

SUMMARY
Neutrophils are recruited from the blood to sites of sterile inflammation, where they are involved in wound healing but can also cause tissue damage. During sterile inflammation, necrotic cells release pro-inflammatory molecules including formylated peptides. However, the signaling pathway triggered by formylated peptides to integrin activation and leukocyte recruitment is unknown. By using spinning-disk confocal intravital microscopy, we examined the molecular mechanisms of leukocyte recruitment to sites of focal hepatic necrosis in vivo. We demonstrated that the Bruton’s tyrosine kinase (Btk) was required for multiple Mac-1 activation events involved in neutrophil recruitment and functions during sterile inflammation triggered by fMLF. The Src family kinase Hck, Wiskott-Aldrich-syndrome protein, and phospholipase Cγ2 were also involved in this pathway required for fMLF-triggered Mac-1 activation and neutrophil recruitment. Thus, we have identified a neutrophil Btk signalosome that is involved in a signaling pathway triggered by formylated peptides leading to the selective activation of Mac-1 and neutrophil recruitment during sterile inflammation.
INTRODUCTION
Neutrophils are key players in acute inflammation. They play an important role in host defense and contribute to inflammation-related tissue damage. Necrotic cell death can induce sterile inflammation characterized by the recruitment of innate immune effector cells into the damaged tissue. The recruited neutrophils contribute to the clearance of debris, but they can also cause profound collateral tissue destruction due to the release of their vast arsenal of hydrolytic, oxidative, and pore-forming molecules (McDonald and Kubes 2012). Excessive neutrophil recruitment during sterile inflammation accounts for the immunopathology observed in many diseases, including trauma, autoimmunity, ischemic injuries, and sterile liver injury (Imaeda et al., 2009; McDonald et al., 2010). Therefore, understanding the mechanisms for neutrophil recruitment is of major physiological and pathophysiological importance.
Several endogenous pro-inflammatory damage-associated molecular patterns (DAMPs), including lipid mediators, N-formylated peptides, and extracellular matrix proteins, are released during cell death by necrosis (McDonald and Kubes 2012; McDonald et al., 2010; Imaeda et al., 2009). Neutrophils express a variety of receptors that recognize N-formylated peptides, including those specific for the prototype ligand formylmethionyl-leucyl-phenylalanine (fMLF). Eliminating one of the receptors for fMLF (Fpr1−/−) results in a reduced neutrophil recruitment into the inflamed lung (Grommes et al., 2014) and reduces neutrophil adhesion in the liver during sterile inflammation (McDonald et al., 2010), highlighting the importance of cell stimulation with N-formylated peptides in innate immunity.
Receptors for fMLF are Gαi-linked receptors that trigger a variety of intracellular signaling pathways (Dorward et al., 2015), provoking different cell responses like neutrophil chemotaxis, respiratory burst, and transcriptional regulation. Activation of phosphoinositide 3-kinase γ (PI3Kγ) and phospholipase C (PLC) isoforms are the predominant signaling events upon fMLF-receptor activation. PI3Kγ induces the conversion of phosphoinositol-4,5-biphosphate to phosphoinositol-3,4,5-triphosphate which is involved in neutrophil cytoskeletal reorganization and chemotaxis. The phospholipase Cβ (PLCβ) isoform is required for the production of diacylglycerol (DAG) and inositol-3,4,5-trisphosphate (IP3), which induces release of intracellular calcium into the cytoplasm (Dorward et al., 2015).
In addition to the activation of PI3K and PLC, fMLF receptors trigger a rapid tyrosine phosphorylation of several signaling molecules in neutrophils, including Src family kinases (SFKs) and Tec family kinases (Zarbock and Ley, 2011; Gilbert et al., 2003; Futosi et al., 2013). The SFKs Fgr, Hck, and Lyn are expressed in neutrophils and are involved in several signaling pathways by promoting phosphorylation of downstream effectors (Thomas and Brugge, 1997; Lowell and Berton, 1999). These SFKs share a high degree of structural homology and possess three major domains: a Src homology 3 (SH3) domain, a SH2 domain, and the tyrosine kinase (SH1) domain (Thomas and Brugge, 1997). SFKs can be activated by several molecules and participate in a variety of cell functions in neutrophils (Zarbock and Ley, 2011; Thomas and Brugge, 1997; Lowell and Berton, 1999). They also modulate the activity of other kinases, including Tec family members as well as FAK and Pyk2.
The Bruton’s tyrosine kinase (Btk), a member of the Tec family kinases, has a unique NH2-terminal region containing a pleckstrin homology (PH) domain and a proline-rich stretch, followed by SH3, SH2, and kinase domains. Deficiency of Btk leads to X-linked agammaglobulinemia in humans (Block and Zarbock, 2012). Btk is expressed in the myeloid lineage, and experiments demonstrate that Btk is activated after selectin or fMLF engagement (Mueller et al., 2010; Gilbert et al., 2003). Studies with gene-deficient mice or inhibitors indicate that Btk in neutrophils is involved in G protein-coupled receptor signaling and other functions (Gilbert et al., 2003). Because Btk is a multidomain protein, it can interact with and activate different molecules, including PLCγ2 and Wiskott-Aldrich-syndrome protein (WASp) (Mueller et al., 2010; Sakuma et al., 2015). A recently published study demonstrates that elimination of Btk abolishes selectin-mediated LFA-1-dependent slow leukocyte rolling (Mueller et al., 2010).
The current study was designed to determine the role of Btk in leukocyte recruitment during sterile inflammation. Using functional ex vivo, in vivo, and in vitro assays, we found that Btk is required for multiple Mac-1 integrin activation events involved in neutrophil recruitment and functions during sterile inflammation triggered by fMLF. Furthermore, we excluded that Btk is involved in chemokine-induced integrin activation. Furthermore, we have identified Hck, WASp, and PLCγ2 as key signaling molecules involved in this specific signaling pathway. In addition, we have shown that Btk is involved in integrin-mediated outside-in signaling and FcRγ-mediated functions. In conclusion, we have demonstrated that the Btk signalosome is indispensable for multiple fMLF-induced Mac-1 activation events required for neutrophil recruitment and activation during sterile inflammation.
RESULTS
Btk Is Required for Neutrophil Recruitment during Sterile Inflammation Induced by Focal Hepatic Necrosis
To investigate the role of Btk in neutrophil recruitment during sterile inflammation, spinning disk confocal intravital microscopy (SD-IVM) of the murine liver was performed after focal hepatic necrosis. Wild-type (WT) and Btk−/− mice expressing enhanced green fluorescent protein under the control of the endogenous lysozyme M promoter (Lyz2-eGFP) were used to display the kinetics of eGFP-expressing neutrophils to the site of injury visualized by propidium iodide. Similar to previous reports (McDonald et al., 2010), we observed that neutrophils adhered to the microvascular endothelium in WT and in Btk−/− mice within 30–60 min after focal hepatic necrosis (Figure 1A and Movies S1 and S2). Although the number of adherent neutrophils in WT mice continued to increase over time, the rate of increase in adherent Btk−/− neutrophils was significantly less. The peak of neutrophil recruitment occurred 4 hr after injury in WT mice, whereas in Btk−/− mice at this time point, there was a significant reduction in the number of neutrophils within the area of injury (Figure 1B and Movies S1 and S2). After inducing focal hepatic necrosis, duration of adhesion before neutrophil detachment (Figure S1A) as well as the total number of adherent neutrophils that chemotaxed toward the necrotic tissue was significantly reduced in Btk−/− neutrophils compared to WT neutrophils (Figure 1C). Btk−/− neutrophils also showed a reduced crawling velocity (Figure 1D), crawled distance (Figure 1E), and migration directionality toward the site of injury compared to neutrophils in WT mice (Figures S1B and S1C). As previously demonstrated, blocking the β2-integrin Mac-1 also reduced neutrophil recruitment after sterile liver injury in WT mice (Figures S1D–S1G). Representative time-lapse images from SD-IVM after thermal injury on the surface of the liver of WT and Btk−/− mice are shown in Figure 1F. To confirm that Btk in neutrophils was responsible for the observed phenotype, reconstitution experiments with isolated WT and Btk−/− bone-marrow-derived neutrophils were performed. Significantly fewer Btk−/− neutrophils infiltrated the site of injury compared to WT neutrophils (Figure S1H). To show that Fpr1 is required for the migration of neutrophils to the site of injury, we also performed reconstitution experiments with WT and Fpr1−/− neutrophils. Significantly fewer Fpr1−/− neutrophils infiltrated the site of injury compared to WT neutrophils (Figure S1I). Using flow cytometry, we also found that neutrophil recruitment into the injured liver was significantly reduced in Btk−/− mice compared to WT mice (Figure 1G). Btk−/− mice also manifested significantly reduced hepatic injury, as judged by release of the liver damage indicators glutamic oxaloacetic transaminase (GOT) and glutamic pyruvate transaminase (GPT) into the serum (Figures 1H and 1I). To show that the liver damage induced by focal hepatic necrosis is neutrophil dependent, we depleted neutrophils by approximately 95% in WT and Btk−/− mice (Figure S1J). Depleting neutrophils in WT mice significantly reduced the levels of GOT and GPT in the serum after injury, whereas depletion of neutrophils in Btk−/− animals had little effect (Figures S1K and S1L). These data indicate that the majority of liver damage in this model is neutrophil dependent and that Btk is required for neutrophil recruitment during sterile inflammation.
Figure 1. Btk Is Required for Neutrophil Recruitment during Sterile Inflammation Induced by Focal Hepatic Necrosis.
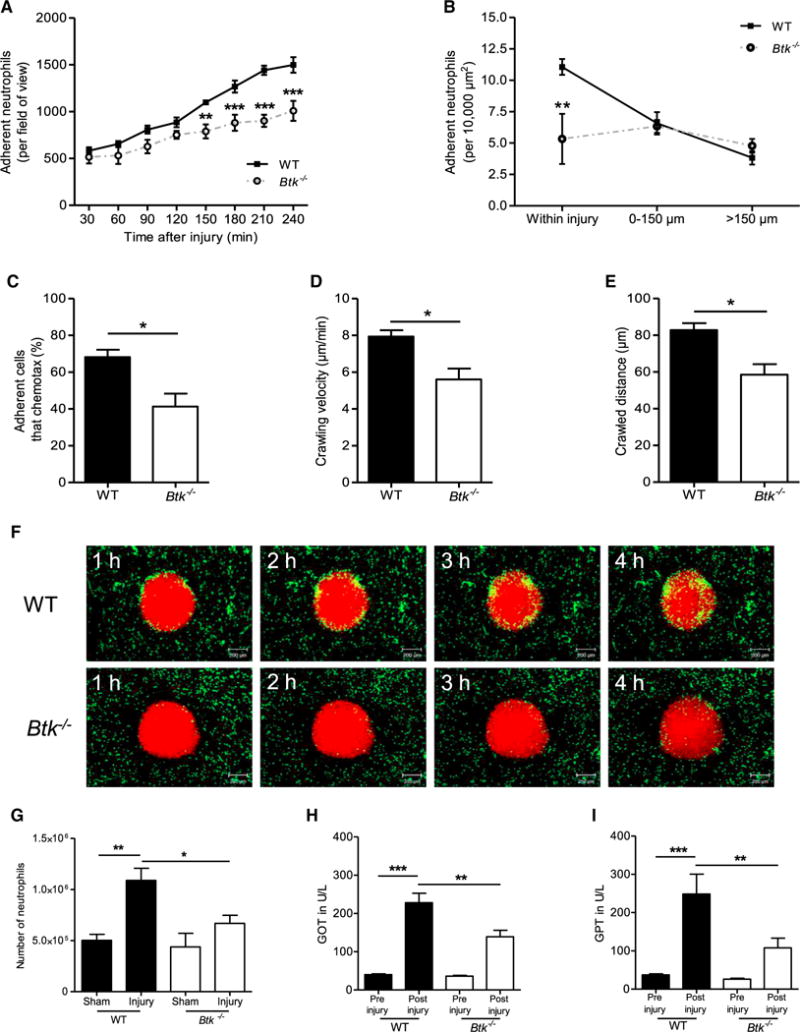
(A) Number of adherent neutrophils per field of view in WT and Btk−/− mice in response to focal hepatic necrosis for all indicated time points (30–240 min).
(B) Number of adherent neutrophils per 10,000 μm2 within indicated regions around foci of necrosis 4 hr after sterile injury in WT and Btk−/− mice.
(C–E) Percentage of neutrophils that chemotax toward the injury site (C), neutrophil crawling velocity (D), and crawled distance 2.5 hr after injury (E).
(F) Representative time-lapse images from SD-IVM demonstrating the recruitment of neutrophils (green) to the injury area (red, propidium iodide) in WT and Btk−/− mice. Scale bars represent 200 μm.
(G) Number of Gr-1-labeled neutrophils per liver derived from WT or Btk−/− mice normalized to the weight of the liver 4 hr after focal hepatic necrosis or sham surgery as determined by flow cytometry.
(H and I) Levels of GOT (H) and GPT (I) in U/l in serum before and 4 hr after liver injury in WT and Btk−/− mice.
Depicted are mean + SEM; n ≥ 3 individual mice/group; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figures S1 and S7 and Movies S1 and S2.
Btk Is Required for fMLF-Mediated Neutrophil Extravasation in the Murine Cremaster Muscle and Chemotaxis In Vitro
To investigate the role of Btk in the different steps of the recruitment cascade in the systemic circulation, we performed intravital microscopy (IVM) of the cremaster muscle 4 hr after in-trascrotal injection of fMLF or the vehicle control. The numbers of adherent and transmigrated neutrophils were significantly higher in WT mice treated with fMLF compared to the vehicle control, whereas Btk−/− mice revealed significantly reduced numbers of adherent and transmigrated neutrophils after fMLF injection compared to WT mice (Figures 2A and 2B), suggesting that Btk is involved in neutrophil recruitment in the systemic circulation during sterile inflammation. Figure 2C shows representative pictures from IVM experiments. Monoclonal antibody-mediated blockade of LFA-1 and/or Mac-1 significantly reduced the numbers of adherent and transmigrated neutrophils in WT mice, indicating that both integrins are important for an efficient neutrophil recruitment (Figures S2A and S2B). In addition, neutrophil adhesion and transmigration was significantly reduced in Fpr1−/− mice after intrascrotal injection of fMLF (Figures S2C and S2D). To examine whether Btk is involved in GPCR-induced arrest, we conducted intravital microscopy of the cremaster muscle before and after injection of fMLF. WT and Btk−/− mice had the same number of adherent neutrophils under baseline conditions and immediately after fMLF injection (Figure 2D). Because the initial neutrophil adhesion is mediated by LFA-1, these data suggest that Btk is not involved in fMLF-induced LFA-1 activation. Whereas the attached neutrophils detached slowly over time in WT mice, Btk−/− neutrophils detached significantly faster after initial adhesion (Figure 2D), suggesting that Btk−/− neutrophils have a defect in Mac-1-dependent post-adhesion strengthening. To confirm that Mac-1-dependent post-adhesion is defective in Btk−/− mice, we repeated the GPCR-induced arrest in WT and Btk−/− mice in the presence of blocking antibodies against LFA-1 or Mac-1 (Figure 2E). Blocking LFA-1 resulted in no neutrophil arrest in WT mice. When Mac-1 was blocked, WT neutrophils adhered directly after fMLF injection, but detachment was significantly accelerated compared to untreated WT mice (Figure 2E). However, blocking Mac-1 in Btk−/− mice did not change the pattern of fMLF-induced adhesion and detachment, suggesting that the lack of Btk in neutrophils affects fMLF-induced Mac-1 function.
Figure 2. Btk Is Required for fMLF-Mediated Neutrophil Extravasation in the Murine Cremaster Muscle and Chemotaxis In Vitro.
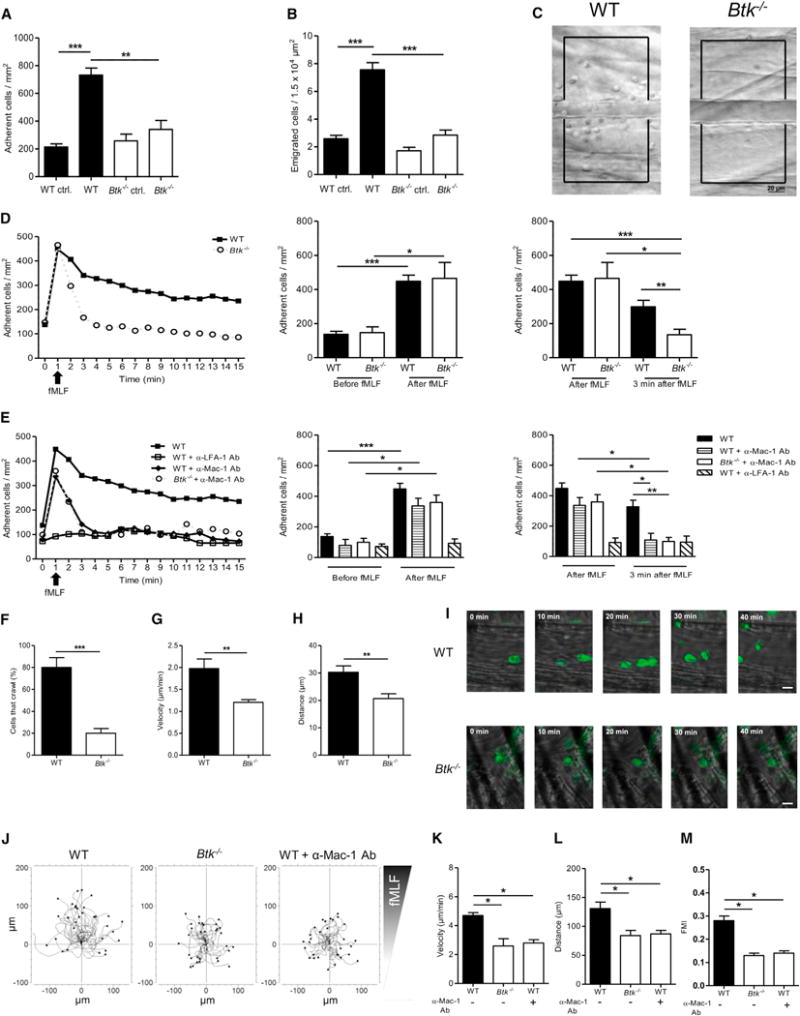
(A–H) Intravital microscopy of postcapillary venules in the murine cremaster was performed in WT and Btk−/− mice.
(A and B) Number of adherent cells per mm2 (A) and number of transmigrated cells per 1.5 × 104 μm2 (B) 4 hr after intrascrotal fMLF or vehicle injection.
(C) Representative images of inflamed WT (left) and Btk−/− (right) cremasteric venules visualized by reflected light oblique transillumination microscopy. Scale bar represents 20 μm.
(D and E) GPCR-induced arrest of neutrophils in postcapillary venules of WT (filled square) and Btk−/− (open circle) mice after fMLF injection (i.v.) (D) or after injection of blocking antibodies against LFA-1 (open square) or Mac-1 (filled diamond) prior to fMLF injection (E). Respective statistics are presented in related bar graphs.
(F–H) Intravascular crawling of Gr-1-labeled neutrophils during superfusion with fMLF. Percentage of adherent cells that crawled (F), crawling velocity (G), and crawled distance (H).
(I) Representative images of extravasated WT and Btk−/−. Scale bars represent 10 μm.
(J) Representative trajectory plots (30 cells from 3 independent experiments) of chemotaxing WT and Btk−/− neutrophils toward indicated fMLF gradient in vitro.
(K–M) Velocity (K), migration distance (L), and forward migration index (FMI) (M) of WT and Btk−/− neutrophils.
Depicted are mean + SEM; n ≥ 3 individual mice/group; *p < 0.05; **p < 0.01; ***p < 0.001 for all panels except (C), (I), and (J). See also Figures S2, S3, and S7 and Movies S3 and S4.
It has been demonstrated that intravascular crawling of neutrophils is also Mac-1 dependent. To examine the role of Btk in this step, intravital microscopy of the cremaster muscle during fMLF superfusion was conducted. Significantly fewer neutrophils crawled in Btk−/− mice compared to WT mice (Figure 2F). Furthermore, Btk−/− neutrophils revealed a significantly reduced crawling velocity (Figure 2G) and crawling distance (Figure 2H) compared to WT neutrophils. Representative time-lapse images of adherent neutrophils in WT and Btk−/− mice that crawled are depicted in Figure 2I (see also Movies S3 and S4).
To determine whether Btk is involved in chemotaxis, we examined the responses of WT and Btk−/− neutrophils toward an fMLF gradient in vitro. The chemotaxis of Btk−/− neutrophils was significantly reduced compared to WT neutrophils (Figures 2J–2M). The requirement of Mac-1 for chemotaxis toward fMLF was demonstrated by blocking Mac-1 in WT neutrophils. All investigated parameters were significantly reduced to the level of Btk−/− neutrophils compared to untreated WT neutrophils.
In order to investigate the potential involvement of Btk in CXCL1-induced integrin activation and leukocyte recruitment, we performed different in vivo and in vitro experiments. Btk−/− neutrophils showed no defects in integrin activation, intravascular adhesion, crawling, or chemotaxis in response to CXCL1 stimulation (Figure S3). However, pharmacologic blockade of CXCR2 completely blocked CXCL1-mediated adhesion in both WT and Btk−/− mice. These results suggest that Btk functions relatively selectively downstream of fRPs but not CXCR2 during neutrophil recruitment.
Hck, but Not Other Src Family Kinases, Is Required for fMLF-Mediated Neutrophil Recruitment
SFKs are involved in several neutrophil functions and it has been shown that these kinases are involved in Btk activation. To investigate whether SFKs are involved in neutrophil recruitment during sterile inflammation, we performed intravital microscopy of the murine cremaster muscle 4 hr after intrascrotal injection of fMLF. The SFK inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine (PP2) or its negative control PP3 were injected intravenously before injecting fMLF intrascrotally. The number of adherent and transmigrated neutrophils at 4 hr after injection of fMLF was significantly reduced in PP2-treated WT mice compared to WT mice treated with PP3 (Figures 3A and 3B), suggesting that SFKs play a role in fMLF-triggered sterile inflammation. To scrutinize which SFK is responsible for the observed effect, intravital microscopy on all three SFK knockout mice (Fgr−/−, Hck−/−, and Lyn−/−) was performed. Whereas the number of adherent and transmigrated neutrophils in Fgr−/− and Lyn−/− mice was similar to WT animals, the number of adherent and transmigrated neutrophils was significantly reduced in Hck−/− mice (Figures 3C and 3D). To examine whether Hck is likewise involved in GPCR-induced arrest and only required for the activation of Mac-1, we examined the early kinetics of neutrophil adhesion in cremaster muscle before and after the intravenous injection of fMLF. WT and Hck−/− mice had the same number of adherent neutrophils under baseline conditions and immediately after fMLF injection (Figure 3E). Like Btk, these data suggest that Hck is not involved in fMLF-induced LFA-1 activation. Whereas the attached neutrophils detached slowly over time in WT mice, Hck−/− neutrophils detached significantly faster after initial adhesion (Figure 3E), suggesting that Hck−/− neutrophils have a defect in Mac-1-dependent post-adhesion strengthening, similar to the defect seen in Btk−/− neutrophils. Likewise, the Hck−/− neutrophils also manifested a defect in intravascular crawling, showing both a reduced percentage of crawling cells with lower velocity and crawled distance (Figures 3F–3H). Finally, mirroring the defect in Btk−/− neutrophils, Hck-deficient cells manifested reduced fMLF-induced chemotaxis on fibrinogen surfaces, showing reduced velocity and directionality (Figures 3I–3L). These results suggest that Hck and Btk operate in the same fMLF-induced signaling pathway leading to Mac-1-mediated functions. Like Btk, Hck−/− neutrophils showed no defects in crawling or chemotaxis in response to CXCL1 stimulation (Figures S3F–S3L).
Figure 3. Hck, but Not Other Src Family Kinases, Is Required for fMLF-Mediated Neutrophil Recruitment.
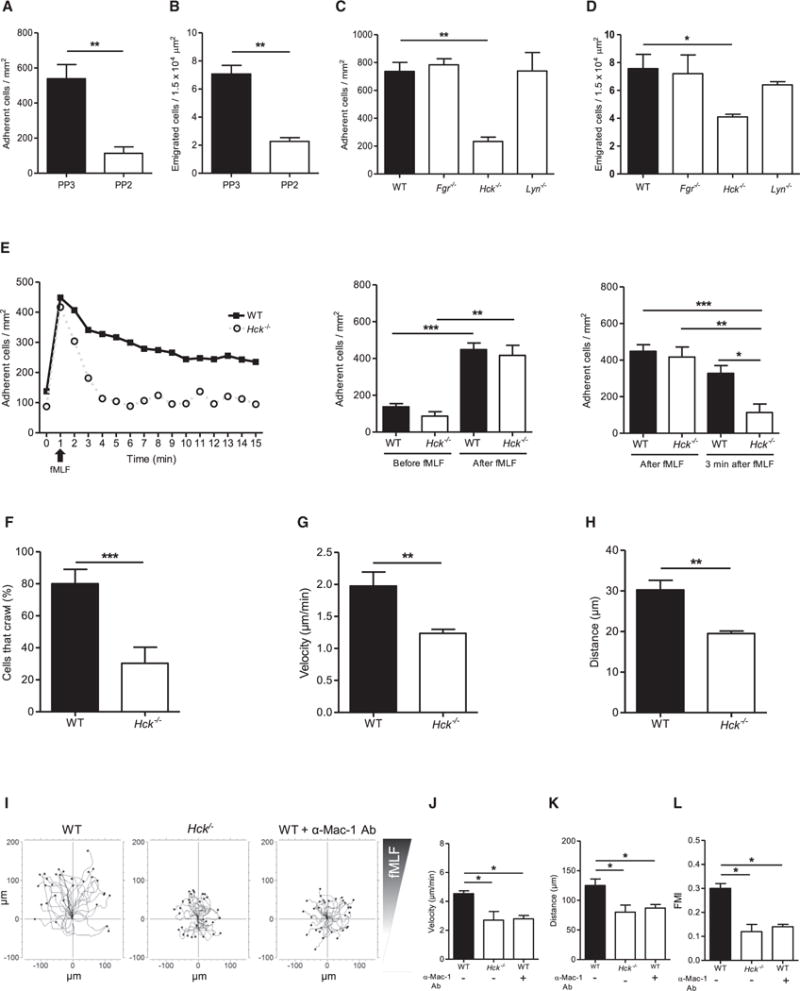
(A–H) Intravital microscopy of murine cremaster muscle venules after treatment with fMLF for 4 hr in WT mice after i.v. injection of the SFK inhibitor (PP2) or the inactive control (PP3) (A, B) and in WT, Fgr−/−, Hck−/−, and Lyn−/− (C, D) mice.
(A–D) Number of adherent cells per mm2 (A, C) and number of transmigrated cells per 1.5 × 104 μm2 (B, D) 4 hr after intrascrotal injection of fMLF.
(E) GPCR-induced arrest of neutrophils in postcapillary venules of WT (filled square) and Hck−/− (open circle) mice after fMLF injection (i.v.). Respective statistics are presented in related bar graphs.
(F–H) Intravascular crawling of Gr-1-labeled neutrophils in WT and Hck−/− mice during superfusion with fMLF. Percentage of adherent cells that crawled (F), crawling velocity (G), and crawled distance (H).
(I) Representative trajectory plots (30 cells from 3 independent experiments) of chemotaxing WT and Hck−/− neutrophils toward indicated fMLF gradient in vitro.
(J–L) Velocity (J), migration distance (K), and forward migration index (FMI) (L) of WT and Hck−/− neutrophils.
Depicted are mean + SEM; n ≥ 3 individual mice/group; *p < 0.05; **p < 0.01; ***p < 0.001 for all panels except (I). See also Figures S3 and S7 and Movies S3 and S4.
WASp and PLCγ2 Are Required for fMLF-Mediated Neutrophil Recruitment
WASp and PLCγ2 have been implicated in different intracellular signaling events in neutrophils and possess the ability to interact with Btk. Therefore, we investigated whether WASp and PLCγ2 are required for neutrophil recruitment during sterile inflammation. In the cremaster model, Was−/− and Plcg2−/− mice had significantly reduced numbers of adherent and extravasated neutrophils at 4 hr after fMLF injection compared to WT mice (Figures 4A and 4B). We noted a defect in Mac-1-dependent post-adhesion strengthening during GPCR-induced arrest and intravascular crawling in Btk−/− and Hck−/− mice, so we examined the early kinetics of intravascular adhesion in Was−/− and Plcg2−/− mice. As seen in Btk−/− and Hck−/− mice, deficiency of WASp or PLCγ2 did not result in a defect in activating LFA-1, because the number of adherent cells was the same as in WT mice immediately after intravenous injection of fMLF, but Was−/− and Plcg2−/− neutrophils failed to activate Mac-1 similar to Btk−/− and Hck−/− neutrophils (Figure 4C). Furthermore, Was−/− and Plcg2−/− mice showed a significantly reduced percentage of crawling cells compared to WT mice (Figure 4D). Crawling velocity (Figure 4E) and crawling distance (Figure 4F) were also significantly reduced in Was−/− and Plcg2−/− mice compared to WT mice. These data suggest that Btk, Hck, WASp, and PLCγ2 are all involved in the same signaling pathway leading to neutrophil recruitment during sterile inflammation.
Figure 4. WASp and PLCγ2 Are Required for fMLF-Mediated Neutrophil Recruitment.
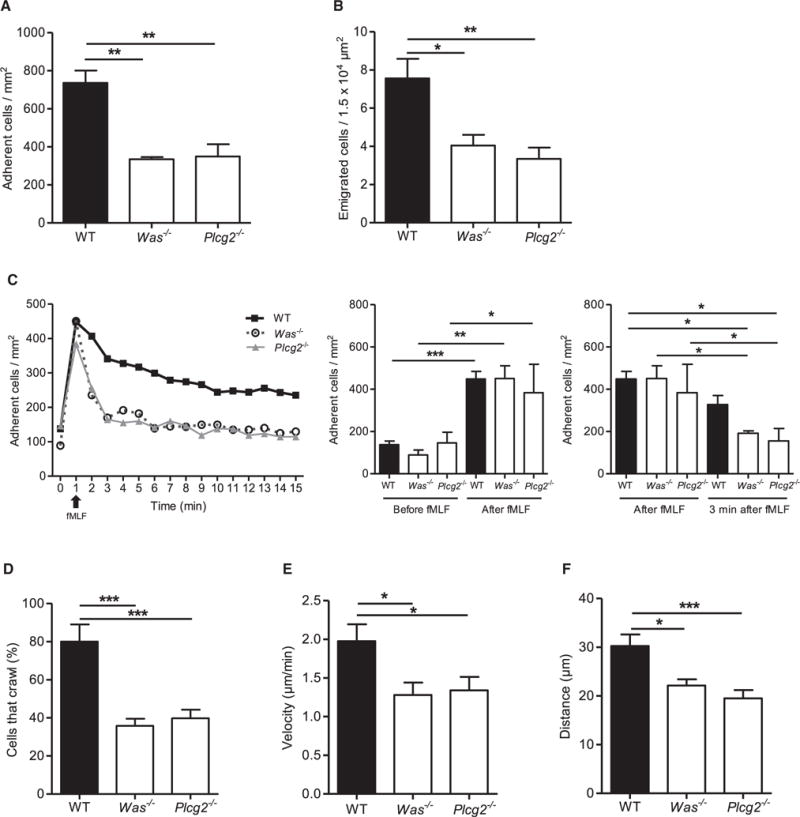
Intravital microscopy of postcapillary venules in the murine cremaster was performed in WT, Was−/−, and Plcg2−/− mice.
(A and B) Number of adherent cells per mm2 (A) and number of transmigrated cells per 1.5 × 104 μm2 (B) 4 hr after intrascrotal fMLF injection.
(C) GPCR-induced arrest of neutrophils in postcapillary venules of WT (filled square), Was−/− (open circle), and Plcg2−/− (filled triangle) after fMLF injection (i.v.). Respective statistics are presented in related bar graphs.
(D–F) Intravascular crawling of Gr-1-labeled neutrophils in WT, Was−/−, and Plcg2−/− mice during superfusion with fMLF. Percentage of adherent cells that crawled (D), crawling velocity (E), and crawled distance (F).
Depicted are mean + SEM; n ≥ 3 individual mice/group; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S7.
Crosstalk between Btk and WASp Is Required for fMLF-Mediated Mac-1, but Not LFA-1, Activation
Hck−/−, Btk−/−, Was−/−, and Plcg2−/− mice had a similar defect in neutrophil recruitment into inflamed tissue. To investigate LFA-1 and Mac-1 activation in Btk−/− and Was−/− neutrophils, we performed soluble ICAM-1- and fibrinogen-binding assays. Under resting conditions, WT, Btk−/−, and Was−/− neutrophils showed almost no ICAM-1 binding. After stimulation with fMLF, WT, Btk−/−, and Was−/− neutrophils showed a significantly increased ICAM-1 binding (Figure 5A), confirming our observation that Btk and WASp are not involved in fMLF-induced LFA-1 activation. We also conducted a fibrinogen-binding assay to investigate Mac-1 activation in these neutrophils. Fibrinogen binding was low in unstimulated WT, Btk−/−, and Was−/− neutrophils. After stimulation with fMLF, fibrinogen binding significantly increased in WT neutrophils, whereas the increase was significantly reduced in Btk−/− and Was−/− neutrophils (Figure 5B). We confirmed that ICAM-1 binding was mediated mainly by LFA-1, whereas fibrinogen binding was via Mac-1, using blocking mAbs for each integrin (Figures S4A and S4B). To ask whether LFA-1-induced signaling leading to Mac-1 activation was also affected in these cells, we directly crosslinked LFA-1 and then examined fibrinogen binding to Mac-1. In these experiments, crosslinking of LFA-1 led to significantly increased fibrinogen binding in WT cells but not in Btk−/− cells (Figure S4C). In addition, we demonstrated that blocking LFA-1 resulted in significantly less specific ICAM-1 binding to Mac-1 in Btk−/− and Was−/− neutrophils compared to WT neutrophils (Figure S4B). To show that fMLF-induced Mac-1 activation is also defective after pharmacologic inhibition of Btk (Btk inhibitor LFM A13) or PLC (PLC inhibitor U73122), we assessed the binding of a reporter antibody that detects only the activated form of MAC-1 in human neutrophils via flow cytometry. After stimulation with fMLF, inhibition of Btk or PLC in primary human neutrophils significantly abolished MAC-1 activation compared with control neutrophils (Figure 5C).
Figure 5. Crosstalk between Btk and WASp Is Required for fMLF-Mediated Mac-1, but Not LFA-1, Activation.
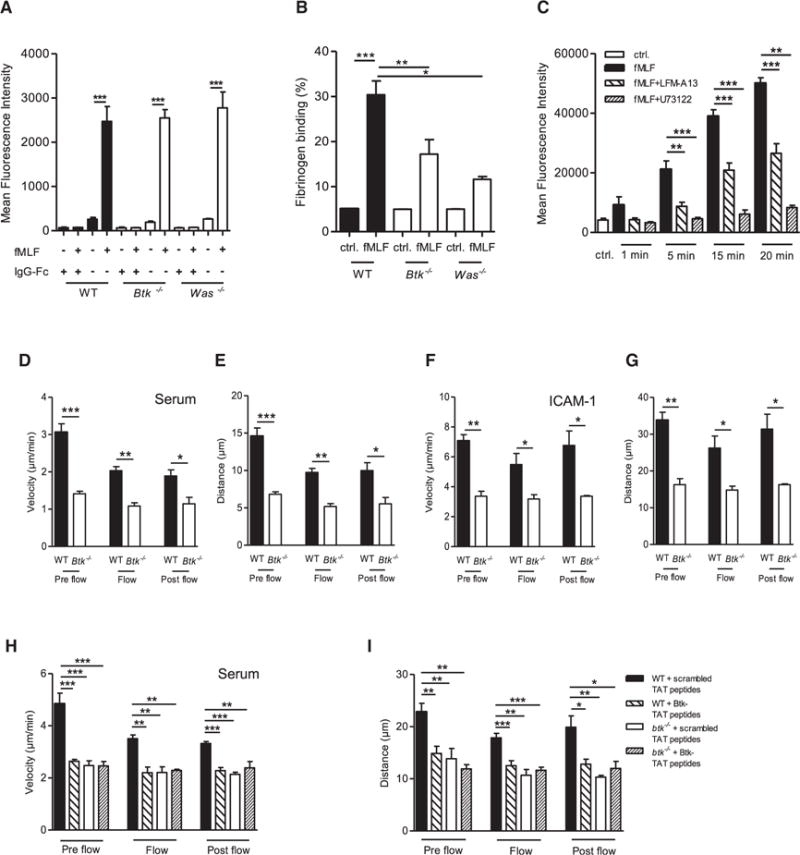
(A) LFA-1 binding of ICAM-1 by unstimulated and fMLF-stimulated WT, Btk−/−, and Was−/− neutrophils and respective IgG-Fc control.
(B) Mac-1-dependent fibrinogen binding in unstimulated and fMLF-stimulated WT, Btk−/−, and Was−/− neutrophils.
(C) Binding of Mac-1 on isolated human neutrophils by an activation-dependent reporter antibody for Mac-1 (CBRM1/5) with or without fMLF stimulation for indicated time points and with or without preincubation with the Btk inhibitor LFM-A13 and the phospholipases C inhibitor U73122.
(D–I) Parallel plate flow chamber crawling assay of WT and Btk−/− neutrophils stimulated with fMLF. Crawling velocities (D, F, H) and accumulated crawled distance (E, G, I) before (pre flow), during (flow), and after (post flow) applying flow (2 dyn/cm2, 5 min) of untreated WT and Btk−/− neutrophils on murine blood serum (D, E) or ICAM-1 (F, G) and scrambled or Btk-TAT peptide-pretreated cells on murine blood serum (H, I).
Depicted are mean + SEM of n ≥ 3 independent performed experiments; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figures S4 and S7.
Furthermore, we analyzed isolated Btk−/− and WT neutrophils in an in vitro crawling assay. The crawling velocity of Btk−/− neutrophils was significantly reduced under non-flow and flow conditions compared with WT neutrophils, on both serum- and ICAM-1-coated surfaces (Figures 5D and 5F). Furthermore, Btk−/− neutrophils had a significantly reduced accumulated distance compared with WT neutrophils (Figures 5E and 5G). By using blocking TAT peptides against the tyrosine 223 of Btk, we demonstrated that phosphorylation of this residue was crucial to ensure crawling in vitro (Figures 5H, 5I, and S4D–S4H). Furthermore, we demonstrated that phosphorylation of WASp in WT neutrophils treated with blocking TAT peptides and/or LFM-A13 was abolished after fMLF stimulation, indicating that WASp is a downstream effector of Btk (Figures S4I–S4K). Incubation of neutrophils with TAT peptides or LFM-A13 did not interfere with unrelated Fpr-mediated ERK1 and 2 activation (Figure S4L).
Btk Regulates fMLF-Triggered Intracellular Signaling
To investigate how these molecules are integrated in the fMLF-mediated signaling pathway leading to integrin activation, we stimulated bone-marrow-derived neutrophils with fMLF and performed biochemistry experiments. Stimulation of WT neutrophils induced phosphorylation of Btk at tyrosine 223 (Figures 6A and S5A) but not at tyrosine 551 (Figure 6A and S5B). The SFKs Fgr, Hck, and Lyn were also phosphorylated at tyrosine 416 within their kinase domains (Figures 6B and S5C–S5E). To demonstrate that Btk is downstream of SFKs, Btk−/− neutrophils were stimulated with fMLF. Phosphorylation of SFKs (Tyr416) was present in WT and Btk−/− neutrophils (Figures 6C and S5F). To investigate whether only Hck is responsible for fMLF-triggered signaling, we measured Btk phosphorylation in neutrophils from WT, Fgr−/−, Hck−/−, and Lyn−/− mice. fMLF stimulation induced phosphorylation of Btk in neutrophils from WT, Fgr−/−, and Lyn−/− (Figures 6D, S5G, and S5I) but not Hck−/− (Figures 6D and S5H) neutrophils. In Btk−/− neutrophils, phosphorylation of PLCγ2 (Tyr1217), Akt (Ser473), p38 MAPK, and WASp as well as activation of Rap1 was abolished (Figures 6E, 6F, and S5J–S5M). Btk is not involved in the fMLF-triggered activation of Erk1 and 2 (Figure 6G). By treatment of WT and Btk−/− neutrophils with pertussis toxin (PTx) prior to stimulation with fMLF, we could demonstrate that phosphorylation of Btk and its target molecules is dependent on Gαi signaling (Figure S6).
Figure 6. Btk Regulates fMLF-Triggered Intracellular Signaling.
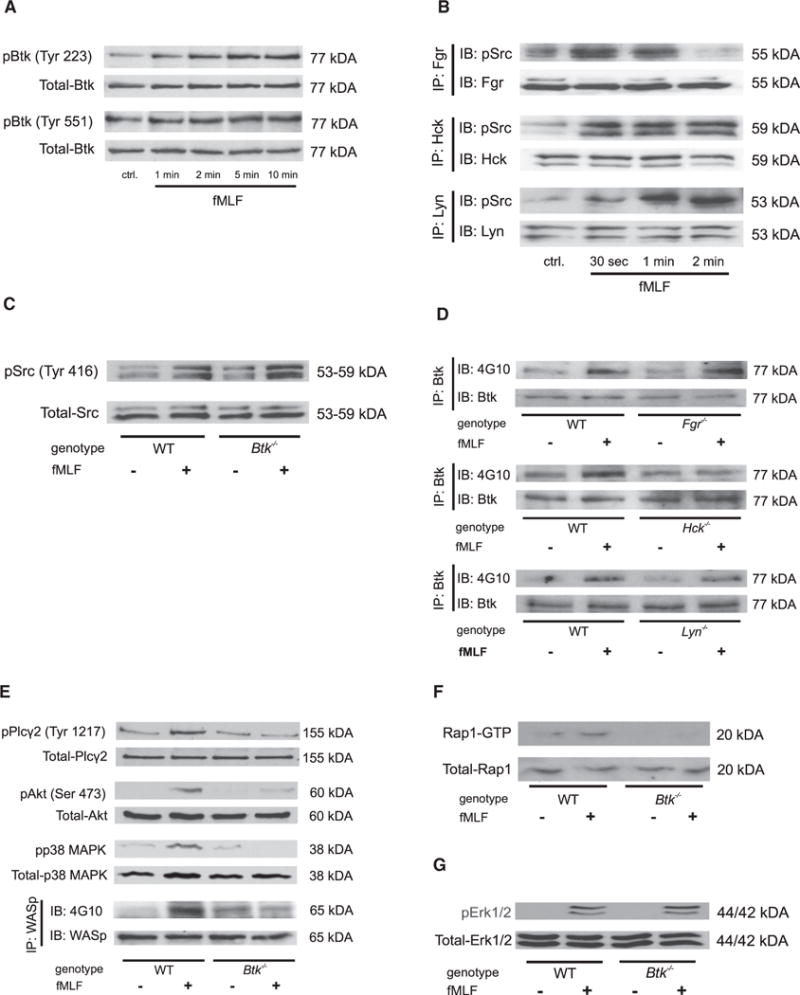
(A) Lysates of WT neutrophils were immunoblotted with a p-Btk (Tyr223 or Tyr551) antibody or total-Btk antibody (n = 4).
(B) Lysates of WT neutrophils were immunoprecipitated (IP) with an antibody against Fgr, Hck, or Lyn followed by immunoblotting with a p-Src (Tyr416) antibody or total Fgr, Hck, or Lyn antibodies.
(C) Lysates of WT and Btk−/− neutrophils were immunoblotted with a p-Src (Tyr416) antibody or total-Src antibody.
(D) Lysates of WT, Fgr−/−, Hck−/−, and Lyn−/− neutrophils were immunoprecipitated (IP) with an antibody against Btk followed by immunoblotting with a general phosphotyrosine (4G10) antibody or total Btk antibody.
(E) Lysates of WT and Btk−/− neutrophils were immunoblotted or immunoprecipitated, demonstrating the phosphorylation of PLCγ2, Akt, p38 MAPK, or WASp. Total lysates were immunoblotted with antibodies to p-PLCγ2 (Tyr1217), total-PLCγ2, p-Akt (Ser473), total-Akt, p-p38 MAPK, or total-p38 MAPK. Lysates were immunoprecipitated with a general phosphotyrosine (4G10) antibody or total-WASp antibody.
(F) GTP-bound Rap1 (Rap1-GTP) was precipitated from whole cell lysates using immobilized GST effector fusion protein. Lysates were immunoblotted with an antibody against Rap1.
(G) Lysates of WT and Btk−/− neutrophils were immunoblotted with a p-p44 and 42 MAPK (Erk1 and 2) antibody or total-p44 and 42 MAPK (Erk1 and 2) antibody.
Depicted are representative immunoblots of n ≥ 3 independent performed experiments. See also Figures S5–S7.
Btk Is Required for Outside-In and FcRγ Signaling in Neutrophils
To investigate the potential role of Btk in other signaling modalities like integrin-mediated outside-in signaling and FcRγ signaling, and to determine whether the fMLF-triggered signaling synergizes with these events, we analyzed the superoxide production in WT and Btk−/− neutrophils. Whereas WT neutrophils produced reactive oxygen species in response to fMLF stimulation (Figure 7A) or after integrin-mediated adhesion on poly-RGD (Figure 7B), Btk−/− neutrophils almost completely failed to produce any superoxide after stimulation. Adhesion on poly-RGD in presence of fMLF further increased the generation of reactive oxygen species in WT but not in Btk−/− neutrophils (Figure 7C), indicating a synergy of both signaling events.
Figure 7. Btk Is Involved in Integrin-Mediated Outside-In Signaling and FcRγ-Mediated Functions.
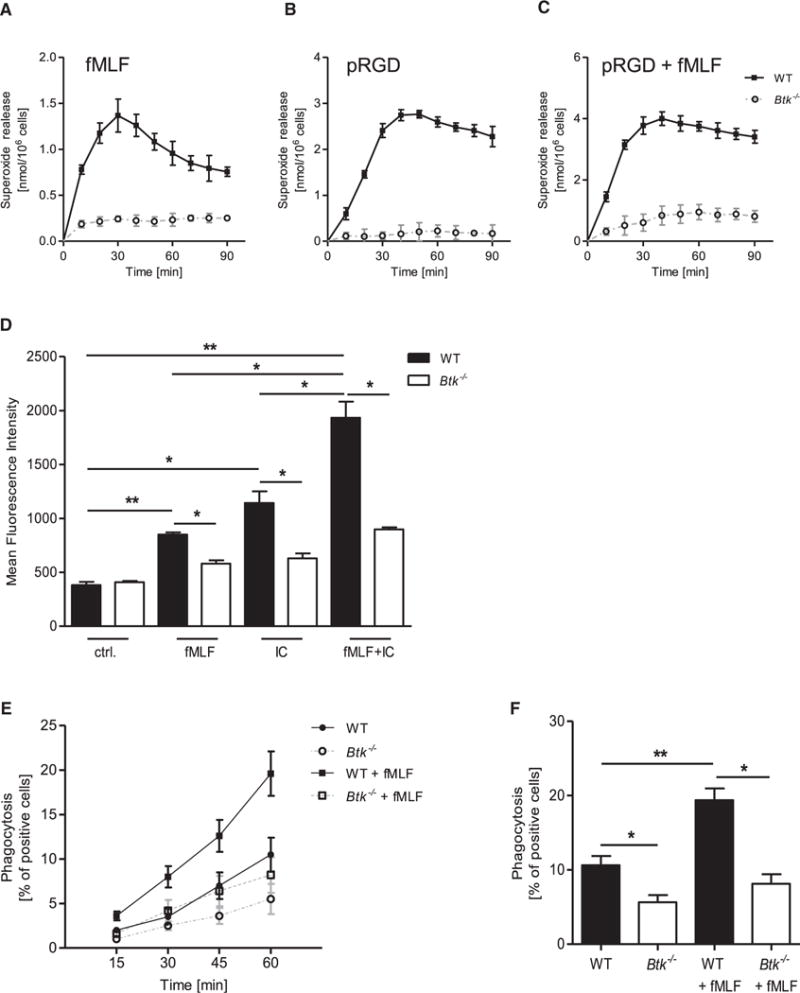
(A–C) Superoxide release of WT and Btk−/− neutrophils stimulated with 3 μM fMLF (A), plated on a polyvalent integrin ligand-coated surface (pRGD) without stimulus (B) or with 3 μM fMLF (C). Control values were subtracted from stimulated values.
(D) Quantitative binding of fibrinogen-coated fluorescent beads to unstimulated WT and Btk−/− neutrophils, stimulated with 1 μM fMLF, 10 μg/ml IgG immune complexes, or a combination of both as determined by flow cytometry.
(E) Phagocytosis of IgG-coated fluorescent beads by WT and Btk−/− neutrophils incubated at 37°C with or without 1 μM fMLF for indicated time points.
(F) Respective statistics of phagocytosis after 1 hr.
Depicted are mean + SEM of n ≥ 3 independent performed experiments; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S7.
To examine whether fMLF-mediated Mac-1 activation synergizes with FcRγ-triggered Mac-1 activation, WT and Btk−/− neutrophils were stimulated with either fMLF, IgG immune complexes, or a combination of both. Mac-1 activation determined by the binding of fibrinogen-coated fluorescent beads to WT neutrophils was significantly increased upon stimulation with fMLF or IgG immune complexes (IC) compared to Btk−/− neutrophils (Figure 7D). In addition, the stimulation of WT neutrophils with fMLF and IC in parallel further enhanced Mac-1 activation in WT but not in Btk−/− neutrophils. A similar effect was observed in FcRγ-mediated phagocytosis of IgG-coated fluorescent beads when neutrophils were co-stimulated with fMLF (Figures 7E and 7F). These results indicate that the synergy between Fpr- and FcRγ-mediated signaling events also requires Btk.
DISCUSSION
Several prior studies have shown that stimulation of neutrophils with fMLF can activate Src and Tec family kinases (Zarbock and Ley, 2011; Gilbert et al., 2003; Futosi et al., 2013; Liao et al., 2015), induce PI3K and Akt, phospholipases C, p38-MAPK, and WASp phosphorylation (Dorward et al., 2015), and trigger leukocyte recruitment (McDonald and Kubes, 2012). However, the signaling pathway linking Fprs with Mac-1 activation and neutrophil recruitment within necrotic regions of sterile inflammation is largely unknown. In this study, we identified the crucial role of Btk in the fMLF-induced Mac-1 activation pathway leading to neutrophil recruitment during sterile inflammation. By contrast, Btk played no role in CXCL1-induced integrin activation. The Src family kinase Hck, WASp, and PLCγ2 were also involved in the fMLF-Mac-1 activation pathway. Disturbing the interaction between Btk and WASp abolished Mac-1-dependent neutrophil migration. Thus, we identified a signalosome consisting of Hck, Btk, WASp, and PLCγ2 that is indispensable for fMLF-induced Mac-1 activation events required for neutrophil recruitment and Mac-1-dependent cell migration during sterile inflammation. In addition, we found that Btk was involved in FcRγ-mediated functions, as well as in the synergistic interaction between fMLF signaling and FcRγ-mediated phagocytosis.
Activation of β2 integrins occurs in a sequential fashion and via different pathways during neutrophil recruitment and is dependent on the stimulus and its respective binding receptor (Herter et al., 2013; Phillipson et al., 2009; Lawson et al., 2011; Heit et al., 2005; McDonald et al., 2010). Rac1 and its specific guanine nucleotide exchanging factor are required for Mac-1 activation, neutrophil crawling, and neutrophil recruitment (Herter et al., 2013). Our data provide insight into the intracellular signaling hierarchy between SFKs, integrin activation, and neutrophil responses triggered by fMLF. We have demonstrated that Btk shares prominent parallels with P-Rex1, having distinct and unique roles in integrin activation. Btk is not involved in LFA-1 activation, but acts like P-Rex1 as a cell-intrinsic positive regulator for Mac-1-mediated steps like in vivo and in vitro crawling via WASp. Moreover, we identified that only Hck and no other SFK is required for fMLF-mediated neutrophil recruitment during sterile inflammation. One can hypothesize that Btk might link Hck to the activation of Rac-GEFs (Futosi et al., 2013; Fumagalli et al., 2007; Lawson et al., 2011), because it is known that Btk can directly interact with Rac-GEFs via its SH3 domain (Guinamard et al., 1997). It is also conceivable that this Btk signalosome might be associated with a specialized adaptor that links it selectively to the αM but not the αL subunit of β2 integrins. We identified WASp and PLCγ2 as essential downstream molecules of Btk required for different neutrophil recruitment steps, so it is possible to speculate that different branches exist in the fMLF-triggered signaling pathway required for well-orchestrated neutrophil responses and integrin activation.
The SFK-Btk-PLC2 pathway has striking similarities to the immunoreceptor and integrin outside-in signaling pathways (Abram and Lowell, 2007). However, there are also differences. Elimination of only one SFK does not affect the inflammatory response in an endotoxic shock model (Lowell and Berton, 1998) or β2-integrin-dependent neutrophil functions (Lowell et al., 1996). At least two SFKs must be eliminated to abolish β2-integrin-dependent neutrophil adhesion and superoxide production (Lowell et al., 1996). However, such a redundancy does not exist in the signaling pathway triggered by fMLF. Elimination of Hck abolishes downstream signaling, including Btk phosphorylation and Mac-1-dependent recruitment steps. This study is one of the few examples demonstrating a specific and non-redundant function for a SFK. In most other signaling pathways investigated, one SFK might be dominant (such as Src in β3-in-tegrin signaling in osteoclasts or Lyn in inhibitory signaling in B lymphocytes) or there might be no apparent kinase selectivity (such as in integrin signaling). The specific role of Hck in neutrophil fMLF-triggered signaling is unique and provides a potential explanation for the evolution of this individual member of the Src kinase family.
The SH3 domain of Btk is described to be crucial for binding to interaction partners and especially for its autophosphorylation (Park et al., 1996). It is well known that SFKs can bind to and activate Btk via its SH3 domain (Afar et al., 1996; Cheng et al., 1994). We found that Btk is phosphorylated upon fMLF stimulation at the tyrosine 223 within the SH3 domain and that Hck—as the one and only SFK—is important for downstream regulation of Btk activity. Moreover, it is well known that an interaction of the cytoskeletal regulator WASp with the SH3 domain of Btk is critical for signaling in macrophages (Sakuma et al., 2012, 2015) and that WASp deficiency in neutrophils leads to impaired firm adhesion during shear flow, highlighting the importance of this molecule in neutrophil responses (Zhang et al., 2006). In line with these findings, we have demonstrated in this study that WASp was required for fMLF-induced Mac-1 activation and neutrophil recruitment. In addition, disruption of the Btk-WASp pathway, either through deletion of Btk or TAT peptide-mediated blockade of Btk phosphorylation, leads to reduced WASp activity and impaired neutrophil crawling. This study is clearly demonstrating the crosstalk between Btk and WASp leading to Mac-1-dependent neutrophil recruitment steps during sterile inflammation. In mast cells, Btk regulates PLCγ2- and Rac-dependent branches, involving both Ca2+ signaling and F-actin reorganization leading to cell migration (Kuehn et al., 2010). This pathway has striking similarities with the fMLF-triggered pathway in neutrophils, because we identified in addition to WASp, that PLCγ2 also acts as a second downstream effector of Btk. Taking these results together, we identified a fMLF-triggered signaling axis consisting of Hck, Btk, WASp, and/or PLCγ2, which specifically regulates Mac-1-mediated neutrophil recruitment events and functions, graphically presented in Figure S7.
Previous studies clearly demonstrated that hepatic injury results in the release of endogenous mitochondrial alarmins including formylated peptides into the circulation (Phillipson and Kubes, 2011; Zhang et al., 2010). These signals induce profound changes of the vascular microenvironment and activate neutrophils resulting in directional crawling toward damaged cells (Phillipson and Kubes, 2011). Furthermore, McDonald et al. (2010) have demonstrated that adhesion, crawling, and chemotaxis around the necrotaxis zone (which is defined as the zone 150 μm around the point of liver injury) is chemokine dependent. MIP2 expression was detectable only outside the necrotaxis area. However, within the necrotaxis zone, the formyl-peptide necrotaxis signal predominates (McDonald et al., 2010; Phillipson and Kubes, 2011). Neutrophils prioritize this signal in an Fpr-dependent manner, allowing them to directly enter into lesion. When Fpr1 signaling is blocked, for example in Fpr1−/− mice, neutrophils are recruited into the injured liver around the necrotaxis zone but do not directly enter the necrotic lesion, which ultimately attenuates the overall severity of liver injury (McDonald et al., 2010; Zhang et al., 2010; McDonald and Kubes, 2012; Phillipson and Kubes, 2011). Here, we have demonstrated that Btk is involved in fMLF-induced Mac-1 activation but not CXCL1-induced integrin activation. As a result, during liver injury, Btk-deficient neutrophils migrate into the injured liver but fail to directly enter the necrotaxis zone, leading to overall reduced hepatic injury in Btk−/− mice.
This study points out a crucial role of Btk in a number of fMLF-triggered neutrophil functions that are active in sterile tissue injury. In addition, this study provides insight in the involvement of Btk in other modalities of neutrophil activation such as integrin-mediated outside-in and FcRγ signaling, which might synergize with Fpr signaling in the extravascular space to modulate appropriate neutrophil responses. Our data suggest that there might be an interplay between multiple signaling pathways that are activated by inside-out and outside-in signals (see Figure S7). The finding that Btk is critical for the specific Mac-1 activation and Mac-1-dependent recruitment steps during sterile inflammation suggests that Btk might be a promising therapeutic target for anti-inflammatory therapy.
EXPERIMENTAL PROCEDURES
Spinning Disk Confocal Intravital Microscopy of the Murine Liver after Focal Hepatic Necrosis
The Animal Care and Use Committees of North Rhine Westphalia (Germany) approved all animal experiments. Mice were anesthetized intraperitoneally with a mixture of ketamine hydrochloride (125 mg/kg, Sanofi Winthrop Pharmaceuticals) and xylazine (12.5 mg/kg, TranquiVed, Phonix Scientific). After anesthesia, mice were placed on a heating pad set to 37°C to maintain the body temperature for the duration of all performed experiments. The left carotid artery was cannulated for administration of additional anesthetics.
For intravital microscopy of the liver, mice were prepared as previously described (McDonald et al., 2010). In brief, a midline laparotomy was performed followed by removal of the skin and abdominal muscle to expose the liver lobe. A thin platinic wire was heated and briefly pressed on the liver to induce a focal injury on the surface of the liver. With a single application of 2 μl of a propidium iodide solution (1.0 mg/ml) diluted 1:25 in PBS to the surface of the liver, necrotic cells were labeled immediately. To avoid dehydration, all exposed tissues were moistened with PBS-soaked tissues. Directly after preparation, the exposed liver was visualized with an upright spinning disc confocal microscope (CellObserver SD, Zeiss) equipped with a 5×/0.25 FLUAR objective and time-lapse Z-stacks were recorded for up to 4 hr after focal injury. The number of adherent neutrophils was determined for different time points per field of view, or after 4 hr after injury within specific regions (within injury, proximal 150 μm around injury, and beyond 150 μm from injury border). Duration of adhesion and neutrophil crawling was determined within the necrotaxis zone 2.5 hr after injury. 30 neutrophils per field of view were randomly selected for tracking, and chemotaxis parameters (migration plots, forward migration index, crawling velocities) were set. All investigated parameters were analyzed with FIJI or Chemotaxis and Migration tool (Ibidi).
The determination of recruited neutrophils to the liver 4 hr after focal injury or sham procedure via flow cytometry was performed as previously described (Rossaint et al., 2014).
The levels of the transaminases GOT and GPT before and 4 hr after focal hepatic necrosis in serum of blood in WT and Btk−/− mice was determined with a high-volume hematology analyzer (ADVIA, Siemens).
Intravital Microscopy of the Murine Cremaster Muscle
Mice were anesthetized with injection of 125 mg/kg ketamine hydrochloride (Sanofi Winthrop Pharmaceuticals) and 12.5 mg/kg xylazine (Tranqui Ved, Phonix Scientific) i.p., and the cremaster muscle was prepared for intravital imaging as previously described (Mueller et al., 2010). Postcapillary venules with a diameter between 20 and 40 μm were investigated. To determine adhesion and transmigration in vivo, mice were injected intrascrotally with 16 μg fMLF (Sigma-Aldrich) 4 hr before preparation of the cremaster muscle. Intravital microscopy was performed on an upright microscope (Axioskop, Carl Zeiss) with a 40×0.75 NA saline immersion objective. Neutrophil adhesion was determined by transillumination intravital microscopy, whereas neutrophil extravasation was investigated by reflected light oblique transillumination microscopy as described previously (Mueller et al., 2010). Recorded images were analyzed off-line with ImageJ (NIH) and AxioVision (Carl Zeiss) software. Emigrated cells were determined in an area 75 × 100 μm to each side of a vessel (representing 1.5 × 104 μm2 tissue area). The microcirculation was recorded with a digital camera (Sensicam QE, Cooke). Blood flow centerline velocity was measured with a dual photodiode sensor system (Circusoft Instrumentation). Centerline velocities were converted to mean blood flow velocities as previously described (Mueller et al., 2010). For GPCR-induced arrest, the carotid artery was cannulated for injection of fMLF (16 μg), CXCL-1 (500 ng), or blocking antibodies. In a representative vessel, the initial number of adherent neutrophils was determined, fMLF was injected, and the vessel was recorded for 15 min. Movies were analyzed with Slidebook software (Intelligent Imaging Innovations). For blocking experiments, monoclonal antibodies against Mac-1 (30 μg/mouse; eBioscience), LFA-1 (30 μg/mouse; eBioscience), or both were injected i.v. immediately before the experiments.
Soluble ICAM-1- and Fibrinogen-Binding Assay
The soluble ICAM-1- and fibrinogen-binding assays were performed as previously described (Lefort et al., 2012). In brief, to assess LFA-1-specific ICAM-1 binding, isolated murine neutrophils were preincubated with a functional blocking anti-CD11b (clone M1/70, 10 μg/ml) antibody to prevent Mac-1-dependent ICAM-1 binding or a blocking anti-CD11a antibody (clone TIB217, 10 μg/ml) to prevent LFA-1-dependent ICAM-1 binding. Afterward, neutrophils were stimulated with fMLF (10 μM) or CXCL-1 (100 ng/ml, 3 min, 37°C) or left unstimulated in the presence of ICAM-1/Fc (20 μg/ml, R&D Systems) or IgG control and APC-conjugated anti-human IgG1 (Fc-specific, Southern Biotechnology). Neutrophils were fixed on ice and stained with FITC-conjugated anti-Ly6G (Biolegend). LFA-1-specific binding to ICAM-1/Fc was measured by flow cytometry. To investigate Mac-1 affinity to fibrinogen, isolated murine neutrophils were incubated for 10 min at 37°C with 150 μg/ml Alexa 647-conjugated fibrinogen (Invitrogen) and stimulated with fMLF or CXCL-1 or were left unstimulated. Neutrophils were treated with EDTA (2 mM) as negative controls. LFA-1 crosslinking to induce Mac-1 activation was performed by incubating murine neutrophils with anti-CD11a (Biolegend, clone M17/4) and an anti-rat secondary antibody (Santa Cruz) for 10 min at 37°C. Fluorescence intensity was measured by flow cytometry. The percentage of neutrophil positive for fibrinogen binding was calculated by defining a threshold of the fluorescence intensity where 95% of neutrophils in the EDTA control were considered as negative.
Supplementary Material
Highlights.
Btk is required for neutrophil recruitment during sterile inflammation
Btk is required for selective fMLF-triggered Mac-1, but not LFA-1, activation
Hck, WASp, and PLCγ2 are key signaling molecules in the fMLF-triggered pathway
Btk is required for integrin-mediated outside-in signaling and FcRγ-mediated functions
Acknowledgments
The authors would like to thank Stefan Volkery for expert technical support. This work was supported by the Deutsche Forschungsgemeinschaft (ZA428/6-1 and SFB1009_A5 to A.Z. and BL-1292/1 to H.B.), IZKF Münster (Za2/001/14 to A.Z.), and Cells-in-Motion Cluster of Excellence EXC 1003-CiM (University of Münster, Germany; to A.Z.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, Supplemental Experimental Procedures, and four movies and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.11.011.
AUTHOR CONTRIBUTIONS
S.V. and H.B. performed experiments, analyzed the data, and wrote the manuscript; M.B. performed experiments, analyzed the data, and contributed to writing the manuscript; C.A.L. interpreted data and contributed to writing the manuscript; and A.Z. conceived of the study, analyzed the data, and wrote the manuscript.
References
- Abram CL, Lowell CA. Convergence of immunoreceptor and integrin signaling. Immunol Rev. 2007;218:29–44. doi: 10.1111/j.1600-065X.2007.00531.x. [DOI] [PubMed] [Google Scholar]
- Afar DE, Park H, Howell BW, Rawlings DJ, Cooper J, Witte ON. Regulation of Btk by Src family tyrosine kinases. Mol Cell Biol. 1996;16:3465–3471. doi: 10.1128/mcb.16.7.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block H, Zarbock A. The role of the tec kinase Bruton’s tyrosine kinase (Btk) in leukocyte recruitment. Int Rev Immunol. 2012;31:104–118. doi: 10.3109/08830185.2012.668982. [DOI] [PubMed] [Google Scholar]
- Cheng G, Ye ZS, Baltimore D. Binding of Bruton’s tyrosine kinase to Fyn, Lyn, or Hck through a Src homology 3 domain-mediated interaction. Proc Natl Acad Sci USA. 1994;91:8152–8155. doi: 10.1073/pnas.91.17.8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. 2015;185:1172–1184. doi: 10.1016/j.ajpath.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli L, Zhang H, Baruzzi A, Lowell CA, Berton G. The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J Immunol. 2007;178:3874–3885. doi: 10.4049/jimmunol.178.6.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futosi K, Fodor S, Mócsai A. Reprint of neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013;17:1185–1197. doi: 10.1016/j.intimp.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Gilbert C, Levasseur S, Desaulniers P, Dusseault AA, Thibault N, Bourgoin SG, Naccache PH. Chemotactic factor-induced recruitment and activation of Tec family kinases in human neutrophils. II. Effects of LFM-A13, a specific Btk inhibitor. J Immunol. 2003;170:5235–5243. doi: 10.4049/jimmunol.170.10.5235. [DOI] [PubMed] [Google Scholar]
- Grommes J, Drechsler M, Soehnlein O. CCR5 and FPR1 mediate neutrophil recruitment in endotoxin-induced lung injury. J Innate Immun. 2014;6:111–116. doi: 10.1159/000353229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R, Fougereau M, Seckinger P. The SH3 domain of Bruton’s tyrosine kinase interacts with Vav, Sam68 and EWS. Scand J Immunol. 1997;45:587–595. doi: 10.1046/j.1365-3083.1997.d01-447.x. [DOI] [PubMed] [Google Scholar]
- Heit B, Colarusso P, Kubes P. Fundamentally different roles for LFA-1, Mac-1 and alpha4-integrin in neutrophil chemotaxis. J Cell Sci. 2005;118:5205–5220. doi: 10.1242/jcs.02632. [DOI] [PubMed] [Google Scholar]
- Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood. 2013;121:2301–2310. doi: 10.1182/blood-2012-09-457085. [DOI] [PubMed] [Google Scholar]
- Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, Rådinger M, Brown JM, Ali K, Vanhaesebroeck B, Beaven MA, Metcalfe DD, Gilfillan AM. Btk-dependent Rac activation and actin rearrangement following FcepsilonRI aggregation promotes enhanced chemotactic responses of mast cells. J Cell Sci. 2010;123:2576–2585. doi: 10.1242/jcs.071043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson CD, Donald S, Anderson KE, Patton DT, Welch HCE. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol. 2011;186:1467–1476. doi: 10.4049/jimmunol.1002738. [DOI] [PubMed] [Google Scholar]
- Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, Critchley DR, Ginsberg MH, Fässler R, Ley K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. 2012;119:4275–4282. doi: 10.1182/blood-2011-08-373118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H-R, Chien C-R, Chen J-J, Lee T-Y, Lin S-Z, Tseng C-P. The anti-inflammatory effect of 2-(4-hydroxy-3-prop-2-enyl-phenyl)-4-prop-2-enyl-phenol by targeting Lyn kinase in human neutrophils. Chem Biol Interact. 2015;236:90–101. doi: 10.1016/j.cbi.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Lowell CA, Berton G. Resistance to endotoxic shock and reduced neutrophil migration in mice deficient for the Src-family kinases Hck and Fgr. Proc Natl Acad Sci USA. 1998;95:7580–7584. doi: 10.1073/pnas.95.13.7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA, Berton G. Integrin signal transduction in myeloid leukocytes. J Leukoc Biol. 1999;65:313–320. doi: 10.1002/jlb.65.3.313. [DOI] [PubMed] [Google Scholar]
- Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B, Kubes P. Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol Pathol. 2012;40:157–165. doi: 10.1177/0192623311427570. [DOI] [PubMed] [Google Scholar]
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, Zarbock A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood. 2010;115:3118–3127. doi: 10.1182/blood-2009-11-254185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, Scharenberg AM, Kinet JP, Witte ON. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 1996;4:515–525. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson M, Heit B, Parsons SA, Petri B, Mullaly SC, Colarusso P, Gower RM, Neely G, Simon SI, Kubes P. Vav1 is essential for mechanotactic crawling and migration of neutrophils out of the inflamed microvasculature. J Immunol. 2009;182:6870–6878. doi: 10.4049/jimmunol.0803414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, Soehnlein O, Zarbock A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood. 2014;123:2573–2584. doi: 10.1182/blood-2013-07-516484. [DOI] [PubMed] [Google Scholar]
- Sakuma C, Sato M, Takenouchi T, Chiba J, Kitani H. Critical roles of the WASP N-terminal domain and Btk in LPS-induced inflammatory response in macrophages. PLoS ONE. 2012;7:e30351. doi: 10.1371/journal.pone.0030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma C, Sato M, Takenouchi T, Kitani H. Specific binding of the WASP N-terminal domain to Btk is critical for TLR2 signaling in macrophages. Mol Immunol. 2015;63:328–336. doi: 10.1016/j.molimm.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Zarbock A, Ley K. Protein tyrosine kinases in neutrophil activation and recruitment. Arch Biochem Biophys. 2011;510:112–119. doi: 10.1016/j.abb.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Zhang H, Schaff UY, Green CE, Chen H, Sarantos MR, Hu Y, Wara D, Simon SI, Lowell CA. Impaired integrin-dependent function in Wiskott-Aldrich syndrome protein-deficient murine and human neutrophils. Immunity. 2006;25:285–295. doi: 10.1016/j.immuni.2006.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.