

Abstract
Background
Risk stratification in patients with type 3 long QT syndrome (LQT3) by clinical and genetic characteristics and effectiveness of β-blocker therapy have not been studied previously in a large LQT3 population.
Methods
The study population included 406 LQT3 patients with 51 different sodium-channel mutations; 391 patients were known to be event free during the first year of life and were the focus of our study. Clinical, electrocardiographic, and genetic parameters were acquired on patients from 7 participating LQT3 registries. Cox regression analysis was used to evaluate the independent contribution of clinical, genetic, and therapeutic factors to the first occurrence of time-dependent cardiac events (CE) from age 1 to 41 years.
Results
118 (30%) patients (41 males) experienced at least one CE (syncope, aborted cardiac arrest [ACA] or LQTS-related sudden death [SD]), and 20% suffered from LQT3-related ACA/SD. The risk of a first CE was directly related to the degree of QTc prolongation. Cox regression analysis revealed that time-dependent β-blocker therapy was associated with an 83% reduction in CE’s in females (p=0.015) but not in males (who had much fewer events), with a significant gender x β-blocker interaction (p=0.04). Each 10ms increase in QTc duration up to 500ms was associated with a 19% increase in CE’s. Prior syncope doubled the risk for life-threatening events (p<0.02).
Conclusions
Prolonged QTc and syncope predispose patients with LQT3 to life-threatening CE’s. β-blocker therapy reduces this risk in females, but efficacy in males could not be conclusively determined due to low number of events.
Keywords: long QT syndrome, SCN5A, genetic testing, sudden cardiac death, arrhythmia, risk stratification
INTRODUCTION
Several large studies have described the clinical course of patients with the long QT syndrome (LQTS).1,2 Initially regarded as one disease entity, it has become increasingly clear that the underlying genetic substrate, now subdividing LQTS into at least 16 genotypes, impacts many aspects of the disease phenotype including prognosis and therapy.3 Approximately two-thirds of all LQTS patients host loss-of-function mutations in one of two potassium channel genes, KCNQ1 (LQT1) or KCNH2 (LQT2).4,5 β-blocker therapy or other anti-adrenergic measures are effective in the majority of these patients.4–8
LQT3 is caused by gain-of-function mutations in the SCN5A-encoded Nav1.5 sodium channel involving a pathological increase in late sodium current, a pathological increase in the “window” current (as one of the mechanisms of the late sodium inward current), or both. LQT3 comprises approximately 5–10% of patients with LQTS. The phenotype differs from the more common potassium channel-mediated forms in various aspects. Cardiac events in LQT3 frequently occur at rest or with inactivity and are less likely to be triggered by adrenergic stress or emotions.7 Compared to LQT1 and LQT2, patients with LQT3 have more marked resting bradycardia and the first cardiac event is more likely to be lethal and seems to occur later in childhood, during or after puberty.9,10 Based on these data and in contrast to the well-established efficacy of β-blockers in LQT1 and LQT2, there is anxiety and fear that β-blockers may not be effective in LQT3,6,7 and might, in fact, be pro-arrhythmic.11 This concern has translated into a relatively high use of prophylactic implantable cardioverter defibrillators (ICDs) in LQT3 patients, even those who are asymptomatic.12,13 Patients with defective Nav1.5 channels may also manifest other arrhythmia phenotypes (Brugada syndrome, progressive cardiac conduction disease, atrial arrhythmias, and sinus node disease), and patients with LQT3 frequently present with associated characteristics including discrete conduction disturbances, bradycardia, atrial arrhythmias, or right precordial ST-elevation.14 Indeed, even patients with the “prototype” LQT3 mutation, p.K1505_Q1507del (ΔKPQ), have notably longer cardiac conduction intervals than patients with either LQT1 or LQT2.15
The current study involves the largest multicenter LQT3 cohort described to date and is designed to identify the risk and therapeutic factors associated with cardiac events in patients with SCN5A-mediated LQT3. The risk factors evaluated include clinical features (age, gender, electrocardiographic measurements), the mutation type and topological location of the mutation in Nav1.5. The therapeutic effects of β-blockers therapy, other medications, and ICD on outcome were also evaluated.
METHODS
Study Population
The study population comprised 406 subjects with LQT3 (90 LQT3 probands and their 316 LQT3-positive family members). The subjects were enrolled from 7 different centers including the U.S. Rochester, NY/Cleveland portion of the International LQTS Registry (n=186), the Dutch (Amsterdam) LQTS Registry (n=75), the Italian (Pavia) LQTS Registry (n=48), the Israeli LQTS Registry (n=30), the Japanese (National Cardiovascular Center) LQTS Registry (n=29), the Mayo Clinic LQTS Registry (n=28), and the Denmark LQTS Registry (n=10). In all centers IRB approval was obtained for this type of study. Not included in the study population were 14 subjects with evidence of mutations involving two or more LQTS genes, and 2 patients with multiple SCN5A mutations. In addition, patients/families with clear evidence of an SCN5A-mediated hybrid/overlapping phenotype (i.e., conduction disease or right precordial ST-elevation, the so-called overlap syndromes as for example the large Dutch SCN5A-1795insD family16) were not included in this study. All subjects or their guardians provided informed consent for the genetic and clinical studies.
Phenotype Characterization
Routine clinical and electrocardiographic parameters were acquired at the time of enrollment in each of the registries. In order to minimize the influence of coronary disease on cardiac events, follow-up was censored at age 41. Measured parameters on the first recorded ECG included PR, QRS, QT, and R-R intervals in milliseconds, with QT corrected for heart rate by Bazett’s formula (QTc). The QTc was expressed in its continuous form and categorized into three appropriate risk levels as described below. Clinical data were collected on prospectively designed forms with information on demographic characteristics, personal and family medical history, electrocardiographic findings, therapy, and end points during long-term follow-up. As in the two previous studies in LQT1 and LQT2 subtypes by our group,4, 5 data on patients with the LQT3 genotype were merged electronically into a common database.
Genotype Characterization
The SCN5A mutations were identified using standard genetic tests performed in either a molecular-genetic research laboratory in a participating center or in one of the commercially available LQTS genetic-testing laboratories. There were 32 subjects who died of sudden unexplained cardiac death who had not been genotyped. We assumed that the SCN5A mutation, that was established as LQT3-associated in their respective families, was also present in these 32 subjects.
Genetic alterations of the amino acid sequence were characterized by location in the Nav1.5 channel protein (subdivided into N-terminus; transmembrane spanning domains DI, DII, DIII, DIV; their respective inter-domain linkers (IDL); C-terminus; and by the type of mutation (missense, frame shift, and in-frame deletions). The different regions of the SCN5A-encoded Nav1.5 channel were defined as the coding sequence involving amino acid (aa) residues from N-terminus (aa1–126), DI (aa127–415), IDL I–II (aa416–711), DII (aa712–939), IDL II–III (aa940–1200), DIII (aa1201–1470), IDL III–IV (aa1471–1523), DIV (aa1524–1772) and C-terminus (aa1773–2016). The genetic mutations are presented in Supplementary Table S1 by coding effect, location, and frequency.
Statistical Analysis
The primary end point was the time from age 1 until the first cardiac event (syncope, aborted cardiac arrest [ACA], or LQT3-related sudden cardiac death [SCD]), censored at loss to follow-up, or age 41, whichever occurred first. The restricted, more severe secondary end point was ACA or LQT3-related SCD, whichever occurred first. All long-term analyses were conditional on cardiac event-free survival to age 1, in order to curtail any potential influence of cardiac events in the first year of life on the model for the remaining 39 years. Accordingly, 391/406 patients (96%) with LQT3 were eligible for long-term analyses.
Clinical characteristics were described via means and standard deviations for continuous variables or proportions and counts for categorical variables. Kaplan-Meier curves were used to estimate distributions of censored time-to-event outcomes, with inference based on the logrank test. A total of 35 of 391 (8.9%) in the study population older than 1 year of age did not have a recorded ECG; 25 of the 35 patients without an ECG died. Missing QTc values were imputed based on gender and mutation, using regression imputation. There were 5 prevalent mutations (N1325S, K1505_Q1507del, I1768V, E1784K, D1790G ), while the other “rare” mutations (< 5% prevalence each, together constituting < 41% of all patients) were pooled together to form a sixth mutation group. Each of the six mutation groups was stratified by gender, resulting in a total of 12 gender-mutation subgroups. Missing QTc values were imputed as the mean of the non-missing QTc values among subjects in the corresponding gender-mutation subgroup.
The Cox model17 was used to evaluate the independent contribution of clinical, therapeutic, and genetic factors to the risk of the first occurrence of time-dependent cardiac events from age 1 year through age 40 years. Cox models were stratified by gender to relax the assumption of proportional hazards by allowing gender-specific nonparametric baseline hazard functions for males and females. The effect of β-blocker therapy was modeled via a time-dependent indicator for being on versus off β-blockers at each point in time, allowing for the fact that subjects may start and stop β-blockers at different ages. Interacting time-dependent β-blocker status with gender allowed estimation of gender-specific hazard ratios for β-blockers, as well as a test of equality of the effect of β-blockers for males versus females.
The effect of syncope on the risk of aborted cardiac arrest or death was similarly modeled via a time-dependent indicator for having had at least one syncopal event. There was insufficient evidence that risk differed for those whose first syncopal event occurred on versus off β-blockers, though power to test this was limited.
Effects of the five most common mutations (each 5–18% prevalent) were modeled by comparing each to the pooled set of all infrequent mutations (<5% prevalent each, but totaling 41% of all subjects), and then further pooling the three non-significantly different common mutations (I1768V, K1505_Q1507del, N1325S) with the infrequent mutations to estimate the hazard ratios for E1784K and D1790G relative to all others.
Effects of QTc and birthdate were modeled via continuous piecewise linear splines to account for their significantly nonlinear effects that could not be well modeled via discrete groups. In particular, the log-hazard increased linearly for QTc up to 500 ms but then leveled off. For Kaplan-Meier curve estimation, QTc was categorized into 3 groups: <450ms, 450–490ms, and ≥500ms. The log-hazard was constant until 1955, but then increased linearly with birthdate after 1955.
Proportional hazards assumptions were tested by interacting predictors with follow-up time, with stratification used to extend the model to remedy violations. All two-way interactions between pairs of predictors in the model were considered for inclusion, one at a time. Frailties (non-significant) and robust group jackknife inference (yielding generally smaller not larger standard errors) for family membership were considered, but were found unnecessary after adjusting for mutation. SAS version 9.3 (SAS Institute Inc, Cary, NC) was used for all analyses, and a 2-sided significance level of 0.05 was used for hypothesis testing. See comments in the Supplementary Appendix regarding relevant interpretation of multivariate Cox model analyses and associated predicted survival analyses for males and females hypothesized to be always on and always off β-blockers.
RESULTS
First Year of Life
The LQT3 study population involved 406 patients. Twelve patients were symptomatic in the first year of their life: 7 had unexplained syncope thought to be related to LQT3, 6 had ACA (4 of them died in the first year of life while receiving what was thought to be appropriate LQTS therapy), and one had documented Torsades de Pointes (published case, ref 18). It is generally appreciated that LQTS patients who are symptomatic in their first year of life have a poor prognosis,19,20 and for this reason we excluded these patients in the subsequent analyses that begin at age 1 year. Three patients had no follow-up after 1 year of age, and they were also excluded from the long-term follow-up study. See Supplementary Appendix Table S2 for clinical and genetic details on these 15 patients.
Study Population Beginning at Age 1 year (n=391)
Baseline Characteristics
The clinical characteristics of the 391 remaining LQT3 subjects are summarized in Table 1. Baseline patient characteristics, electrocardiographic parameters, and genetic variables were similar in the three geographic sources of the subjects. The use of β-blockers was less frequent in Japan (p<0.01), but other modalities of treatment were similar by geographic region. For the 82 LQT3 probands and their 309 mutation-positive family members, 51 distinct LQT3-associated mutations were identified including 47 missense mutations in 322 patients (82%) and 4 deletion/insertion/frameshift mutations in 69 patients (18%). Overall, 275 patients (70%) had mutations located in either the transmembrane spanning domains or the interdomain linkers, 115 patients (29%) had mutations located in the C-terminus, and only 1 patient had a mutation located in the N-terminus region.
Table 1.
Clinical characteristics of 391 LQT3 patients, event-free at age 1 year. Abbreviations: QTp: QT peak interval, LCSD: Left cardiac sympathetic denervation, ICD: Internal Cardiac Defibrillator. Information on the use of mexiletine and flecainide was not uniformly collected in Europe and Japan.
| Characteristics | US | Europe | Japan | missing | Total |
|---|---|---|---|---|---|
| No of Patients | 208 | 155 | 28 | - | 391 |
| Male, #(%) | 85(41) | 72(46) | 17(61) | - | 174(45) |
| Proband #(%) | 37(18) | 34(22) | 11(39) | - | 82(21) |
| Age at ECG, yr | 26±19 | 31±20 | 25±19 | 33 | 28±20 |
| ECG, mean±sd | |||||
| RR, msec | 865±240 | 896±206 | 946±213 | 33 | 884±225 |
| PR, msec | 159±36 | 162±28 | 165±30 | 84 | 161±33 |
| QRS, msec | 83±13 | 104±112 | 87±16 | 35 | 92±73 |
| QTp, msec | 353±78 | 362±70 | 387±81 | 36 | 359±75 |
| QT, msec | 442±87 | 443±84 | 473±89 | 35 | 445±86 |
| QTc, msec | 479±50 | 471±63 | 487±62 | 35 | 476±57 |
| QTc Males, msec | 487±52 | 475±60 | 487±69 | 9 | 482±57 |
| QTc Females, msec | 473±48 | 466±67 | 486±51 | 26 | 471±56 |
| Treatment, #(%) | |||||
| β-blockers | 77(38) | 31(21) | 3(11) | 8 | 111(29) |
| LCTSD | 1(0) | 5(3) | 0(0) | 3 | 6(2) |
| Pacemaker | 13(6) | 6(4) | 0(0) | 3 | 19(5) |
| ICD | 49(24) | 16(10) | 4(14) | - | 69(18) |
| Location, #(%) | |||||
| N-Terminus | 1(0) | 0(0) | 0(0) | - | 1(0) |
| Transmembrane | 77(37) | 77(50) | 15(54) | - | 169(43) |
| C-Term | 59(28) | 44(28) | 12(43) | - | 115(29) |
| Intra | 71(34) | 34(22) | 1(4) | - | 106(27) |
| Mutation Type, #(%) | |||||
| Missense | 153(74) | 147(95) | 22(79) | - | 322(82) |
| Deletions | 55(26) | 8(5) | 6(21) | - | 69(18) |
| E1784K | 47(23) | 10(6) | 12(43) | - | 69(18) |
| D1790G | 0(0) | 29(19) | 0(0) | - | 29(7) |
| First Cardiac Event | |||||
| Syncope | 51(25) | 28(18) | 7(25) | - | 86(22) |
| ACA | 4(2) | 3(2) | 0(0) | - | 7(2) |
| Sudden Cardiac Death | 19(9) | 6(4) | 0(0) | - | 25(6) |
| Ever Cardiac Events | |||||
| Syncope | 51(25) | 28(18) | 7(25) | 1 | 86(22) |
| ACA | 8(4) | 8(5) | 6(21) | - | 22(6) |
| Sudden Cardiac Death | 27(13) | 11(7) | 2(7) | 1 | 40(10) |
| Appropriate Shock | 4(2) | 0(0) | 1(4) | 1 | 5(1) |
Clinical Outcome: Univariate Analyses (unadjusted)
One hundred eighteen (41 males, 77 females) patients (30%) experienced at least one suspected LQT3-triggered cardiac event of syncope, ACA, or SCD by age 40. The cumulative probabilities of a first cardiac event for syncope, ACA, or SCD; for ACA or SCD; and for SCD as the first event as a function of age for the 391 subjects are presented in Figure 1, with cumulative event rates of 38%, 20%, and 14%, respectively. The risk of a first cardiac event was related directly to the degree of QTc prolongation, at least between the ages of 16–26 (Figure 2). Females had a higher probability of a first cardiac event than males, especially in the 30 to 40-year age range (Figure 3). The duration of the QRS interval was not associated with an increased probability of cardiac events (data not shown). Removing patients with imputed data, including the 25 individuals who died without known QTc, did not essentially change the obtained results (data not shown).
Figure 1.
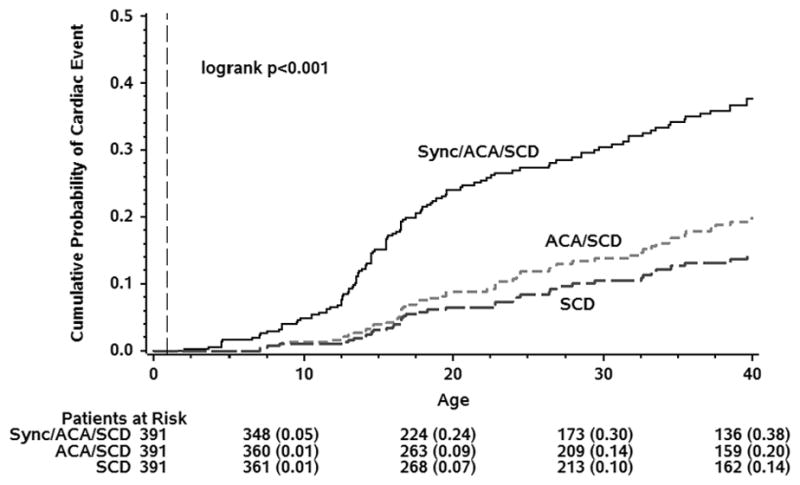
Kaplan-Meier Cumulative Probability of First LQT3-Triggered Cardiac Event for Combinations of Syncope, Aborted Cardiac Arrest (ACA), and Sudden Cardiac Death (SCD), Conditional on Event-Free Survival to Age 1-year.
Figure 2.

Kaplan-Meier Cumulative Probability of Cardiac Events (Syncope/Aborted Cardiac Arrest/LQT3-related Sudden Cardiac Death, whichever comes first) for Three QTc Ranges, Conditional on Event-Free Survival to Age 1-year.
Figure 3.
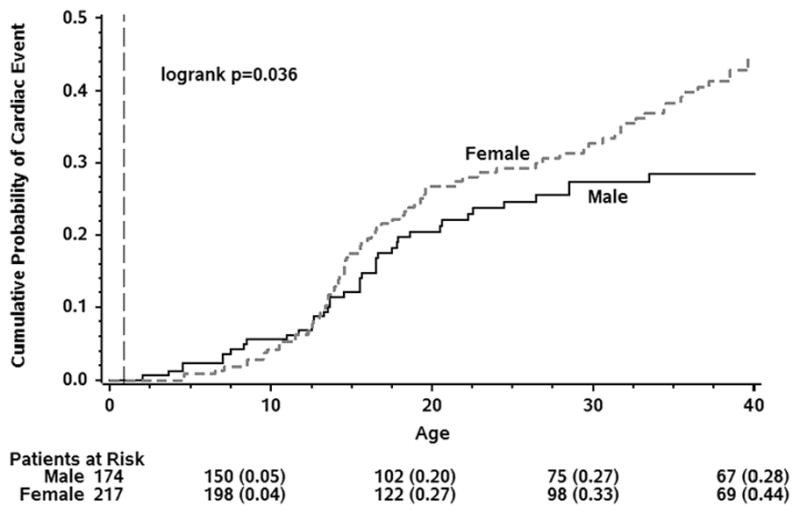
Kaplan-Meier Cumulative Probability of Cardiac Events (Syncope/Aborted Cardiac Arrest/LQT3-related Sudden Cardiac Death, whichever comes first) for Females and Males, Conditional on Event-Free Survival to Age 1-year.
Various therapies were used in the treatment of these LQT3 subjects. There were 69 patients (17.6%) who received an ICD during follow-up. Unfortunately, we have only limited documentation about the indication for ICD implantation or the frequency of interrogation-recorded ICD therapy for ventricular tachyarrhythmic events. No patient who had received an ICD died during a median follow-up of 36 months. A total of 111 patients (28.3%) were started on β-blockers, and because of the time-dependent use of β-blockers, i.e., when they were on and off of β-blockers, and the influence of various risk factors influencing β-blocker efficacy, the appropriate effectiveness of β-blockers can only be evaluated in the multivariate analyses. The efficacy of other therapies (flecainide, mexiletine, ranolazine, and left cardiac sympathetic denervation) could not be judged because of the relatively small number of patients so treated (in addition to lack of data) and few events.
Clinical Outcome: Multivariate Analyses
Findings from the multivariate Cox regression analyses for the end point of syncope, ACA, or SCD, whichever came first, are presented in Table 2. Contrary to the speculated pro-arrhythmic risk of β-blocker therapy in LQT3, time-dependent β-blocker use was associated with a reduction in cardiac events of 83% in females whereas in males a non-significant 6% decrease was observed (Table 2). Hence, there appeared to be a significant gender x β-blocker interaction (p<0.04). Each 10ms increase in QTc duration up to 500ms was associated with a 19% increase in the risk for cardiac events, and patients born after 1955 had a 5% annual increase in the risk for cardiac events. There is no apparent increase in risk for QT when greater than 500ms. It appears that this is due, at least in part, to the difference between the adjusted versus unadjusted effects of QTc. In our multivariable Cox models, there is insufficient evidence that risk increases with QTc beyond about 500ms (see Methods for the multivariable Cox model approach). Two mutations, E1784K and D1790G, were relatively benign with hazard ratios for cardiac events significantly less than 1.0.
Table 2.
Multivariate Cox Model Analyses for Risk of Cardiac Events: First Cardiac Event (Syncope, Aborted Cardiac Arrest [ACA], or LQT3 related Sudden Cardiac Death [SCD]).
| Endpoint = Cardiac Event (118 CE: 25 SCD + 7 ACA + 86 syncope) | ||||
|---|---|---|---|---|
| Hazard | 95% Conf Int | |||
| Parameter | p-value | Ratio | LCL | UCL |
| β-blockers among females* | 0.014 | 0.17 | 0.04 | 0.70 |
| β-blockers among males* | 0.895 | 0.94 | 0.40 | 2.21 |
| E1784K mutation | < 0.001 | 0.35 | 0.19 | 0.62 |
| D1790G mutation | 0.007 | 0.32 | 0.14 | 0.73 |
| QTc per 10 ms (up to 500 ms) | <0.0001 | 1.18 | 1.11 | 1.26 |
| Year of birth (>1955) | <0.0001 | 1.05 | 1.04 | 1.07 |
| *Test for β-blockers x sex interaction: | ||||
| β-blockers for males vs females | 0.039 | |||
When the end point was restricted to ACA or SCD (Table 3), time-dependent syncope doubled the risk for the more malignant cardiac events (p=0.02), while the other risk variable effects were similar to those presented in Table 2. β-blockers reduced the risk of ACA/SCD by 80% among females (p=0.03) and by 49% among males (NS).
Table 3.
Multivariate Cox Model Analyses for Risk of Cardiac Events: First Aborted Cardiac Arrest [ACA] or LQT3 related Sudden Cardiac Death [SCD].
| Endpoint = ACA/SCD (56 ACA/SCD: 34 SCD + 22 ACA) | ||||
|---|---|---|---|---|
| Hazard | 95% Conf Int | |||
| Parameter | p-value | Ratio | LCL | UCL |
| Syncope | 0.023 | 2.03 | 1.10 | 3.72 |
| β-blockers among females* | 0.032 | 0.20 | 0.05 | 0.87 |
| β-blockers among males* | 0.308 | 0.51 | 0.14 | 1.88 |
| E1784K mutation | 0.001 | 0.09 | 0.02 | 0.37 |
| D1790G mutation | 0.049 | 0.30 | 0.09 | 0.99 |
| QTc per 10 ms (up to 500 ms) | <0.001 | 1.33 | 1.19 | 1.48 |
| Year of birth (after 1955) | <0.001 | 1.06 | 1.03 | 1.09 |
| *Test for β-blockers x sex interaction: | ||||
| β-blockers for males vs females | 0.353 | |||
The numbers of patients with cardiac events by time-dependent β-blocker status are provided in Supplementary Table S3, and the numbers of subjects with cardiac events while on β-blocker therapy are provided in Table 4. All together during a median FU of over 7 years 5 patients developed life threatening arrhythmias on β-blocker therapy (3 died). Figures 4A and 4B show Cox model-based predicted distributions of the age at first ACA or SCD, conditional on event-free survival to age 1, by β-blocker status and gender for asymptomatic subjects (no prior syncope) born in 1971 (median) with a QTc of 470ms (median risk, Figure 4A) and a QTc of 500ms (high risk, Figure 4B) and neither lower risk mutation (not E1784K nor D1790G). As is shown, β-blockers appear clearly effective in females. In males, the number of cardiac events is much lower precluding the ability to detect a further attenuation of risk by β-blocker therapy. However, a pro-arrhythmic signal is absent. Again, the importance of baseline QTc is evident.
Table 4.
Numbers of Subjects and Events While on Beta-Blocker Therapy.
| Beta-blocker Therapy | Number of Patients Treated with β-blockers | Follow-up Duration in Months After Treatment 25th–75th Quartile; median | Syncope, Aborted Cardiac Arrest, or Death # (% of treated) | Death # (% of treated) | ACA/SCD # (% of treated) |
|---|---|---|---|---|---|
| All patients | 111 | 36–161; 87 | 15 (14) | 3 (3) | 5 (5) |
| Male patients | 51 | 40–180; 92 | 8 (16) | 2 (4) | 2 (4) |
| Female patients | 60 | 34–144; 86 | 7 (12) | 1 (2) | 3 (5) |
Figure 4.
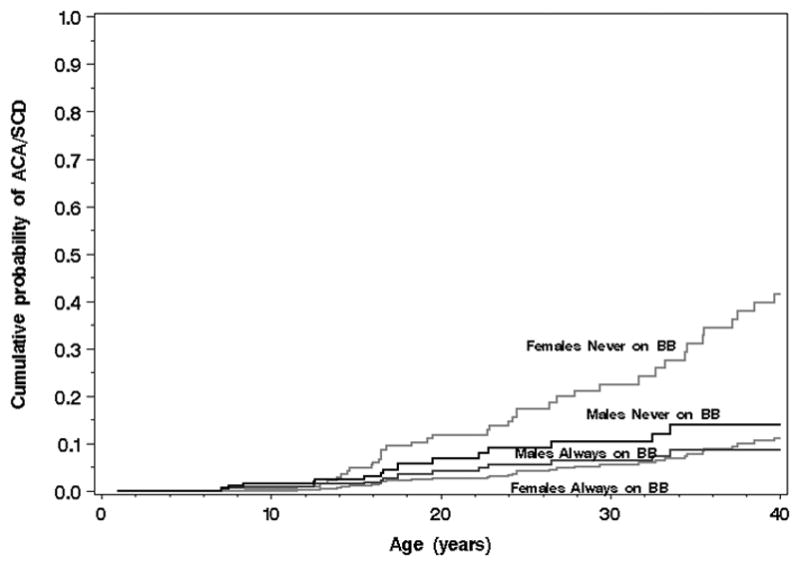

Figure 4A. (Plot for Medium-Risk Patients). Cox model-based predicted distributions of the age at first aborted cardiac arrest (ACA) or sudden cardiac death (SCD), conditional on event-free survival to age 1, by β-blocker status (BB) and gender for asymptomatic subjects (no prior syncope) born in 1971 (median) with a QTc of 470 ms (median) and neither lower risk mutation (not E1784K nor D1790G). See supplement for additional explanation.
Figure 4B. (Plot for High-Risk Patients). Cox model-based predicted distributions of the age at first aborted cardiac arrest (ACA) or sudden cardiac death (SCD), conditional on event-free survival to age 1, by β-blocker status (BB) and gender for asymptomatic subjects (no prior syncope) born in 1971 (median) with a QTc ≥ 500 ms and neither lower risk mutation (not E1784K nor D1790G).
See supplement for additional explanation.
As one may argue that PM/ICD therapy may protect against the possible detrimental effects of β-blockers we ran additional analyses where we censored patients at the time of pacemaker or ICD implantation, and found similar results for β-blocker efficacy among those without a pacemaker or ICD: HR = 0.22 (p = 0.035) and 0.25 (p = 0.059) for females, and HR = 1.03 (p = 0.94) and 0.60 (p = 0.52) for males.
DISCUSSION
We studied 391 LQT3 patients, asymptomatic during the first year of life, and found that the degree of QT prolongation and history of syncope were the major risk factors for an LQT3-related cardiac event including ACA or SCD. The risk for a first event increases rapidly during adolescence and continued to increase in both genders, though more slowly, during the adult years. β-blocker therapy significantly reduced the risk for cardiac events in treated individuals, in particularly in females.
Clinical risk factors
In the present study, we were able to confirm the age-dependent occurrence of cardiac events; with the exception of a few events in very young infants ≤1 year of age; these 12 patients were excluded in the long-term follow-up analyses. The number of individuals who experienced a sentinel cardiac event increased rapidly between age 10 and age 20, and by the age of 40 almost 40% of patients had experienced a first cardiac event. Among those experiencing a LQT3-related cardiac event, about 50% were ACA or SCD (Fig. 1). As in other LQTS subtypes,4,5 probands were at higher risk than family members (not shown), probably reflecting a referral bias based on earlier or more severe first symptoms than in affected family members and also the fact that the proband’s QTc values were greater than the QTc values of the affected family members since the length of the QTc was an independent predictor of cardiac events (Figure 2). Females had a greater risk than males, especially in the older age group (Figure 3). However, unlike in LQT1 and LQT2 where the risk in males is predominantly pre-puberty, the risk of an LQT3-triggered event in males shifts to post-puberty and continues throughout adulthood. Unfortunately, no 24-hr Holter monitoring data were available for analysis, nor were data on the circumstances leading to cardiac events.
Mutation type and mutation location
Mutation type and mutation location did not have a significant effect on outcome, although patients with some particular mutations (E1784K, D1790G; Tables 2 and 3) had a rather benign clinical course.
Therapy for LQT3
β-blocker therapy reduces the risk for cardiac events in patients with LQT1 by >95% and in patients with LQT2 by 70–80%.4,5 In contrast, the early genotype-phenotype studies showed no demonstrable β-blocker efficacy for LQT3.6,7 Subsequent cellular in vitro studies raised concerns regarding the possible pro-arrhythmic effect of β-blockers for LQT3,11 which got translated prematurely to the bed side with an unproven view that β-blockers might be contraindicated in LQT3. For the last decade this notion has resulted in a fairly high rate of prophylactic ICD therapy in LQT3.12,13 The theoretical arguments, based on just a few cases, suggested that β-blockers should not be used in LQT3, especially in patients with longer QT intervals at low heart rates with associated events during sleep or rest.7 Atrioventricular block,21 bradycardia,22 sinus pauses,23 and sinus arrest were thought to be possible mechanisms of death in LQT3.23,24
Our large study provides evidence that β-blocker therapy is not pro-arrhythmic. In contrast, a clear and significant protective effect for cardiac events was demonstrated. The effect was clear in females, whereas in males, their lower event rate precluded a demonstration of efficacy. However, a detrimental effect of β-blocker therapy in males with LQT3 is absent (Tables 2 and 3, suppl. Figures 4A and 4B). Only 3 patients (3%) died on β-blocker therapy during a median FU of over 7 years (table 4).
The absolute risk of dying from LQT3-related arrhythmias (in individuals asymptomatic up till age 1) is <15% by the age of 40 (Figure 1). QTc and the presence of symptoms are strong modifiers of this risk (Tables 2 and 3), and it is likely that high-risk patients with prior syncope or ACA and/or QTc in the 500 ms range may require adjunctive therapy such as left cardiac sympathetic denervation,25–27 ICD,12,13,28 or LQT3-directed pharmacotherapy with medications like mexiletine, flecainide, and more specific late sodium current blockers including ranolazine and some experimental drugs.29–34 However, the current study cannot address precisely when these therapies should be used. Treatment in high-risk patients requires clinical judgment with balance of the disease risk versus the risk/benefit related to the selected therapy in each patient based on age, gender, QTc duration, and prior symptoms as well as tolerance and clinical response to β-blocker therapy.
Study Limitations
Although this is the largest study for this 3rd most common subtype (LQT3) of LQTS, an inherent limitation of this study is still the relatively small number of cardiac events, in particular in males, and the small number of patients receiving therapies despite an international collaboration. The assumption that the deceased young individuals carried the familial mutation is reasonable but not certified. Yet removing them from the analysis did not change the results. In addition, the generalizability even within LQT3 is limited somewhat as the study population was dominated by 5 specific mutations. Although an attempt was made to exclude families with obvious evidence of an overlap syndrome, any of the 36 functionally uncharacterized SCN5A mutations (out of the 51 LQT3-associated mutations represented in this study) might potentially exhibit an expressed phenotype of overlap if the families were large enough or followed for longer durations. The E1784K mutation is an example of this,35 but we would like to stress that patients with an overt overlap syndrome (i.e. signs of right precordial ST-elevation) have been excluded. Another limitation was the non-randomized use of β-blockers and that less than one-third of the patients in this cohort were ever treated with β-blockers. Finally, follow-up was censored at age 41, and cardiac events may continue later in life in LQT3, especially if the patient acquires concomitant coronary artery disease.
Conclusions
Patients with LQT3 can be stratified as to their risk of life-threatening cardiac events based on clinical and genetic characteristics. A high-risk subpopulation of LQT3 patients with QTc ≥500 ms and a history of syncope can be identified, and this population may require adjunctive therapy. β-blocker therapy significantly reduced the risk for cardiac events.
Supplementary Material
Clinical Perspective.
What is new?
Long QT syndrome type 3 (LQT3) is caused by gain-of-function mutations in the SCN5A-encoded Nav1.5 sodium channel. The phenotype differs from the more common potassium channel-mediated forms, among other characteristics, by a more lethal course. Risk stratification in LQT3 is not well defined and the effectiveness of β-blocker therapy has not been studied in a large LQT3 cohort.
In a study of almost 400 LQT3 patients (1–40 years of age) it was demonstrated that the risk of a cardiac event was directly related to the degree of QTc prolongation. In addition, the presence of syncope doubled the risk for future life threatening events. β-blocker therapy significantly reduced the cardiac event rate in females but in males this effect could not conclusively be determined due to the low number of events in males.
What are the clinical implications?
The clinical implications of this study are that LQT3 patients can be stratified as to their risk of life-threatening cardiac events based on clinical and genetic characteristics. A high-risk subpopulation of LQT3 patients with QTc ≥ 500 ms and a history of syncope can be identified, and this population may require adjunctive therapy. β-blocker therapy significantly reduced the risk for cardiac events.
Acknowledgments
Funding Sources: Supported in part by: 1) the health sciences research grant (H18 - Research on Human Genome - 002) and the Research Grant for the Cardiovascular Diseases (21C-8, 22-4-7, H24-033, H26-040) from the Ministry of Health, Labour and Welfare, Japan (WS); 2) Research grants HL-33843, HL-51618, and HL-123483 from the National Institutes of Health, Bethesda, Maryland, USA (AJM); 3) Grant 2000.059 and 2005T024 from the Nederlandse Hartstichting, The Netherlands (AAMW& CRB), and the Cardiovascular Onderzoek Nederland and Netherlands Heart Foundation project PREDICT (AAMW&CRB), NIH grant HL68880 and Telethon grant GGP09247 (PJS and LC), and the Windland Smith Rice Comprehensive Sudden Cardiac Death Program (MJA).
Footnotes
Conflict of Interest Disclosures: Dr. Wilde serves on the scientific advisory board of Lilanova. Dr. Ackerman is a consultant for Boston Scientific, Gilead Sciences, Medtronic, and St. Jude Medical. Dr. Ackerman and Mayo Clinic receive royalties from Transgenomic for their FAMILION-LQTS and FAMILION-CPVT genetic tests. None of these entities provided financial support for this study. The other authors report no conflicts.
References
- 1.Hobbs JB, Peterson DR, Moss AJ, McNitt S, Zareba W, Goldenberg I, Qi M, Robinson JL, Sauer AJ, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Towbin JA, Vincent GM, Zhang L. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–54. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- 2.Sauer AJ, Moss AJ, McNitt S, Peterson DR, Zareba W, Robinson JL, Qi M, Goldenberg I, Hobbs JB, Ackerman MJ, Benhorin J, Hall WJ, Kaufman ES, Locati EH, Napolitano C, Priori SG, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–37. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 3.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRS expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association. Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 4.Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–9. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu W, Moss AJ, Wilde AA, Towbin JA, Ackerman MJ, January CT, Tester DJ, Zareba W, Robinson JL, Qi M, Vincent GM, Kaufman ES, Hofman N, Noda T, Kamakura S, Miyamoto Y, Shah S, Amin V, Goldenberg I, Andrews ML, McNitt S. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol. 2009;54:2052–62. doi: 10.1016/j.jacc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, Moncalvo C, Tulipani C, Veia A, Bottelli G, Nastoli J. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–4. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 8.Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, Crotti L, Piippo K, Lupoglazoff JM, Villain E, Priori SG, Napolitano C, Zhang L. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–21. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]
- 9.Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, Schwartz PJ, Vincent GM, Priori SG, Benhorin J, Towbin JA, Robinson JL, Andrews ML, Napolitano C, Timothy K, Zhang L, Medina A. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol. 2003;42:103–9. doi: 10.1016/s0735-1097(03)00554-0. [DOI] [PubMed] [Google Scholar]
- 10.Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–5. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–86. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, Gasparini M, Wilde AA, Knops RE, Denjoy I, Toivonen L, Mönnig G, Al-Fayyadh M, Jordaens L, Borggrefe M, Holmgren C, Brugada P, De Roy L, Hohnloser SH, Brink PA. Who are the long QT syndrome patients who receive an implantable cardioverter defibrillator and what happens to them? Data from the European LQTS ICD Registry. Circulation. 2010;122:1272–82. doi: 10.1161/CIRCULATIONAHA.110.950147. [DOI] [PubMed] [Google Scholar]
- 13.Horner JM, Kinoshita M, Webster TL, Haglund CM, Friedman PA, Ackerman MJ. Implantable cardioverter defibrillator therapy for congenital long QT syndrome: a single centre experience. Heart Rhythm. 2010;7:1616–22. doi: 10.1016/j.hrthm.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 14.Wilde AAM, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circulation Research. 2011;108:884–897. doi: 10.1161/CIRCRESAHA.110.238469. [DOI] [PubMed] [Google Scholar]
- 15.Zareba W, Sattari MN, Rosero S, Couderc JP, Moss AJ. Altered atrial, atrioventricular, and ventricular conduction in patients with the long QT syndrome caused by the DeltaKPQ SCN5A sodium channel gene mutation. Am J Cardiol. 2001;88:1311–4. doi: 10.1016/s0002-9149(01)02097-5. [DOI] [PubMed] [Google Scholar]
- 16.Bezzina C, Veldkamp MW, van den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 17.Cox DR. Regression models and life-tables. J Stat Soc [B] 1972;34:187–220. [Google Scholar]
- 18.Valdivia CR, Ackerman MJ, Tester DJ, Wada T, McCormack J, Ye B, Makielski JC. A novel SCN5A arrhythmia mutation, M1766L, with expression defect rescued by mexiletine. Cardiovasc Res. 2002;55:279–289. doi: 10.1016/s0008-6363(02)00445-5. [DOI] [PubMed] [Google Scholar]
- 19.Spazzolini C, Mullally J, Moss AJ, Schwartz PJ, McNitt S, Ouellet G, Fugate T, Goldenberg I, Jons C, Zareba W, Robinson JL, Ackerman MJ, Benhorin J, Crotti L, Kaufman ES, Locati EH, Qi M, Napolitano C, Priori SG, Towbin JA, Vincent GM. Clinical implications for patients with Long QT Syndrome who experience a cardiac event during infancy. J Am Coll Cardiol. 2009;54:832–7. doi: 10.1016/j.jacc.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD True or false? Heart Rhythm. 2009;6:113–120. doi: 10.1016/j.hrthm.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 21.Miura M, Yamagishi H, Morikawa Y, Matsuoka R. Congenital long QT syndrome and 2:1 atrioventricular block with a mutation of the SCN5A gene. Pediatr Cardiol. 2003;24:70–72. doi: 10.1007/s00246-002-0169-5. [DOI] [PubMed] [Google Scholar]
- 22.Veldkamp MW, Wilders R, Baartscheer A, Bezzina CR, Zegers JG, Wilde AAM. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circulation Research. 2003;92:976–983. doi: 10.1161/01.RES.0000069689.09869.A8. [DOI] [PubMed] [Google Scholar]
- 23.Medina A, Corcos AP, Lysy J, Benhorin J, Tzivoni D. Cardiac standstill as a cause of death in long QT syndrome. Isr J Med Sci. 1987;23:302–4. [PubMed] [Google Scholar]
- 24.van den Berg MP, Wilde AA, Viersma TJW, Brouwer J, Haaksma J, van der Hout AH, Stolte-Dijkstra I, Bezzina TCR, Van Langen IM, Beaufort-Krol GC, Cornel JH, 2nd, Crijns HJ. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:630–6. doi: 10.1046/j.1540-8167.2001.00630.x. [DOI] [PubMed] [Google Scholar]
- 25.Moss AJ, McDonald J. Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N Engl J Med. 1970;285:903–4. doi: 10.1056/NEJM197110142851607. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, Bloise R, De Ferrari GM, Klersy C, Moss AJ, Zareba W, Robinson JL, Hall WJ, Brink PA, Toivonen L, Epstein AE, Li C, Hu D. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. 2004;109:1826–33. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 27.Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752–9. doi: 10.1016/j.hrthm.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 28.Zareba W, Moss AJ, Daubert JP, Hall WJ, Robinson JL, Andrews M. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003;14:337– 41. doi: 10.1046/j.1540-8167.2003.02545.x. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS, Colatsky TJ. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–86. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 30.Benhorin J, Taub R, Goldmit M, Kerem B, Kass RS, Windman I, Medina A. Effects of flecainide in patients with new SCN5A mutation: Mutation-specific therapy for long-QT syndrome? Circulation. 2000;101:1698–1706. doi: 10.1161/01.cir.101.14.1698. [DOI] [PubMed] [Google Scholar]
- 31.Windle JR, Geletka RC, Moss AJ, Zareba W, Atkins DL. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A: deltaKPQ mutation. Ann Noninvasive Electrocardiol. 2001;6:153–8. doi: 10.1111/j.1542-474X.2001.tb00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moss AJ, Windle JR, Hall J, Zareba W, Robinson JL, McNitt S, Severski P, Rosero S, Daubert JP, Qi M, Cieciorka M, Manalan AS. Safety and efficacy of flecainide in subjects with Long QT-3 Syndrome (ΔKPQ Mutation): A randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol. 2005;10(Suppl):59–66. doi: 10.1111/j.1542-474X.2005.00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to Type-3 Long-QT Syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC. Cardiac late Na+ current: pro arrhythmic effects, roles in long QT syndromes, and pathological relatiohsip to CAMKII and oxidative stress. Heart Rhythm. 2015;12:440–8. doi: 10.1016/j.hrthm.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, Schulze-Bahr E, Fukuhara S, Mochizuki N, Makiyama T, Itoh H, Christiansen M, McKeown P, Miyamoto K, Kamakura S, Tsutsui H, Schwartz PJ, George AL, Jr, Roden DM. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–29. doi: 10.1172/JCI34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.