

Abstract
Polyketides and nonribosomal peptides are two important classes of natural products that are produced by many species of bacteria and fungi, but are exceedingly rare in metazoans. Here, we elucidate the structure of a hybrid polyketide-nonribosomal peptide from Caenorhabditis elegans that is produced in the CAN neurons and promotes survival during starvation-induced larval arrest. Our results uncover a novel mechanism by which animals respond to nutrient fluctuations to extend survival.
Polyketides and nonribosomal peptides represent two of the most important classes of natural products used in modern medicine. They include avermectin, whose derivative ivermectin is used to treat parasitic worms in over 300 million people annually, the antibiotic vancomycin, which is used to treat life-threating infections by gram-positive bacteria, and the immunosuppressant FK506, an essential drug after organ transplantation1. These natural products are biosynthesized by polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs), modular megasynthases that function in either an assembly-line or iterative manner2. Although PKS and NRPS genes are commonly found in many bacterial and fungal species, only simple, single-module PKSs and NRPSs are present in a few animal species3,4. Thus, it is quite remarkable that the genome of the nematode C. elegans encodes a huge (865 kDa), multi-module hybrid PKS/NRPS on the X chromosome (PKS-1) and a large (333 kDa), multi-module NRPS on chromosome III (NRPS-1)5,6. Homologs of PKS-1 and NRPS-1 are present in most nematode species, including parasitic ones (Supplementary Results, Supplementary Fig. 1)5. Here, we elucidate the chemical structure of the polyketide-nonribosomal peptide produced by PKS-1/NRPS-1 and show that this natural product promotes recovery from and survival during starvation-induced larval arrest.
We used comparative, untargeted metabolomics to identify the masses of the natural products that PKS-1 and NRPS-1 produce. Extracts from worms and from conditioned culture medium were generated from mixed-stage cultures of wild-type worms, pks-1 mutant worms, and nrps-1 mutant worms. The metabolites in the extracts were analyzed by HR-LC-MS and compared using XCMS (Fig. 1a,b)7. Two peaks (m/z 757.3866 and 755.3700), termed nemamide A (1) and nemamide B (2), respectively, were present in wild-type worm extracts and completely absent in both mutant worm extracts (Supplementary Fig. 2). Thus, PKS-1 and NRPS-1 likely work together to make a hybrid polyketide-nonribosomal peptide. Nemamide A and B are associated with the worm body rather than secreted into the culture medium, as the molecules were not detected by HR-LC-MS in the culture medium extracts.
Figure 1. Discovery and biosynthesis of the nemamides.

(a,b) Comparison of average peak areas for metabolite features in wild-type worms versus pks-1 mutant worms (a) and in wild-type worms versus nrps-1 mutant worms (b), with nemamide A and B highlighted in red. In a and b, extracts from three separate cultures were analyzed for each strain, and P values were calculated in XCMS using a Welch’s t-test. (c) Chemical structures of nemamide A and B. (d) Proposed biosynthetic assembly line for the nemamides. Domain abbreviations: acyl transferase (AT), acyl carrier protein (ACP), ketosynthase (KS), ketoreductase (KR), dehydratase (DH), methyltransferase (MT), aminotransferase (AMT), adenylation (A), peptidyl carrier protein (PCP), condensation (C), and thioesterase (TE). Domains labeled with an asterisk are predicted to be inactive based on the nemamide structures. The KR and A domains are labeled with numbers for further discussion in Supplementary Figure 13 and Supplementary Table 4. KR1 is predicted to be B-type, while KR2 and KR3 are neither A- nor B-type (Supplementary Fig. 13). Given that the nemamides contain four amino acids and that PKS-1 and NRPS-1 have only three A domains, A2 may act twice to incorporate two Asn residues.
To purify enough of the nemamides to identify their structures by NMR spectroscopy, wild-type worms were grown in an axenic, semi-defined medium8 that gives a much higher density of worms than bacteria-fed worm cultures. We grew ~50L of worm culture, and ultimately, we estimate that we purified only ~70 μg of nemamide A and less of nemamide B. These compounds were extracted from freeze-dried worms and purified using a short silica gel column, followed by an HP-20 column, a Sephadex LH-20 column, and then HPLC. The extraction and purification process had to be completed for small batches of worms (from ~2L of culture) within 1–2 days to prevent degradation of the nemamides. The exact masses of nemamide A and B indicated the molecular formulas C34H54N8O10 and C34H52N8O10, respectively. NMR spectra, including dqf-COSY, TOCSY, HSQC, HMBC, and ROESY spectra, were obtained for nemamide A and used to determine its molecular connectivity (Supplementary Table 1, Supplementary Fig. 3 and 4).
Marfey’s method9 was used to establish that nemamide A contains one L-Asn and two D-Asn (Online Methods). The relative configurations of the six stereocenters in nemamide A were determined using coupling constants and ROESY correlations (Supplementary Figs. 5–7). The absolute configurations of the four stereocenters in the macrolactam ring were determined by chemically synthesizing three model cyclic peptides with the three possible configurations (2S,6R,10R,18R, 2R,6S,10R,18S, and 2R,6R,10S,18S) and comparing their NMR spectra to that of nemamide A (Supplementary Fig. 8, Supplementary Tables 2 and 3). Additional support for the absolute configurations of the stereocenters at C-20 and C-22 was obtained through comparison of the observed CD spectrum of nemamide A to a predicted CD spectrum (Supplementary Fig. 9). Additional support was also provided by analysis of the ketoreductase (KR) domain responsible for installing the C-22 stereocenter (see discussion of biosynthesis below). Thus, we propose that the absolute configuration of nemamide A is 2S,6R,10R,18R,20R,22S (Fig. 1c). In comparison to nemamide A, nemamide B has one additional double bond, based on its NMR spectra, HR-MS, MS/MS, and UV spectrum (Fig. 1c, Supplementary Fig. 10–12).
Although the nemamide structures could not be predicted from the protein sequences of PKS-1 and NRPS-1, they are largely consistent with the domain architectures of the megasynthases. Biosynthesis begins on PKS-1, which initially extends the growing natural product through six iterative cycles, then uses two additional PKS modules to further extend the polyketide in an assembly-line manner, and then uses the C-terminal NRPS module to incorporate β-Ala (Fig. 1d). Next, the growing natural product is passed to NRPS-1, which sequentially adds D-Asn, D-Asn, and L-Asn and then forms the macrolactam ring (Fig. 1d). The biosynthetic pathway, however, has several non-canonical features, including (1) KR domains (specifically, KR2 and KR3) that cannot be classified as A-type or B-type, which would enable their stereospecificities to be predicted (Fig. 1d, Supplementary Fig. 13)10, (2) Missing enzymatic domains, such as methyltransferase and aminotransferase domains, that are likely encoded elsewhere in the C. elegans genome (Fig. 1d)11, (3) Adenylation domains with protein sequences that diverge significantly from those of bacterial and fungal adenylation domains (Fig. 1d, Supplementary Table 4)12, (4) The absence of any obvious epimerase domains2 despite the presence of D-Asn in the nemamides (Fig. 1d), and (5) A chain-terminating thioesterase domain present not only at the C-terminus of NRPS-1, but also, unusually, at the C-terminus of PKS-1 (Fig. 1d, Supplementary Figs. 14 and 15)2,13.
Using transcriptional reporter strains, we showed that pks-1 and nrps-1 are expressed during all larval stages and the adult stage specifically in the CAN neurons, two essential neurons with a poorly defined role that extend the length of the worm and are closely associated with the excretory canals (Fig. 2a,b)14. Given that pks-1 and nrps-1 are expressed neuronally, we speculated that the nemamides might play a signaling role in development. With sufficient food (bacteria), C. elegans will progress from the egg, through four larval stages (L1–L4) to the adult. However, if C. elegans eggs hatch to L1 larvae in the complete absence of food, the L1 larvae will arrest, but then resume development upon addition of food15. The pks-1 and nrps-1 arrested L1s recovered much slower than wild-type arrested L1s when placed on food (Fig. 2c). The mutants progress from the egg to the L4 stage at the same rate as wild-type worms (Supplementary Fig. 16), and thus, do not have a general defect in larval progression or development, but rather a specific defect in recovery from L1 arrest. Additionally, the pks-1 and nrps-1 mutants enter and recover from the dauer larval stage as well as wild type (Supplementary Fig. 17). Like wild-type worms, the pks-1 and nrps-1 mutants maintain proper somatic progenitor cell and germline arrest during L1 arrest (Supplementary Fig. 18 and 19). Thus, although the pks-1 and nrps-1 mutants are defective in recovery from L1 arrest, they are not defective in L1 arrest initiation and maintenance16.
Figure 2. Site of expression and biological role of the nemamides.
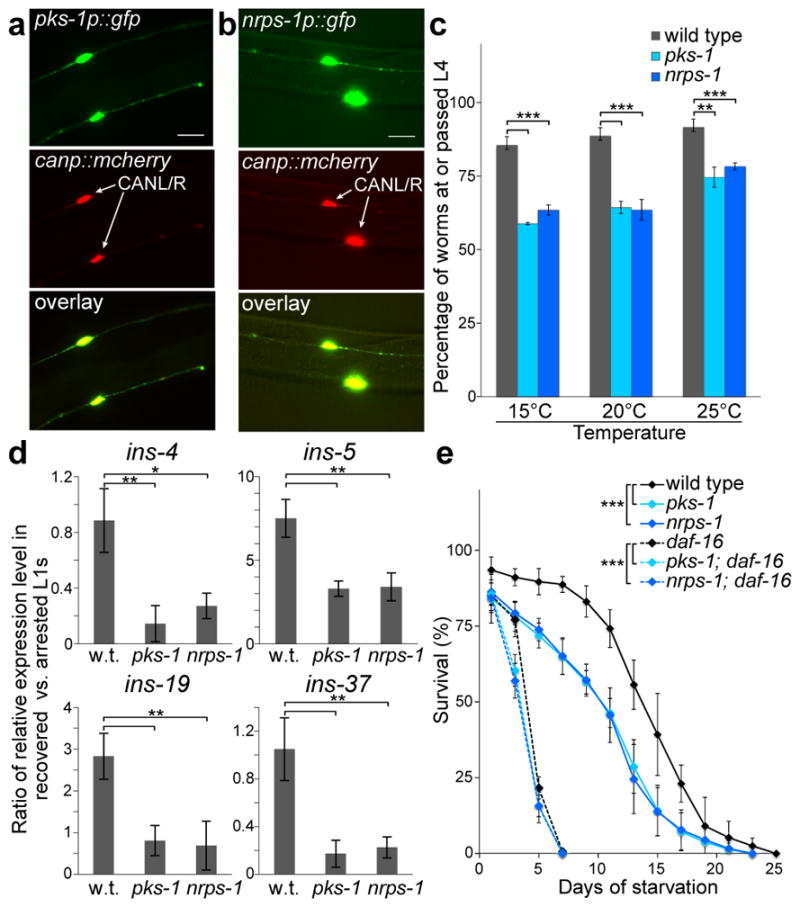
(a,b) Expression of the transcriptional reporters pks-1p::gfp (a) or nrps-1p::gfp (b), as well as canp::mcherry (marker for CAN neurons), in transgenic worms. Scale bar, 20 μm. We obtained similar results for pks-1p::gfp-pest and nrps-1p::gfp-pest reporters, in which GFP undergoes rapid turnover20. (c) Recovery of arrested L1s and development to the L4 stage for wild-type, pks-1, and nrps-1 worms at different temperatures. (d) Expression of specific insulins in recovered versus arrested wild-type, pks-1, and nrps-1 L1s, as determined by qRT-PCR. (e) Survival of wild-type, pks-1, and nrps-1 arrested L1s over time. The insulin/IGF-1 pathway controls L1 survival in a manner dependent on the downstream daf-16/foxo transcription factor15,21. The poor survival of daf-16/foxo (mu86 null) was slightly enhanced by the pks-1 and nrps-1 mutations. Mean survival (days ± SE): 14.3±0.2 for wild type, 11.3±0.3 for pks-1, 11.0±0.3 for nrps-1, 4.4±0.1 for daf-16, 3.7±0.1 for pks-1; daf-16, and 3.5±0.1 for nrps-1; daf-16. In c–e, the data represent the mean ± SD of three independent experiments, and two-tailed, unpaired t-tests were applied (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
The insulin/IGF-1 pathway is an important regulator of L1 arrest and recovery, and specific insulins are down-regulated upon L1 arrest and then up-regulated following food addition15,17,18. To determine whether the nemamides affect insulin expression, we profiled the expression of all 40 C. elegans insulins by qRT-PCR during L1 arrest and recovery. In the pks-1 and nrps-1 arrested L1s, ins-4, ins-5, ins-19, and ins-37 are expressed at higher levels than in wild-type arrested L1s (Supplementary Fig. 20). Furthermore, unlike in wild type, in the pks-1 and nrps-1 backgrounds, ins-5 and ins-19 are not induced during L1 recovery and ins-4 and ins-37 are down-regulated during L1 recovery (Fig. 2d, Supplementary Fig. 21). The production of the nemamides decreases during L1 recovery (Supplementary Fig. 22). Thus, our data suggest that the nemamides are negative regulators of the expression of specific insulins, such that expression of these insulins increases as nemamide levels decrease during L1 recovery.
The pks-1 and nrps-1 mutants show reduced survival during prolonged L1 arrest (Fig. 2e, Supplementary Fig. 23). Although it has been shown that L1 survival is density-dependent and that L1s secrete unidentified small molecules that increase survival19, the nemamides are unlikely to be a component of this pheromone, since the mutants showed reduced survival relative to wild type, regardless of worm density (Supplementary Fig. 24). C. elegans mutants which eat less display reduced survival during L1 arrest15. However, the pks-1 and nrps-1 mutants are not defective in bacterial food consumption or pharynx pumping (Supplementary Figs. 25 and 26), and thus, their reduced survival is not simply due to reduced nutrient stores. Because the insulin/IGF-1 pathway regulates L1 survival, we investigated the genetic interactions between this pathway and the nemamide pathway. Performing the survival assay in insulin/IGF-1 pathway mutant backgrounds suggests that although nemamide signaling regulates insulin expression, it functions at least partially independently of the insulin/IGF-1 pathway (Fig. 2e, Supplementary Fig. 27)15.
The mechanisms by which animals control their development and physiology in response to nutrient fluctuations are poorly understood. The nemamides could potentially serve as a chemical tool with which to dissect this process. We show that the nemamides are important for survival during and recovery from starvation-induced larval arrest. The nemamides likely influence larval development in C. elegans in part by modulating insulin signaling (Supplementary Fig. 28). The nemamides represent the first polyketide-nonribosomal peptides biosynthesized in an assembly-line manner in a metazoan. Their discovery will enable the exploration of polyketide and nonribosomal peptide biosynthesis in the context of a complex animal system. Nemamide biosynthesis likely requires additional enzymes that act in trans. Future studies of the site of expression and regulation of these enzymes could potentially provide additional insights into the biological role and site of action of the nemamides. As the nemamide biosynthetic genes are found in most nematode species, including parasitic ones, the role of the nemamides in larval development is likely conserved across nematode evolution.
Online Methods
Strains and culture methods
Worms were maintained on E. coli OP50 according to standard methods. Strains used in this study include wild type (N2), pks-1(ttTi24066), pks-1(ok3769) (this allele was only used in Supplementary Fig. 23), nrps-1(ttTi45552), pks-1(ttTi24066); nrps-1(ttTi45552), ayIs7[hlh-8p::gfp], pks-1(ttTi24066); ayIs7, nrps-1(ttTi45552); ayIs7, unc-31(e928), unc-31(e928); pks-1(ttTi24066), unc-31(e928); nrps-1(ttTi45552), daf-16(mu86), daf-16(mu86); pks-1(ttTi24066), and daf-16(mu86); nrps-1(ttTi45552). The pks-1(ttTi24066), nrps-1(ttTi45552), and pks-1(ok3769) strains were backcrossed two, four, and four times, respectively. The double mutants were constructed from single mutants using standard genetic methods and the presence of alleles was verified by PCR (Supplementary Table 5).
Generation of worm extracts for metabolomic analysis
The wild-type, pks-1, and nrps-1 strains were each grown at room temperature on two NGM agar plates (10 cm) spread with 0.75 mL 25X OP50 until the food on the plates was almost gone. Then, the worms were transferred to 1 L Erlenmeyer flasks containing S medium (350 mL). The worm cultures were grown at 22.5°C for 3 d and were fed with 3.5 mL of 25X OP50 every day. For sample collection, the culture flasks were placed in an ice-bath for 30 min to 1 h to settle the worms. Then, the worms were transferred from the bottom of the flasks to a 50 mL centrifuge tube and were centrifuged (1000 rpm for 5 min) to separate the worms from the worm medium. After the centrifugation, the supernatant was combined with the worm medium. The process was repeated until most of the worms were removed from the flasks. The collected worms were washed with water three times and centrifuged (1000 rpm for 5 min), and then they were soaked in 10 mL of water for 1 h in a shaking incubator (22.5°C, 225 rpm) to remove bacteria from their digestive tract. The worms were collected by centrifugation and were freeze-dried. The dried worm pellets were ground with sea sand (2 g sand per 800–900 mg dried worms) using a mortar and pestle. The ground worms were extracted with 50 mL of ethanol for 1 h, and the extract was filtered through filter paper. The fitrate was collected and dried using a rotovap. The collected worm medium was filtered using Celite and then using a 0.2 μm filter. The medium was extracted with ethyl acetate, and the ethyl acetate layer was then dried using a rotovap. The dried worm and worm media samples were each resuspended in 125 μL of 50% (vol/vol) ethanol in water, sonicated (if needed), and centrifuged (15000 rpm for 1 min) before analysis by HR-LC-MS. In later experiments to attempt to detect the nemamides in culture medium, the culture medium was instead freeze-dried and extracted with 190 proof ethanol.
General methods for chemical analysis
HR-LC-MS analysis was performed on an Agilent 1200 high performance liquid chromatography (HPLC) system equipped with a UV-Vis diode array detector and a 6220 TOF MS using positive ESI in both profile and centroid modes. NMR spectra were recorded on a Varian INOVA 600 equipped with a 1.5 mm microcryoprobe22, except for an additional HMBC spectrum of nemamide A, which was recorded on a Bruker Avance 800 spectrometer equipped with a 1.7 mm microcryoprobe (Supplementary Fig. 3i). CD spectra were obtained on an AVIV-202 CD spectrometer.
Metabolomic analysis
The worm and conditioned medium samples for each worm strain (N2, pks-1, and nrps-1) each consisted of three biological replicates and two technical replicates. 5 μL of each sample was injected. LC separation was achieved on two Onyx Monolithic C18 (Phenomenex, 100 × 4.6 mm, 5 μm) columns in series using a solvent gradient of 1% acetic acid in water (solvent A) and 1% acetic acid in acetonitrile (solvent B) at a flow rate of 0.33 mL / min. The solvent flow was maintained at 5% B for 6.5 min, then ramped to 100% B over 19 min, then maintained at 100% B for 9.5 min, then ramped to 5% B over 10 min, and maintained at 5% B for 5 min. The mass spectrometer settings include a drying gas flow of 10 L / min, a gas temperature of 325 °C, a nebulizer pressure of 50 psi, a capillary voltage of 4000 V, a fragmentor voltage of 180 V, and a skimmer voltage of 60 V. Worm samples were run in a random order to minimize the impact of mass or retention time shift on the analyses. Data files were converted to mzXML format in centroid mode using ProteoWizard software23. Metabolomic comparisons were performed using XCMS Online7 using the default parameters for HPLC-Q-TOF, except that the “Matched Filter” feature detection method and the “Peak Groups” retention time correction method (with nonlinear alignment) were used.
Purification and characterization of nemamides
Wild-type worms were shaken at 225 rpm for 7 d at 22.5 °C in 2.8 L baffled flasks containing 500 mL of CeHR medium8 with 20% cow’s milk. Worms were collected by centrifugation, washed with water, shaken in water for 30 min to clear their intestines, and washed again with water. Worms were stored frozen at −20 °C until needed. For extraction and fractionation process, worms from 2 L-worth of culture were processed at a time. After freeze drying, worms were ground for 15 min with 70 g of sand using a mortar and pestle. The pulverized worms were transferred to a 1 L Erlenmeyer flask, and 700 mL of 190 proof ethanol was added to the flask. The flask was shaken at 300 rpm for 3.5 h. The extract was filtered using a Buchner funnel and filter paper and evaporated with a rotovap at 27 °C. The extract was then subjected to silica gel chromatography and eluted with a gradient of ethyl acetate/methanol (1:0, 9:1, 1:1, 0:1) to give four fractions (A – D). Fraction D was evaporated with a rotovap at 27 °C, redissolved in 12 mL of methanol, and centrifuged at 3500 rpm for 10 min. The supernatant was dried and dissolved in 10 mL 70% methanol/water. Fraction D was then applied to an HP-20 column, eluting with MeOH/H2O (7:3 to 9:1) to give four subfractions (D1 – D4). Subfraction D3 was applied to a Sephadex LH-20 column, eluting with methanol to give seven subfractions (D3a – D3g). Fraction D3g, which contained both nemamide A and B based on LC-MS analysis, was further fractionated by HPLC (eclipse XDB-C18 column, 150 × 4.6 mm, 5 μm), using a gradient of methanol and water (ramping from 10% to 100% methanol over 30 min, holding at 100% methanol for 6 min, then returning to 10% methanol over 4 min; flow rate 1 mL / min; UV detection at 280 nm), to obtain purified nemamide A and B. Nemamide A: For NMR spectra and 1H and 13C NMR data of nemamide A, see Supplementary Figure 3 and Supplementary Table 1; UV (methanol): λmax 258, 269, 279 nm; HR-ESIMS (m/z): [M+Na]+ calcd. for C34H54N8O10Na 757.3861, found 757.3866. Nemamide B: For NMR spectra, see Supplementary Figure 12; UV (methanol): λmax 286, 301, 315 nm; HR-ESIMS (m/z): [M+Na]+ calcd. for C34H52N8O10Na 755.3704, found 755.3700.
Marfey’s analysis
Nemamide A (purified from worms from 2.5 L of culture) was hydrolyzed with 200 μL of 6 N HCl at 110 °C for 12 h. The reaction was then dried down by rotovap, and the residue was dissolved in 50 μL of water. 50 mM stock solutions of the amino acid standards (L-Asp, D-Asp, L-Asn, D-Asn) were made in water. 20 μL of 1 M NaHCO3 and 100 μL of 1-fluoro-2,4-dinitrophenyl-5-L-alaninamide (L-FDAA, Marfey’s reagent; 1% w/v in acetone) were added to 50 μL of the sample or the amino acid standards. After heating at 37 °C for 60 min, reactions were quenched by addition of 20 μL of 1 N HCl. The sample reaction was diluted with 100 μL of acetonitrile while the reactions of the amino acid standards were diluted with 810 μL of acetonitrile. The reactions of the sample and standards were subjected to LC-MS analysis (Phenomenex Luna C18, 4.6 × 100 mm, 5 μm) using a linear gradient of water with 0.1% formic acid and acetonitrile with 0.1% formic acid (holding at 10% acetonitrile for 5 min, then ramping to 50% acetonitrile over 30 min; flow rate, 0.7 mL/min; UV and ESI-MS detection, 340 nm and negative ion mode). Analysis with both Asn and Asp amino acids indicated the conversion of Asn to Asp during the acid hydrolysis step for both the sample and the Asn amino acid standards. Retention times for L-FDAA-L-Asp and L-FDAA-D-Asp were 22.0 and 22.7 min, respectively. The extracted ion chromatogram (m/z 384) for the sample indicated the presence of L-FDAA-L-Asp and L-FDAA-D-Asp in a 1:2.16 ratio.
Cyclic peptide synthesis
H-β-Ala-2-ClTrl resin was purchased from Novabiochem, and Fmoc-L-Asn(Trt)-OH, Fmoc-D-Asn(Trt)-OH, (R)-3-(Fmoc-amino)butyric acid, and (S)-3-(Fmoc-amino)butyric acid were purchased from Chem-Impex International. Compound 3 was made through the sequential coupling to the resin of (R)-3-(Fmoc-amino)butyric acid, Fmoc-L-Asn(Trt)-OH, Fmoc-D-Asn(Trt)-OH, and Fmoc-D-Asn(Trt)-OH, followed by cleavage from the resin and cyclization. Compound 4 was made through the sequential coupling to the resin of (S)-3-(Fmoc-amino)butyric acid, Fmoc-D-Asn(Trt)-OH, Fmoc-L-Asn(Trt)-OH, and Fmoc-D-Asn(Trt)-OH, followed by cleavage from the resin and cyclization. Compound 5 was made through the sequential coupling to the resin of (S)-3-(Fmoc-amino)butyric acid, Fmoc-D-Asn(Trt)-OH, Fmoc-D-Asn(Trt)-OH, and Fmoc-L-Asn(Trt)-OH, followed by cleavage from the resin and cyclization. Solid-phase peptide synthesis was conducted in 10 mL BD Luer-Lok syringes. For deprotection, H-β-Ala-2-ClTrl resin (0.25 g, 0.375 mmol/g) was swelled in dry CH2Cl2 for 20 min. The resin was treated with 20% (v/v) piperidine / DMF (5 mL) for 30 min and then washed with DMF (3 × 5 mL). For amino acid coupling, a solution of protected amino acid (5 eq.) in DMF, HOBt (5.5 eq.) in DMF and DIC (5.5 eq.) together with 5 mL CH2Cl2 was added to the resin. After 4 h, the resin was washed with DMF (5 mL), CH2Cl2 (5 mL), and DMF (5 mL). The reaction was monitored using the ninhydrin test. When the ninhydrin test was negative, deprotection and coupling to the next amino acid was carried out. For cleavage of the linear peptide, the resin was treated with a 5 mL solution of 1,1,1,3,3,3-hexafluoro-2-propanol in CH2Cl2 (1:4, v/v) for 30 min. This process was repeated two additional times, and the combined cleavage solution was concentrated in vacuum. For cyclization of the linear peptide, the crude linear peptide (100 mg, 0.08 mmol) was dissolved in 80 mL DMF, and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate (DMTMM+ BF4−, 54 mg, 0.16 mmol) and iPr2NEt (28 μL, 158.6 μmol) was added to the solution. The solution was stirred overnight and then concentrated under reduced pressure using a rotovap. For deprotection of the trityl groups, 10 mL of a solution of TFA, triisopropylsiline, and water (95:2.5:2.5, v/v/v) was added for 2 h, and then concentrated to give the crude cyclic peptide. For purification of the cyclic peptide, the crude cyclic peptide was initially purified on a C18 column (50g Octadecyl-functionalized silica gel, 3.5 cm × 50 cm), eluted with 10% methanol, and then further purified using reversed-phase HPLC (0–5 min: 2% acetonitrile, 5–15 min: 2%–30% acetonitrile, 15–20 min: 30% acetonitrile) to afford pure cyclic peptide.
Construction of reporter strains
The PEST sequence from pAF20720 (gift of Alison Frand) was subcloned into pPD114.108 at the XhoI/EcoRI sites to generate pPD114.108-gfp::pest. 4.563 kb of the pks-1 promoter and 3 kb of the nrps-1 promoter were amplified from C. elegans genomic DNA (Supplementary Table 5). The pks-1 and nrps-1 promoters were inserted into the SalI/NotI and AscI/NotI sites, respectively, of pPD114.108 or pPD114.108-gfp::pest to obtain pks-1p::gfp and nrps-1p::gfp or pks-1p::gfp-pest and nrps-1p::gfp-pest, respectively. For the canp::mcherry reporter, the CAN-specific promoter was cut from canp::yfp24 (gift of Nadeem Moghal) at two SphI sites and inserted into pMC10 (a gift of Piali Sengupta). 50 ng/μL of the transgenes, along with 50 ng/μL of the co-injection marker unc-122p::DsRed (gift of Piali Sengupta), were injected into wild-type worms. At least three independent transgenic strains were analyzed. Imaging was conducted on a Zeiss Axiovert. A1 microscope equipped with ZEN lite 2012 camera.
Cyclic peptide 3 (2S,6R,10R,18R)
1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 8.63 (brs, 10-NH), 8.20 (brd, J6,6-NH = 8.8 Hz, 6-NH), 7.80 (brs, 8-NH2b), 7.49 (brd, J2,2-NH = 8.1 Hz, 2-NH), 7.43 (brs, 12-NH2b), 7.36 (brs, 15-NH), 7.32 (brs, 8-NH2a), 7.11 (brs, 4-NH2b), 6.96 (brs, 12-NH2a), 6.95 (brd, J18,18-NH = 7.9 Hz, 18-NH), 6.82 (brs, 4-NH2a), 4.48 (m, H-2), 4.41 (m, H-6), 4.25 (m, H-10), 3.96 (m, H-18), 3.42 (m, H-15b), 3.18 (m, H-15a), 2.98 (dd, J7a,7b = 16.4 Hz, J6,7b = 4.7 Hz, H-7b), 2.62 (dd, J3a,3b = 19.3 Hz, J2,3b = 6.5 Hz, H-3b), 2.58 (brd, J7a,7b = 17.2 Hz, H-7a), 2.57 (overlap, H-14b), 2.51 (overlap, H-11a, 11b), 2.46 (overlap, H-17b), 2.43 (dd, J3a,3b = 19.4 Hz, J2,3a = 12.7 Hz, H-3a), 2.42 (overlap, H-14a), 2.15 (brd, J17a,17b = 12.0 Hz, H-17a), 0.98 (d, J18,19 = 5.9 Hz, H-19); 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 173.6 (C-8, 12), 173.3 (C-13), 171.2 (C-4), 171.1 (C-9), 170.7 (C-16), 170.5 (C-5), 169.7 (C-1), 52.1 (C-10), 49.9 (C-2), 49.2 (C-6), 43.0 (C-18), 41.4 (C-17), 36.8 (C-3), 35.6 (C-11), 35.4 (C-7), 34.8 (C-15), 33.5 (C-14), 20.7 (C-19); ESIMS m/z 499.3 [M+H]+, m/z 497.3 [M-H]−.
Cyclic peptide 4 (2R,6S,10R,18S)
1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 8.39 (brd, J10,10-NH = 7.4 Hz,10-NH), 7.92 (brs, 8-NH2b), 7.85 (brd, J2,2-NH = 8.8 Hz, 2-NH), 7.81 (brd, J6,6-NH = 8.8 Hz, 6-NH), 7.47 (brs, 8-NH2a), 7.38 (brs, 4-NH2b), 7.33 (brs, 12-NH2b), 7.25 (brs, 15-NH), 7.08 (overlap, 18-NH), 6.95 (brs, 12-NH2a), 6.85 (brs, 4-NH2a), 4.49 (m, H-2), 4.46 (m, H-10), 4.40 (m, H-6), 3.97 (m, H-18), 3.55 (overlap, H-15b), 3.14 (overlap, H-15a), 3.05 (dd, J7a,7b = 17.2 Hz, J6,7b = 3.8 Hz, H-7b), 2.69 (dd, J3a,3b = 15.8 Hz, J2,3b = 5.6 Hz, H-3b), 2.65 (dd, J11a,11b = 15.7 Hz, J10,11b = 6.8 Hz, H-11b), 2.54 (overlap, H-7a), 2.51 (overlap, H-17b), 2.48 (overlap, H-11a, 14b), 2.43 (dd, J3a,3b = 15.8 Hz, J2,3a = 6.9 Hz, H-3a), 2.36 (m, H-14a), 2.11 (brd, J17a,17b = 11.3 Hz, H-17a), 0.97 (d, J18,19 = 5.9 Hz, H-19); 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 173.8 (C-8), 172.6 (C-13), 172.3 (C-12), 171.8 (C-4), 170.3 (C-5), 170.2 (C-9, 16), 169.6 (C-1), 49.7 (C-2), 49.3 (C-10), 48.7 (C-6), 43.1 (C-18), 41.5 (C-17), 35.4 (C-11), 35.3 (C-7), 35.1 (C-3), 34.7 (C-14), 34.3 (C-15), 20.9 (C-19); ESIMS m/z 499.3 [M+H]+, m/z 497.2 [M-H]−.
Cyclic peptide 5 (2R,6R,10S,18S)
1H NMR (600 MHz, dimethyl sulfoxide-d6) δ 8.89 (brd, J6,6-NH = 5.2 Hz, 6-NH), 8.48 (brs, 10-NH), 7.49 (brd, J2,2-NH = 7.9 Hz, 2-NH), 7.40 (brs, 12-NH2b), 7.33 (brs, 8-NH2b), 7.27 (brs, 4-NH2b), 7.00 (brs, 15-NH), 6.98 (brs, 12-NH2a), 6.89 (brs, 8-NH2a), 6.88 (brs, 4-NH2a), 6.66 (brd, J18,18-NH = 7.7 Hz, 18-NH), 4.40 (m, H-2, 10), 4.33 (m, H-6), 3.97 (m, H-18), 3.33 (overlap, H-15a,15b), 2.60 (overlap, H-7b), 2.56 (overlap, H-3a, 3b), 2.56 (overlap, H-14b), 2.49 (overlap, H-11b), 2.48 (overlap, H-7a), 2.36 (brd, J17a,17b = 13.1 Hz, H-17b), 2.31 (brd, J11a,11b = 16.1Hz, H-11a), 2.23 (m, H-14a), 2.06 (brd, J17a,17b = 13.0 Hz, J17a,18 = 9.4 Hz, H-17a), 1.04 (d, J18,19 = 6.5 Hz, H-19); 13C NMR (125 MHz, dimethyl sulfoxide-d6) δ 173.9 (C-9), 172.3 (C-13), 171.2 (C-4), 170.9 (C-8), 170.7 (C-5), 170.3 (C-12), 170.1 (C-1), 170.0 (C-16), 50.9 (C-2, 6, 10), 42.6 (C-18), 41.7 (C-17), 36.5 (C-3), 35.6 (C-11), 35.4 (C-7, 15), 34.0 (C-14), 20.1 (C-19); ESIMS m/z 499.3 [M+H]+, m/z 497.3 [M-H]−.
L1 recovery, dauer formation, and dauer recovery assays
For L1 recovery assays, eggs were isolated from well-fed gravid worms using alkaline bleach treatment, diluted to 4–6 eggs/ μL in M9 buffer, and shaken for 24 h at 22.5 °C and 225 rpm. Approximately 80–120 synchronized L1s were placed onto a 3 cm NGM plate with OP50 at 15 °C, 20 °C, or 25 °C. After a certain period of time (40 h at 25 °C, 48 h at 20 °C, and 80 h at 15 °C), the percentage of worms at or passed the L4 stage was determined. For each experiment, five plates were analyzed for each strain, the percentage of worms at or passed the L4 stage was calculated for each plate, and the percentages for each strain were averaged. Dauer formation assays were performed for wild-type, pks-1, and nrps-1 with vehicle control or 1 μM asc-C6-MK at 25 °C as described26. Dauer recovery assays were performed by taking dauers from dauer formation assay plates and moving them to a lawn of bacteria for 24 h at 20 °C before scoring for recovery.
Egg to L4 development assay
Worms were maintained at 15 °C, 20 °C, or 25 °C for 2–3 generations. For egg lay assay, L4s were moved onto a new plate one day before the beginning of the experiment, and the next day 8 adults were used to perform a 1 h egg lay for each 3 cm NGM plate with OP50. Alternatively, for egg prep experiment, eggs were isolated from well-fed gravid worms using alkaline bleach treatment, washed, and added to 3 cm NGM plates with OP50. In both egg lay and egg prep assays, eggs were then incubated at 15 °C, 20 °C, or 25 °C, and after a certain period of time (40 h at 25 °C, 48 h at 20 °C and 80 h at 15 °C), the percentage of worms at or passed the L4 stage was determined. For each experiment, five plates were analyzed for each strain, and the percentages for each strain were averaged.
Analysis of expression of insulins using qRT-PCR
Well-fed gravid adults were collected from multiple 10 cm NGM plates and eggs were isolated by using alkaline bleach treatment, diluted to 4–6 eggs / μL in M9 buffer, and shaken for 24 h at 21 °C, 225 rpm. Then 25 mg/mL of OP50 was supplied to initiate recovery. Worms at 0 h and 6 h after feeding were collected by washing with cold M9 buffer, flash-frozen and stored at −80 °C before qRT-PCR. Total RNA extraction, an on-column DNase treatment, and cDNA generation were performed as described26, except that 0.25 μg of total RNA was used for reverse transcription. All primers for insulin genes were as described27, except for the ones for ins-3, ins-26, and ins-31, which were as described here16, and the ones for ins-11, which were designed using the Real-time PCR Primer Design Tool (IDT) and are listed in Supplementary Table 5. qPCR was performed using SYBR Green select Master Mix (Life Technologies) on a 7500 Fast Real-Time PCR system (Applied Biosystems) using the standard mode. PCR parameters include a holding stage at 50 °C for 2 min and another holding stage at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10s, 57 °C for 20s and 72 °C for 30s. Relative expression levels for recovered versus arrested L1s were determined using the ΔΔCt method28, and normalized to the expression levels of endogenous control genes act-1, pmp-3 and Y45F10D.426. Ratio of relative expression level for each strain was calculated by comparing the relative expression level at 6 h after feeding with the level at 0 h.
Nemamide production
Eggs were isolated from well-fed gravid worms using alkaline bleach treatment, diluted in S basal in a 125 mL flask, and shaken for 24 h at 20 °C and 200 rpm. Then, the synchronized L1s were inoculated into 2.8 L flasks with the density adjusted to 6 L1s / μL in 200 mL S medium (starved L1s) or 190 mL S medium plus 10 mL concentrated OP50 (fed L1s). Starved and fed L1 worms were cultured at 20 °C and 150 rpm for 6 h. L1s were then harvested by washing three times with S basal. L1s were freeze-dried, ground with sand, and extracted with ethanol, as described above, and then the nemamides were analyzed by LC-MS.
M cell division assay
For the ayIs7[hlh-8p::gfp], pks-1; ayIs7, and nrps-1; ayIs7 strains, eggs were isolated from well-fed gravid worms using alkaline bleach treatment, diluted to 4–6 eggs/ μL in M9 buffer with 0.08% ethanol, and shaken for 7d at 22.5 °C and 225 rpm. Worms were examined for M cell division using a fluorescent microscope. > 50 worms were examined for each genotype.
Fertility and brood size upon recovery from L1 arrest
For the wild-type, pks-1, and nrps-1 strains, eggs were isolated from well-fed gravid worms using alkaline bleach treatment, diluted to 4–6 eggs/ μL in M9 buffer, and shaken for 5d at 22.5 °C and 225 rpm. Worms were then moved to a lawn of bacteria, allowed to develop to the L4 stage, and then singled. Survival and fertility were examined 2–3d later.
L1 survival assay
Eggs were isolated from well-fed gravid worms using alkaline bleach treatment, diluted in M9 buffer in a 125 mL flask, and shaken for 24 h at 20 °C and 200 rpm. Then, the density of the synchronized L1s was adjusted to 4–6 L1s / μL (Fig. 2e) or 20–25 L1s / μL (Supplementary Fig. 24, high density) or 0.5–0.8 L1s / μL (Supplementary Fig. 24, low density) in M9 buffer. Every other day, 20 μL starved L1 samples were seeded onto 3 cm NGM plate with OP50 at 20 °C, and three plates were seeded for each strain. The plated worms were scored after 2–3 d, and worms at or passed the L2 stage were scored as surviving worms. For each experiment, three plates were analyzed for each strain, the percentage of surviving worms was calculated for each plate, and the percentages for each strain were averaged. Survival curves in Figure 2e and Supplementary Figure 24 and Supplementary Figure 27 were statistically analyzed as previously described29.
Pumping assay
At least 30 young adults for each strain (wild-type, pks-1, and nrps-1) were analyzed while on a lawn of OP50 by counting the number of pharynx pumps per 30s under the dissecting microscope at room temperature.
Phylogeny and domain analysis
Phylogenetic trees were generated in MEGA 6, and domain analysis was performed using antiSMASH 3.0.30,31 Protein sequences were retrieved from NCBI or Wormbase Parasite.
Supplementary Material
Acknowledgments
We thank Andy Fire, Alison Frand, Nadeem Moghal, and Piali Sengupta for plasmids, Jim Rocca for help with NMR acquisition, Jodie Johnson for MS-MS analysis, Yousong Ding and Yi Zhang for help with Sybyl software, Steve Hagan, Gail Fanucci, and Zhanglong Liu for help with CD spectroscopy, and Yu Zhu for help with calculating CD spectra. We acknowledge the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), and the NemaGENETAG consortium for providing strains. This work was supported by funds to R.A.B. from the NIH (GM118775), the NSF (1555050), the Ellison Medical Foundation (AG-NS-0963-12), the Alfred P. Sloan Foundation (BR2014-071), and the National High Magnetic Field Laboratory, which is supported by NSF Cooperative Agreement No. DMR-1157490 and the State of Florida.
Footnotes
Author Contributions
Q.S. purified and structurally characterized the nemamides. L.F. generated transgenic worm strains and performed biological assays. Y.L. performed metabolomic analyses. J.H. generated extracts. J.K.N. analyzed nemamide stability. Q.S., L.F., Y.L., D.H.P., and R.A.B. analyzed the data. R.A.B., Q.S., and L.F. wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Omura S, Crump A. Ivermectin: panacea for resource-poor communities? Trends Parasitol. 2014;30:445–455. doi: 10.1016/j.pt.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 3.Castoe TA, Stephens T, Noonan BP, Calestani C. A novel group of type I polyketide synthases (PKS) in animals and the complex phylogenomics of PKSs. Gene. 2007;392:47–58. doi: 10.1016/j.gene.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Hojo M, et al. Unexpected link between polyketide synthase and calcium biomineralization. Zoological Letters. 2015;1:1–16. doi: 10.1186/s40851-014-0001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Brien RV, Davis RW, Khosla C, Hillenmeyer ME. Computational identification and analysis of orphan assembly-line polyketide synthases. J Antibiot (Tokyo) 2014;67:89–97. doi: 10.1038/ja.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc Natl Acad Sci U S A. 2014;111:9259–9264. doi: 10.1073/pnas.1401734111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gowda H, et al. Interactive XCMS Online: simplifying advanced metabolomic data processing and subsequent statistical analyses. Anal Chem. 2014;86:6931–6939. doi: 10.1021/ac500734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nass R, Hamza I. The nematode C. elegans as an animal model to explore toxicology in vivo: solid and axenic growth culture conditions and compound exposure parameters. Curr Protoc Toxicol. 2007;Chapter 1(Unit 1):9. doi: 10.1002/0471140856.tx0109s31. [DOI] [PubMed] [Google Scholar]
- 9.Bhushan R, Brückner H. Marfey’s reagent for chiral amino acid analysis: a review. Amino Acids. 2004;27:231–247. doi: 10.1007/s00726-004-0118-0. [DOI] [PubMed] [Google Scholar]
- 10.Kwan DH, Schulz F. The stereochemistry of complex polyketide biosynthesis by modular polyketide synthases. Molecules. 2011;16:6092–6115. doi: 10.3390/molecules16076092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aron ZD, Dorrestein PC, Blackhall JR, Kelleher NL, Walsh CT. Characterization of a new tailoring domain in polyketide biogenesis: the amine transferase domain of MycA in the mycosubtilin gene cluster. J Am Chem Soc. 2005;127:14986–14987. doi: 10.1021/ja055247g. [DOI] [PubMed] [Google Scholar]
- 12.Rottig M, et al. NRPSpredictor2--a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du L, Lou L. PKS and NRPS release mechanisms. Nat Prod Rep. 2010;27:255–278. doi: 10.1039/b912037h. [DOI] [PubMed] [Google Scholar]
- 14.Forrester WC, Perens E, Zallen JA, Garriga G. Identification of Caenorhabditis elegans genes required for neuronal differentiation and migration. Genetics. 1998;148:151–165. doi: 10.1093/genetics/148.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baugh LR. To grow or not to grow: nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics. 2013;194:539–555. doi: 10.1534/genetics.113.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuyama M, Kontani K, Katada T, Rougvie AE. The C elegans Hypodermis Couples Progenitor Cell Quiescence to the Dietary State. Curr Biol. 2015;25:1241–1248. doi: 10.1016/j.cub.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 17.Lee BH, Ashrafi K. A TRPV channel modulates C elegans neurosecretion, larval starvation survival, and adult lifespan. PLoS Genet. 2008;4:e1000213. doi: 10.1371/journal.pgen.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Baugh LR. Ins-4 and daf-28 function redundantly to regulate C elegans L1 arrest. Dev Biol. 2014;394:314–326. doi: 10.1016/j.ydbio.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Artyukhin AB, Schroeder FC, Avery L. Density dependence in Caenorhabditis larval starvation. Sci Rep. 2013;3:2777. doi: 10.1038/srep02777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frand AR, Russel S, Ruvkun G. Functional genomic analysis of C elegans molting. PLoS Biol. 2005;3:e312. doi: 10.1371/journal.pbio.0030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baugh LR, Sternberg PW. DAF-16/FOXO regulates transcription of cki-1/Cip/Kip and repression of lin-4 during C elegans L1 arrest. Curr Biol. 2006;16:780–785. doi: 10.1016/j.cub.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 22.Ramaswamy V, et al. Development of a 13C-optimized 1.5-mm high temperature superconducting NMR probe. J Magn Reson. 2013;235:58–65. doi: 10.1016/j.jmr.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chambers MC, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butcher RA, Fujita M, Schroeder FC, Clardy J. Small-molecule pheromones that control dauer development in Caenorhabditis elegans. Nat Chem Biol. 2007;3:420–422. doi: 10.1038/nchembio.2007.3. [DOI] [PubMed] [Google Scholar]
- 25.Modzelewska K, Lauritzen A, Hasenoeder S, Brown L, Georgiou J, Moghal N. Neurons refine the Caenorhabditis elegans body plan by directing axial patterning by Wnts. PLoS Biol. 2013;11:e1001465. doi: 10.1371/journal.pbio.1001465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, et al. Acyl-CoA oxidase complexes control the chemical message produced by Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2015;112:3955–3960. doi: 10.1073/pnas.1423951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ritter AD, et al. Complex expression dynamics and robustness in C elegans insulin networks. Genome Res. 2013;23:954–965. doi: 10.1101/gr.150466.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Zabinsky R, Teng Y, Cui M, Han M. microRNAs play critical roles in the survival and recovery of Caenorhabditis elegans from starvation-induced L1 diapause. Proc Natl Acad Sci U S A. 2011;108:17997–18002. doi: 10.1073/pnas.1105982108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber T, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.