

Abstract
CD1d-restricted T (natural killer T [NKT]) cells are important for controlling a herpes simplex virus (HSV) infection. One of the mechanisms of immune evasion by HSV is to downregulate CD1d-mediated activation of NKT cells. VP22 is an HSV-1-encoded protein responsible for reorganizing the host cell's cytoskeletal network and viral spreading. We have previously shown that modification of the cytoskeleton can alter CD1d-mediated antigen presentation. In this study, we found that an HSV-1 lacking VP22 (ΔUL49) was impaired in its ability to inhibit CD1d-mediated antigen presentation compared with the wild-type (WT) virus; this was reversed by a repair virus (UL49R) in CD1d-expressing cells. We further demonstrated that CD1d recycling was inhibited by infection with WT and UL49R, but not the ΔUL49 virus. Ectopic expression of VP22 in CD1d-expressing cells complemented the VP22-deficient virus in inhibiting antigen presentation. Moreover, inhibiting viral protein synthesis rescued VP22-dependent inhibition of CD1d antigen presentation. In conclusion, our findings suggest that VP22 is required (but not sufficient) for the inhibition of CD1d-mediated antigen presentation by an HSV-1 infection.
Introduction
Host cells have the capacity to recognize danger signals during a viral infection. As a result, an immune response by the host can limit the infection. As viruses that can infect a wide variety of mammalian species, the α herpesvirus subfamily member herpes simplex virus (HSV), has evolved many ways to escape from the host's immune response. Some HSV-encoded viral proteins, such as γ34.5, ICP0, US3, and US11, counteract the host's innate antiviral immune response (21,22,24,27). On the other hand, HSV can also evade the adaptive immune response. The HSV-1-encoded molecule, ICP47, specifically binds to the transporter associated with antigen processing, blocks an antigenic peptide from translocating into the endoplasmic reticulum for antigen loading onto MHC class I molecules, and allows the infected cells to hide from immune recognition by cytotoxic T lymphocytes (1,16,20,41,44). An HSV-1 infection also suppresses MHC class II-mediated antigen representation, by downregulating the level of invariant chain in antigen-presenting lymphoid cells (30), and the HSV-1-encoded protein gB binds to (and causes the release of) HLA-DR into the exosomal pathway (30,40). HSV-encoded proteins can also inhibit the cross-presentation of viral peptides by dendritic cells to CD8+ T cells (31).
Natural killer T (NKT) cells are a subpopulation of T cells that recognize CD1d, an MHC class I-like molecule that mediates lipid antigen presentation to NKT cells (7,18). NKT cells have been reported to play important roles in protecting a host from an HSV infection. For example, NKT cell-deficient mice are less able to clear an infection by HSV-1 or HSV-2 (3,19), although there have been reports that conflict with these observations (8). Nonetheless, in humans, two clinical cases of a life-threatening infection with the vaccine strain of varicella zoster virus (another member of the α herpesviridae subfamily), were related to a deficiency in NKT cells and/or diminished CD1d expression (5,23); these reports strongly suggest that a deficiency in the NKT cell/CD1d system is associated with increased susceptibility to herpesvirus infections. In addition, the number of NKT cells has recently been correlated with the level of HSV-1-specific antibodies in humans (34). We and others have shown that HSV-1 can inhibit CD1d-mediated antigen presentation to NKT cells by suppressing CD1d recycling (6,25,33,45). This inhibition of NKT cell function by HSV-1 is dependent on cell-to-cell contact of infected APCs and NKT cells, but not due to infection of NKT cells (6).
VP22, encoded by the UL49 gene in HSV-1, is a major tegument protein in the virion (15). A VP22-deficient virus (ΔUL49) has a defect in cell-to-cell spreading, likely due to reduced virion release (10). VP22 is also required for viral protein synthesis at late times during infection (10), and the defect in late viral protein synthesis in ΔUL49 can be rescued by secondary mutations in vhs (virion host shutoff) (28,37). During infection, VP22 is transported between cells and is necessary for the stabilization and hyperacetylation of microtubules in host cells (12,13). It forms multimers with the HSV-1-encoded proteins, V16 and VHS, and is phosphorylated by host casein kinase II and the HSV-1 viral kinase, UL13 (2,14). Furthermore, two di-leucine motifs in VP22 are important for its ability to regulate the intracellular distribution of multiple viral and cellular proteins (39).
We have previously shown that disruption of the cytoskeletal network increases CD1d-mediated antigen presentation (17). Since VP22 has an impact on the host cell's cytoskeletal network, we investigated the contribution of VP22 in the inhibition of CD1d-mediated antigen presentation following an HSV-1 infection. Our results suggest that VP22 is required (but not sufficient) for HSV-1 in the decrease of antigen presentation by CD1d postinfection.
Materials and Methods
Cell culture, constructs, and viral infections
HEK293 cells expressing wild-type (WT) CD1d and T322A/S323A mutant have been previously described (25,26). HEK293 cells were kindly provided by Prof. Philip Cohen (MRC Protein Phosphorylation Unit, University of Dundee, Scotland, United Kingdom). HeLa.CD1d cells were obtained from Dr. Janice Blum (Indiana University, Indianapolis, IN), with permission from Dr. Peter Cresswell (Yale University, New Haven, CT). Cells were cultured in DMEM supplemented with 10% FBS and 2 mM l-glutamine (Lonza).
To generate a VP22-expressing vector, two primers, 5′-AGTCAGATCTATGACCTCTCGCCGCTCCGTG-3′ and 5′-ATCGGGATCCTCACTCGACGGGCCGTCTGG-3′, flanked with Bgl II/BamH I (as underlined), were used for polymerase chain reaction (PCR) amplification of VP22 cDNA from an Apo I-BamH I DNA fragment of HSV-1 (bps 104768-107027) in pUC19. The PCR products were gel purified, digested with Bgl II and BamH I, and subcloned into an IRES-containing bicistronic pIRES2-AcGFP1 vector (Clontech). The resulting plasmid was digested with Bgl II and Not I, and then the cDNA fragment encoding VP22 and GFP, each flanking the IRES, was inserted into the pCDNA3.1-hygro-based vector (Invitrogen). The pcDNA3.1-hygro vector containing IRES and GFP alone was used as a control and both vectors were used to stably transfect HEK293-CD1d cells as described previously (26).
Cells were infected with the HSV-1 F strain WT virus, a VHS-deficient HSV-1 (ΔVHS), a VP22-deficient HSV-1 (ΔUL49), and rescue virus UL49R (10), for the indicated periods of time at 37°C.
Antibodies and related reagents
A pan-HSV-1- and 2-specific mAb (clone 4F10.3) was purchased from Millipore. Phycoerythrin (PE)-conjugated and purified versions of the human CD1d-specific 42.1 mAb were purchased from BD Biosciences. The pan-HLA class I-specific mAb HB95 hybridoma was kindly provided by J. Yewdell and J. Bennink (Laboratory of Viral Diseases, NIAID, NIH). The anti-CD1d free heavy chain (HC)-specific mAb clone C3D5 was from Santa Cruz Biotechnology. Recombinant cytokines (human IL-2, IL-4, and GM-CSF) were from PeproTech, Inc. Antibody pairs for the human IL-4 and GM-CSF ELISA assays were purchased from BD-Biosciences and BioLegend, respectively.
Western blotting
VP22/GFP- or GFP-expressing HEK293-CD1d cells were lysed in lysis buffer [10 mM Tris (pH 7.4), 150 mM NaCl, 0.5 mM EDTA, 2% CHAPS] and resolved on a 10% SDS-PAGE gel. The proteins were then transferred onto a PVDF membrane (Millipore). To detect VP22 expression, the membrane was incubated with a VP22-specific chicken IgY (9), followed by incubation with an HRP-conjugated anti-IgY Ab (Sigma). The membrane was then developed using chemiluminescence before exposure on film.
Flow cytometry
Cells were first washed in PBS and then fixed in 1% paraformaldehyde. To stain for surface CD1d expression, the cells were washed in Hank's balanced salt solution with 0.1% bovine serum albumin (HBSS/BSA) three times and then incubated with the 42.1 mAb. A PE-conjugated rabbit anti-mouse Ig antiserum was added as the secondary antibody. Intracellular staining with specific mAbs was performed in HBSS/BSA in the presence of 0.1% saponin. Thus, the cells were permeabilized in HBSS/BSA with 0.1% saponin and then incubated with the C3D5 mAb for detecting the CD1d HC or an HSV-specific mAb (clone 4F10.3), followed by a PE-conjugated rabbit anti-mouse Ig antiserum. All stained cells were analyzed by flow cytometry as previously described (36).
NKT cell stimulation assays
Human NKT cells were isolated from deidentified peripheral blood from normal human volunteers obtained from the Indiana Blood Center (Indianapolis, IN). The NKT cells were expanded ex vivo as described (26). Antigen-presenting cells were infected with WT or mutant HSV-1 at the indicated multiplicities of infection (MOI) and incubation time. For cycloheximide treatment, cells were treated with medium control or cycloheximide (25 μg/mL) for 30 min. The cells were then infected with the viruses as indicated in the presence or absence of cycloheximide and subsequently fixed in 0.05% paraformaldehyde, washed three times with RPMI medium (Lonza) supplemented with 10% FBS, and cocultured with human NKT cells as previously described (25). Cytokine (IL-4 and GM-CSF) production by NKT cells was measured by ELISA.
Recycling assay
The recycling assay was performed as previously described (25,45). Briefly, 5 million HEK293-CD1d WT cells were infected with WT or mutant HSV-1 (MOI = 2) for 4 h at 37°C. One hour before harvest, cyclohexamide (25 μg/mL) was added. Purified 42.1 mAb was added to block surface CD1d. After washing, the cells were kept in IMDM medium (Lonza) containing cyclohexamide for different time intervals at 37°C. At the indicated time points, an aliquot was placed in a well of an ice-cold 96-well plate. All cells were stained with a PE-labeled CD1d-specific mAb (42.1) on ice. After washing three times in ice-cold PBS, the cells were fixed in 1% paraformaldehyde and resuspended in HBSS/BSA buffer until data acquisition using flow cytometry.
Statistical analysis
Graphs were made and statistics analyzed using GraphPad Prism 5 (GraphPad Software). Each experiment was performed at least three times and one representative assay is shown. The error bars represent the standard error of the mean (SEM) of triplicate samples. A two-way ANOVA, one-way ANOVA with Bonferroni's post-test, or Student's t-test was used as appropriate. A p-value of less than 0.05 was considered significant.
Results
VP22 is required for the inhibition of antigen presentation following an HSV-1 infection
Previous work from our laboratory has demonstrated that the disruption of actin filaments increases CD1d-mediated antigen presentation (17). Conversely, TGF-β treatment of CD1d-expressing cells, which induces the formation of actin stress fibers, inhibits antigen presentation by murine (4) and human CD1d (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/vim). VP22, a protein encoded by the HSV-1 UL49 gene, can reorganize the host cell's cytoskeletal network and facilitate viral spreading (10,13,42). To determine whether VP22 is important for inhibiting CD1d-mediated antigen presentation upon an HSV-1 infection, CD1d-expressing human HEK293 (HEK293-CD1d) cells were infected with a VP22-deficient HSV-1 (ΔUL49). Expression of VP22 in HSV-1 WT or the VP22-rescued (UL49R) virus-infected cells was confirmed by western blotting analysis at 4 h postinfection (Fig. 1A). By staining host cells with an HSV-specific mAb, we found that the ΔUL49 virus was as infectious as WT and UL49R viruses at 4 h postinfection (Fig. 1B). There was a slight reduction in surface CD1d expression when HEK293-CD1d cells were infected with WT or mutant HSV-1 viruses, as compared with mock-infected cells (Fig. 1C); however, consistently, CD1d-mediated antigen presentation by cells infected with ΔUL49 was less impaired than cells infected with WT or UL49R virus (Fig. 1D). Similar results were observed in a different CD1d-expressing cell line, HeLa.CD1d (45) (Fig. 1E). When infected with WT HSV-1 and the mutant viruses, HeLa.CD1d cells infected with ΔUL49 stimulated human NKT cells to a higher degree when compared with cells that were infected with either UL49R or WT HSV-1 (Fig. 1E). As with the HEK293-CD1d cells above, comparable infectivity of these viruses in HeLa.CD1d cells was again observed by flow cytometry using an HSV-specific antibody (Supplementary Fig. S2A). Additionally, an HSV-1 infection in HeLa.CD1d cells did not cause the slight reduction in CD1d surface expression observed in HEK293-CD1d (Supplementary Fig. S2B), but did, nonetheless, produce an impairment in antigen presentation by CD1d (Fig. 1E). We have previously shown that another surface protein, MHC class I molecule (MHC-I), unlike CD1d, is not downregulated by an HSV-1 infection within the time frame (25). In the current study, MHC I surface expression was also not affected by HSV-1 WT or mutant viruses (data not shown). Overall, these data suggest that VP22 is required for the inhibition of CD1d-mediated antigen presentation following an HSV-1 infection.
FIG. 1.
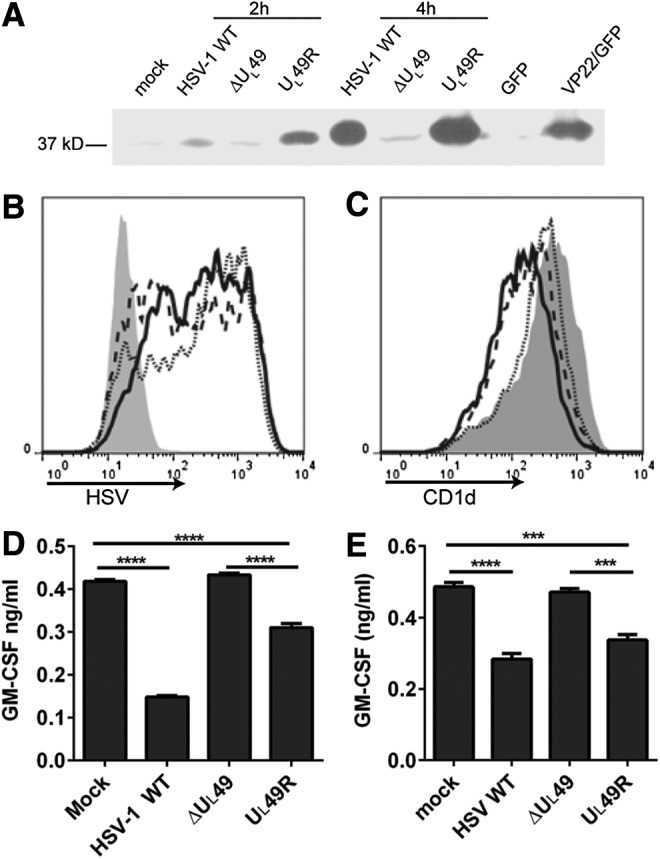
VP22 is required for the inhibition of CD1d-mediated antigen presentation to NKT cells. (A) Human CD1d-expressing HEK293 cells were either mock treated or infected (MOI = 2) for 2 or 4 h with WT HSV-1, or ΔUL49 and UL49R mutant HSV-1. These cells, along with HEK293-CD1d cells that ectopically expressed VP22 (VP22/GFP) or the vector control (GFP), were lysed. The lysates were resolved by 10% SDS-PAGE. Expression of VP22 was detected by western blotting analysis using an anti-VP22 antibody. (B) Human CD1d-expressing HEK293 cells were mock treated (gray-filled histogram) or infected (MOI = 2) for 4 h with WT HSV-1 (solid line), ΔUL49 (dotted line), or UL49R (dashed line). The cells were fixed, permeabilized with 0.1% saponin, stained with an anti-HSV antibody, and analyzed by flow cytometry. (C) The same mock-treated or virus-infected cells were also fixed and stained with the CD1d-specific mAb 42.1 and analyzed by flow cytometry. (D) Aliquots of the cells used in (A, B) were fixed and cocultured with human NKT cells (105/well, E:T = 1:1) for 48 h. The supernatants were analyzed for GM-CSF production by ELISA. (E) HeLa.CD1d cells were mock treated or infected with WT HSV-1 and mutant viruses as indicated. The cells were fixed and cocultured with human NKT cells (105/well, E:T = 1:1) for 48 h. The supernatants were analyzed for GM-CSF production by ELISA. Data shown are representative of three independent experiments. ***p < 0.001; ****p < 0.0001 (one-way ANOVA). HSV, herpes simplex virus; MOI, multiplicities of infection; NKT, natural killer T; WT, wild type.
VP22 is important for inhibiting CD1d recycling upon an HSV-1 infection
It was previously reported that an HSV-1 infection inhibits CD1d-mediated antigen presentation, likely by inhibiting CD1d molecules from recycling back to the cell surface (25,45). VP22 is known to modify the cytoskeletal network in host cells (13), which may cause changes in the endocytic trafficking of cellular proteins. To determine whether CD1d trafficking is affected by VP22, recycling assays were performed on HEK293-CD1d cells that were infected for 4 h with WT HSV-1, ΔUL49, or UL49R. The infected cells expressed similar levels of HSV viral proteins (Fig. 2A). Consistent with previous reports (6,25,33,45), an HSV-1 infection reduced the ability of CD1d molecules to recycle back to the cell surface (Fig. 2B). In cells infected with ΔUL49, CD1d recycled at a similar rate as those in mock-treated cells, and therefore, much faster than CD1d in cells infected with either UL49R or the WT HSV-1 (Fig. 2B). Thus, these data confirm the observation that CD1d molecules are impaired in recycling back to the surface following an HSV-1 infection and moreover, show that this is VP22 dependent.
FIG. 2.

VP22 is important for inhibiting CD1d recycling upon an HSV-1 infection. HEK293-CD1d cells were mock treated (gray-filled histogram) or infected with WT HSV-1 (solid line), ΔUL49 (dotted line) or UL49R (dashed line) viruses at an MOI of 2 for 4 h (A). The cells were fixed, permeabilized with 0.1% saponin, stained with an anti-HSV antibody, and analyzed by flow cytometry. (B) The same cells that were mock treated (open circles), or infected with WT HSV-1 (open triangles), ΔUL49 (solid circles), and UL49R (solid triangles), were blocked with purified anti-CD1d (42.1) and then cultured at 37°C as described in the Materials and Methods section. At each indicated time point, a cell aliquot was harvested and stained with PE-labeled 42.1 mAb. The level of recycled CD1d is shown and was calculated by subtracting the mean fluorescence intensity (MFI) at each time point from time 0. ns, not significant; data shown are representative of three independent experiments. ****p < 0.0001 (two-way ANOVA). PE, phycoerythrin.
VP22 is not sufficient for inhibiting CD1d-mediated antigen presentation postinfection
To determine if VP22 is sufficient for the inhibition of CD1d-mediated antigen presentation that was observed post-HSV infection, VP22 cDNA was stably transfected into HEK293-CD1d cells using a GFP-tagged bicistronic IRES construct (HEK293-CD1d.VP22/GFP cells). The ectopic expression of VP22 in HEK293-CD1d.VP22/GFP cells was confirmed by western blot analysis (Fig. 3A). We found that CD1d expression in both control and VP22-expressing HEK293-CD1d cells was comparable (Fig. 3B). The cells were also similar in terms of antigen presentation to NKT cells (Fig. 3C). These results suggest that the ectopic expression of VP22 is not sufficient to inhibit CD1d-mediated antigen presentation to NKT cells, as we observed following infection with WT HSV-1.
FIG. 3.

VP22 is not sufficient to inhibit antigen presentation by CD1d following an HSV-1 infection. (A) VP22/GFP- or control GFP-expressing HEK293-CD1d cells were lysed and resolved by SDS-PAGE. The expression of VP22 was detected by western blot analysis using a VP22-specific IgY antibody. The arrow points to the expected VP22 band. (B) VP22/GFP-expressing HEK293-CD1d (solid line) or GFP-expressing HEK293-CD1d cells (dotted line) were stained for intracellular CD1d HC and the surface CD1d-β2m complex as indicated, and analyzed by flow cytometry. (C) VP22-expressing HEK293-CD1d and control cells were cocultured with human NKT cells for 48 h. The supernatants were harvested for a GM-CSF ELISA. The level of GM-CSF production by the human NKT cells is shown. Data shown are representative of at least three independent experiments. HC, heavy chain.
Ectopic expression of VP22 complements the ΔUL49 virus in inhibiting CD1d-mediated antigen presentation
Since ectopic expression of VP22 did not, by itself, inhibit CD1d-mediated antigen presentation (Fig. 3C), we hypothesized that other viral proteins (in the context of the whole virion) contribute to the inhibition of antigen presentation observed postinfection. To test this hypothesis, HEK293-CD1d.VP22/GFP cells and control cells were infected with the VP22-deficient (ΔUL49) or VP22-rescued (UL49R) HSV-1. For cells infected with the ΔUL49 virus, ectopic expression of VP22 resulted in a significantly greater decrease in antigen presentation by CD1d, as compared with cells expressing a control vector (Fig. 4C). For cells infected with the UL49R virus, VP22 expression in host cells also caused a further inhibition in antigen presentation (Fig. 4C). Similar viral infectivity in these cells was confirmed by flow cytometry using an HSV-specific antibody (Fig. 4A). Furthermore, we did not observe any noticeable changes in CD1d surface expression postinfection (Fig. 4B). Therefore, these data suggest that VP22 requires coexpression of other HSV-1-encoded proteins in the inhibition of antigen presentation by CD1d postinfection.
FIG. 4.
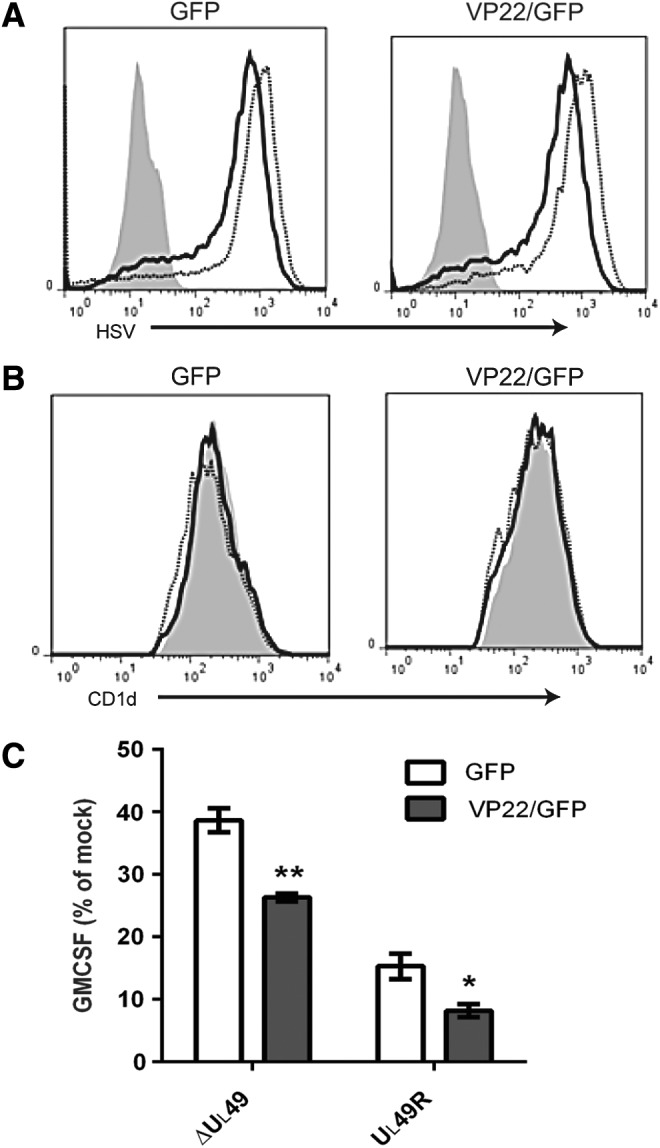
Ectopic expression of VP22 complements ΔUL49 virus in the inhibition of antigen presentation following an HSV-1 infection. (A) VP22-expressing HEK293-CD1d (VP22/GFP) and control (GFP) cells were mock infected (gray-filled histogram), or infected with ΔUL49 (solid line) or UL49R (dotted line) for 6 h at an MOI of 5. The cells were fixed, permeabilized with 0.1% saponin, stained with an anti-HSV antibody, and analyzed by flow cytometry. (B) The same mock-treated or virus-infected cells were also fixed, stained with the CD1d-specific mAb 42.1 and analyzed by flow cytometry. (C) Aliquots of cells used in (A, B) were fixed and cocultured with human NKT cells (105/well, E:T = 1:1) for 48 h. Data are shown as the percentage of GM-CSF production compared with mock (set at 100%). Representative data from three independent experiments are shown. *p < 0.05; **p < 0.01 (Student's t-test).
Viral protein synthesis is important for VP22-mediated inhibition of antigen presentation
To determine whether other viral proteins are important for the observed VP22-dependent inhibition of CD1d Ag presentation, we utilized the protein synthesis inhibitor, cycloheximide. We found that cycloheximide treatment by itself had a minimal effect on CD1d-mediated antigen presentation in HEK293-CD1d cells (data not shown). Interestingly, cycloheximide treatment in WT HSV-1-infected HEK293-CD1d.GFP cells rescued CD1d-mediated antigen presentation (Fig. 5), suggesting that viral protein synthesis inhibits CD1d Ag presentation. On the other hand, HEK293-CD1d.VP22/GFP cells had similar CD1d Ag presentation as control cells when cells were infected with HSV-1 WT virus, with or without cycloheximide treatment. When HEK293-CD1d.VP22/GFP cells were infected with the ΔUL49 HSV-1 virus, they were less able to activate NKT cells compared with control cells (Fig. 5). In contrast, when HEK293-CD1d.VP22/GFP cells were treated with cycloheximide and infected with ΔUL49, there was enhanced CD1d-mediated antigen presentation compared with control cells. Therefore, our data suggest that the synthesis of other viral proteins is also important for the VP22-mediated inhibition of antigen presentation by CD1d.
FIG. 5.
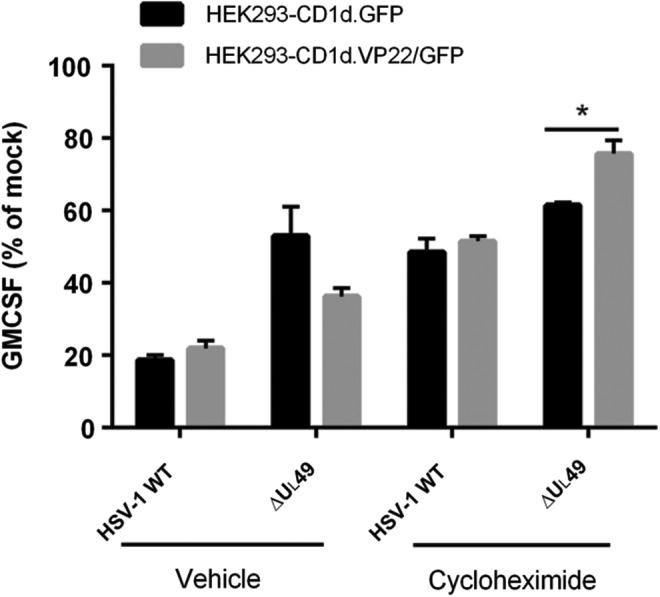
Viral protein synthesis is important for VP22-mediated inhibition of CD1d-mediated antigen presentation. HEK293-CD1d (VP22/GFP) and control (GFP) cells were mock infected or infected with ΔUL49 for 6 h at an MOI of 5, with or without cycloheximide. The cells were subsequently fixed and cocultured with human NKT cells for 48 h. Data are presented as the percentage of GM-CSF produced by NKT cells relative to mock treatment (set at 100%). Representative data from three independent experiments are shown. *p < 0.05 (Student's t-test).
Discussion
VP22 is a major tegument protein in the HSV-1 virion and has many important functions that are critical for HSV-1 replication and pathogenicity (39). In the current study, we have shown that VP22 is necessary (but not sufficient) for inhibiting CD1d-mediated antigen presentation following an HSV-1 infection. It is likely that VP22 requires the presence of other HSV-1-encoded proteins to be functional. For example, in HSV-1-infected cells, VP22 forms homotypic complexes, which then associate with the HSV-1-encoded proteins, VP16 and UL13 (29). VP22 also forms a complex with VP16, which interacts with VHS; the presence of both VP22 and VP16 is required for the accumulation of VHS (38). It is known that in the absence of VP22, the expression of other viral proteins is also impaired. For example, less expression of ICP0, VP16, VHS, glycoprotein D, and gE has been reported in VP22-null mutant viruses (11). VP22 is also required for optimal expression of the HSV-1-encoded protein, gB (39). Interestingly, gB has been shown to bind CD1d and alter CD1d-mediated antigen presentation postinfection (35). Nonetheless, gB itself is not sufficient to inhibit CD1d-mediated antigen presentation (35). A recent publication indicates that KIF3A, a subunit of kinesin-2 motor protein that transports protein complexes along microtubule tracks, is required for CD1d surface expression (43). KIF3A can be phosphorylated by the viral kinase US3 and downregulates CD1d surface expression (43). This is consistent with our discovery that VP22, a viral protein modifies host cell microtubule network, is important in inhibiting CD1d-mediated antigen presentation.
VP22 is highly phosphorylated and it can be phosphorylated by both viral and host kinases (14). Although the phosphorylation profiles of VP22 synthesized during transfection and infection are the same, ectopically expressed VP22 lacks the nonphosphorylated form of VP22, which exists in infected cells. Nonphosphorylated VP22 is the major form of VP22 in virions (14), and in infected cells it exhibits enhanced binding to microtubule bundles and efficiently interacts with VP16 (32). It is possible that the nonphosphorylated form of VP22, absent in VP22-transfected cells, is the functional form that inhibits CD1d recycling and antigen presentation. This may explain why ectopic expression of VP22 in HEK293-CD1d cells does not inhibit CD1d-mediated antigen presentation. Inhibiting viral protein synthesis using cycloheximide abolishes the inhibition of CD1d-mediated antigen presentation by ectopically expressed VP22 (Fig. 5), suggesting other viral proteins cooperate with (or assist) VP22, allowing for its effect on CD1d. Further investigations are needed to determine whether the different forms of VP22 are functionally distinct in host cells.
A VP22-deficient virus (ΔUL49) has been reported to produce a smaller plaque and there is reduced virion release in infected cells (10). One may argue that the reduced inhibition in antigen presentation observed following infection with ΔUL49 is due to its impaired infectivity. However, the ΔUL49 virus did not show a significant difference from WT HSV-1 or a VP22-rescued virus (UL49R) in single cycle growth analyses when cells were infected at a high multiplicity of infection (10). Indeed, our data also demonstrated that the ΔUL49 virus was just as infectious as WT and UL49R viruses 4 h postinfection (Fig. 1A). In addition, infection of HEK293-CD1d cells with another HSV-1 mutant virus (ΔUS3/ΔUL13), which also produces very small plaques, inhibited CD1d-mediated antigen presentation at a similar level as WT HSV-1 (25). Consistently, infection with the ΔUS3/ΔUL13 virus also inhibited CD1d recycling in HEK293-CD1d cells (data not shown). Therefore, the reduced ability of the ΔUL49 virus to inhibit CD1d-mediated antigen presentation is not due to reduced infectivity.
We and others have reported that an HSV-1 infection prevents intracellular CD1d from recycling back to the cell surface (25,45). Our study has found that VP22 is required for the inhibition of CD1d recycling postinfection. To the best of our knowledge, it had not been previously shown that VP22 is directly involved in regulating subcellular vesicular trafficking. However, VP22 has been reported to bind to microtubule bundles and myosin HC 9, the latter being a component of the actin cytoskeleton (13,32). Similarly, substitution of the CD1d residues T322 and S323 with Ala (T322A/S323A) prevents their phosphorylation and also increases the CD1d recycling rate postinfection (25). In a previous study, we speculated that viral infections may activate host Ser/Thr kinases and thereby phosphorylate the cytoplasmic tail of CD1d, causing lysosomal targeting and degradation of CD1d (25). Further studies will be important to determine how VP22 impacts these activities post-HSV-1 infection.
In summary, our current study has demonstrated that following an HSV-1 infection, VP22 is required (but not sufficient) for the inhibition of CD1d recycling and antigen presentation to NKT cells. Future studies will be focused on how VP22 may interact with other viral proteins and the host cell's cytoskeletal network and, as a result, regulate the functional expression of CD1d.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. P. Cohen (University of Dundee), P. Cresswell (Yale University), and J. Blum (Indiana University) for critical reagents, and technical assistance from the Flow Cytometry Resource Facility, Indiana University School of Medicine. This work was supported by the National Institutes of Health grants R01 AI46455 and P01 AI056097 (to R.R.B.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ahn K, Meyer TH, Uebel S, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. Embo J 1996;15:3247–3255 [PMC free article] [PubMed] [Google Scholar]
- 2.Asai R, Ohno T, Kato A, et al. Identification of proteins directly phosphorylated by UL13 protein kinase from herpes simplex virus 1. Microbes Infect 2007;9:1434–1438 [DOI] [PubMed] [Google Scholar]
- 3.Ashkar AA, and Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol 2003;77:10168–10171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey JC, Iyer AK, Renukaradhya GJ, et al. Inhibition of CD1d-mediated antigen presentation by the TGF-beta/Smad signaling pathway. Immunology 2014;143:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banovic T, Yanilla M, Simmons R, et al. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis 2011;204:1893–1901 [DOI] [PubMed] [Google Scholar]
- 6.Bosnjak L, Sahlstrom P, Paquin-Proulx D, et al. Contact-dependent interference with invariant NKT cell activation by herpes simplex virus-infected cells. J Immunol 2012;188:6216–6224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brigl M, and Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol 2004;22:817–890 [DOI] [PubMed] [Google Scholar]
- 8.Cornish AL, Keating R, Kyparissoudis K, et al. NKT cells are not critical for HSV-1 disease resolution. Immunol Cell Biol 2006;84:13–19 [DOI] [PubMed] [Google Scholar]
- 9.Dewberry EJ, Dunkerley E, and Duffy C. Purification of full-length VP22 from cells infected with HSV-1: a two-pronged approach for the solubilization and purification of viral proteins for use in biochemical studies. J virol Methods 2012;183:180–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffy C, Lavail JH, Tauscher AN, et al. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J Virol 2006;80:8664–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duffy C, Mbong EF, and Baines JD. VP22 of herpes simplex virus 1 promotes protein synthesis at late times in infection and accumulation of a subset of viral mRNAs at early times in infection. J Virol 2009;83:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott G, and O'Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997;88:223–233 [DOI] [PubMed] [Google Scholar]
- 13.Elliott G, and O'Hare P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J Virol 1998;72:6448–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott G, O'Reilly D, and O'Hare P. Phosphorylation of the herpes simplex virus type 1 tegument protein VP22. Virology 1996;226:140–145 [DOI] [PubMed] [Google Scholar]
- 15.Elliott GD, and Meredith DM. The herpes simplex virus type 1 tegument protein VP22 is encoded by gene UL49. J Gen Virol 1992;73 (Pt 3):723–726 [DOI] [PubMed] [Google Scholar]
- 16.Fruh K, Ahn K, Djaballah H, et al. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995;375:415–418 [DOI] [PubMed] [Google Scholar]
- 17.Gallo RM, Khan MA, Shi J, et al. Regulation of the actin cytoskeleton by rho kinase controls antigen presentation by CD1d. J Immunol 2012;189:1689–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godfrey DI, MacDonald HR, Kronenberg M, et al. NKT cells: what's in a name? Nat Rev Immunol 2004;4:231–237 [DOI] [PubMed] [Google Scholar]
- 19.Grubor-Bauk B, Simmons A, Mayrhofer G, et al. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant Vα14-Jα281 TCR. J Immunol 2003;170:1430–1434 [DOI] [PubMed] [Google Scholar]
- 20.Hill A, Jugovic P, York I, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995;375:411–415 [DOI] [PubMed] [Google Scholar]
- 21.Koelle DM, and Corey L. Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med 2008;59:381–395 [DOI] [PubMed] [Google Scholar]
- 22.Leib DA. Counteraction of interferon-induced antiviral responses by herpes simplex viruses. Curr Top Microbiol Immunol 2002;269:171–185 [DOI] [PubMed] [Google Scholar]
- 23.Levy O, Orange JS, Hibberd P, et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis 2003;188:948–953 [DOI] [PubMed] [Google Scholar]
- 24.Lin R, Noyce RS, Collins SE, et al. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol 2004;78:1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Glosson NL, Du W, et al. A Thr/Ser dual residue motif in the cytoplasmic tail of human CD1d is important for the down-regulation of antigen presentation following a herpes simplex virus 1 infection. Immunology 2013;140:191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Shaji D, Cho S, et al. A threonine-based targeting signal in the human CD1d cytoplasmic tail controls its functional expression. J Immunol 2010;184:4973–4981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Y, and He B. Recognition of herpes simplex viruses: toll-like receptors and beyond. J Mol Biol 2014;426:1133–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mbong EF, Woodley L, Dunkerley E, et al. Deletion of the herpes simplex virus 1 UL49 gene results in mRNA and protein translation defects that are complemented by secondary mutations in UL41. J Virol 2012;86:12351–12361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouzakitis G, McLauchlan J, Barreca C, et al. Characterization of VP22 in herpes simplex virus-infected cells. J Virol 2005;79:12185–12198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann J, Eis-Hubinger AM, and Koch N. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J Immunol 2003;171:3075–3083 [DOI] [PubMed] [Google Scholar]
- 31.Nopora K, Bernhard CA, Ried C, et al. MHC class I cross-presentation by dendritic cells counteracts viral immune evasion. Front Immunol 2012;3:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potel C, and Elliott G. Phosphorylation of the herpes simplex virus tegument protein VP22 has no effect on incorporation of VP22 into the virus but is involved in optimal expression and virion packaging of ICP0. J Virol 2005;79:14057–14068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raftery MJ, Winau F, Kaufmann SH, et al. CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol 2006;177:6207–6214 [DOI] [PubMed] [Google Scholar]
- 34.Raftery MJ, Wolter E, Fillatreau S, et al. NKT cells determine titer and subtype profile of virus-specific IgG antibodies during herpes simplex virus infection. J Immunol 2014;192:4294–4302 [DOI] [PubMed] [Google Scholar]
- 35.Rao P, Pham HT, Kulkarni A, et al. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol 2011;85:8093–8104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts TJ, Sriram V, Spence PM, et al. Recycling CD1d1 molecules present endogenous antigens processed in an endocytic compartment to NKT cells. J Immunol 2002;168:5409–5414 [DOI] [PubMed] [Google Scholar]
- 37.Sciortino MT, Taddeo B, Giuffre-Cuculletto M, et al. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J Virol 2007;81:10924–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taddeo B, Sciortino MT, Zhang W, et al. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc Natl Acad Sci U S A 2007;104:12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka M, Kato A, Satoh Y, et al. Herpes simplex virus 1 VP22 regulates translocation of multiple viral and cellular proteins and promotes neurovirulence. J Virol 2012;86:5264–5277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Temme S, Eis-Hubinger AM, McLellan AD, et al. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J Immunol 2010;184:236–243 [DOI] [PubMed] [Google Scholar]
- 41.Tomazin R, Hill AB, Jugovic P, et al. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. Embo J 1996;15:3256–3266 [PMC free article] [PubMed] [Google Scholar]
- 42.van Leeuwen H, Elliott G, and O'Hare P. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J Virol 2002;76:3471–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong R, Rao P, Kim S, et al. Herpes simplex virus 1 US3 phosphorylates cellular KIF3A to downregulate CD1d expression. J Virol 2015;89:6646–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.York IA, Roop C, Andrews DW, et al. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994;77:525–535 [DOI] [PubMed] [Google Scholar]
- 45.Yuan W, Dasgupta A, and Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol 2006;7:835–842 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.