

Abstract
The shikimate pathway of bacteria, fungi, and plants generates chorismate, which is drawn into biosynthetic pathways that form aromatic amino acids and other important metabolites, including folates, menaquinone, and siderophores. Many of the pathways initiated at this branch point transform chorismate using an MST enzyme. The MST enzymes (menaquinone, siderophore, and tryptophan biosynthetic enzymes) are structurally homologous and magnesium-dependent, and all perform similar chemical permutations to chorismate by nucleophilic addition (hydroxyl or amine) at the 2-position of the ring, inducing displacement of the 4-hydroxyl. The isomerase enzymes release isochorismate or aminodeoxychorismate as the product, while the synthase enzymes also have lyase activity that displaces pyruvate to form either salicylate or anthranilate. This has led to the hypothesis that the isomerase and lyase activities performed by the MST enzymes are functionally conserved. Here we have developed tailored pre-steady-state approaches to establish the kinetic mechanisms of the isochorismate and salicylate synthase enzymes of siderophore biosynthesis. Our data are centered on the role of magnesium ions, which inhibit the isochorismate synthase enzymes but not the salicylate synthase enzymes. Prior structural data have suggested that binding of the metal ion occludes access or egress of substrates. Our kinetic data indicate that for the production of isochorismate, a high magnesium ion concentration suppresses the rate of release of product, accounting for the observed inhibition and establishing the basis of the ordered-addition kinetic mechanism. Moreover, we show that isochorismate is channeled through the synthase reaction as an intermediate that is retained in the active site by the magnesium ion. Indeed, the lyase-active enzyme has 3 orders of magnitude higher affinity for the isochorismate complex relative to the chorismate complex. Apparent negative-feedback inhibition by ferrous ions is documented at nanomolar concentrations, which is a potentially physiologically relevant mode of regulation for siderophore biosynthesis in vivo.
Introduction
Menaquinone, siderophore, and tryptophan (MST) biosynthetic enzymes are magnesium-dependent and transform chorismate1 by rearrangement, alteration, and/or elimination of ring substituents. These transformations are observed in biosynthetic pathways for the formation of a variety of siderophores, specific redox cofactors (menaquinone and folates), and tryptophan. The rearrangements include C4 substituent interconversion (hydroxyl to amine) or elimination of the C4 hydroxyl by addition of a hydroxyl or amine at C2 (designated herein as “isomerase” activity), and these processes may or may not be coupled to elimination of pyruvate (“lyase” activity) (Figure 1A). When the two activities are coupled within a single active site (isomerase and lyase), the products are an ortho-substituted benzoic acid (salicylate or anthranilate) and pyruvate (designated herein as “synthase” activity). The chemical mechanism(s) of these reactions have been the subject of conjecture for almost three decades. The original hypotheses for isomerase activity were based on the idea that the magnesium plays a direct catalytic role (involved in the transition state),2 but the first structures indicated that the magnesium ion chelates the substrate’s or product’s benzylic carboxylate.3−8 As a consequence of these structures, more recent mechanistic hypotheses have suggested that the metal ion facilitates nucleophilic attack by enhancing the electrophilicity of the ring carbon C2,1 allowing alteration of the ring substituents, with the nucleophile being either an active-site lysine or an ammonia or water molecule.1,3,4,9,10 Despite the observation of significant magnesium ion inhibition of turnover,11 the influence of the magnesium on the kinetics of the reaction has been mostly overlooked.
Figure 1.

Reactions and representative conformational states of MST enzymes. (A) MST enzymes that isomerize chorismate to isochorismate (isomerase activity) and enzymes that eliminate the pyruvyl enol substituent (lyase activity). Synthase activity refers to enzymes that perform isomerase activity and subsequently perform lyase activity. The color scheme introduced here will be maintained throughout: PchA = red; EntC = green; Irp9 = blue; MenF = purple. PchB (black) is not an MST enzyme; however, PchB performs the lyase reaction and is used in this work to generate the fluorescent salicylate for isomerase-only enzymes. (B) Open (right, PDB ID 3BZM) and closed (left, PDB ID 3BZN) forms of MenF. The closed form has a magnesium ion (gray sphere), but the isochorismate (cyan sticks) was modeled by superposition of the EntC structure (PDB ID 3HWO). In both structures, the amino acids that serve as metal ligands are depicted as green sticks. It should be noted that in the closed structure, the passageway between the active site and the solvent is occluded by the presence of the magnesium ion. In other words, there is no portal for substrate or product entry or egress without dissociation of the catalytic magnesium. The gray mesh shown here represents the surface topology, including cavities, pockets, and voids, as calculated by CASTp.
The MST enzymes have highly similar structural topologies and conserved active-site residues.12 The enzymes are composed of a functional domain with the active site in the pocket formed at the interface of two structural domains. Some of these enzymes contain allosteric binding sites that provide a negative-feedback mechanism for the biosynthetic product of the pathway in which they function. An example is anthranilate synthase (TrpE), the initial enzyme in the pathway to generate tryptophan, for which the activity is downmodulated by binding of tryptophan distant from the active site.13,14 Members of this structural class of enzymes may also work in concert with other proteins, frequently for the generation of nucleophiles. For example, aminodeoxychorismate synthase (PabB) of folate biosynthesis requires the transient formation of a heterocomplex. PabB interacts with the glutamine amidotransferase, PabA, to obtain the ammonia nucleophile.5,15,16 Here we focus on the simplest enzymes of this class, which lack an apparent binding site for negative-feedback inhibition, do not require additional proteins for physiological activity, and use water as a nucleophile. Specifically, we are studying enzymes that isomerize chorismate to isochorismate, the isochorismate synthases, and enzymes that perform this isomerization and then subsequently eliminate the pyruvyl enol substituent, the salicylate synthases (Figure 1A). These include (1) the isochorismate synthase from Pseudomonas aeruginosa (PchA) involved in production of the siderophore pyochelin, (2) the isochorismate synthase from Escherichia coli involved in the production of the siderophore enterobactin (EntC), and (3) the salicylate synthase from Yersinia enterocolitica involved in the production of the siderophore yersiniabactin (Irp9).
Examination of the available structures for the isochorismate4,6,8 and salicylate synthases,1,17−19 and indeed all of the MST enzymes, suggests that these enzymes exist in at least two states: an open form and a closed form. These two conformations are most easily visualized using the structures of the isochorismate synthase from E. coli for production of menaquinone (MenF)6 (Figure 1B). The active site is shaped like a pocket with the magnesium ion serving as the button that fastens the opening, sealing the active site for catalysis and preventing association or egress of substrates and products. This arrangement would imply both ordered association of substrate and metal ion and ordered dissociation of the cofactor and product(s). While the literature has most often concluded that the acquisition of substrates is random,4,20 ordered binding has also been reported.21 Regardless, the metal ion has been typically added to experiments at saturating concentrations.1,3,8−10,18,22−28 The uncertainty of the dynamic interaction of magnesium with MST enzymes and the complications arising from the use of excess metal ion in experiments has potentially undermined prior conclusions drawn from studies of these enzymes. Here we sought to develop new methods to study the chemistry of MST enzymes and provide a comprehensive investigation of the kinetic mechanisms. We also provide evidence that Fe(II) is a tight-binding direct negative-feedback modulator of the activity of these enzymes, which is consistent with their roles in siderophore biosynthesis.
Materials and Methods
Preparation of Overexpression Plasmids
The wild-type pchA(9) and irp9(27) genes were cloned as previously described. The wild-type entc gene was amplified from E. coli strain JM109 genomic DNA by polymerase chain reaction using Vent DNA Polymerase (New England Biolabs). The forward primer (5′-CGA CCG CATATG GAT ACG TCA CTG GCT GAG-3′) includes an NdeI site (underlined), and the reverse primer (5′-GCG CTCGAG TTA ATG CAA TCC AAA AAC GTT C-3′) contains an XhoI site (underlined). The amplified 1 176 base pair fragment was digested with NdeI and XhoI and ligated into the pET15b plasmid (Novagen), modified to replace the thrombin cleavage site to a tobacco etch virus (TEV) protease cleavage site,29 digested with the same enzymes. The expression construct thus encodes a five residue N-terminal histidine tag with a TEV cleavage site. Variant enzymes were produced with QuikChange XL (Agilent) using the wild-type plasmid as the template. The forward primers used are shown in Table 1 and were used in conjunction with complementary reverse primers. The plasmids contained only the desired mutation relative to the wild-type plasmids.
Table 1. Primers Used To Generate Enzyme Variants.
| protein | variant | primer |
|---|---|---|
| PchA | G220D | 5′-CAG GGA CGC TTC GAC AAG GTC GTG CTG-3′ |
| G220V | 5′-CAG GGA CGC TTC GTC AAG GTC GTG CTG-3′ | |
| Irp9 | V192D | 5′-CGC CGT GGC GAG TAT GAC AAA GTG ATT GTC TCG-3′ |
| V192G | 5′-CGC CGT GGC GAG TAT GGC AAA GTG ATT GTC TCG-3′ | |
| EntC | D146G | 5′-CAC GCC GCA GGT CGG CAA AGT GGT GTT GT-3′ |
| D146V | 5′-CAC GCC GCA GGT CGT CCA AGT GGT GTT-3′ |
Protein Overexpression and Purification
Wild-type and variant Irp9,27 wild-type and variant PchA,9 and PchB30 were purified as previously described. BL21 (DE3) E. coli containing the EntC wild-type or variant expression plasmid was grown in LB medium containing 200 μg/mL ampicillin at 37 °C with shaking (250 rpm) until an optical density at 600 nm (OD600) of ∼0.8 was reached. Protein production was induced in the culture with the addition of 1 mM isopropyl β-d-thiogalactopyranoside, and the temperature was reduced to 16 °C. The cells were harvested by centrifugation (6 000g, 10 min, 4 °C) after 20–22 h. The cell pellet was resuspended in 10 mL of buffer A (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, 150 mM NaCl, 10 mM imidazole, 0.2 mM tris(2-carboxyethyl)phosphine (TCEP)) per liter of culture. Cells were disrupted using a French pressure cell (35 000 psi), and cellular debris was removed by centrifugation (12 000g, 45 min, 4 °C). The supernatant was applied to a chelating Sepharose fast-flow column (GE Healthcare) charged with nickel chloride and pre-equilibrated in buffer A. EntC protein was eluted with a linear gradient of 10–300 mM imidazole in buffer A. The pooled fractions were applied to a Superdex 200 size-exclusion column (GE Healthcare) equilibrated with 50 mM HEPES (pH 8) containing 150 mM NaCl and 0.2 mM TCEP. The fractions containing EntC were pooled and concentrated using an Amicon stirred cell with a YM-30 membrane to 5–25 mg/mL as determined by the Bradford assay.
EntC protein used for crystallization had the histidine tag removed by cleavage with TEV protease. EntC was eluted from the nickel column and incubated with TEV protease at a 50:1 ratio and dialyzed overnight in 50 mM HEPES (pH 7.5) containing 100 mM NaCl, 0.5 mM ethylenediaminetetraacetic acid (EDTA), and 0.2 mM TCEP at 4 °C. The cleaved protein was reapplied to the nickel column, and the fractions that did not bind to the column were concentrated and applied to the Superdex 200 size-exclusion column equilibrated with 50 mM HEPES (pH 8) containing 150 mM NaCl and 0.2 mM TCEP. The fractions containing EntC were pooled and concentrated, and the protein concentration was determined by the Bradford assay. The concentrated protein was stored at −80 °C.
Preparation of Chorismate and Isochorismate
Chorismate was isolated from Klebsiella pneumoniae 62-1 as previously described31 with a few modifications.9 Isochorismate was isolated from the same strain harboring the entC plasmid pKS3-0232 with only minor changes.33
Steady-State Activity Assays
The standard assay buffer contained 50 mM Tris-HCl (pH 7.5), 10% glycerol, and various concentrations of magnesium chloride (0–20 mM). Chorismate isomerase activity was determined for PchA or EntC with PchB in 100-fold excess and chorismate as the substrate, ensuring that any isochorismate produced by PchA or EntC would be instantaneously converted to salicylate by PchB for detection by fluorescence. Salicylate synthase activity was determined for Irp9 using chorismate as the substrate and monitoring salicylate formation by fluorescence. All of the isochorismate isomerase and salicylate synthase assays were performed on a TgK Scientific stopped-flow device operated at 25 °C using a mercury–xenon lamp with the monochromator set to 310 nm (excitation), detecting all light (beyond a 360 nm cutoff filter) emitted perpendicular to the light source. Enzyme in standard assay buffer was mixed in the stopped-flow apparatus in a 1:1 volume ratio with reaction buffer containing chorismate. For the magnesium ion inhibition experiments, the final concentration of chorismate was saturating at 20 μM for the PchA and Irp9 assays and 200 μM for the EntC assays with magnesium concentrations varying from 0 to 20 mM. For assays varying the chorismate concentration, the final concentrations of chorismate were 0–20 μM for the PchA and Irp9 assays and 0–200 μM for the EntC assays at 0.5 and 10 mM magnesium chloride. The final enzyme concentrations used for the chorismate isomerase assays were 100 nM for wild-type and variant PchA and EntC and 10 μM for PchB (∼300-fold excess activity). The final enzyme concentrations for Irp9 salicylate synthase assays were 100 nM for the wild type and variants. For assays measuring PchB lyase activity, the PchB concentration was 100 nM with isochorismate concentrations ranging from 0 to 25 μM at 0.5 and 10 mM MgCl2. It should be noted that PchB does not bind and is not inhibited by magnesium. For iron inhibition experiments, the magnesium concentration was 0.5 mM, and the ferrous ammonium sulfate concentration was varied from 0 to 160 μM. The enzyme and chorismate concentrations were the same as those used for magnesium inhibition experiments. Steady-state kinetic data were fit to the Michaelis–Menten equation. Magnesium and iron inhibition data were fit to the general form of the substrate inhibition equation (eq 1):
| 1 |
Ligand Dissociation Constants by Perturbation of Intrinsic Fluorescence
The binding of chorismate, isochorismate, salicylate, pyruvate, magnesium, and iron to MST enzymes was observed by perturbation of the intrinsic fluorescence of each enzyme with the addition of ligand. For each titration, PchA (0.75 μM), EntC (0.75 μM), or Irp9 (0.1 μM) was prepared in 50 mM Tris buffer (pH 7.5) containing 10% glycerol (100 μM EDTA was added for titration of substrate ligands). Emission spectra from 300 to 500 nm were recorded using a Cary Eclipse fluorescence spectrometer (Varian) with a temperature controller. Incident light was set at 280 nm, producing an emission maximum at ∼335 nm. For each ligand the equilibrium fluorescence decrease that occurred upon binding was recorded. Ligands were titrated to approximate saturation (0–60 μM for chorismate, isochorismate, salicylate and pyruvate; 0–25 mM for magnesium chloride; 0–50 μM for ferrous ammonium sulfate). After correction for dilution, the change in amplitude at 335 nm was plotted against the total ligand concentration. Magnesium ion binding was fit to the hyperbolic form of the single-site binding isotherm equation (eq 2):
| 2 |
where f is the fractional saturation and [Mg] is the total magnesium concentration (assuming that the correction for the fraction bound is negligible).
Chorismate, isochorismate, salicylate, and pyruvate (ligands, denoted as L) were fit to the quadratic form of the single-site ligand binding equation added to a straight line (eq 3):
| 3 |
In this equation, the slope of the linear term (M) accounts for incident-light inner-filter effects that arise from overlapping absorption spectral contributions of chorismate, isochorismate, salicylate, or pyruvate. Ferrous ion binding was also best fit to eq 3, with the linear term required to account for apparent signal alterations that occur at relatively high concentrations of ferrous ions.
Ligand Binding Rates
The rates of binding of chorismate, isochorismate, and magnesium to the MST enzymes were determined using stopped-flow spectroscopy at 25 °C. The change in intrinsic tryptophan fluorescence was monitored utilizing a 320 nm cutoff filter upon excitation at 280 nm with a mercury–xenon lamp. In each case, the enzyme was prepared in 50 mM Tris buffer (pH 7.5) containing 10% glycerol, with the addition of 200 μM EDTA to chelate trace magnesium from the solution, and mixed against two concentrations of ligand, subsaturating and saturating. The enzyme final concentrations were 0.75 μM for PchA, 0.75 μM for EntC, and 0.1 μM for Irp9, and the final ligand concentrations were 0.5 or 5 μM for chorismate, 0.5 or 5 μM for isochorismate, and 0.3125 or 1.25 mM for magnesium. The chorismate and isochorismate binding rates were determined on two separate days, with three shots per trace on each day. The magnesium binding rates were determined on three separate days, with three shots per trace on each day. Representative traces are shown in Figure 6D.
Figure 6.
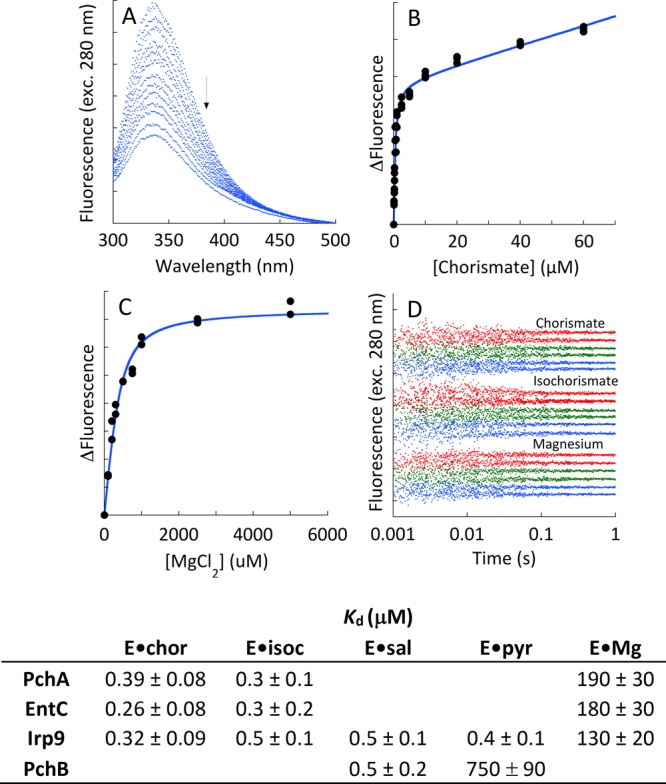
Dissociation constants of ligands from MST enzymes. (A) The titration depicts the perturbation of intrinsic tryptophan fluorescence that is observed when chorismate is titrated to Irp9. The arrow denotes increasing chorismate concentration. (B) The fit of the change in fluorescence to a single binding isotherm plus a linear term that accounts for chorismate inner filter, as described by eq 3. (C) Titration of Irp9 with magnesium showing the fit of the change in fluorescence to a single binding isotherm. (D) Kinetics of ligand binding. EntC (green, 0.75 μM), PchA (red, 0.75 μM), and Irp9 (blue, 0.1 μM) when mixed with chorismate (0.5 μM upper trace, 5 μM lower trace), isochorismate (0.5 μM upper trace, 5 μM lower trace), and magnesium (0.310 mM upper trace, 1.25 mM lower trace). For each ligand set, pairs of traces have been separated for clarity. The table includes dissociation constants of all of the native ligands from each of the enzymes studied as measured by comparable methods.
Single-Turnover Experiments
Single turnover of chorismate for PchA and EntC and chorismate and isochorismate for Irp9 was achieved by double-mixing stopped-flow spectrophotometry at 25 °C. In each case, the E·S complex was formed by mixing enzyme (20 μM, with 400 μM EDTA) with substrate (2 μM). EDTA was added to the enzyme to chelate trace magnesium from the solution prior to mixing; after the double mix, the EDTA concentration was 100 μM. The E·S complex was aged for 0.5 s and mixed with a variety of magnesium concentrations (0–3.6 mM for chorismate reactions and 0–300 μM for isochorismate reactions, which also included excess PchB (50 μM final) for PchA and EntC reactions). The data obtained monitored total salicylate fluorescence measured perpendicular to the light source (utilizing a 360 nm cutoff filter) with excitation at 310 nm provided by a mercury–xenon lamp. The data were fit to eq 4, an expression that describes a monophasic first order reaction:
| 4 |
The dependence of the observed rate constant on the concentration of magnesium was fit to eq 3 (without the added linear term, M[L]) to calculate the limiting rate for the catalytic step(s) (where klimit and kobs were substituted for the [E] and [EL] terms, respectively) and the dissociation constant of magnesium from the E·S·Mg complex (KL).
The Irp9 single-turnover data obtained with isochorismate as a substrate were fit to a comprehensive kinetic model to obtain an estimate of the high-affinity dissociation constant of magnesium from the Irp9·isochorismate·Mg complex. This model included all of the steps depicted in the lyase reaction of Scheme 1 and an EDTA·Mg equilibrium. In this model, each of the ligand-binding equilibrium constants for Irp9 and EDTA was defined by a fixed ratio of rate constants according to known or measured values (data herein). Global fitting numerical integration was used to optimize only the ratio of rate constants defining the dissociation constant of magnesium from the Irp9·isochorismate·Mg complex and the value of the rate constant for the lyase chemical reaction.
Scheme 1. Overall Kinetic Model for MST Enzymes.

Single-turnover experiments were performed on three separate days (Figure 7A–C), or two separate days (Figure 7D). Each trace was measured two or three times on each day. Representative families of curves are shown in Figure 7.
Figure 7.

Single-turnover reactions of Irp9, EntC, and PchA. Single-turnover conditions were established on the basis of the Kd values for substrates obtained by titration of each enzyme’s intrinsic fluorescence (Figure 6). (A, C, D) Chorismate or (B) isochorismate was added to an enzyme concentration sufficient to provide greater than 90% substrate bound. This complex was prepared in a buffer containing EDTA (100 μM final concentration after double mix) to ensure that no turnover occurred prior to mixing with magnesium ions. The E·S complex was then mixed with pseudo-first-order concentrations of Mg(II). The Irp9 reactions are shown in (A) chorismate and (B) isochorismate. The (C) EntC and (D) PchA reactions with chorismate included excess PchB in the second mix. These conditions approximate first-order conditions under the assumption that the release of Mg(II) and products are fast relative to reversible catalytic steps (Figure 6D). The data were fit to single-exponential events (eq 4), and the dependence of the observed rate constant is shown in the inset of each plot.
Suppression of the Isochorismate Release Rate by Exogenous Magnesium
Modulation of the rate of release of isochorismate from PchA and EntC was determined by double-mixing stopped-flow spectrophotometry at 25 °C. The data obtained monitored total salicylate fluorescence measured perpendicular to the light source (utilizing a 360 nm cutoff filter) upon excitation at 310 nm with a mercury–xenon lamp. In each case, 20 μM enzyme with 400 μM EDTA was mixed with 2 μM isochorismate, and the mixture was allowed to age for 0.5 s before subsequent mixing with PchB and various concentrations of magnesium. The postmixing concentrations used were 5 μM for PchA and EntC, 5 μM for PchB, 0.5 μM for isochorismate, and 0–20 mM for magnesium. In these experiments, the isochorismate release was registered as salicylate formation via the activity of PchB. The isochorismate release data obtained were fit to a comprehensive kinetic model to obtain the dissociation constant of magnesium from the E·isochorismate·Mg complex on the basis of the dependence of the suppression of the observed isochorismate release rate. This model included all of the steps depicted in the isomerase reaction of Scheme 1, an EDTA·Mg equilibrium, and the PchB catalytic cycle. In this model, each of the ligand-binding equilibrium constants for E·S and EDTA was defined by a fixed ratio of rate constants according to known or measured values (data herein). Rate constants for catalytic steps for EntC and PchA were also fixed to the values obtained from single-turnover reactions. Global fitting numerical integration was used to optimize only the ratio of rate constants defining the dissociation constant of magnesium from the E·isochorismate·Mg complex and the value of the rate constant for the PchB lyase chemical reaction.
Experiments on the suppression of isochorismate release were performed on four separate days. Each trace was measured two or three times on each day. Representative families of curves are shown in Figure 8.
Figure 8.
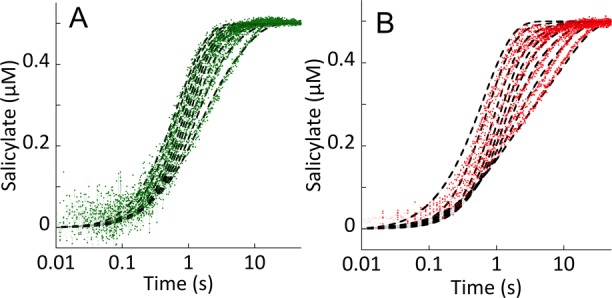
Suppression of the rate of isochorismate release with exogenous magnesium. (A) EntC (5 μM final concentration in the presence of 100 μM EDTA final concentration) was mixed with 2 μM isochorismate (0.5 μM final concentration), and the mixture was allowed to age for 0.5 s before subsequent mixing with 0–20 mM magnesium (increasing Mg concentration left to right) and PchB (5 μM final concentration). (B) PchA (5 μM final concentration in the presence of 100 μM EDTA final concentration) was mixed with 2 μM isochorismate (0.5 μM final concentration), and the mixture was allowed to age for 0.5 s before subsequent mixing with 0–20 mM magnesium (increasing Mg concentration left to right) and PchB (5 μM final concentration). For both enzymes, isochorismate release was visualized by the increase in fluorescence due to the activity of PchB. The data were modeled and fit using the numerical integration routine available within KinTek Explorer (dashed black lines). The model included all of the relevant kinetic steps, including those for PchB and EDTA. All known equilibrium constants and rate constants were fixed, and only the steps required to form or dissociate the E·isochorismate·Mg complex were optimized to obtain the fits.
Kinetic Analysis
Nested sets of primary kinetic traces were fit globally to a single model using KinTek Explorer (KinTek Corporation).34,35 Concentration dependence data were fit using KaleidaGraph graphing software (Synergy Software). All of the signals were calibrated to the salicylate concentration using a standard curve constructed by exciting at 310 nm and recording the total fluorescence emission collected at 90° with respect to the light source utilizing a 360 nm cutoff filter.
Crystallization of EntC
Native EntC crystallization was carried out by the microbatch method at 25 °C. Drops containing 1.3 μL of purified EntC protein in 10 mM HEPES (pH 8.0) containing 150 mM NaCl, 0.2 mM TCEP, and 1.5 mM chorismate were mixed with equal volumes of reservoir solution composed of 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) (pH 6.0), 63% poly(ethylene glycol) (PEG) 200, and 0.05 M ammonium chloride. Large diamond crystals (500 μm × 300 μm × 300 μm) formed after 3 days. The low-magnesium crystals were initially intended to provide an inhibitor-bound structure and as such were soaked with 16 mM 4,6-dinitro-2-oxo-1,3-benzoxathiol-5-yl methyl carbonate (inhibitor 10 in ref (36)) for 45 min before flash cooling at −160 °C; however, none of the compound was evident in the electron density maps. High-magnesium EntC crystals were grown similarly to the low-magnesium crystals in a solution containing 0.1 M MES (pH 6.2), 50% PEG 200, and 0.05 M ammonium chloride. The crystals were soaked in mother liquor supplemented with 50 mM MgCl2 for 15 min and then flash-cooled at −160 °C. Iron-bound EntC crystals were grown similarly to low-magnesium crystals in a solution containing 0.1 M MES (pH 6.0), 60% PEG 200, and 0.05 M ammonium chloride. The crystals were soaked with 16 mM ferrous ammonium sulfate for 45 min and then flash-cooled at −160 °C.
Data Collection and Structure Solution for EntC
Native EntC diffraction data (0.2° oscillation images for a total of 180°) were collected at the Stanford Synchrotron Radiation Laboratory (SSRL) (Stanford, CA) on beamline 7-1 at a wavelength of 1.100 Å at 100 K. The exposure time per frame was 7.94 s with 0% attenuation and a crystal to detector distance of 225.0 mm. The data were indexed and scaled with XDS37 to 1.88 Å. The crystals were assigned to space group P41212 with unit cell dimensions a = b = 80.21 Å and c = 272.20 Å. Molecular replacement calculations were performed using the program PHASER38 in Phenix.39 Molecule A of the published EntC structure (PDB ID 3HWO) with the isochorismate, magnesium, and waters removed was used as a search model (for this and all subsequent structures of EntC) and yielded a clear solution (log likelihood gain of 13 074) with two protein molecules in the asymmetric unit. The map generated with this solution indicated clear density for the ligand in the active site. Model building and refinement were performed using Coot40 and Phenix Refine.41 Waters were added automatically in Phenix, and the positions were verified following a refinement cycle. Molecule A contains residues 15–106 and 110–390 (of 391), and molecule B contains residues 14–107, 112–253, 256–285, and 288–390. Ramachandran analysis as calculated by MolProbity42 showed a good geometry with 97.81% of the residues in the favored region and no residues in the outlier region. The structure contains one magnesium ion per active site. While the crystals were grown in the presence of chorismate, the electron density is clearly indicative of a mixture of substrate and product. Partial occupancies were refined, with 0.54 for chorismate and 0.46 for isochorismate in molecule A and 0.56 for chorismate and 0.44 for isochorismate in molecule B, in close agreement with the solution equilibrium values for this enzyme.2,27 The model includes 269 water molecules.
High-Mg EntC diffraction data (0.15° oscillation images for a total of 102°) were collected on SSRL beamline 12-2 at a wavelength of 0.979 Å at 100 K. The exposure time per frame was 0.2 s with 80% attenuation and a crystal to detector distance of 350.0 mm. The data were indexed and scaled with XDS to 2.11 Å. The crystals were assigned to space group P41212 with unit cell dimensions a = b = 80.91 Å and c = 265.23 Å. Molecular replacement calculations were performed using the program PHASER in Phenix, yielding a clear solution with a log likelihood gain of 8 378. The map generated with this solution indicated clear density for the ligand and magnesium ion in the active site. Model building and refinement were performed using Coot and Phenix Refine. Waters were added automatically in Phenix, and the positions were verified following a refinement cycle. Molecule A contains residues 15–389, and molecule B contains residues 15–107 and 111–391. Cysteine35 of the A monomer was modeled as sulfenic acid because of electron density consistent with oxidation. Ramachandran analysis as calculated by MolProbity showed a good geometry with 95.29% of the residues in the favored region and 0.13% of the residues in the outlier region. The structure contains one magnesium ion per active site. The ligand was modeled only as isochorismate and the model has 142 water molecules.
Iron-bound EntC single anomalous dispersion diffraction data (0.2° oscillation images for a total of 180°) were collected on SSRL beamline 7-1 at a wavelength of 1.739 Å at 100 K. The exposure time per frame was 14.71 s with 0% attenuation and a crystal to detector distance of 225.0 mm. The data were indexed and scaled with XDS to 2.94 Å. The crystals were assigned to space group P41212 with unit cell dimensions a = b = 81.381 Å and c = 262.28 Å. Molecular replacement calculations were performed using the program PHASER in CCP4,43 yielding a clear solution with a log likelihood gain of 5 958. The map generated with this solution had a clear peak for iron in place of each active-site magnesium and two iron peaks on the protein periphery (anomalous map contoured at 5σ). Model building and refinement were performed using Phenix Refine. The final Fe-bound EntC model contains two monomers per asymmetric unit. The structure contains four Fe and two chorismate molecules. The resolution was insufficient for reliable placement of water molecules. Molecule A contains residues 16–106 and 114–390, and molecule B contains residues 16–105, 113–205, and 207–390. Ramachandran analysis as calculated by MolProbity showed a good geometry with 92.97% of the residues in the favored region and 0.14% of the residues in the outlier region.
For detailed analysis of the published electron density, structure factors and coordinates for the deposited EntC structure (PDB ID 3HWO(8)) were downloaded. The crystals that produced these data were grown under different conditions than those documented above (0.1 M MES (pH 6.5) with 12% PEG 20 000 using the hanging drop method), and the data belong to space group P21212 with unit cell dimensions a = 62.47 Å, b = 104.79 Å, and c = 140.01 Å.8 The waters and ligands were removed from the coordinates, and the protein component was subjected to a cycle of simulated annealing in Phenix Refine. The map generated indicated clear density for the ligand and magnesium ion in the active site, but the second magnesium was not readily evident in the second site of monomer A (loop 141-ATPQVDK-147). Monomer B had poor density for the same loop. The structure was then re-refined with cycles of model building in Coot and refinement with Phenix Refine. Waters were added automatically in Phenix, and the positions were verified following a refinement cycle. Molecule A contains residues 14–391, and molecule B contains residues 15–391. Ramachandran analysis as calculated by MolProbity showed a good geometry with 94.14% of the residues in the favored region and 0.13% of residues in the outlier region. The structure contains one magnesium ion per monomer in the active site. The ligand was modeled as isochorismate and has 78 water molecules.
Crystallization of Iron-Bound Irp9
Irp9 crystallization was carried out by the hanging drop method at 18 °C using the previously published conditions.3 Drops containing 1.5 μL of purified Irp9 protein containing 2.5 mM chorismate were mixed with equal volumes of reservoir solution composed of 0.1 M MES (pH 6.5), 14% PEG 8 000, and 0.25 M magnesium acetate. Large crystals (300 μm × 300 μm × 300 μm) formed after 5 days. The crystals were soaked with reservoir solution supplemented with 15 mM ferrous ammonium sulfate for 5 min, serially washed in reservoir solution supplemented with 1.25 mM ferrous ammonium sulfate and 26% (v/v) ethylene glycol as a cryoprotectant, and flash-cooled at −160 °C.
Data Collection and Structure Solution for Iron-Bound Irp9
Iron-bound Irp9 single anomalous dispersion diffraction data (0.2° oscillation images for a total of 344°) were collected on SSRL beamline 7-1 at a wavelength of 1.739 Å at 100 K. The exposure time per frame was 4.14 s with 0% attenuation and a crystal to detector distance of 153.8 mm. The data were indexed and scaled with XDS to 2.16 Å. The crystals were assigned to space group P21 with unit cell dimensions a = 56.39 Å, b = 145.35 Å, c = 58.44 Å, and β = 108.02°. Molecular replacement calculations were performed using the program PHASER in CCP4. Molecule A of the salicylate-, pyruvate-, and magnesium-bound Irp9 structure (PDB ID 2FN1) with the ligands, metal, and waters removed was used as a search model, yielding a clear solution with a log likelihood gain of 11 112. The map generated with this solution had a clear peak for iron (anomalous map contoured at 5σ) in place of each active-site magnesium. Model building and refinement were performed using Phenix Refine. Waters were added automatically in Phenix, and the positions were verified following a refinement cycle. The final Fe-bound Irp9 model contains two monomers per asymmetric unit. The structure contains one Fe2+ (at the location of the active-site magnesium ion) per monomer, two acetates bound in the active site of molecule A, one sulfate and one acetate in the active site of molecule B, and 155 water molecules. Molecule A contains residues 5–8, 16–139, and 162–434 (of 434), and molecule B contains residues 2–9, 12–153, and 161–434. Ramachandran analysis as calculated by MolProbity showed a good geometry with 95.57% of the residues in the favored region and 0.12% of the residues in the outlier region.
Structural Analysis
Data collection and refinement statistics for the native, high-Mg, and re-refined 3HWO structures can be found in Table 2, whereas the statistics for the iron-anomalous structures of EntC and Irp9 are found in Table 3. Structural comparisons (including root-mean-square deviation calculations) were conducted using PDBeFold.44 Protein structure figures were generated using PyMOL.45 The surface topology (including cavities, pockets, and voids) was calculated using CASTp.46 The atomic coordinates and structure factors have been deposited in the Protein Data Bank (Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ) as entries 5JXZ, 5JY4, 5JY8, 5JY9, and 5JZD.
Table 2. EntC Data Collection and Refinement Statistics.
| low-Mg | high-Mg | 3HWO-re-refined | |
|---|---|---|---|
| Data Collectiona | |||
| wavelength (Å) | 1.100 | 0.979 | |
| space group | P41212 | P41212 | |
| cell dimensions | |||
| a (Å) | 80.21 | 80.91 | |
| b (Å) | 80.21 | 80.91 | |
| c (Å) | 272.20 | 265.23 | |
| resolution (Å) | 39.49–1.88 (1.92–1.88) | 38.90–2.11 (2.17–2.11) | |
| Rsymb | 0.081 (0.751) | 0.074 (1.031) | |
| Rpim | 0.046 (0.451) | 0.031 (0.433) | |
| total observations | 526488 (28537) | 384657 (30695) | |
| total unique observations | 73403 (4306) | 51991 (4113) | |
| mean (I/σ(I)) | 14.2 (2.0) | 14.5 (2.0) | |
| completeness (%) | 99.8 (96.4) | 99.7 (97.8) | |
| redundancy | 7.2 (6.6) | 7.4 (7.5) | |
| Refinement | |||
| resolution (Å) | 39.49–1.88 (1.93–1.88) | 38.90–2.11 (2.16–2.11) | 41.95–2.30 (2.36–2.30) |
| Rcrystc | 18.71 (25.90) | 20.07 (25.90) | 19.53 (28.84) |
| Rfree | 22.91 (31.13) | 24.83 (31.08) | 25.56 (33.21) |
| total unique observations | 73207 (4840) | 51881 (3424) | 41374 (2462) |
| no. of non-hydrogen atoms | |||
| protein | 5712 | 5788 | 5843 |
| ligand | 64 | 32 | 32 |
| metal | 2 | 2 | 2 |
| water | 269 | 142 | 78 |
| bond rmsd (Å) | 0.013 | 0.014 | 0.013 |
| angle rmsd (deg) | 1.118 | 1.273 | 1.287 |
| overall mean B factor (Å2) | 24.61 | 41.71 | 42.07 |
| Ramachandran plot analysisd | |||
| most favored regions | 97.81 | 95.29 | 94.14 |
| additionally allowed regions | 2.19 | 4.58 | 5.73 |
| disallowed regions | 0 | 0.13 | 0.13 |
Data were indexed and scaled with XDS.
Rsym = ∑h|Ih – ⟨I⟩|/ΣhIh, where Ih is the intensity of reflection h and ⟨I⟩ is the mean intensity of all symmetry-related reflections.
Rcryst = ∑||Fo| – |Fc||/∑|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes. Five percent of the relections were reserved for the calculation of Rfree.
Calculated with MolProbity.
Table 3. Iron-Bound EntC and Irp9 Data Collection and Refinement Statistics.
| Fe-EntC | Fe-Irp9 | |
|---|---|---|
| Data Collectiona | ||
| wavelength (Å) | 1.739 | 1.739 |
| space group | P41212 | P21 |
| cell dimensions | ||
| a (Å) | 81.38 | 56.39 |
| b (Å) | 81.38 | 145.35 |
| c (Å) | 262.28 | 58.44 |
| β (deg) | – | 108.02 |
| resolution (Å) | 38.86–2.94 (3.12–2.94) | 39.12–2.16 (2.23–2.16) |
| Rsymb | 0.167 (0.754) | 0.096 (0.729) |
| Rpim | 0.044 (0.204) | 0.039 (0.355) |
| total observations | 541745 (82062) | 641609 (33764) |
| total unique observations | 19603 (3001) | 47172 (3604) |
| mean (I/σ(I)) | 21.6 (5.6) | 18.3 (3.0) |
| completeness (%) | 99.4 (96.7) | 98.9 (88.1) |
| redundancy | 27.6 (27.3) | 13.6 (9.4) |
| anomalous completeness (%) | 99.4 (96.5) | 98.0 (79.3) |
| anomalous redundancy | 15.2 (14.6) | 6.8 (5.0) |
| DelAnom correlation between half-setsc | 0.128 (−0.017) | –0.056 (0.014) |
| mid-slope of anom normal probabilityd | 1.039 | 0.945 |
| Refinement | ||
| resolution (Å) | 38.86–2.94 (3.02–2.94) | 36.52–2.16 (2.21–2.16) |
| Rcryste | 18.00 (22.88) | 17.20 (22.47) |
| Rfree | 26.67 (37.91) | 23.48 (29.15) |
| total unique observations | 19521 (1152) | 47138 (2205) |
| no. of non-hydrogen atoms | ||
| protein | 5663 | 6419 |
| ligand | 32 | 17 |
| metal | 4 | 2 |
| water | 0 | 155 |
| bond rmsd (Å) | 0.03 | 0.013 |
| angle rmsd (deg) | 1.471 | 1.214 |
| overall mean B factor (Å2) | 47.68 | 29.16 |
| Ramachandran plot analysisf | ||
| most favored regions | 93.24 | 95.57 |
| additionally allowed regions | 6.48 | 4.31 |
| disallowed regions | 0.28 | 0.12 |
Data were indexed and scaled with XDS.
Rsym = ∑h|Ih – ⟨I⟩|/∑hIh, where Ih is the intensity of reflection h and ⟨I⟩ is the mean intensity of all symmetry-related reflections.
Anomalous correlation between half-sets = ∑||FPH ± FP| – FHcalc|/∑|FPH| reported for all centric reflections.
Mid-slope of anomalous probability = ⟨|FH|/||FP + FH| – |FPH||⟩ reported for all reflections.
Rcryst = ∑||Fo| – |Fc||/∑|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes. Five percent of the relections were reserved for the calculation of Rfree.
Calculated with MolProbity.
Results
Isochorismate Synthases Are Inhibited by Magnesium in the Steady State
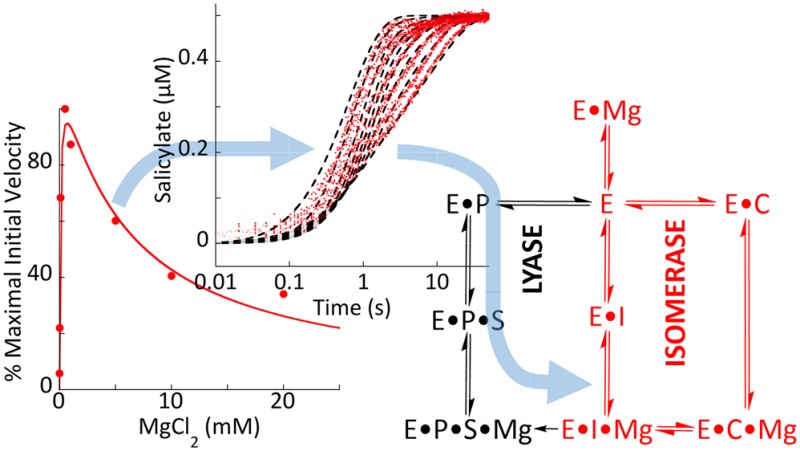
Michaelis–Menten analyses were performed for the chorismate isomerase enzymes, PchA and EntC. When the magnesium ion concentration was varied, the curve obtained was not hyperbolic but showed a shape indicative of magnesium enhancement of the reaction at low concentration and inhibition at high concentration (Figure 2A). For both enzymes, the maximal velocity was observed at approximately 0.5 mM magnesium. In contrast, the salicylate synthase, Irp9, showed a normal hyperbolic shape for the same range of magnesium ion concentrations. Michaelis–Menten analyses at 0.5 mM (solid lines) and 10 mM (dashed lines) magnesium were compared with chorismate as the varied factor for all three enzymes (Figure 2B). The value of vmax is 1.3- to 3-fold greater at lower magnesium concentrations (Figure 2B is normalized for vmax), but Km for chorismate is decreased by 5- to 9-fold (Figure 2C). This dictates that the overall specificity constant for chorismate improves by 5- to 14-fold with a 20-fold increase in magnesium concentration. These initial velocity data suggest that all three enzymes acquire their substrate and cofactor by a common kinetic mechanism and that the isochorismate synthases are susceptible to an additional magnesium binding process that results in suppression of the rate of turnover that is not experienced by the salicylate synthase, Irp9.
Figure 2.

Influence of magnesium ions on the steady-state kinetics of MST enzymes. (A) Plots of percent of maximal velocity vs MgCl2 concentration for EntC (circles, green line), PchA (squares, red line), and Irp9 (diamonds, blue line). The effect of the magnesium ion on vmax is apparent in this panel. It should be noted that magnesium-dependent inhibition is evident for the isochorismate synthases (isomerases) (EntC and PchA) but not for the salicylate synthase (Irp9). Inset: an enlargement of the upper left-hand corner of the plot. (B) Plots of percent of maximal velocity vs chorismate concentration at 0.5 mM MgCl2 for EntC (circles, solid green line), PchA (squares, solid red line), and Irp9 (diamonds, solid green line) and at 10 mM MgCl2 for EntC (circles, dashed green line), PchA (squares, dashed red line), and Irp9 (diamonds, dashed blue line). The decrease in the chorismate Km with increasing magnesium concentration is easily recognizable in this panel. (C) Table of steady-state kinetic values derived from (B).
A Potential Second Metal Binding Site
The structure of EntC (PDB ID 3HWO) has magnesium and isochorismate bound in the active site (the gray sphere labeled “1” in Figure 3A).8 Each monomer also has a second magnesium modeled in the structure, in the loop that precedes the strand containing the general base lysine 147 (gray sphere labeled “2” in Figure 3A). This magnesium is chelated by backbone carbonyls and the side-chain carboxylate of aspartic acid 146. There is also the potential for the helix dipole to assist in the binding interaction, since this loop is at the C-terminus of a helix. In PchA, the residue comparable to Asp146 in EntC is a glycine, whereas in Irp9 this residue is a valine (Figure 3B). Since one potential model for the observed magnesium inhibition is binding at a second site, we generated interconverting variants of this proposed chelating residue (Figure 3C–E). In measurements of Michaelis–Menten kinetics with magnesium as the varied factor, the EntC variants (Figure 3D) and PchA variants (Figure 3E) did not abolish or significantly diminish the magnesium ion inhibition. In contrast, the V192G-Irp9 variant gave a curve with a shape indicative of magnesium inhibition (Figure 3C).
Figure 3.

Proposed second magnesium binding site. (A) The magnesium binding sites in EntC (PDB ID 3HWO). The catalytic magnesium is labeled 1, and the additional site is labeled 2. Isochorismate is shown in cyan sticks. The general base (K147) and general acid (E197) are shown in yellow. The residues of the turn that bind the secondary magnesium are shown in green. This turn connects an α-helix to the β-strand that is initiated at the general base. (B) Sequence alignment for the loop highlighted in (A), comparing PchA, EntC, and Irp9. It should be noted that the residue side chain (colored) that chelates the second magnesium in the 3HWO structure is not conserved among the three proteins. The lysine in bold is the general base residue. (C) Steady-state magnesium dependence for three forms of Irp9: WT (circles), V192D (squares), and V192G (triangles). (D) Steady-state magnesium dependence for three forms of EntC: WT (circles), D146G (squares), and D146V (triangles). (E) Steady-state magnesium dependence for three forms of PchA: WT (circles), G220D (squares), and G220V (triangles).
The EntC Structure Has an Equilibrium Mixture of Chorismate and Isochorismate in the Active Site
We were unsuccessful in producing EntC crystals using the published vapor diffusion conditions with PEG 20 000 as the precipitant.8 A new crystallization protocol was developed that uses a microbatch method and PEG 200 as the precipitant. These crystals belong to a different space group: P41212, as opposed to the previously determined P21212. The structure was easily determined by molecular replacement using the previously determined 3HWO structure as the model, with two monomers in the asymmetric unit. Analysis in PDBeFold calculated a root-mean-square deviation of 0.50–0.52 Å for 371–373 α-carbons, depending on the monomer, illustrating that the structures are highly similar and that there are no major conformational changes to loops or deviations in fold, with the exception of one disordered loop in monomer A at residues 107–109 and three minor chain breaks in monomer B at residues 108–111, 254–255, and 286–287. In other words, the structure has the expected α+β fold of chorismate-utilizing enzymes, as shown in Figure 4A, and is highly similar to the previously determined structure.
Figure 4.

Low-magnesium EntC structure. (A) The overall topology of the EntC structures presented here is not changed from the previously determined structure. A cartoon of the fold is shown in pale green, with the catalytic magnesium ion shown as a gray sphere. Isochorismate (the product) is shown in pale cyan, whereas chorismate (the substrate) is shown in deep teal. (B) A closeup of the active site for the low-magnesium structure is shown, with colors as in (A). In addition, the general acid (E197) and general base (K147) are shown as yellow sticks, and the magnesium-ligand residues (E241 and E376) are shown in pale-green sticks. Two water molecules also act as ligands of the magnesium (red spheres), and one of the residues that holds these waters in place is visible in this view (D238). A simulated-annealing omit map contoured at 3σ surrounds the magnesium ion, chorismate, and isochorismate (the parts of the structure omitted during the calculation).
The structure reported here is at 1.88 Å resolution, while the previously published structure is reported at 2.3 Å resolution. The previous structure was crystallized from a protein purification buffer containing 5 mM magnesium chloride, but there is no mention of whether chorismate or isochorismate was added to the crystallization conditions.8 However, chorismate was used in the enzymatic assays, so we assume that chorismate was also added for crystallization. The final modeled structure for 3HWO shows a product complex with isochorismate chelating to the magnesium ion. Despite that no additional magnesium ions were added to the protein purification buffer or mother liquor for our low-magnesium structure, a magnesium ion was readily apparent in the active site that was presumably acquired from buffers during the purification protocol. We crystallized EntC in the presence of chorismate. Interestingly, this higher-resolution structure is best modeled with a mixture of chorismate and isochorismate in the active site (Figure 4B) with refined occupancies that reflect the measured equilibrium of the isochorismate synthase reaction.2,27
Evaluation of the Potential Second Metal Binding Site in Three EntC Structures
The possibility of magnesium binding at the potential second site (Figure 3) was investigated with three structures. First, the low-magnesium structure, described in the previous section, was analyzed for metal binding at this site. Second, the possibility exists that the second binding site has a lower affinity than the catalytic binding site and that the lack of density for magnesium ion at the second site is simply a result of this site not being populated. Therefore, low-magnesium crystals were soaked in mother liquor containing 50 mM MgCl2 for 15 min and then flash-cooled, after which the structure was determined; this structure is designated as the high-magnesium structure. Finally, the structure factors for the original 3HWO structure were re-refined with careful attention paid to avoiding model bias at this site.
For each of the three structures, the monomer with the better density for this loop is shown in Figure 5. Since water and magnesium ions are of similar electron density, the crystallographic evidence for placing a magnesium ion instead of a water comes from the coordination, magnesium ions being preferentially octahedrally coordinated. The density at this site in all three structures is at most three-coordinate. For the low-magnesium EntC structure, the monomer shown has a water coordinated (Figure 5A) whereas the opposing monomer has no density at this site. Neither monomer of the high-magnesium structure (Figure 5B) has sufficient electron density to justify placement of a water or magnesium ion bound in this loop. Finally, the re-refined 3HWO structure can be modeled with a water bound to this loop in one monomer (Figure 5C), but the loop of the opposing monomer has poorly defined electron density.
Figure 5.
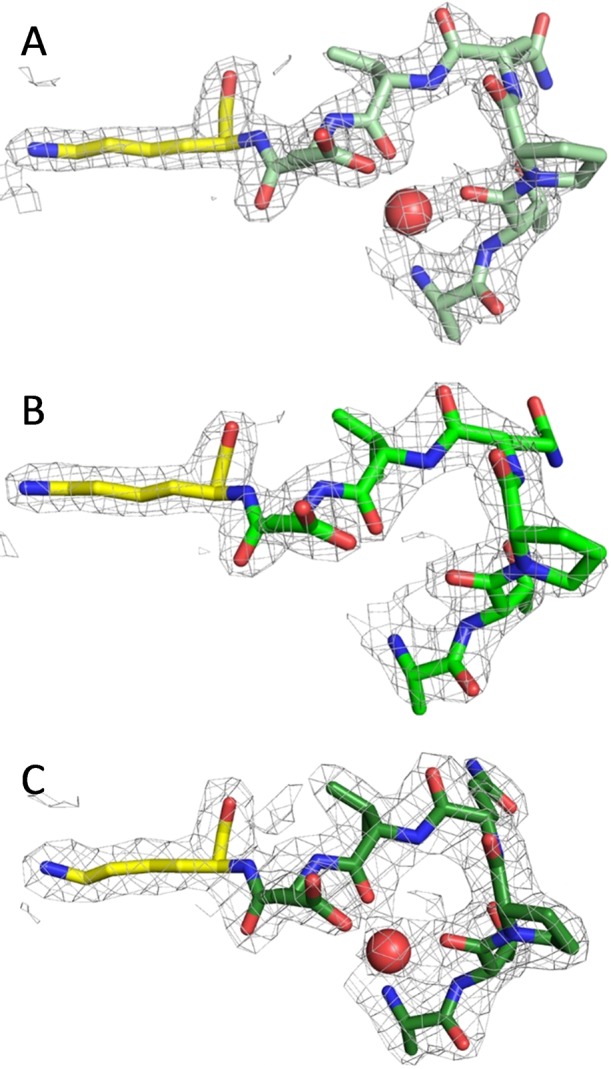
Structural evidence for the proposed second metal site. Three structures of EntC are shown: (A) the low-Mg structure, as also shown in Figure 4; (B) the high-Mg structure; and (C) the re-refined 3HWO structure. The maps shown here are 2Fo – Fc maps contoured at 1.5σ. The monomer in the asymmetric unit with the best density for the loop preceding the general base lysine (yellow) is shown in shades of green. Water molecules are shown as red spheres. This site was hypothesized to bind magnesium (see Figure 3), but that could not be confirmed with any of the structures evaluated.
Dissociation Constants of Ligands from MST Enzymes
Intrinsic tryptophan fluorescence proved to be a sensitive probe for measuring ligand binding for all three enzymes. When each enzyme was excited at 280 nm and the emission spectrum was recorded from 300 to 500 nm, an ∼40–60% decrease in fluorescence intensity was observed with ligand binding. Figure 6A shows a representative set of emission spectra for the binding of chorismate to Irp9 in the presence of the metal chelator EDTA, which was added to prevent turnover from trace magnesium ions. As representative titrations, the binding isotherms of chorismate and magnesium ions to Irp9 are shown in Figure 6B,C (all of the remaining binding isotherms can be found in the Supplemental Figure). The data in Figure 6B were fit to the quadratic form of the single-site binding equation with an added linear term (M[L]) to account for the chorismate inner-filter effect (eq 3). This equation was used for all of the organic ligands, whereas the magnesium isotherms were fit to eq 2, as in Figure 6C. All of the titrations, for organic ligands and magnesium ion, showed unimodal binding. The apparent unimodal binding of magnesium to PchA and EntC is most significant because this is consistent with a single metal ion binding site. The values of the dissociation constants are shown in the table in Figure 6.
The kinetics of binding of magnesium ions, chorismate, and isochorismate to all three enzymes is rapid. Figure 6D shows that for each ligand, equilibrium binding is attained within the dead time of the stopped-flow instrument. On the basis of the ∼1.0 ms dead time, an observed rate constant of greater than 1600 s–1 is required to obscure all evidence of association, suggesting association rate constants on the order of 108 M–1 s–1.
Single-Turnover Experiments
Approximately first-order single-turnover conditions were established for each enzyme utilizing the measured dissociation constants for chorismate and isochorismate (Figure 6). In these experiments, the substrate was first saturated (∼90–94% bound) by being mixed with excess enzyme in the presence of EDTA (used to suppress activity from trace magnesium). The E·S complex was then mixed with pseudo-first-order concentrations of magnesium to induce activity (Figure 7). For each enzyme, the production of the product was registered as a nearly monophasic accumulation of salicylate fluorescence. This was achieved directly for Irp9 and via a coupled reaction for EntC and PchA using a vast excess of PchB. This method assumes that the binding and release of magnesium and substrate/products is rapid, consistent with the observed equilibrium complexation for magnesium, chorismate, and isochorismate, which occurs within the dead time of the stopped-flow instrument for each enzyme (Figure 6D).
For Irp9, the production of salicylate from chorismate (Figure 7A) or isochorismate (Figure 7B) could be observed directly as a monophasic increase in fluorescence emission. The observed rate constants derived from the fits to the individual traces indicated an asymptotic magnesium dependence. For chorismate, this value (0.09 s–1; Figure 7A inset) is the net rate constant for the chemistry that transforms chorismate to salicylate and pyruvate and thus includes the potentially reversible conversion of chorismate to isochorismate. The fit of the magnesium dependence predicts a binding constant for the Irp9·chorismate·Mg complex of ∼0.3 mM, a value that is likely artificially elevated by the presence of EDTA (0.1 mM final concentration after double mix). For isochorismate reacting with Irp9, the limit of the asymptotic dependence (0.15 s–1; Figure 7B inset) represents only the rate constant for irreversible lyase reaction. The dependence suggests that the magnesium binds to the Irp9·isochorismate complex with much higher affinity than to the Irp9·chorismate complex. Qualitative evidence of this can be seen in Figure 7B, where in the absence of added magnesium the preformed Irp9·isochorismate complex is able to compete with the added EDTA and sequester magnesium from the buffer during mixing and turnover to some extent, which is not observed for the E·chorismate single-turnover reactions in Figure 7A,C,D.
Measurement of the dissociation constant for the Irp9·isochorismate·Mg complex by fitting the observed rate dependence is hampered by the relatively large concentration of EDTA. This is manifest in the Figure 7B inset as a distinctly sigmoidal shape for the magnesium dependence. To obtain an estimate of the dissociation constant for the Irp9·isochorismate·Mg complex, the kinetic data in Figure 7B were fit using a comprehensive kinetic model that included all of the steps of the lyase reaction with all of the known equilibrium constants fixed, including the EDTA·Mg equilibrium (Kd = 2 nM) and the rate constants determined in other experiments herein (see the lyase catalytic cycle in Scheme 1). The dashed lines shown in Figure 7B are the global fit to this kinetic model, in which only the rate constants for association of magnesium with the Irp9·isochorismate complex and dissociation of magnesium from the Irp9·isochorismate·Mg complex and the partitioning rate constants for the chemical steps that take isochorismate back to chorismate and chorismate forward to isochorismate were optimized. This model predicts a dissociation constant of magnesium from this complex of ∼2 nM. This very tight binding establishes a model for Irp9 synthase chemistry in which isochorismate is a genuine intermediate species that is unable to appreciably dissociate from the Irp9·isochorismate·Mg complex in turnover despite a dissociation constant and rapid binding equilibria comparable to those observed in PchA and EntC for the E·isochorismate complex (Figure 6). Interestingly, the data could not be fit assuming comparable forward and reverse reaction rate constants for the conversion of isochorismate to chorismate, suggesting that the free energy of isochorismate in the Irp9·isochorismate·Mg complex is lower than that of chorismate bound to either the Irp9, EntC, or PchA·chorismate·Mg complex. We assert that the high affinity for magnesium ions at this stage of catalysis prevents egress of isochorismate from the active site (consistent with an ordered mechanism for the two ligands) and that the complex promotes lyase chemistry over the back-reaction to form chorismate.
Single-turnover reactions for PchA and EntC reacting with chorismate are shown in Figure 7C,D. These reactions approximate first-order conditions by utilizing enzyme:substrate concentration ratios that bind all substrate (based on measured Kd values) and by taking advantage of the high exchange rates of substrates, metal ion, and products that permit the large excess of PchB activity to pull the reaction forward, ensuring that the reverse reaction does not come into play significantly (Figure 6). For this reason, the kinetic traces in Figure 7C,D were fit to individual single exponentials and the limiting rate constant and magnesium ion dissociation constant were derived from the fit of the dependence to the quadratic form of the single-site binding equation (eq 3 without the added linear term, M[L]). For both enzymes, the asymptote of the magnesium ion dependence of the observed rate constants derived from the fits to individual kinetic traces indicates that the isomerase chemistry is slow compared to binding events (ca. 0.3 s–1). The equilibrium constant for isochorismate and chorismate interconversion is approximately 1,2 indicating that the reverse reaction (E·isochorismate·Mg to E·chorismate·Mg) must have a comparable rate constant. This analysis gave values of 0.6 and 1.3 mM for the dissociation constants of magnesium ions from the E·chorismate·Mg complexes of EntC and PchA, respectively.
Magnesium Suppression of the Rate of Isochorismate Release
Whereas all three enzymes form E·Mg complexes with modest affinity (Kd = 0.13–0.19 mM; Figure 6), only EntC and PchA exhibit magnesium ion inhibition in turnover (Figure 2). Figure 7B indicates that retention of magnesium by Irp9 in the presence of isochorismate in part facilitates the lyase chemistry by preventing the dissociation of the isochorismate intermediate. If magnesium occludes the association or dissociation of substrates and products (Figure 1), the relatively low affinity of PchA and EntC for magnesium in the presence of isochorismate could account for the inhibition observed in the steady state (Figure 2). The E·isochorismate·Mg complex would be readily repopulated in the presence of elevated exogenous magnesium ions, thereby hindering isochorismate release and slowing the accumulation of unbound isochorismate. This would only be observed to influence the steady-state isochorismate formation rate if the rate of isochorismate release could be suppressed by magnesium ions to the extent that it became comparable to the rate of the catalytic chemistry (∼0.3 s–1; Figure 7C,D).
In order to test this hypothesis, the E·isochorismate complexes of EntC and PchA (prepared with a 0.1 mM EDTA final concentration post double mix) were formed by mixing excesses of the enzymes with a limiting concentration of isochorismate. These complexes were mixed with a range of concentrations of magnesium ions in the presence of an excess of PchB activity. Therefore, the release of isochorismate is reported by fluorescence of salicylate that is produced by PchB. Figure 8 indicates that elevated magnesium ions can dramatically hinder the rate of isochorismate release. The data for both EntC and PchA show progressively slower release of isochorismate as the magnesium ion concentration increases.
The reaction conditions do not approximate a first-order processes. Instead, the model must include the complexity of the reverse reaction that forms chorismate, which is then reinducted to form isochorismate. In other words, at low magnesium concentration, the isochorismate is readily released and converted to salicylate by PchB. At high magnesium concentrations, the rate of release of isochorismate is limited by two related processes: first, population of the E·isochorismate·Mg complex, in which the magnesium ion occludes isochorismate dissociation (Figure 1), and second, initiation of the reverse reaction that forms chorismate, which then must pass through the forward reaction (chorismate to isochorismate) before release and conversion to salicylate by PchB. Therefore, the data were fit to an inclusive kinetic model that included all of the steps of the isomerase catalytic cycle (Scheme 1). In these fits, all of the equilibrium constants and rate constants were fixed to the values determined in this study with the exception of the two rate constants that define the dissociation constant of the E·isochorismate·Mg complex. The dashed lines of fit shown in Figure 8 are the global fit to the isomerase catalytic cycle shown in Scheme 1 and yielded dissociation constants of 30 and 6 μM for the EntC and PchA E·isochorismate·Mg complexes, respectively. We therefore conclude both that release of chorismate and isochorismate is dependent on the dissociation of magnesium and that the repopulation of the E·isochorismate·Mg complex is the cause of the magnesium ion inhibition observed in the steady state for the isomerase enzymes, EntC and PchA. The salicylate synthase enzyme, Irp9, is immune from this mode of suppression of activity, as the magnesium ion (and hence isochorismate) is retained to promote the ensuing lyase chemistry (Figure 2).
Evaluating Ferrous Ions as a Potential Negative-Feedback Regulator of MST Enzymes of Siderophore Biosynthesis
Ferrous ammonium sulfate was soaked into crystals of EntC (grown as above) and Irp9 (grown using the published conditions3), and the diffraction data were collected at the iron anomalous edge (1.739 Å). A strong anomalous signal allowed for the generation of an experimental map pinpointing the location of the iron in the structures with peaks greater than 5σ (Figure 9A,B). In both cases, the iron is bound in the site of the catalytic magnesium ion, and no anomalous signal is found at the second potential metal binding site in the loop preceding the general base residue. Two additional iron peaks are found at surface residues in EntC, one bound between residues Glu259 and His261 of monomer A and the second bound between Asp40 and Glu41 of monomer B. The Fe-EntC structure has low resolution (2.94 Å), but the density was sufficient for placement of an organic ligand in the active site, modeled as a chorismate. Fe-Irp9 crystals diffracted to higher resolution (2.16 Å) and were likewise grown with chorismate, but the electron density does not justify the placement of substrate or products. Instead, the model includes a sulfate (likely derived from ferrous ammonium sulfate) bound to the iron in monomer A, where the salicylate would be bound in a product structure.3 Monomer B has an acetate at this site from the crystallization solution at this site. Both monomers have density most simply modeled as an acetate (pink sticks) at the pyruvate binding site of Irp9.
Figure 9.

Binding of iron to EntC and Irp9. Experimental anomalous difference maps contoured at 5σ (orange cages) show that ferrous ions bind at the catalytic magnesium site. (A) Fe-EntC. Chorismate is shown in pale-cyan sticks. The general base (K147) and general acid (E197) are shown in yellow. The loop preceding the general base (141-ATPQVD-146) is shown in lime-green sticks. It should be noted that there is no iron anomalous signal at this loop. (B) Fe-Irp9. A sulfate (gold sticks) is bound to the iron in monomer A, whereas an acetate (pink sticks) has been modeled at the pyruvate binding site. The general base (K193) and general acid (E240) are shown in yellow. The loop preceding the general base (187-RRGEYV-192) is shown in marine-blue sticks. (C) Inhibition by iron in the steady state. The upper Michaelis–Menten curve (circles) was obtained in the absence of iron, whereas the lower curve (squares) was obtained in the presence of 125 μM ferrous ammonium sulfate. The table shows dissociation constants for Fe(II) binding, measured by the change in intrinsic tryptophan fluorescence fit to a single binding isotherm plus a linear term.
Steady-state kinetic experiments in the presence of 0.5 mM magnesium ions showed significant inhibition in the presence of ferrous ammonium sulfate (Figure 9C). Measurement of dissociation constants by monitoring the decrease in intrinsic tryptophan fluorescence showed very tight binding of iron in an apparent unimodal fashion, with nanomolar Kd values (Figure 9 table; binding isotherms can be seen in the Supplemental Figure), potentially providing a direct negative-feedback mechanism for iron sequestered by the action of siderophores.
Discussion
The MST enzymes have been investigated previously, with considerable interest as to their promise in the development of new antimicrobials. There is a wealth of literature describing the development of inhibitors, both designed rationally (as substrate and potential transition state analogues)17,19,20,47−52 and by high-throughput screening methodologies.36,53 While some inhibitors have been developed with micro- to nanomolar affinities, the failures to some extent highlight our lack of understanding of the kinetic and chemical mechanisms of these enzymes. Mechanistic studies of these enzymes have been limited primarily to mutational analysis and structural biology approaches. These methods have been successful in identifying residues that are likely to be the general acid/base residues and nucleophiles for ring addition/displacement reactions (described herein as isomerization chemistry). It is generally accepted that the isomerization-type reactions are likely carried out by a conserved mechanism,23,54 the only difference being the identity of the nucleophile (water, ammonia, lysine). Likewise, the lyase activities are hypothesized to be conserved, even if the mechanism itself is debated (pericyclic versus acid/base).1,3,12,22
While the philosophy exists in the literature that the kinetic mechanisms by which the reactions are carried out by the MST enzymes are likely conserved, interpretation of the available data has not resulted in accord. As examples, the isochorismate synthase MenF,4 the salicylate synthase Irp9,20 and anthranilate synthase20 have been described as equilibrium-random, whereas aminodeoxychorismate synthase has been described as ordered-sequential with chorismate binding before magnesium.21 The interpretation of the double-reciprocal plots that led to these conclusions is complicated by the observed inhibition of the enzymes at high magnesium concentrations. Indeed, the analysis of most of the kinetic data for the MST enzymes has disregarded the complexity of magnesium ion acquisition.1,3,8−10,18,22−28
Using the isochorismate synthase enzymes PchA and EntC, which isomerize chorismate to isochorismate and perform the reverse reaction with similar efficiency, we noted magnesium inhibition at concentrations over 0.5 mM (Figure 2). However, the salicylate synthase Irp9, which performs first the isomerization reaction and then the lyase reaction to eliminate the pyruvyl enol substituent, producing salicylate, did not exhibit the same inhibition. It should be noted that magnesium ion concentrations in the range of 1 to 10 mM are physiologically relevant for the bacteria using these enzymes.55−57 This observation led us to consider the modes of inhibition by magnesium ions. Several models were considered. First, a potential second binding site had been identified in EntC, in the loop that precedes the general base residue (Figure 3A).8 Alteration of this site by mutational analysis did not produce a predictive change in the steady-state profile that would be indicative of generation or elimination of a metal binding site (Figure 3B). Further crystallographic analysis provided an intriguing view of the active site of EntC, with chorismate and isochorismate bound in an equilibrium structure that mimics the equilibrium profile exhibited in solution for this reaction (Figure 4). However, analysis of this structure, a structure soaked with a high concentration of magnesium, and re-refinement of the original EntC structure (3HWO) does not support the inclusion of a second metal binding site in these structures (Figure 5). Finally, direct measurement of dissociation constants for magnesium binding show apparent unimodal binding (Figure 6). In other words, the data indicate either a single magnesium binding site per monomer or that there are two (or more) binding sites with similar dissociation constants. In light of the crystallographic evidence, the simplest answer is that there is only one magnesium binding site in MST enzymes.
If a second metal binding site is not involved, then modes of inhibition by magnesium are dependent on interactions at the primary, catalytic magnesium ion binding site. These modes of inhibition are possible only if the reaction is ordered-sequential, whereby chorismate binds first followed by magnesium, chorismate is converted to isochorismate, and magnesium then departs followed by isochorismate. This model is supported by all of the crystallographic structures, in which the magnesium is the button that closes the active-site pocket, as illustrated in Figure 1B. However, as commented above, there are differing interpretations derived almost exclusively from patterns of double-reciprocal plots in the literature. To approach this question using different and potentially more definitive experiments, we designed experiments in the steady and transient states. First, if an ordered-sequential binding model is correct, we would expect that magnesium must be released with every catalytic step. Our data indicate that the binding and release of all ligands is vastly more rapid than the chemical transformation steps, dictating that magnesium dissociates with the completion of each catalytic cycle (Figure 6D). Moreover, the observation that magnesium can suppress the rate of isochorismate release (Figure 8) indicates that this substrate/product cannot dissociate when magnesium is bound, an observation consistent only with ordered addition.
Magnesium binds to the free enzyme, and the extent of population of the E·Mg complex is detected as a perturbation of the enzyme’s intrinsic tryptophan fluorescence, which has a dissociation constant on the order of ∼150 μM (Figure 6). Single-turnover experiments were designed to measure both the rate of catalytic chemistry directly and the affinities of magnesium ions for the various E·substrate complexes (Figure 7). These double mix experiments required a low concentration of EDTA in the first mix that forms E·S to prevent turnover from adventitious magnesium prior to the second mix that forms E·S·Mg. All three E·chorismate·Mg complexes displayed magnesium ion dissociation constants of a similar order as those observed for the E·Mg complexes. However, the Irp9·isochorismate complex competes successfully with EDTA for magnesium binding, and a global fit of the data estimates a low-nanomolar binding constant. Since this value is specific for the lyase reaction, these data provide the first clue about the mode of magnesium inhibition documented in Figure 2. For the isomerase reaction, magnesium is inhibitory at high concentrations, but for the lyase reaction, magnesium does not readily dissociate and instead promotes the ensuing chemistry. These data are supported by the observation that isochorismate cannot be detected in Irp9-catalyzed experiments25,27 and corroborate the previous in vivo work showing that Irp9 cannot complement EntC-deficient E. coli, which requires the isochorismate intermediate, not the salicylate product.58
If we accept that there is a single magnesium binding site and that magnesium is bound and released with each catalytic cycle, then there are two possible modes of inhibition to be considered that are unique to the isomerase reaction. First, it is possible that formation of the E·Mg complex prevents binding of substrate and thus that this is a dead-end complex. Second, it is possible that at relatively high concentrations of magnesium ions, the E·isochorismate·Mg complex remains populated, both preventing dissociation of the product and promoting the facile reverse reaction. Isochorismate release from PchA and EntC is suppressed by increasing magnesium concentration, as shown in Figure 8. It should be recalled that magnesium binding to Irp9 promotes the lyase reaction at a rate that exceeds that of the isomerase reaction in all of the enzymes. Therefore, population of the catalytic magnesium site at high concentrations is the source of inhibition for the isomerase reaction, which is not evident if the enzyme also has lyase activity. Furthermore, global modeling indicates that the major contributor to the observed inhibition at high magnesium ion concentrations is not the formation of the E·Mg complex but is instead predominantly the result of promotion of the reverse reaction.
The mechanism of PchA has been previously defined as dependent on reverse protonation states of the active-site general acid and general base residues on the basis of pH rate profiles, the absence of a solvent kinetic isotope effect, and the observation that the rate of the reaction is diffusion-controlled by microviscogens.9 The data here align well with those previous conclusions. First, we have shown that ligand binding is very rapid, coming to equilibrium within the dead time of the stopped-flow instrument. Substrate binding and product release, as well as magnesium binding and release, will be markedly slowed by the addition of microviscogens. Second, the pH rate profiles indicated that residues with pKa values matching a lysine and a glutamic acid are involved in catalysis. Here we have shown that chemistry is rate-limiting and for the first time have measured the rate constants of the chemical steps. A likely hypothesis is that the rate-limiting step is the reversal of the protonation states in the active-site residues from the zwitterionic form that is most prevalent in the free enzyme. A steady-state solvent kinetic isotope effect of unity had been previously reported,9 but the effect was likely obscured by the elevated magnesium concentration used, where the isochorismate release step had become largely rate-limiting. The importance of magnesium for closing the active site and its requirement in catalysis leads one to wonder whether magnesium binding is required for formation of the uncharged lysine and glutamate residues for catalysis. With the development of single-turnover methods that report the catalytic step, we are poised to determine the chemical mechanism of MST enzymes using specific isotopic insertions targeted to alter the rates of the isomerase and lyase activities.
The first description of the isochorismate synthase PchA59 indicated that mechanisms of feedback inhibition had been tested, with no inhibition by pyochelin, aromatic amino acids, or metabolites of pyochelin production (cysteine). The authors also reported that ferrous iron at a concentration of 100 μM did not alter PchA activity. The assays of that article were more cumbersome than the continuous coupled assay used herein, as they included stopping the reaction with acid and extracting the products with ethyl acetate before detection by fluorescence. This work proposed that regulation of pyochelin biosynthesis by P. aeruginosa was directly influenced by the concentration of PchA in the cell. This assertion was in contrast to the chorismate-utilizing enzyme of tryptophan biosynthesis (TrpE), which is allosterically downregulated by the final metabolite of the pathway, tryptophan.13,14
The cellular concentration of proteins of siderophore biosynthesis in the cell is regulated by Fur, the transcriptional repressor that in the presence of iron downregulates the expression of the positive regulatory factors PchR and PvdS, which in turn regulate the production of the pyochelin and pyoverdin biosynthesis genes.60−62 The concentration of total iron in an E. coli is proposed to be 200 μM to 2 mM on the basis of calculation from dry weight,63−65 whereas the concentration of “loosely bound” iron is ∼10 μM.66 Fur demonstrates a Kd for Fe(II) binding of 1.2 μM. By comparison, PchA, EntC, and Irp9 have nanomolar (30–180 nM) E·Fe(II) dissociation constants. Therefore, the data presented here indicate that the chorismate-utilizing enzymes of siderophore biosynthesis are directly inhibited by iron at a concentration that is 1 to 2 orders of magnitude lower than would promote downregulation of their genes by Fur. This seems to be a novel mode of regulation since the regulator of activity is not the final compound of the biosynthetic pathway (the siderophore) but is instead the nutrient that is scavenged by the siderophore.
Conclusions
Metal ion binding to MST enzymes prevents the exchange of substrates and products. As such, catalysis occurs only when the enzyme has first bound the substrate and the active site has then been capped by a magnesium ion. In the presence of excess magnesium, two modes of inhibition are possible, but only one mode contributes to lower rates of catalysis and only for the isochorismate synthase enzymes. Free MST enzymes bind magnesium ions to form dead-end complexes that prevent the association of substrates. However, this is not the prominent mode of inhibition, as substrates bind with considerably higher affinity, effectively negating the influence of this inhibitory mechanism. Moreover, lyase-active enzymes form the same E·Mg complex with affinity comparable to that of the isomerase enzymes but do not exhibit magnesium ion inhibition at physiologically relevant concentrations of the metal. Susceptibility to the second mode of inhibition occurs only with the isochorismate synthase (isomerase) enzymes, as these enzymes retain isochorismate at high magnesium concentrations when the E·isochorismate·Mg complex is repopulated by exogenous metal ions. The lyase reaction does not have this susceptibility to magnesium inhibition because magnesium is retained by these enzymes with extremely high affinity when isochorismate is present, promoting the subsequent lyase reaction. Ferrous ions also bind to the catalytic metal site with high affinity, occluding exchange of ligands and/or suppressing the chemistry. This observation suggests a novel form of negative-feedback inhibition for a biosynthetic pathway whereby the first enzyme in the pathway is inhibited not by the biosynthetic product but rather by the nutrient that is scavenged by export and reimport of the iron-loaded siderophore.
Acknowledgments
This publication was made possible by funds from NIH Grants R01 AI77725 and K02 AI093675 from the National Institute of Allergy and Infectious Diseases (A.L.L.), NIH Grant P20 RR016475 from the INBRE Program of the National Center for Research Resources (A.L.L.), NIH Grant R01 GM116957 from the National Institute of General Medical Sciences (A.M.G.), National Science Foundation Grants CHE-1402475 (G.R.M.) and CHE-1403293 (A.L.L.), University of Kansas 2015 General Research Fund Grant 230189 (A.L.L.), and a UWM Research Growth Initiative Grant (G.R.M.). We are grateful to A. S. Chilton for technical expertise in protein and substrate preparation. Diffraction data were collected at the Stanford Synchrotron Radiation Laboratory, a National User Facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. We thank the staff at the Stanford Synchrotron Radiation Laboratory for their support and assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b05134.
Binding isotherms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zwahlen J.; Kolappan S.; Zhou R.; Kisker C.; Tonge P. J. Biochemistry 2007, 46, 954. 10.1021/bi060852x. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Liu J.; Rusnak F.; Sakaitani M. Chem. Rev. 1990, 90, 1105. 10.1021/cr00105a003. [DOI] [Google Scholar]
- Kerbarh O.; Chirgadze D. Y.; Blundell T. L.; Abell C. J. Mol. Biol. 2006, 357, 524. 10.1016/j.jmb.2005.12.078. [DOI] [PubMed] [Google Scholar]
- Kolappan S.; Zwahlen J.; Zhou R.; Truglio J. J.; Tonge P. J.; Kisker C. Biochemistry 2007, 46, 946. 10.1021/bi0608515. [DOI] [PubMed] [Google Scholar]
- Parsons J. F.; Jensen P. Y.; Pachikara A. S.; Howard A. J.; Eisenstein E.; Ladner J. E. Biochemistry 2002, 41, 2198. 10.1021/bi015791b. [DOI] [PubMed] [Google Scholar]
- Parsons J. F.; Shi K. M.; Ladner J. E. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008, 64, 607. 10.1107/S0907444908005477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spraggon G.; Kim C.; Nguyen-Huu X.; Yee M. C.; Yanofsky C.; Mills S. E. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 6021. 10.1073/pnas.111150298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan S.; Howard N.; Kerbarh O.; Blaszczyk M.; Abell C.; Blundell T. L. J. Mol. Biol. 2010, 397, 290. 10.1016/j.jmb.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Meneely K. M.; Luo Q.; Dhar P.; Lamb A. L. Arch. Biochem. Biophys. 2013, 538, 49. 10.1016/j.abb.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebart K. T.; Toney M. D. Biochemistry 2010, 49, 2851. 10.1021/bi100021x. [DOI] [PubMed] [Google Scholar]
- Daruwala R.; Bhattacharyya D. K.; Kwon O.; Meganathan R. J. Bacteriol. 1997, 179, 3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb A. L. Biochemistry 2011, 50, 7476. 10.1021/bi2009739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri M. G.; Bauerle R. J. Biol. Chem. 1991, 266, 8328. [PubMed] [Google Scholar]
- Morollo A. A.; Eck M. J. Nat. Struct. Biol. 2001, 8, 243. 10.1038/84988. [DOI] [PubMed] [Google Scholar]
- Rayl E. A.; Green J. M.; Nichols B. P. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1996, 1295, 81. 10.1016/0167-4838(96)00029-5. [DOI] [PubMed] [Google Scholar]
- Roux B.; Walsh C. T. Biochemistry 1992, 31, 6904. 10.1021/bi00145a006. [DOI] [PubMed] [Google Scholar]
- Chi G.; Manos-Turvey A.; O’Connor P. D.; Johnston J. M.; Evans G. L.; Baker E. N.; Payne R. J.; Lott J. S.; Bulloch E. M. Biochemistry 2012, 51, 4868. 10.1021/bi3002067. [DOI] [PubMed] [Google Scholar]
- Harrison A. J.; Yu M.; Gardenborg T.; Middleditch M.; Ramsay R. J.; Baker E. N.; Lott J. S. J. Bacteriol. 2006, 188, 6081. 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manos-Turvey A.; Bulloch E. M.; Rutledge P. J.; Baker E. N.; Lott J. S.; Payne R. J. ChemMedChem 2010, 5, 1067. 10.1002/cmdc.201000137. [DOI] [PubMed] [Google Scholar]
- Ziebart K. T.; Dixon S. M.; Avila B.; El-Badri M. H.; Guggenheim K. G.; Kurth M. J.; Toney M. D. J. Med. Chem. 2010, 53, 3718. 10.1021/jm100158v. [DOI] [PubMed] [Google Scholar]
- Dixon S.; Ziebart K. T.; He Z.; Jeddeloh M.; Yoo C. L.; Wang X.; Lehman A.; Lam K. S.; Toney M. D.; Kurth M. J. J. Med. Chem. 2006, 49, 7413. 10.1021/jm0609869. [DOI] [PubMed] [Google Scholar]
- Culbertson J. E.; Chung D.; Ziebart K. T.; Espiritu E.; Toney M. D. Biochemistry 2015, 54, 2372. 10.1021/acs.biochem.5b00013. [DOI] [PubMed] [Google Scholar]
- He Z.; Stigers Lavoie K. D.; Bartlett P. A.; Toney M. D. J. Am. Chem. Soc. 2004, 126, 2378. 10.1021/ja0389927. [DOI] [PubMed] [Google Scholar]
- Kerbarh O.; Bulloch E. M.; Payne R. J.; Sahr T.; Rebeille F.; Abell C. Biochem. Soc. Trans. 2005, 33, 763. 10.1042/BST0330763. [DOI] [PubMed] [Google Scholar]
- Kerbarh O.; Ciulli A.; Howard N. I.; Abell C. J. Bacteriol. 2005, 187, 5061. 10.1128/JB.187.15.5061-5066.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Quinn N.; Berchtold G. A.; Walsh C. T. Biochemistry 1990, 29, 1417. 10.1021/bi00458a012. [DOI] [PubMed] [Google Scholar]
- Meneely K. M.; Luo Q.; Lamb A. L. Arch. Biochem. Biophys. 2013, 539, 70. 10.1016/j.abb.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino L.; Reimmann C.; Baur H.; Beyeler M.; Visca P.; Haas D. Mol. Gen. Genet. 1995, 249, 217. 10.1007/BF00290369. [DOI] [PubMed] [Google Scholar]
- Kapust R. B.; Tozser J.; Fox J. D.; Anderson D. E.; Cherry S.; Copeland T. D.; Waugh D. S. Protein Eng., Des. Sel. 2001, 14, 993. 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Zaitseva J.; Lu J.; Olechoski K. L.; Lamb A. L. J. Biol. Chem. 2006, 281, 33441. 10.1074/jbc.M605470200. [DOI] [PubMed] [Google Scholar]
- Rieger C. E.; Turnbull J. L. Prep. Biochem. Biotechnol. 1996, 26, 67. 10.1080/10826069608000051. [DOI] [PubMed] [Google Scholar]
- Schmidt K.; Leistner E. Biotechnol. Bioeng. 1995, 45, 285. 10.1002/bit.260450402. [DOI] [PubMed] [Google Scholar]
- Luo Q.; Olucha J.; Lamb A. L. Biochemistry 2009, 48, 5239. 10.1021/bi900456e. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Anal. Biochem. 2009, 387, 30. 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Anal. Biochem. 2009, 387, 20. 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Meneely K. M.; Luo Q.; Riley A. P.; Taylor B.; Roy A.; Stein R. L.; Prisinzano T. E.; Lamb A. L. Bioorg. Med. Chem. 2014, 22, 5961. 10.1016/j.bmc.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. J. Appl. Crystallogr. 2007, 40, 658. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 352. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2256. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.PyMOL; DeLano Scientific: San Carlos, CA, 2002; www.pymol.org.
- Dundas J.; Ouyang Z.; Tseng J.; Binkowski A.; Turpaz Y.; Liang J. Nucleic Acids Res. 2006, 34, W116. 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski M. C.; Bartlett P. A. J. Am. Chem. Soc. 1991, 113, 5897. 10.1021/ja00015a073. [DOI] [Google Scholar]
- Kozlowski M. C.; Tom N. J.; Seto C. T.; Sefler A. M.; Bartlett P. A. J. Am. Chem. Soc. 1995, 117, 2128. 10.1021/ja00113a002. [DOI] [Google Scholar]
- Liu Z.; Liu F.; Aldrich C. C. J. Org. Chem. 2015, 80, 6545. 10.1021/acs.joc.5b00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne R. J.; Bulloch E. M.; Kerbarh O.; Abell C. Org. Biomol. Chem. 2010, 8, 3534. 10.1039/c004062b. [DOI] [PubMed] [Google Scholar]
- Payne R. J.; Bulloch E. M.; Toscano M. M.; Jones M. A.; Kerbarh O.; Abell C. Org. Biomol. Chem. 2009, 7, 2421. 10.1039/b901694e. [DOI] [PubMed] [Google Scholar]
- Payne R. J.; Kerbarh O.; Miguel R. N.; Abell A. D.; Abell C. Org. Biomol. Chem. 2005, 3, 1825. 10.1039/b503800f. [DOI] [PubMed] [Google Scholar]
- Vasan M.; Neres J.; Williams J.; Wilson D. J.; Teitelbaum A. M.; Remmel R. P.; Aldrich C. C. ChemMedChem 2010, 5, 2079. 10.1002/cmdc.201000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbarh O.; Ciulli A.; Chirgadze D. Y.; Blundell T. L.; Abell C. ChemBioChem 2007, 8, 622. 10.1002/cbic.200700019. [DOI] [PubMed] [Google Scholar]
- Froschauer E. M.; Kolisek M.; Dieterich F.; Schweigel M.; Schweyen R. J. FEMS Microbiol. Lett. 2004, 237, 49. 10.1111/j.1574-6968.2004.tb09677.x. [DOI] [PubMed] [Google Scholar]
- Hurwitz C.; Rosano C. L. J. Biol. Chem. 1967, 242, 3719. [PubMed] [Google Scholar]
- Tyrrell J.; McGinnis J. L.; Weeks K. M.; Pielak G. J. Biochemistry 2013, 52, 8777. 10.1021/bi401207q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelludat C.; Brem D.; Heesemann J. J. Bacteriol. 2003, 185, 5648. 10.1128/JB.185.18.5648-5653.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaille C.; Reimmann C.; Haas D. J. Biol. Chem. 2003, 278, 16893. 10.1074/jbc.M212324200. [DOI] [PubMed] [Google Scholar]
- Hassett D. J.; Sokol P. A.; Howell M. L.; Ma J. F.; Schweizer H. T.; Ochsner U.; Vasil M. L. J. Bacteriol. 1996, 178, 3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner U. A.; Vasil A. I.; Vasil M. L. J. Bacteriol. 1995, 177, 7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner U. A.; Vasil M. L. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 4409. 10.1073/pnas.93.9.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Tehrani H.; Hudson A. J.; Chang Y.-S.; Timms A. R.; Hawkins C.; Williams J. M.; Harrison P. M.; Guest J. R.; Andrews S. C. J. Bacteriol. 1999, 181, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. C.; Robinson A. K.; Rodriguez-Quinones F. FEMS Microbiol Rev. 2003, 27, 215. 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- Rouf M. A. J. Bacteriol. 1964, 88, 1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyer K.; Imlay J. A. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13635. 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.