients (from E) reclassified by the MDS-PGx score.
We next compared this MDS-PGx signature to known prognostic variables, including the well-established IPSS risk score. According to the assigned IPSS scores, 29 MDS patients in the intermediate-1 (int-1) and 25 patients in the intermediate-2 (int-2) groups of our cohort were not well separated (Fig. 6B). In clinical practice, a distinction is often made between lower risk MDS patients, who are typically treated with growth factors and supportive care, and higher risk MDS patients, who are typically treated with more intensive options, such as hypomethylating agents, chemotherapy, or stem cell transplantation. Application of the MDS-PGx classifier to the 54 IPSS int-1 and int-2 patients identified 14 (26%) of them as good risk patients, and 24 (44%) as poor risk patients (Fig. 6C). Interestingly, some patients with lower IPSS risk (low and Int-1) could be reclassified using the MDS-PGx to higher risk (P < 0.001; Supplemental Fig. S8A). Similarly, in patients with higher IPSS risk (Int-2 and high), MDS-PGx could identify a subset of those patients to belong to a lower than perceived risk (P = 0.011; Supplemental Fig. S8B). These observations suggest a potential value of the MDS-PGx in clinical applications.
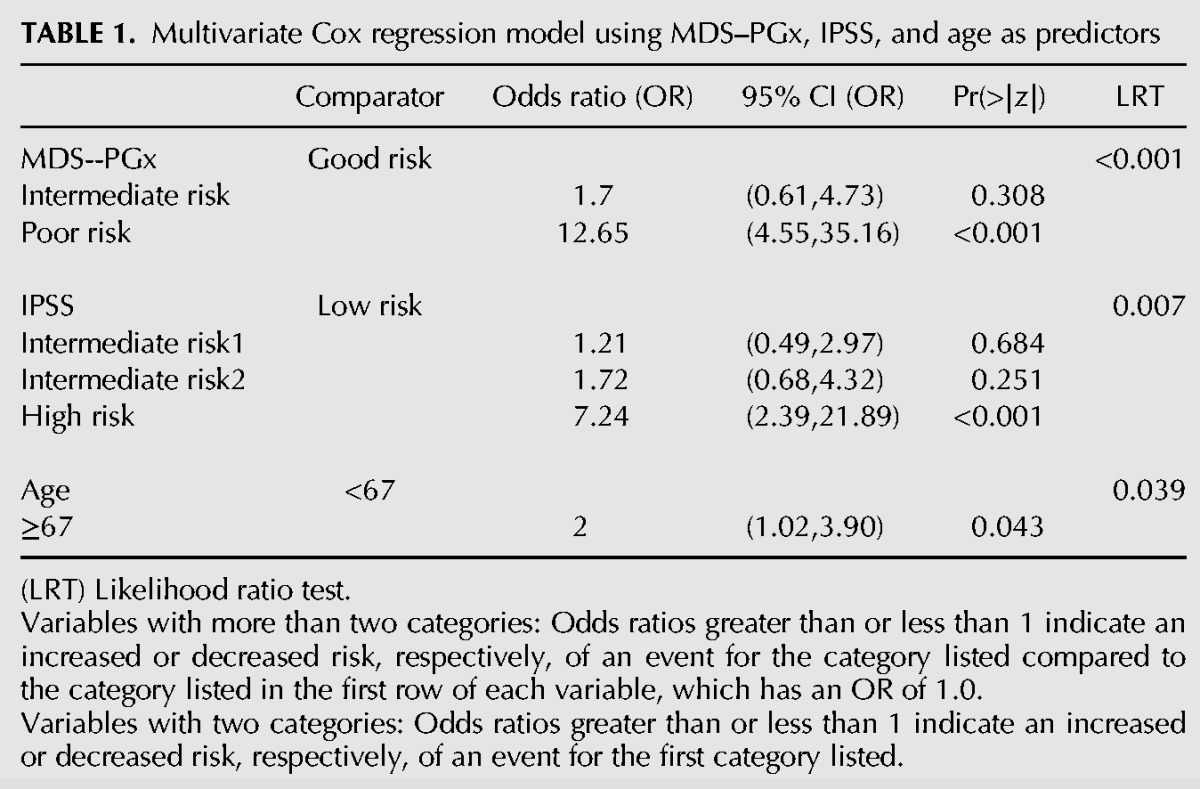
Demographic and clinical characteristics were compared between the 3 MDS-PGx score tertiles (Supplemental Table S15). While bone marrow blasts and cytogenetic risk groups were distributed evenly between MDS-PGx tertiles, lower IPSS risk groups were significantly more frequent in patients from the good risk MDS-PGx tertile compared with those in the poor risk MDS-PGx tertile (Supplemental Table S15). Patients in the good risk MDS-PGx tertile were younger compared to the MDS–PGx intermediate and poor risk tertiles, while Hgb levels were higher in patients from MDS-PGx good risk tertile compared to those in the MDS–PGx intermediate and poor risk tertiles (Supplemental Table S15). To evaluate the degree to which the MDS-PGx risk score has independent prognostic power, we performed multivariate analysis (Table 1) by including all risk factors that were significant in univariate analysis (MDS-PGx, IPSS, age; Supplemental Table S16). This analysis showed that, despite its association with known prognostic features, the MDS-PGx is a prognostic indicator independent of IPSS and age.
TABLE 1.
Multivariate Cox regression model using MDS–PGx, IPSS, and age as predictors
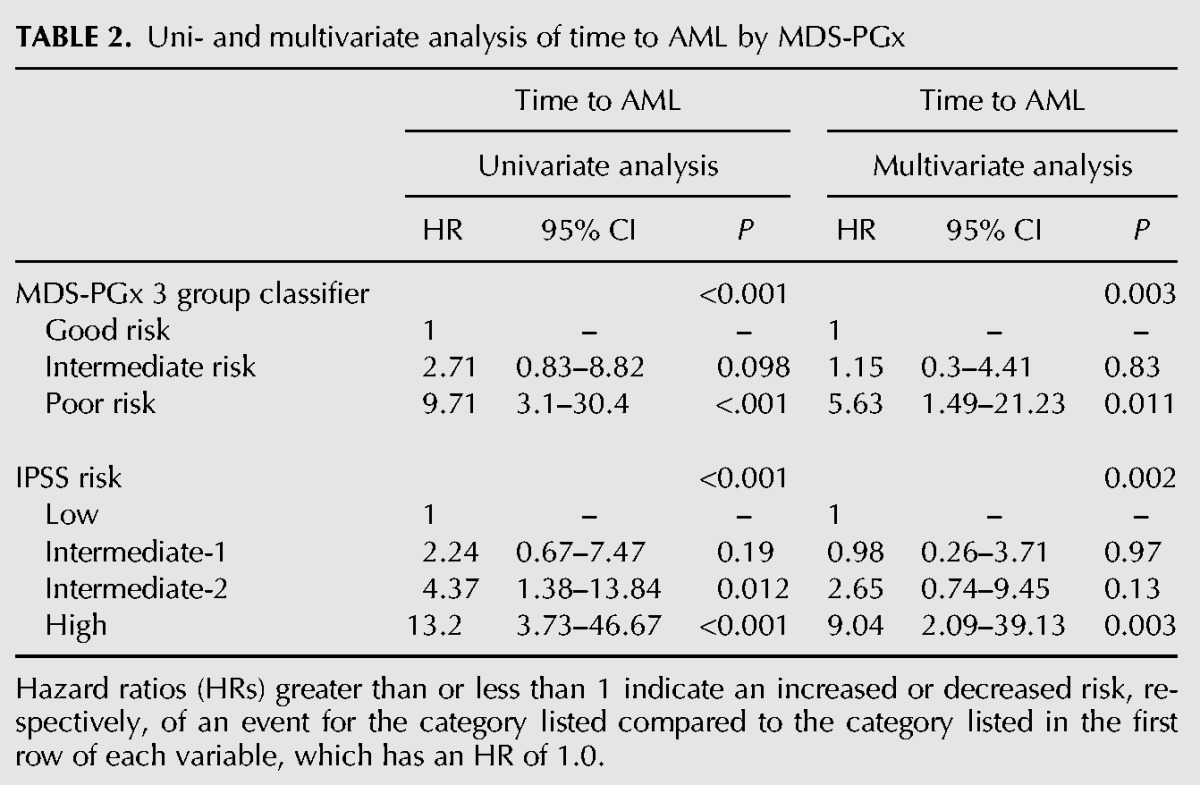
Finally, we examined the performance of the MDS-PGx classifier on its ability to predict progression to acute myeloid leukemia (AML), a relevant biologic characteristic for which the classifier was not specifically selected. In our cohort, patients within each MDS-PGx tertile had significantly different rates of transformation to AML (P = 0.0011; Fig. 6D). The 1-yr AML progression rate was 10%, 25%, and 64% for MDS-PGx good, intermediate, and poor risk patients, respectively (Fig. 6D). Similarly, IPSS risk score also identified groups with differences in rates of AML progression (P < 0.001, Fig. 6E). However, application of the MDS-PGx score to the 54 IPSS int-1 and int-2 patients could further stratify this intermediate risk subset, identifying 14 patients with good risk of AML transformation and 24 with poor risk of AML transformation (P = 0.0262, Fig. 6F). In patients with lower risk IPSS (low and int-1), the MDS-PGx classifier differentiated those with poor risk for AML transformation (Supplemental Fig. S8C). The 1-yr AML progression rate in this subgroup was 4%, 16%, and 47% for MDS-PGx good, intermediate, and poor risk tertiles, respectively (P = 0.002). In patients with higher risk IPSS (int-2 and high), the MDS-PGx classifier identified patients with lower than predicted risk for AML transformation (Supplemental Fig. S8D). The 1-yr AML progression rate in this subgroup was 36%, 36%, and 80% for MDS-PGx good, intermediate, and poor risk groups, respectively (P = 0.022). In multivariate analysis, again, the MDS-PGx classifier remains a significant predictor for AML progression independent of the IPSS risk score (Table 2). We lastly compared the baseline and genetic characteristics between the three groups of the MDS-PGx score. The good risk MDS-PGx group had more patients with lower risk MDS, which, from a clinical point of view, is closer to normal hematopoietic cells than high-risk MDS cells (Supplemental Table S17). This is also supported by clustering analysis based on MDS-Dx (Fig. 3B) where more patients clustered with healthy samples belonged to the low-risk group (Supplemental Fig. S9). Together, these data strongly suggest that the splicing signature derived from the current cohort contains prognostic information in MDS patients, which may be further developed as a biomarker for risk and treatment stratification.
TABLE 2.
Uni- and multivariate analysis of time to AML by MDS-PGx
DISCUSSION
We have used a target-specific global approach to characterize the splicing program in MDS in comparison with samples from healthy individuals. In the past two decades, gene expression profiling has been a powerful tool for studying diseases by determining changes in transcriptomes, which has also been applied to MDS (Pellagatti et al. 2013). However, altered mRNA isoform expression has been recognized to have the potential to more robustly characterize specific disease states (Zhang et al. 2013), which is particularly relevant to MDS because of prevalent mutations in specific splicing factors found in the disease. This has prompted the identification of specific mRNA isoforms associated with MDS, and RNA-seq appears uniquely suited for this purpose. However, published results to date using this approach have only yielded a small number of disease-linked mRNA isoforms, and in most cases, there is limited quantitative information that can be used to classify MDS.
In the present study, we took advantage of the RASL-seq technology developed in our laboratory, which is specifically designed to interrogate mRNA isoforms, even those from low-expressed transcripts. Thus, all reads are related to specific targets under survey. Our previous studies demonstrated that the approach is of high sensitivity and can well tolerate partially degraded RNA. We assume that, by targeting >5000 annotated events, which contain numerous disease (including cancer)-linked events documented in the literature, we have sufficient power for global comparison. The cost-effectiveness of this tool coupled with the quantitative information obtained thus significantly offsets the limitation of RNA-seq-based approaches.
To efficiently dissect the splicing landscape of MDS, we recognize a challenging problem particularly relevant to studying hematological malignancies, which is the highly heterogeneous cell population in both healthy and disease samples. Thus, a putative signature may reflect changes in the population of cells or within specific cell types or both. We thus developed a strategy to first identify cell type-specific alternative splicing events among sorted cells from healthy individuals. The signature we obtained (Hemo-SP) can clearly differentiate cell types in different blood lineages. Interestingly, pathway analysis of the panel showed enrichment of genes in cell differentiation, indicating their contribution to hematopoiesis. Filtering out genes in the Hemo-SP panel enabled us to use relatively cell type-independent alternative splicing events to characterize MDS, leading to the MDS-Dx panel that could efficiently distinguish between healthy and MDS samples. The altered splicing events in this panel are enriched in genes involved in cell cycle control, apoptosis, and DNA damage responses, strongly arguing for their direct contribution to the MDS disease phenotype.
As MDS contain prevalent mutations in specific splicing factors, one of the most pressing questions is whether these mutations affect a common set of splicing events that may be underlying MDS because SRSF2, U2AF1, and SF3B1 have convergent functions in the 3′ splice site definition, yet puzzling is the observation that the mutations in these splicing factors are divergently associated with prognosis (Papaemmanuil et al. 2011; Makishima et al. 2012). By identifying specific splicing signatures associated with MDS samples containing individual splicing factor mutations, we found that each signature appears to be largely confined to a unique set of genes, suggesting that mutations in each of these splicing factors affect a unique spectrum of splicing events in MDS patients. Interestingly, however, alternatively spliced genes that are associated with mutations in different splicing factors appear to converge to several common pathways, such as those involved in cell cycle control and DNA damage response/repair. This finding suggests that alterations in those key pathways likely contribute to MDS.
It is also important to point out that all splicing factors may also have independent splicing functions. For example, it has been demonstrated that SRSF2 plays a critical role in maintaining genome stability (Xiao et al. 2007), and recent studies also showed its direct activity in transcriptional control (Mo et al. 2013). A recent study also revealed that U2AF1 and SF3B1 are part of the BRCA-DNA damage response complex (Savage et al. 2014). These observations raise the possibility that mutations in these splicing factors may employ both splicing-dependent and -independent mechanisms to cause MDS.
Interestingly, RASL-seq identified that the U2AF1 mutation program has the same consensus sequence change at the 3′ acceptor site as the changed consensus sequence in U2AF1 mutated AML cohorts detected by RNA-seq, even though the individual events identified in the two studies are different. We also confirmed the previous results on cellular and animal models that mutations in SRSF2 altered its RNA binding preference for CCNG and GGNG motifs (Kim et al. 2015; Zhang et al. 2015). These findings further validate RASL-seq as an effective tool for analyzing functional alternative splicing in patients.
The newly developed Coxnet approach enables the identification of critical events associated with disease prognosis (Pellagatti et al. 2013). We applied this bioinformatics approach in the current study to identify a panel of 11 events (MDS-PGx) associated with clinical outcomes of the disease. As a prognostic feature independent of the IPSS, the MDS-PGx classifier efficiently differentiated patients into good, intermediate, and poor prognosis tertiles, but also improved prognostication of patients in the IPSS int-1 and int-2 groups, which may further improve treatment allocation for these patients. As MDS-PGx is derived from a training model, which utilizes patient survival as the endpoint, there is a risk of overfitting its prognostic power to our cohort. To examine the performance of the MDS-PGx on a context for which it was not specifically selected, we applied the classifier to another disease-related and biologically relevant endpoint, the MDS-to-AML transformation rate. The MDS-PGx characterized several patients as having higher and lower risk with regard to AML transformation, even when the IPSS predicted them to have lower and higher risk, respectively. This argues that MDS-PGx is not grossly overfit to a single disease feature. However, the classifier requires validation in an independent cohort.
The performance of the MDS-PGx may reflect a pathogenic role in disease progression for the underlying splicing events measured in the signature. Six out of the 11 events in MDS-PGx are either out-of-frame or in-frame within a functional protein domain (Supplemental Table S14). Included in the 11 events that form the MDS-PGx are BCAS3, which is related to progression of other tumor types (Gururaj et al. 2006), PROM1, which is involved in stem cell maintenance (Sompallae et al. 2013), MBTD1 and CDCA2, which regulate chromosome structure, while CDCA2 also served as a prognostic marker for synovial sarcomas (Lagarde et al. 2013; Luo et al. 2013). ABI2 and TAF4B are known to regulate hematopoietic cell function (Dai and Pendergast 1995). CSNK1E is a member of the casein kinase I protein family, whose members have been implicated in the control of cytoplasmic and nuclear processes, including DNA replication and repair. The stabilization of components of cytokines and Wnt signaling by CSNK1E might be critical for hematopoietic cell self-renewal (Okamura et al. 2004). Interestingly, mutations in a related casein kinase, CSNK1A1, are prevalent in MDS with del(5q), suggesting a role of this gene family in MDS pathogenesis (Schneider et al. 2014; Heuser et al. 2015). This functional information suggests that RASL-seq has captured splicing events with discriminatory power as well as clinical significance with novel insights into the pathogenic mechanisms underlying the development of MDS.
MATERIALS AND METHODS
Patient samples
Bone marrow (BM) and/or peripheral whole blood (PB) samples were collected from 112 MDS patients at the time of enrollment in clinical trials at Hannover Medical School (Hannover, Germany), investigating the efficacy of all-trans-retinoic acid, antithymocyte globulin, deferasirox, lenalidomide, or thalidomide for treatment of MDS. Healthy blood and BM donors also provided cells for RNA extraction: peripheral blood mononuclear cells from 18 blood donors, sorted cell populations from PB from five blood donors (CD66b + CD15+ granulocytes, CD14 + CD66b-CD3-CD56- monocytes, CD19+ B-cells, CD8-CD56-CD3 + CD4+ T-cells), BM mononuclear cells from six donors and two BM RNAs purchased from Biochain and Clontech, CD34+ cells from six donors, common myeloid progenitor cells (CMP) (Lin-CD34 + CD38 + CD123lowCD45Ra-) from two donors, and stem cell (Lin-CD34 + CD38−) from one donor (Fig. 1A; Supplemental Table S1). Cell samples were collected and clinical data were recorded after MDS patients and healthy donors were given informed consent in accordance with the Declaration of Helsinki and with the Institutional Review Board (IRB) approval (ethical vote 2467).
Cytogenetic and molecular analysis
Cytogenetic analysis was performed by G- and R-banding. Mutational analysis was performed as described previously (Damm et al. 2010). Mononuclear cells from patient samples were enriched by Ficoll density gradient centrifugation and stored in liquid nitrogen until use. Genomic DNA was extracted from each sample using the All Prep DNA/RNA Kit (Qiagen). Mutational analysis of each sample was performed for ASXL1, DNMT3A, IDH1, IDH2, RUNX1, NPM1, SF3B1, SRSF2, U2AF1, and ZRSR2 as described previously (Thol et al. 2012). PCR fragments were sequenced by Sanger sequencing and analyzed using Mutation Surveyor software (SoftGenetics, State College, PA).
Isolation of lineage-specific cells by flow cytometry
Lineage-specific cells, including CD34+ BM, CMP, granulocytes, monocytes, B cells, and T cells, were purified by flow cytometry according to the markers and related antibodies listed in Supplemental Table S1. The CD34 microbead kit was purchased from Miltenyi Biotech (Bergisch-Gladbach, Germany). All antibodies used were from BD Biosciences (Heidelberg, Germany).
RASL-seq profiling of alternative splicing and data analysis
Total RNAs were purified from collected cells by using the RNeasy Kit (Qiagen) according to the manufacturer's instructions. For RASL-seq, a pool of oligonucleotides was prepared, which targets 5502 alternative splicing events in the human genome, as previously described (Pandit et al. 2013). The pool interrogates a variety of splicing modes, including alternative transcription start, alternative transcription termination, cassette exon (single or multiple), mutually exclusive exons, alternative 5′ splice sites, and alternative 3′ splice sites (Supplemental Table S2). For each splicing event, probe sets were designed to specifically target two (or more than two in certain cases) annotated isoforms with unique exon sequences (Li et al. 2012).
We initially designed oligos to include alternative splicing events conserved in human and mouse based on Yeo and Burge (Yeo et al. 2005) and Ares and Haussler (Sugnet et al. 2004). In addition, we searched PubMed using key words, aberrant/abnormal splicing, splicing signature, tissue-specific splicing, and human disease, to identify reported disease (including cancer)-, tissue-, and differentiation-associated splicing events, resulting in a total number of 995. Together, the current RASL oligo pool contains 5502 annotated alternative events from 3758 genes plus 19 internal controls (total = 5521).
By using specific oligonucleotides to target junction sequences (step 1), paired oligonucleotides annealed on mRNA can be selected by biotinylated oligo-dT immobilized on beads (step 2). Upon selection and ligation, only specifically targeted oligonucleotide pairs can be converted to amplicons (step 3), and upon PCR using a pair of universal primers, the products from each sample are bar-coded (step 4). We routinely pool up to 30 RASL-seq libraries for deep sequencing in one lane of an Illumina HiSeq2500 sequencer (step 5). The sequencing information permits assigning reads to anticipated pairs of oligonucleotides on specific mRNA isoforms and we require a higher than 70% accurate mapping rate for each sample. As not all genes or isoforms are sufficiently expressed in a given cell type, we require a minimum of five counts per isoform in both isoforms from a gene to compute the isoform ratio, and derive the ratio change according to a pipeline that has been detailed in our recently published studies (Zhou et al. 2012b; Pandit et al. 2013).
Because not all mRNAs or their isoforms were expressed in all cell types, we first filtered detectable splicing events by requiring the sequencing reads for both expressed isoforms in each event to be present in at least one-third of samples in our cohort. A total of 1956 splicing events met such criteria in the current study. To define the change in each splicing event, a splicing index was determined as the ratio of read counts between short and long isoforms. Such a splicing index for each event was scaled according to the average index of all samples and log2 transformed. This sample isoform ratio versus average isoform ratio (across all samples) approach eliminates intrinsic biases of oligonucleotide probes in hybridization and ligation.
where SSi is the splicing score for the splicing event in sample i; CSi, CLi are read counts of short isoform, long isoform in sample i; ASi, ALi are oligo annealing coefficient of short isoform, long isoform in sample i; n is total number of samples.
To search for potential splicing signatures associated with different cell types from healthy individuals and patients with MDS, we first reduced the dimensionality of splicing features in a specific cohort into ten major components by principal component analysis (PCA), as previously described (Zhou et al. 2012a). We next employed a supervised multiple logistic regression model with lasso penalty as a classification machine to train samples with interesting labels. After 10-fold cross-validation, a robust regression model for the classification could be established. By summarizing the rotation matrix and coefficient matrix of the model, we finally selected top-ranked contributors (P < 0.05) of splicing events as a signature for each classification model. Hierarchical clustering was performed as previously described (Khan et al. 2001).
For hierarchical clustering, we used 1-PCC (Pearson's correlation coefficient) as the distance metric and the ward's method in R's hclust package to calculate the clustering linkage.
To determine the potential splicing signatures correlated with overall survival of MDS patients, we established a survival model based on all the splicing events by the Coxnet algorithm as previously described (Pellagatti et al. 2013) with minor modifications. Briefly, the Coxnet predictor was established by supervised lasso penalized Cox proportional hazards regression based on all the splicing events and overall survival years of MDS samples. After 10-fold cross validation, a converged model with a stable subset of 11 splicing events was identified.
RT-PCR validation
Total RNA (5–10 ng) from individual healthy or patient samples was used to perform RT-PCR using the One-Step RT-PCR kit (Qiagen). Primers used for validation are listed in Supplemental Table S3. RT-PCR products were resolved on a 2% agarose gel and signals analyzed by ImageJ64.
Statistical analysis of clinical characteristics
Overall survival (OS) end points, measured from the date of initial sample collection, were death (failure) or alive at the last follow-up (censored). Time to AML progression was measured from the date of initial sample collection to the time of AML diagnosis. Progression to AML was defined according to the 2008 WHO classification. Primary analysis was performed on OS and time to AML progression. The Kaplan–Meier method, log-rank test, and Cox proportional hazards models were used to estimate the distribution of OS and time to AML progression and to compare differences between survival curves, respectively. Pairwise comparisons were performed by median test or the Student's t-test for continuous variables and by two-sided χ2 tests for categorical variables. Variables considered for model inclusion were International Prognostic Scoring System (IPSS) risk score, transfusion dependence, age (below versus above median), sex, hemoglobin levels (<8 g/dL versus 8 to <10 g/dL versus ≥10 g/dL), bone marrow blasts (<5% versus 5%–10% versus >10%–20%), cytogenetic risk according to IPSS (low versus intermediate versus high), mutation status in genes ASXL1, RUNX1, IDH1, IDH2, DNMT3A, SF3B1, SRSF2, U2AF1, and ZRSR2. The two-sided level of significance was set at P < 0.05. The uni- and multivariate statistical analyses were performed with the statistical software package SPSS Version 22.0 (IBM Corporation, Armonk, NY).
Analysis of pathways, functions, and protein domains
Pathway analysis was performed using Ingenuity Pathways Knowledge Base-v8.8 (Ingenuity Systems, content version 17199142, release date September 17, 2013). We used 3182 expressed genes (reads detectable in at least 20% samples) as the background control for IPA analysis. Protein interaction networks were constructed using String9.1 and Cytoscape. In- and out-of-frame analysis was performed on cassette exons based on the length of the cassette exon dividable by three. The location of a cassette exon relative to a known protein domain was manually curated on the UCSC Genome Browser, NCBI Refseq, and EMBL-EBI's InterPro.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to all patients and contributing doctors. We acknowledge assistance of the Cell Sorting Core Facility of the Hannover Medical School supported in part by the Braukmann-Wittenberg-Herz-Stiftung and the Deutsche Forschungsgemeinschaft. This study was supported by grants 110284, 110287, 110292, and 111267 from Deutsche Krebshilfe; grant DJCLS R13/14 from the Deutsche José Carreras Leukämie-Stiftunge.V; the German Federal Ministry of Education and Research grant 01EO0802 (IFB-Tx); DFG grants HE 5240/5-1 and HE 5240/6-1; grants from Dieter-Schlag Stiftung to M.H., and National Institutes of Health grant DK098808 to D.Z. and X.D.F.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.056101.116.
REFERENCES
- Adamia S, Bar-Natan M, Haibe-Kains B, Pilarski PM, Bach C, Pevzner S, Calimeri T, Avet-Loiseau H, Lode L, Verselis S, et al. 2014. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): new potential targets in AML. Blood 123: 2816–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. 2011. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hackett CS, Zhang S, Song YK, Bell RJ, Molinaro AM, Quigley DA, Balmain A, Song JS, Costello JF, et al. 2015. The genetics of splicing in neuroblastoma. Cancer Discov 5: 380–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Pendergast AM. 1995. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev 9: 2569–2582. [DOI] [PubMed] [Google Scholar]
- Damm F, Heuser M, Morgan M, Yun H, Grosshennig A, Gohring G, Schlegelberger B, Dohner K, Ottmann O, Lubbert M, et al. 2010. Single nucleotide polymorphism in the mutational hotspot of WT1 predicts a favorable outcome in patients with cytogenetically normal acute myeloid leukemia. J Clin Oncol 28: 578–585. [DOI] [PubMed] [Google Scholar]
- Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, Przychodzen B, Sahgal N, Kanapin AA, Lockstone H, et al. 2015. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 29: 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswaran J, Horvath A, Godbole S, Reddy SD, Mudvari P, Ohshiro K, Cyanam D, Nair S, Fuqua SA, Polyak K, et al. 2013. RNA sequencing of cancer reveals novel splicing alterations. Sci Rep 3: 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feero WG, Guttmacher AE, Collins FS. 2010. Genomic medicine—An updated primer. N Engl J Med 362: 2001–2011. [DOI] [PubMed] [Google Scholar]
- Font P, Loscertales J, Benavente C, Bermejo A, Callejas M, Garcia-Alonso L, Garcia-Marcilla A, Gil S, Lopez-Rubio M, Martin E, et al. 2013. Inter-observer variance with the diagnosis of myelodysplastic syndromes (MDS) following the 2008 WHO classification. Ann Hematol 92: 19–24. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, Shan J, Faderl S, Cortes J, Ravandi F, Borthakur G, Wierda WG, Pierce S, Estey E, Liu J, et al. 2008. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia 22: 538–543. [DOI] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. 2012. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44: 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, Bennett JM, Bowen D, Fenaux P, Dreyfus F, et al. 2012. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 120: 2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururaj AE, Holm C, Landberg G, Kumar R. 2006. Breast cancer-amplified sequence 3, a target of metastasis-associated protein 1, contributes to tamoxifen resistance in premenopausal patients with breast cancer. Cell Cycle 5: 1407–1410. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. 2014. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser M, Meggendorfer M, Cruz MMA, Fabisch J, Klesse S, Köhler L, Göhring G, Ganster C, Shirneshan K, Gutermuth A, et al. 2015. Frequency and prognostic impact of casein kinase 1A1 (CSNK1A1) mutations in MDS patients with deletion of chromosome 5q. Leukemia 29: 1942–1945. [DOI] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2014. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson L, Larsson J. 2012. Normal hematopoietic stem cell function in mice with enforced expression of the Hippo signaling effector YAP1. PLoS One 7: e32013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, et al. 2001. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 7: 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mtations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer cell 27: 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscielny S. 2010. Why most gene expression signatures of tumors have not been useful in the clinic. Sci Transl Med 2: 14ps2. [DOI] [PubMed] [Google Scholar]
- Lagarde P, Przybyl J, Brulard C, Perot G, Pierron G, Delattre O, Sciot R, Wozniak A, Schoffski P, Terrier P, et al. 2013. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31: 608–615. [DOI] [PubMed] [Google Scholar]
- Li H, Qiu J, Fu XD. 2012. RASL-seq for massively parallel and quantitative analysis of gene expression. Curr Protoc Mol Biol 4: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Ochs MF, Ahn SM, Hennessey P, Tan M, Soudry E, Gaykalova DA, Uemura M, Brait M, Shao C, et al. 2014. Expression microarray analysis reveals alternative splicing of LAMA3 and DST genes in head and neck squamous cell carcinoma. PLoS One 9: e91263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, McCleland M, Stawiski EW, Gnad F, Mayba O, Haverty PM, Durinck S, Chen YJ, Klijn C, Jhunjhunwala S, et al. 2014. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat Commun 5: 3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo YB, Ma JY, Zhang QH, Lin F, Wang ZW, Huang L, Schatten H, Sun QY. 2013. MBTD1 is associated with Pr-Set7 to stabilize H4K20me1 in mouse oocyte meiotic maturation. Cell Cycle 12: 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. 2015. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6: 6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. 2012. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119: 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, Giagounidis A, Hildebrandt B, Bernasconi P, Knipp S, et al. 2007. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 25: 3503–3510. [DOI] [PubMed] [Google Scholar]
- Malouf GG, Su X, Yao H, Gao J, Xiong L, He Q, Comperat E, Couturier J, Molinie V, Escudier B, et al. 2014. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clin Cancer Res 20: 4129–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misquitta-Ali CM, Cheng E, O'Hanlon D, Liu N, McGlade CJ, Tsao MS, Blencowe BJ. 2011. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol 31: 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Fujibuchi W, Sasaki I. 2011. Alternative pre-mRNA splicing in digestive tract malignancy. Cancer Sci 102: 309–316. [DOI] [PubMed] [Google Scholar]
- Mo S, Ji X, Fu XD. 2013. Unique role of SRSF2 in transcription activation and diverse functions of the SR and hnRNP proteins in gene expression regulation. Transcription 4: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura A, Iwata N, Nagata A, Tamekane A, Shimoyama M, Gomyo H, Yakushijin K, Urahama N, Hamaguchi M, Fukui C, et al. 2004. Involvement of casein kinase Iε in cytokine-induced granulocytic differentiation. Blood 103: 2997–3004. [DOI] [PubMed] [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. 2014. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29: 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenn GS, Yocum AK, Menon R. 2010. Alternative splice variants, a new class of protein cancer biomarker candidates: findings in pancreatic cancer and breast cancer with systems biology implications. Dis Markers 28: 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. [DOI] [PubMed] [Google Scholar]
- Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M Jr, Fu XD. 2013. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell 50: 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. 2011. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. 2013. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122: 3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton JE, Grieselhuber NR, Chang LW, Murakami M, Geiss GK, Link DC, Nagarajan R, Watson MA, Ley TJ. 2009. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest 119: 1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellagatti A, Benner A, Mills KI, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, Jadersten M, Verma A, et al. 2013. Identification of gene expression-based prognostic markers in the hematopoietic stem cells of patients with myelodysplastic syndromes. J Clin Oncol 31: 3557–3564. [DOI] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. 2013. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 122: 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage KI, Gorski JJ, Barros EM, Irwin GW, Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ, McCluggage WG, et al. 2014. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol Cell 54: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RK, Adema V, Heckl D, Jaras M, Mallo M, Lord AM, Chu LP, McConkey ME, Kramann R, Mullally A, et al. 2014. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 26: 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PA, Burge CB. 1997. Classification of introns: U2-type or U12-type. Cell 91: 875–879. [DOI] [PubMed] [Google Scholar]
- Sompallae R, Hofmann O, Maher CA, Gedye C, Behren A, Vitezic M, Daub CO, Devalle S, Caballero OL, Carninci P, et al. 2013. A comprehensive promoter landscape identifies a novel promoter for CD133 in restricted tissues, cancers, and stem cells. Front Genet 4: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugnet CW, Kent WJ, Ares M Jr, Haussler D. 2004. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac Symp Biocomput 2004: 66–77. [DOI] [PubMed] [Google Scholar]
- Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, et al. 2015. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6: 6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. 2008. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, 4th ed International Agency for Research on Cancer (IARC) Press 2008, Lyon, France. [Google Scholar]
- Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, et al. 2012. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 119: 3578–3584. [DOI] [PubMed] [Google Scholar]
- Tress ML, Martelli PL, Frankish A, Reeves GA, Wesselink JJ, Yeats C, Olason PI, Albrecht M, Hegyi H, Giorgetti A, et al. 2007. The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci 104: 5495–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530–536. [DOI] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, et al. 2009. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 16: 670–676. [DOI] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, et al. 2012. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 366: 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI. 1991. Characterization of G-protein α subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci 88: 10049–10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, Erlandsson R, Ngara M, Anderson K, Deng Q, et al. 2014. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 25: 794–808. [DOI] [PubMed] [Google Scholar]
- Xiao R, Sun Y, Ding JH, Lin S, Rose DW, Rosenfeld MG, Fu XD, Li X. 2007. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol 27: 5393–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB. 2005. Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci 102: 2850–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pal S, Bi Y, Tchou J, Davuluri R. 2013. Isoform level expression profiles provide better cancer signatures than gene level expression profiles. Genome Med 5: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. 2015. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci 112: E4726–E4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yang L, Li S, Huang J, Chen H, Hou L, Wang J, Green CD, Yan Z, Huang X, et al. 2012a. Midlife gene expressions identify modulators of aging through dietary interventions. Proc Natl Acad Sci 109: E1201–E1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. 2012b. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.