

Abstract
Comparative protein structure modeling predicts the three-dimensional structure of a given protein sequence (target) based primarily on its alignment to one or more proteins of known structure (templates). The prediction process consists of fold assignment, target-template alignment, model building, and model evaluation. This unit describes how to calculate comparative models using the program MODELLER and how to use the ModBase database of such models, and discusses all four steps of comparative modeling, frequently observed errors, and some applications. Modeling lactate dehydrogenase from Trichomonas vaginalis (TvLDH) is described as an example. The download and installation of the MODELLER software is also described.
Keywords: comparative modeling, ModBase, MODELLER, protein fold, protein structure, structure prediction
INTRODUCTION
Functional characterization of a protein sequence is one of the most frequent problems in biology. This task is usually facilitated by an accurate three-dimensional (3-D) structure of the studied protein. In the absence of an experimentally determined structure, comparative or homology modeling often provides a useful 3-D model for a protein that is related to at least one known protein structure (Marti-Renom et al., 2000; Fiser, 2004; Misura and Baker, 2005; Petrey and Honig, 2005; Misura et al., 2006). Comparative modeling predicts the 3-D structure of a given protein sequence (target) based primarily on its alignment to one or more proteins of known structure (templates).
Comparative modeling consists of four main steps (Marti-Renom et al., 2000; Fig. 5.6.1): (i) fold assignment, which identifies similarity between the target and at least one known template structure; (ii) alignment of the target sequence and the template(s); (iii) building a model based on the alignment with the chosen template(s); and (iv) predicting model errors.
Figure 5.6.1.
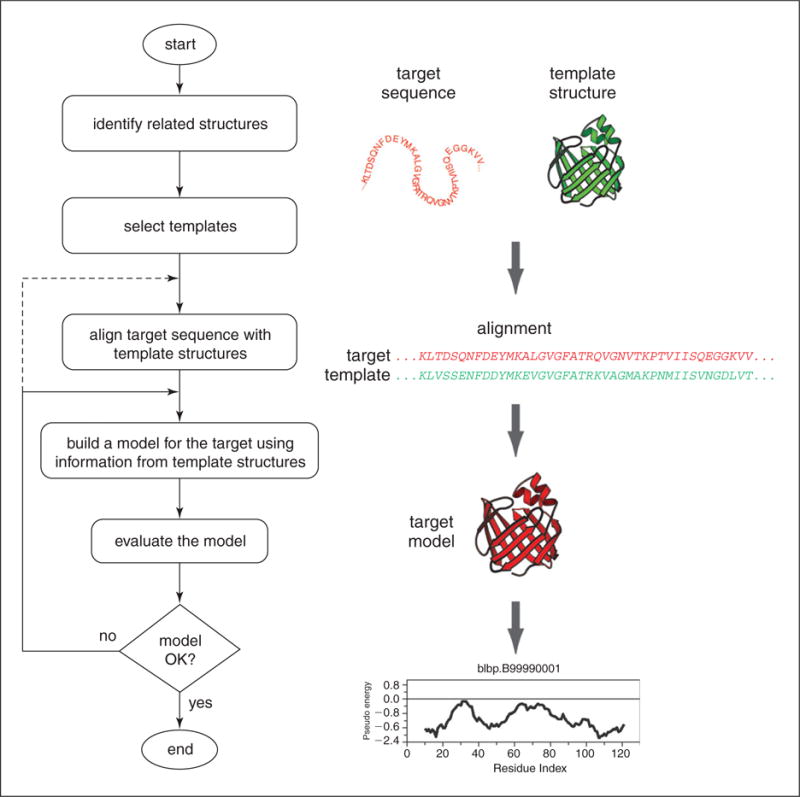
Steps in comparative protein structure modeling. See text for details.
There are several computer programs and Web servers that automate the comparative modeling process (Table 5.6.1). The accuracy of the models calculated by many of these servers is evaluated by CAMEO (Haas et al., 2013) and the biannual CASP (Critical Assessment of Techniques for Proteins Structure Prediction; Moult, 2005; Moult et al., 2009) experiment.
Table 5.6.1.
Programs and Web Servers Useful in Comparative Protein Structure Modeling
While automation makes comparative modeling accessible to both experts and nonspecialists, manual intervention is generally still needed to maximize the accuracy of the models in the difficult cases. A number of resources useful in comparative modeling are listed in Table 5.6.1.
This unit describes how to calculate comparative models using the program MODELLER (Basic Protocol). The Basic Protocol goes on to discuss all four steps of comparative modeling (Fig. 5.6.1), frequently observed errors, and the ModBase database and associated Web services. The Support Protocol describes how to download and install MODELLER.
BASIC PROTOCOL: MODELING LACTATE DEHYDROGENASE FROM TRICHOMONAS VAGINALIS (TvLDH) BASED ON A SINGLE TEMPLATE USING MODELLER
MODELLER is a computer program for comparative protein structure modeling (Sali and Blundell, 1993; Fiser et al., 2000). In the simplest case, the input is an alignment of a sequence to be modeled with the template structures, the atomic coordinates of the templates, and a simple script file. MODELLER then automatically calculates a model containing all non-hydrogen atoms, within minutes on a modern PC and with no user intervention. Apart from model building, MODELLER can perform additional auxiliary tasks, including fold assignment, alignment of two protein sequences or their profiles (Marti-Renom et al., 2004), multiple alignment of protein sequences and/or structures (Madhusudhan et al., 2006; Madhusudhan et al., 2009), calculation of phylogenetic trees, and de novo modeling of loops in protein structures (Fiser et al., 2000).
NOTE: Further help for all the described commands and parameters may be obtained from the MODELLER Web site (see Internet Resources).
Necessary Resources
Hardware
A computer running RedHat Linux (PC, Opteron, or EM64T/Xeon64 systems) or other version of Linux/Unix (x86/x86_64 Linux), Apple Mac OS X (10.6 or later), or Microsoft Windows (XP or later)
Software
The MODELLER 9.15 program, downloaded and installed from http://salilab.org/modeller/download_installation.html (see Support Protocol)
Files
All files required to complete this protocol can be downloaded from http://salilab.org/modeller/tutorial/basic-example.tar.gz (Unix/Linux) or http://salilab.org/modeller/tutorial/basic-example.zip (Windows)
Background to TvLDH
A novel gene for lactate dehydrogenase (LDH) was identified from the genomic sequence of Trichomonas vaginalis (TvLDH). The corresponding protein had higher sequence similarity to the malate dehydrogenase of the same species (TvMDH) than to any other LDH. The authors hypothesized that TvLDH arose from TvMDH by convergent evolution relatively recently (Wu et al., 1999). Comparative models were constructed for TvLDH and TvMDH to study the sequences in a structural context and to suggest site-directed mutagenesis experiments to elucidate changes in enzymatic specificity in this apparent case of convergent evolution. The native and mutated enzymes were subsequently expressed and their activities compared (Wu et al., 1999).
Searching structures related to TvLDH
Conversion of sequence to PIR file format
It is first necessary to convert the target TvLDH sequence into a format that is readable by MODELLER (file TvLDH.ali; Fig. 5.6.2). MODELLER uses the PIR format to read and write sequences and alignments. The first line of the PIR-formatted sequence consists of >P1; followed by the identifier of the sequence. In this example, the sequence is identified by the code TvLDH. The second line, consisting of ten fields separated by colons, usually contains details about the structure, if any. In the case of sequences with no structural information, only two of these fields are used: the first field should be sequence (indicating that the file contains a sequence without a known structure) and the second should contain the model file name ( TvLDH in this case). The rest of the file contains the sequence of TvLDH, with an asterisk (*) marking its end. The standard uppercase single-letter amino acid codes are used to represent the sequence.
Figure 5.6.2.

File TvLDH.ali. Sequence file in PIR format.
Searching for suitable template structures
A search for potentially related sequences of known structure can be performed using the profile.build () command of MODELLER (file build_profile.py). The command uses the local dynamic programming algorithm to identify related sequences (Smith and Waterman, 1981). In the simplest case, the command takes as input the target sequence and a database of sequences of known structure (file pdb_95.pir) and returns a set of statistically significant alignments. The input script file for the command is shown in Figure 5.6.3.
Figure 5.6.3.

File build_profile.py. Input script file that searches for templates against a database of nonredundant PDB sequences.
The script, build_profile.py, does the following:
Initializes the “environment” for this modeling run by creating a new environ object (called env here). Almost all MODELLER scripts require this step, as the new object is needed to build most other useful objects.
Creates a new sequence_db object, calling it sdb, which is used to contain large databases of protein sequences.
Reads a file, in text format, containing nonredundant PDB sequences, into the sdb database. The sequences can be found in the file pdb_95.pir. This file is also in the PIR format. Each sequence in this file is representative of a group of PDB sequences that share 95% or more sequence identity to each other and have less than 30 residues or 30% sequence length difference.
Writes a binary machine-independent file containing all sequences read in the previous step.
Reads the binary format file back in for faster execution.
Creates a new “alignment” object ( aln), reads the target sequence TvLDH from the file TvLDH.ali, and converts it to a profile object ( prf). Profiles contain similar information to alignments, but are more compact and better for sequence database searching.
prf.build() searches the sequence database ( sdb) with the target profile ( prf). Matches from the sequence database are added to the profile.
prf.write() writes a new profile containing the target sequence and its homologs into the specified output file (file build_profile.prf; Fig. 5.6.4). The equivalent information is also written out in standard alignment format.
Figure 5.6.4.
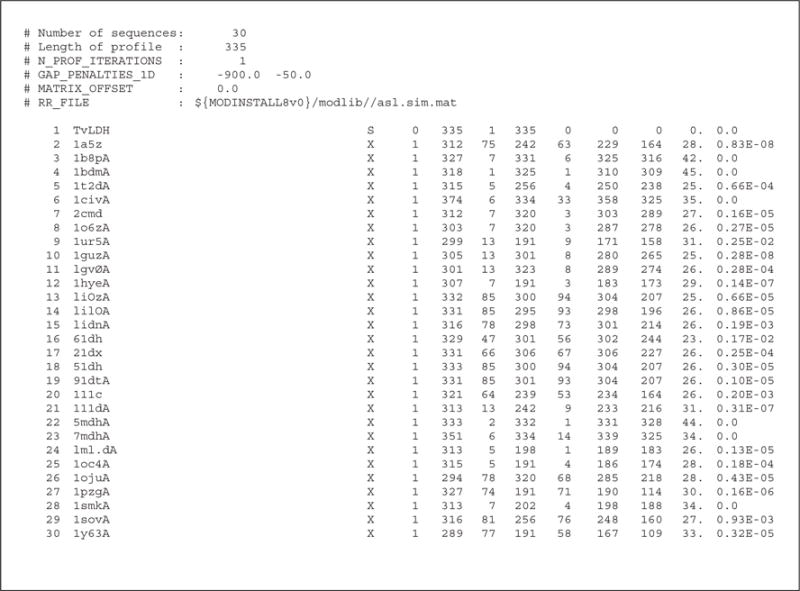
An excerpt from the file build_profile.prf. The aligned sequences have been removed for clarity.
The profile.build() command has many options (see Internet Resources for MODELLER Web site). In this example, rr_file is set to use the BLOSUM62 similarity matrix (file blosum62.sim.mat provided in the MODELLER distribution). Accordingly, the parameters matrix_offset and gap_penalties_1d are set to the appropriate values for the BLOSUM62 matrix. For this example, only one search iteration is run, by setting the parameter n_prof_iterations equal to 1. Thus, there is no need to check the profile for deviation ( check_profile set to False). Finally, the parameter max_aln_evalue is set to 0.01, indicating that only sequences with E-values smaller than or equal to 0.01 will be included in the output.
Execute the script using the command
-
python build_profile.py > build_profile.log
(or, if Python is not installed on the machine, with mod9.15 build_profile.py). At the end of the execution, a log file is created ( build_profile.log). MODELLER always produces a log file. Errors and warnings in log files can be found by searching for the _E> and _W> strings, respectively.
Selecting a template
An extract (omitting the aligned sequences) from the file build_profile.prf is shown in Figure 5.6.4. The first six commented lines indicate the input parameters used in MODELLER to create the alignments. Subsequent lines correspond to the detected similarities by profile.build(). The most important columns in the output are the second, tenth, eleventh, and twelfth columns. The second column reports the code of the PDB sequence that was aligned to the target sequence. The eleventh column reports the percentage sequence identities between TvLDH and the PDB sequence normalized by the length of the alignment (indicated in the tenth column). In general, a sequence identity value above ~25% indicates a potential template, unless the alignment is too short (i.e., <100 residues). A better measure of the significance of the alignment is given in the twelfth column by the E-value of the alignment (lower the E-value the better).
In this example, six PDB sequences show very significant similarities to the query sequence, with E-values equal to 0. As expected, all the hits correspond to malate dehydrogenases (1bdm:A, 5mdh:A, 1b8p:A, 1civ:A, 7mdh:A, and 1smk:A). To select the appropriate template for the target sequence, the alignment.compare_structures() command will first be used to assess the sequence and structure similarity between the six possible templates (file compare.py; Fig. 5.6.5).
Figure 5.6.5.

Script file compare.py.
In compare.py, the alignment object aln is created and MODELLER is instructed to read into it the protein sequences and information about their PDB files. The command malign () calculates their multiple sequence alignment, which is subsequently used as a starting point for creating a multiple structure alignment by malign3d (). Based on this structural alignment, the compare_structures() command calculates the RMS and DRMS deviations between atomic positions and distances, differences between the main-chain and side-chain dihedral angles, percentage sequence identities, and several other measures. Finally, the id_table () command writes a file ( family.mat) with pairwise sequence distances that can be used as input to the dendrogram () command (or the clustering programs in the PHYLIP package; Felsenstein, 1989). dendrogram () calculates a clustering tree from the input matrix of pairwise distances, which helps visualizing differences among the template candidates. Excerpts from the log file ( compare.log) are shown in Figure 5.6.6.
Figure 5.6.6.
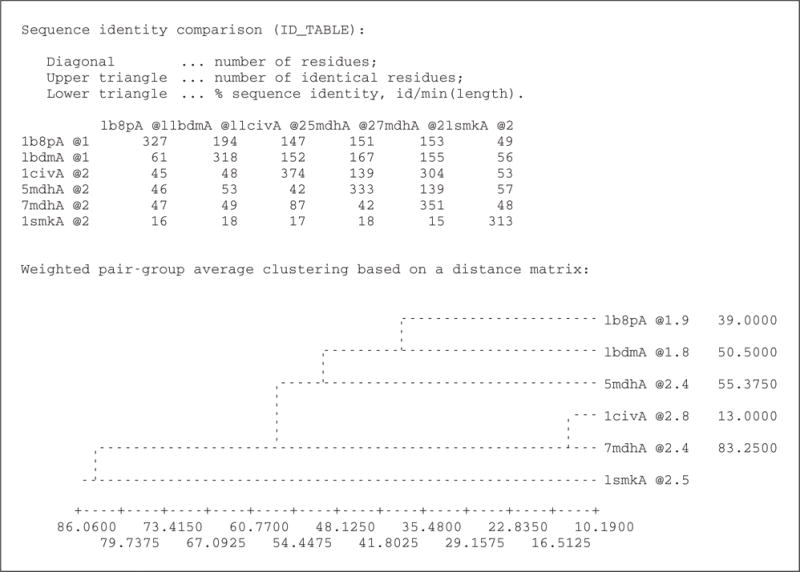
Excerpts from the log file compare.log.
The objective of this step is to select the most appropriate single template structure from all the possible templates. The dendrogram in Figure 5.6.6 shows that 1civ:A and 7mdh:A are almost identical, both in terms of sequence and structure. However, 7mdh:A has a better crystallographic resolution than 1civ:A (2.4 Å versus 2.8 Å). From the second group of similar structures (5mdh:A, 1bdm:A, and 1b8p:A), 1bdm:A has the best resolution (1.8 Å). 1smk:A is most structurally divergent among the possible templates. However, it is also the one with the lowest sequence identity (34%) to the target sequence ( build_profile.prf). 1bdm:A is finally picked over 7mdh:A as the final template because of its higher overall sequence identity to the target sequence (45%).
Aligning TvLDH with the template
One way to align the sequence of TvLDH with the structure of 1bdm:A is to use the align2d () command in MODELLER (Madhusudhan et al., 2006). Although align2d () is based on a dynamic programming algorithm (Needleman and Wunsch, 1970), it is different from standard sequence-sequence alignment methods because it takes into account structural information from the template when constructing an alignment. This task is achieved through a variable gap penalty function that tends to place gaps in solvent-exposed and curved regions, outside secondary structure segments, and between two positions that are close in space. In the current example, the target-template similarity is so high that almost any alignment method with reasonable parameters will result in the same alignment.
The MODELLER script shown in Figure 5.6.7 aligns the TvLDH sequence in file TvLDH.ali with the 1bdm:A structure in the PDB file 1bdm.pdb ( file align2d.py). In the first line of the script, an empty alignment object aln, and a new model object mdl, into which chain A of the 1bmd structure is read, are created. append_model() transfers the PDB sequence of this model to aln and assigns it the name of 1bdmA ( align_codes). The TvLDH sequence, from file TvLDH.ali, is then added to aln using append (). The align2d () command aligns the two sequences and the alignment is written out in two formats, PIR ( TvLDH-1bdmA.ali) and PAP ( TvLDH-1bdmA.pap). The PIR format is used by MODELLER in the subsequent model-building stage, while the PAP alignment format is easier to inspect visually. In the PAP format, all identical positions are marked with a * (file TvLDH-1bdmA.pap; Fig. 5.6.8). Due to the high target-template similarity, there are only a few gaps in the alignment.
Figure 5.6.7.

The script file align2d.py, used to align the target sequence against the template structure.
Figure 5.6.8.

The alignment between sequences TvLDH and 1bdmA, in the MODELLER PAP format. File TvLDH-1bmdA.pap.
Model building
Once a target-template alignment is constructed, MODELLER calculates a 3-D model of the target completely automatically, using its automodel class. The script in Figure 5.6.9 will generate five different models of TvLDH based on the 1bdm:A template structure and the alignment in file TvLDH-1bdmA.ali (file model-single.py).
Figure 5.6.9.

Script file, model-single.py, that generates five models.
The first line (Fig. 5.6.9) loads the automodel class and prepares it for use. An automodel object is then created and called “a” and parameters are set to guide the model-building procedure. alnfile names the file that contains the target-template alignment in the PIR format. knowns defines the known template structure(s) in alnfile ( TvLDH-1bdmA.ali) and sequence defines the code of the target sequence. starting_model and ending_model define the number of models that are calculated (their indices will run from 1 to 5). The last line in the file calls the make method that actually calculates the models. The most important output files are model-single.log, which reports warnings, errors, and other useful information including the input restraints used for modeling that remain violated in the final model, and TvLDH.B9999000[1–5].pdb, which contain the coordinates of the five produced models, in the PDB format. The models can be viewed by any program that reads the PDB format, such as Chimera (http://www.cgl.ucsf.edu/chimera/) or RasMol (http://www.rasmol.org).
Evaluating a model
If several models are calculated for the same target, the best model can be selected by picking the model with the lowest value of the MODELLER objective function or the DOPE (Shen and Sali, 2006) or SOAP (Dong et al., 2013) assessment scores, which are reported at the end of the log file. (To calculate the SOAP score, download the SOAP-Protein library file from http://salilab.org/SOAP/ and uncomment the two SOAP-related lines in model-single.py by removing the ‘#’ characters.) In this example, the second model ( TvLDH.B99990002.pdb) has the lowest objective function and is selected. None of these scores are absolute measures, in the sense that they can only be used to rank models calculated from the same alignment.
Once a final model is selected, there are many ways to further assess it. In this example, the DOPE potential in MODELLER is used to evaluate the fold of the selected model. Links to other programs for model assessment can be found in Table 5.6.1. However, before any external evaluation of the model, one should check the log file from the modeling run for runtime errors ( model-single.log) and restraint violations (see the MODELLER manual for details).
The script, evaluate_model.py (Fig. 5.6.10) evaluates the model with the DOPE potential. In this script, the atomic coordinates of the PDB file are read in (using complete_pdb ()) to a model object, mdl. This is necessary for MODELLER to correctly calculate the energy, and additionally allows for the possibility of the PDB file having atoms in a nonstandard order, or having different subsets of atoms (e.g., all atoms including hydrogens, while MODELLER uses only heavy atoms, or vice versa). The DOPE energy is then calculated using assess_dope(). An energy profile is additionally requested, smoothed over a 15-residue window, and normalized by the number of restraints acting on each residue. This profile is written to a file TvLDH.profile, which can be used as input to a graphing program such as GNUPLOT.
Figure 5.6.10.
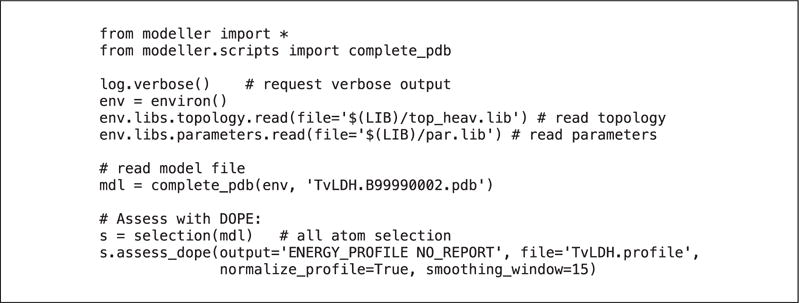
File evaluate_model.py, used to generate a pseudo-energy profile for a single model.
Similarly, the profile can be calculated for the template structure (see the scripts evaluate_template.py and plot_profiles.py in the zipfile). A comparison of the two profiles is shown in Figure 5.6.11. It can be seen that the DOPE score profile shows clear differences between the two profiles for the long active-site loop between residues 90 and 100 and the long helices at the C-terminal end of the target sequence. This long loop interacts with region 220 to 250, which forms the other half of the active site. This latter region is well resolved in both the template and the target structure. However, probably due to the unfavorable nonbonded interactions with the 90 to 100 region, it is reported to be of high energy by DOPE. It is to be noted that a region of high energy indicated by DOPE may not always necessarily indicate actual error, especially when it highlights an active site or a protein-protein interface. However, in this case, the same active-site loops have a better profile in the template structure, which strengthens the argument that the model is probably incorrect in the active-site region. Resolution of such problems is beyond the scope of this unit, but is described in a more advanced modeling tutorial available at http://salilab.org/modeller/tutorial/advanced.html.
Figure 5.6.11.
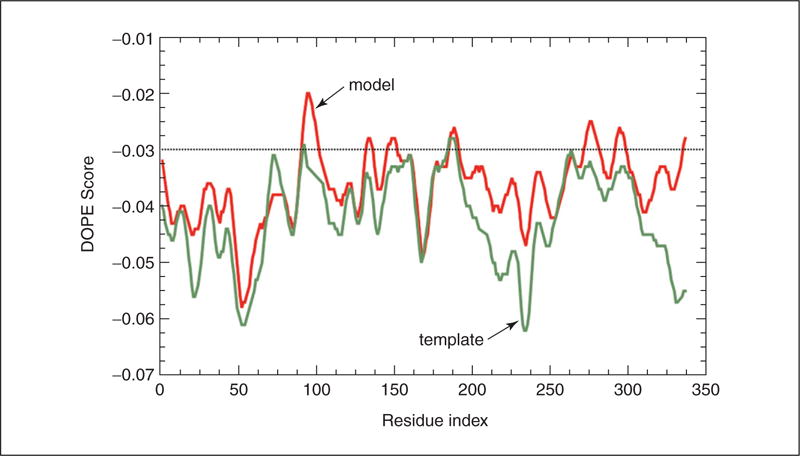
A comparison of the pseudo-energy profiles of the model (red) and the template (green) structures.
Searching for existing models in the ModBase database
ModBase (http://salilab.org/modbase/; Pieper et al., 2014) is our database of annotated comparative protein structure models. These models are constructed using ModPipe (Eswar et al., 2003), a pipeline that automates the entire process of template selection, alignment, model building, and evaluation described earlier. In addition to the basic sequence-sequence template search employed above, it conducts a more thorough sequence-profile and profile-profile search, leveraging PSI-BLAST (Altschul et al., 1997), HHBlits (Remmert et al., 2012), HHSearch (Soding, 2005), and Modeller’s own functionality. Alignments created by any of these methods can cover the complete target sequence, or only a segment of it, depending on the availability of suitable PDB templates.
Models in ModBase are organized in datasets. Because of the rapid growth of public sequence databases, efforts are concentrated on adding datasets that are useful for specific projects, rather than attempting to model all known protein sequences based on all detectably related known structures. Currently, ModBase includes a model dataset for each of 65 complete genomes, as well as datasets for all sequences in the Structure Function Linkage Database (SFLD; Pegg et al., 2006), and for the complete SwissProt/TrEMBL database as of 2005. As of 2015, ModBase contains almost 35 million reliable models for domains in 5.8 million unique protein sequences. Thus, for a sequence of interest, it is possible that models already exist in this database.
The ModBase database can be searched in many ways, e.g., by amino acid sequence, annotation keywords, the template used for modeling, accession number (such as from UniProt; Bairoch et al., 2005; Benson et al., 2013), gene name, or organism. It is also accessible from the Protein Model Portal (http://proteinmodelportal.org; Arnold et al., 2009; Haas et al., 2013) and is cross-linked to many other databases, such as UniProt.
ModBase can be searched for the TvLDH sequence, which was modeled above (Fig. 5.6.2), from the main ModBase search page (http://salilab.org/modbase/), by selecting “Sequence Similarity (Blast)” from the “Search type” drop-down menu, selecting the “100 % Sequence identity” button, and then pasting the raw TvLDH sequence (without the FASTA header) into the search box. On pressing the Search button, the ModBase Sequence Overview page is obtained (Fig. 5.6.12). On clicking the coverage sketch (the blue bar on the left side of that page), the Model Details page is displayed (Fig. 5.6.13).
Figure 5.6.12.

Excerpt of ModBase Sequence Overview page for TvLDH. For this sequence, coverage is shown (the fraction of the sequence for which a model is available, and its quality) together with any annotations available. In this case, the entire sequence was modeled with a good quality (>=30% sequence identity) template.
Figure 5.6.13.

Excerpt of ModBase Model Details page for TvLDH. Metadata about the model are shown on the left side of the page; these data include the part of the sequence that was modeled, the template used, the date when the modeling was performed, and a set of assessment scores. The actual 3-D models are shown on the right side of the page. The “Perform action on this model” menu allows for the models themselves or modeling alignments to be downloaded.
At the time of this writing, ModBase contains two models for the exact TvLDH sequence used here. These models can be selected by clicking on the small protein images on the right side of the Model Details page. The models are similar, differing only in the template used; one model uses the same template (1bdmA) that was chosen for the Modeller run, and the other uses 5mdhA. A key feature of ModPipe is that the validity of sequence–structure relationships is not pre-judged at the fold-assignment stage; instead, sequence-structure matches are assessed after the construction of the models and their evaluation. This approach enables a thorough exploration of fold assignments, sequence–structure alignments, and conformations, with the aim of finding the model with the best evaluation score at the expense of increasing the computational time significantly; for some sequences, a few thousand models can be calculated. In this case, the model built using 5mdhA actually has slightly better assessment scores than that using 1bdmA, even though 1bdmA appeared earlier to be a better-quality template.
The Model Details page also displays basic information about each model, such as the template used, the portion of the sequence that was aligned, the date it was created, and a variety of assessment scores. These scores include a normalized (z-score) version of DOPE as above; the GA341 score (Melo and Sali, 2007); ModPipe’s own quality score (MPQS; Pieper et al., 2011), which is a linear combination of DOPE, GA341, and other scores; and a prediction from TSVMod (Eramian et al., 2008) of the Cα root-mean-squared deviation (RMSD) and native overlap (the fraction of Cα atoms within 3.5 Å of their native positions). Finally the “Perform action on this model” drop-down menu allows the alignment used in modeling and the models to be downloaded.
Adding new models to ModBase
If a sequence does not yet have a model in ModBase, the ModWeb (http://salilab.org/modweb/; Eswar et al., 2003) Web server can be used to model it. ModWeb is a front end to ModPipe and is simple to use; a user needs to provide only an amino acid sequence to model. The entire ModPipe pipeline then runs automatically, and any models generated are uploaded into ModBase where they can be viewed or downloaded in the same way as any other ModBase model. By default, such models are added to the public datasets so that other users of ModBase can see them too; alternatively, the model dataset can be made private, or the models can be e-mailed to the user rather than uploaded into ModBase.
If a sequence already has models in ModBase, but they were generated some time ago, the Model Details page allows the user to request an update. This action rebuilds the models, potentially using any newer templates that have been deposited in the PDB since the last calculation. For example, for TvLDH, new structures (4UUM and 4UUN) that are almost 100% identical in sequence were recently deposited (in Aug 2015), and would almost certainly yield better models.
Other Web tools for model evaluation, validation, refinement, and analysis
ModWeb is one of the Web services associated with the ModBase database. A number of other such Web services exist. These services generally take as input one or more PDB files, so they can be used with models extracted from ModBase, atomic structures from the PDB itself, or models manually generated with MODELLER or another Web service. A selection of these servers is outlined here.
The ModEval server (http://salilab.org/modeval/; Pieper et al., 2011) takes as input a protein structure, an alignment in the PIR format, and the sequence–template sequence identity. The modeling alignment and sequence identity are optional, but should be provided if available as they result in more accurate assessment scores. In the TvLDH case above, the modeling alignment is available in Fig. 5.6.8 and the sequence identity can be read from the header of each model PDB file. The server then computes the TSVMod scores, the DOPE score and profile, and the GA341 score.
ModLoop (http://salilab.org/modloop/; Fiser and Sali, 2003b) takes as input a protein structure and one or more residue ranges. It then applies MODELLER’s loop modeling protocol to the selected residues to generate a set of candidate “loop” models, and returns the single model with the best-scoring loop conformation. The server can be particularly helpful for regions of the structure that have no templates. For example, in the TvLDH model, residues 94 to 102 do not align with the template (Fig. 5.6.8), and while MODELLER generates a stereochemically reasonable structure in this region, its conformation is unlikely to be close to native. The MODELLER model generated earlier ( TvLDH.B99990002.pdb) can thus be uploaded to ModLoop and 94::102:: given as the loop segments, to generate loop models. The resulting models can then be evaluated with the same Python scripts that were used to evaluate the MODELLER models, or with the ModEval server.
The AllosMod Web server (http://salilab.org/allosmod/; Weinkam et al., 2012) predicts conformational differences that may occur in the native ensemble in solution, such as those representing allosteric conformational transitions. The input is one or more macromolecular coordinate files (including DNA, RNA, and sugar molecules) and the corresponding sequence(s). The output is a set of molecular dynamics trajectories based on a simplified energy landscape. Biased energy landscapes result in efficient molecular dynamics sampling at constant temperatures, thereby providing a more ergodic sampling of the conformational space than standard molecular dynamics simulations.
FoXS (http://salilab.org/foxs/; Schneidman-Duhovny et al., 2010) calculates a Small Angle X-ray Scattering (SAXS) profile for an uploaded protein structure and compares it with an experimental profile. SAXS is a common structural characterization technique that is performed with the protein sample in solution, and usually takes only a few seconds on a well-equipped synchrotron beamline (Hura et al., 2009). Models generated with MODELLER can thus be evaluated with FoXS if a SAXS profile is available, or even used in modeling a flexible or multi-modular protein or assembling a macromolecular complex from its subunits.
For a full list of other Web services, see http://salilab.org.
SUPPORT PROTOCOL: OBTAINING AND INSTALLING MODELLER
MODELLER is written in Fortran 90 and uses Python for its control language. All input scripts to MODELLER are, hence, Python scripts. While knowledge of Python is not necessary to run MODELLER, it can be useful in performing more advanced tasks. Pre-compiled binaries for MODELLER can be downloaded from http://salilab.org/modeller.
Necessary Resources
Hardware
A computer running RedHat Linux (PC, Opteron, or EM64T/Xeon64 systems) or other version of Linux/Unix (x86/x86_64 Linux, AIX), Apple Mac OS X (10.6 or later), or Microsoft Windows (XP or later)
Software
An up-to-date Internet browser, such as Internet Explorer (http://www.microsoft.com/ie); Chrome (https://www.google.com/chrome/); Firefox (http://www.mozilla.org/firefox); or Safari (http://www.apple.com/safari)
Installation
The steps involved in installing MODELLER on a computer depend on its operating system. The following procedure describes the steps for installing MODELLER on a generic x86 PC running any Unix/Linux operating system. The procedures for other operating systems differ slightly. Detailed instructions for installing MODELLER on machines running other operating systems can be found at http://salilab.org/modeller/release.html. In particular, installer packages are available for Windows, Mac, RedHat Linux, and Debian/Ubuntu Linux operating systems, and also for the Homebrew and Anaconda Python environments.
Point browser to http://salilab.org/modeller/download_installation.html.
On the page that appears, download the distribution by clicking on the link entitled “Other Linux/Unix” under “Available downloads… ”.
A valid license key, distributed free of cost to academic users, is required to use MODELLER. To obtain a key, go to the URL http://salilab.org/modeller/registration.html, fill in the simple form at the bottom of the page, and read and accept the license agreement. The key will be e-mailed to the address provided.
Open a terminal or console and change to the directory containing the downloaded distribution. The distributed file is a compressed archive file called modeller-9.15.tar.gz.
- Unpack the downloaded file with the following commands:
- gunzip modeller-9.15.tar.gz
- tar -xvf modeller-9.15.tar
- The files needed for the installation can be found in a newly created directory called modeller-9.15. Move into that directory and start the installation with the following commands:
- cd modeller-9.15
- ./Install
- The installation script will prompt the user with several questions and suggest default answers. To accept the default answers, press the Enter key. The various prompts are briefly discussed below:
-
For the prompt below, choose the appropriate combination of the machine architecture and operating system. For this example, choose the default answer by pressing the Enter key.The currently supported architectures are as follows:
- Linux x86 PC (e.g., RedHat, SuSe).
- IBM AIX OS.
- x86_64 (Opteron/EM64T) box (Linux).
- Alternative Linux x86 PC binary (e.g., for FreeBSD).
Select the type of your computer from the list above [1]: - For the prompt below, tell the installer where to install the MODELLER executables. The default choice will place it in the directory indicated, but any directory to which the user has write permissions may be specified.
- Full directory name for the installed MODELLER9.15 [<YOUR-HOME-DIRECTORY>/bin/modeller9.15]:
- For the prompt below, enter the MODELLER license key obtained in step 3.
- KEY_MODELLER9v15, obtained from our academic license server at http://salilab.org/modeller/registration.html:
-
The installer will now confirm the answers to the above prompts. Press Enter to begin the installation. The mod9.15 script installed in the chosen directory can now be used to invoke MODELLER. The installer will also provide information on how to set up MODELLER to work with your operating system’s copy of Python.
Other resources
-
9The MODELLER Web site provides links to several additional resources that can supplement the tutorial provided in this unit, as follows.
- News about the latest MODELLER releases can be found at http://salilab.org/modeller/news.html.
- There is a discussion forum, operated through a mailing list, devoted to providing tips, tricks, and practical help in using MODELLER. Users can subscribe to the mailing list at http://salilab.org/modeller/discussion_forum.html. Users can also browse through or search the archived messages of the mailing list.
- The documentation section of the Web page contains links to Frequently Asked Questions (FAQ; http://salilab.org/modeller/FAQ.html), tutorial examples (http://salilab.org/modeller/tutorial), an online version of the manual (http://salilab.org/modeller/manual), and user-editable Wiki pages (http://salilab.org/modeller/wiki/) to exchange tips, scripts, and examples.
COMMENTARY
Background Information
As stated earlier, comparative modeling consists of four main steps: fold assignment, target-template alignment, model building, and model evaluation (Marti-Renom et al., 2000; Fig. 5.6.1).
Fold assignment and target-template alignment
Although fold assignment and sequence-structure alignment are logically two distinct steps in the process of comparative modeling, in practice, almost all fold-assignment methods also provide sequence-structure alignments. In the past, fold-assignment methods were optimized for better sensitivity in detecting remotely related homologs, often at the cost of alignment accuracy. However, recent methods simultaneously optimize both the sensitivity and alignment accuracy. Therefore, in the following discussion, fold assignment and sequence-structure alignment will be treated as a single procedure, explaining the differences as needed.
Fold assignment
The primary requirement for comparative modeling is the identification of one or more known template structures with detectable similarity to the target sequence. The identification of suitable templates is achieved by scanning structure databases, such as PDB (Berman et al., 2000), SCOP (Andreeva et al., 2004), DALI [UNIT 5.5 (Holm et al., 2006); also see Dietmann et al. (2001)], and CATH (Pearl et al., 2005), with the target sequence as the query. The detected similarity is usually quantified in terms of sequence identity or statistical measures such as E-value or z-score, depending on the method used.
Three regimes of the sequence-structure relationship
The sequence-structure relationship can be subdivided into three different regimes in the sequence similarity spectrum: (i) the easily detected relationships, characterized by >30% sequence identity; (ii) the “twilight zone” (Rost, 1999), corresponding to relationships with statistically significant sequence similarity, with identities in the 10% to 30% range; and (iii) the “midnight zone” (Rost, 1999), corresponding to statistically insignificant sequence similarity.
Pairwise sequence alignment methods
For closely related protein sequences with identities higher than 30% to 40%, the alignments produced by all methods are almost always largely correct. The quickest way to search for suitable templates in this regime is to use simple pairwise sequence alignment methods such as SSEARCH (Pearson, 1994), BLAST (Altschul et al., 1997), and FASTA (Pearson, 1994). Brenner et al. (1998) showed that these methods detect only ~18% of the homologous pairs at less than 40% sequence identity, while they identify more than 90% of the relationships when sequence identity is between 30% and 40% (Brenner et al., 1998). Another benchmark, based on 200 reference structural alignments with 0% to 40% sequence identity, indicated that BLAST is able to correctly align only 26% of the residue positions (Sauder et al., 2000).
Profile-sequence alignment methods
The sensitivity of the search and accuracy of the alignment become progressively difficult as the relationships move into the twilight zone (Saqi et al., 1998; Rost, 1999). A significant improvement in this area was the introduction of profile methods by (Gribskov et al., 1987). The profile of a sequence is derived from a multiple sequence alignment and specifies residue-type occurrences for each alignment position. The information in a multiple sequence alignment is most often encoded as either a position-specific scoring matrix (PSSM; Henikoff and Henikoff, 1994; Altschul et al., 1997) or as a Hidden Markov Model (HMM; Krogh et al., 1994; Eddy, 1998). In order to identify suitable templates for comparative modeling, the profile of the target sequence is used to search against a database of template sequences. The profile-sequence methods are more sensitive in detecting related structures in the twilight zone than the pairwise sequence-based methods; they detect approximately twice the number of homologs under 40% sequence identity (Park et al., 1998; Lindahl and Elofsson, 2000; Sauder et al., 2000). The resulting profile-sequence alignments correctly align approximately 43% to 48% of residues in the 0% to 40% sequence identity range (Sauder et al., 2000; Marti-Renom et al., 2004); this number is almost twice as large as that of the pairwise sequence methods. Frequently used programs for profile-sequence alignment are PSI-BLAST (Altschul et al., 1997), SAM (Karplus et al., 1998), HMMER (Eddy, 1998), HHsearch (Soding, 2005), HHBlits (Remmert et al., 2012), and BUILD_PROFILE (part of MODELLER; Sali and Blundell, 1993).
Profile-profile alignment methods
As a natural extension, the profile-sequence alignment methods have led to profile-profile alignment methods that search for suitable template structures by scanning the profile of the target sequence against a database of template profiles as opposed to a database of template sequences. These methods have proven to include the most sensitive and accurate fold assignment and alignment protocols to date (Edgar and Sjolander, 2004; Marti-Renom et al., 2004; Ohlson et al., 2004; Wang and Dunbrack, 2004). Profile-profile methods detect ~28% more relationships at the superfamily level and improve the alignment accuracy for 15% to 20%, compared to profile-sequence methods (Marti-Renom et al., 2004; Zhou and Zhou, 2005). There are a number of variants of profile-profile alignment methods that differ in the scoring functions they use (Pietrokovski, 1996; Rychlewski et al., 1998; Yona and Levitt, 2002; Panchenko, 2003; Sadreyev and Grishin, 2003; von Ohsen et al., 2003; Edgar, 2004; Marti-Renom et al., 2004; Zhou and Zhou, 2005). However, several analyses have shown that the overall performances of these methods are comparable (Edgar and Sjolander, 2004; Marti-Renom et al., 2004; Ohlson et al., 2004; Wang and Dunbrack, 2004). Some of the programs that can be used to detect suitable templates are FFAS (Jaroszewski et al., 2005), SP3 (Zhou and Zhou, 2005), SALIGN (Marti-Renom et al., 2004), HHBlits (Remmert et al., 2012), HHsearch (Soding, 2005), and PPSCAN, part of MODELLER (Sali and Blundell, 1993).
Sequence-structure threading methods
As the sequence identity drops below the threshold of the twilight zone, there is usually insufficient signal in the sequences or their profiles for the sequence-based methods discussed above to detect true relationships (Lindahl and Elofsson, 2000). Sequence-structure threading methods are most useful in this regime, as they can sometimes recognize common folds even in the absence of any statistically significant sequence similarity (Godzik, 2003). These methods achieve higher sensitivity by using structural information derived from the templates. The accuracy of a sequence-structure match is assessed by the score of a corresponding coarse model and not by sequence similarity, as in sequence-comparison methods (Godzik, 2003). The scoring scheme used to evaluate the accuracy is either based on residue substitution tables dependent on structural features such as solvent exposure, secondary structure type, and hydrogen-bonding properties (Shi et al., 2001; Karchin et al., 2003; McGuffin and Jones, 2003; Zhou and Zhou, 2005) or on statistical potentials for residue interactions implied by the alignment (Sippl, 1990; Bowie et al., 1991; Sippl, 1995; Skolnick and Kihara, 2001; Xu et al., 2003). The use of structural data does not have to be restricted to the structure side of the aligned sequence-structure pair. For example, SAM-T08 makes use of the predicted local structure for the target sequence to enhance homolog detection and alignment accuracy (Karplus et al., 2003). Commonly used threading programs are GenTHREADER (Jones, 1999; McGuffin and Jones, 2003), 3D-PSSM (Kelley et al., 2000), FUGUE (Shi et al., 2001), SP3 (Zhou and Zhou, 2005), SAM-T08 multi-track HMM (Karchin et al., 2003; Karplus et al., 2003), and MUSTER (Wu and Zhang, 2008).
Iterative sequence-structure alignment and model building
Yet another strategy is to optimize the alignment by iterating over the process of calculating alignments, building models, and evaluating models. Such a protocol can sample alignments that are not statistically significant and identify the alignment that yields the best model. Although this procedure can be time consuming, it can significantly improve the accuracy of the resulting comparative models in difficult cases (John and Sali, 2003).
Importance of an accurate alignment
Regardless of the method used, searching in the twilight and midnight zones of the sequence-structure relationship often results in false negatives, false positives, or alignments that contain an increasingly large number of gaps and alignment errors. Improving the performance and accuracy of methods in this regime remains one of the main tasks of comparative modeling today (Moult, 2005). It is imperative to calculate an accurate alignment between the target-template pair, as comparative modeling can almost never recover from an alignment error (Sanchez and Sali, 1997a).
Template selection
After a list of all related protein structures and their alignments with the target sequence have been obtained, template structures are prioritized depending on the purpose of the comparative model. Template structures may be chosen based purely on the target-template sequence identity, or on a combination of several other criteria, such as experimental accuracy of the structures (resolution of X-ray structures, number of restraints per residue for NMR structures), conservation of active-site residues, holo-structures that have bound ligands of interest, and prior biological information that pertains to the solvent, pH, and quaternary contacts. It is not necessary to select only one template. In fact, the use of several templates approximately equidistant from the target sequence generally increases the model accuracy (Srinivasan and Blundell, 1993; Sanchez and Sali, 1997b).
Model building
Modeling by assembly of rigid bodies
The first and still widely used approach in comparative modeling is to assemble a model from a small number of rigid bodies obtained from the aligned protein structures (Browne et al., 1969; Greer, 1981; Blundell et al., 1987). The approach is based on the natural dissection of the protein structures into conserved core regions, variable loops that connect them, and side chains that decorate the backbone. For example, the following semiautomated procedure is implemented in the computer program COMPOSER (Sutcliffe et al., 1987). First, the template structures are selected and superposed. Second, the “framework” is calculated by averaging the coordinates of the Cα atoms of structurally conserved regions in the template structures. Third, the main-chain atoms of each core region in the target model are obtained by superposing the core segment, from the template whose sequence is closest to the target, on the framework. Fourth, the loops are generated by scanning a database of all known protein structures to identify the structurally variable regions that fit the anchor core regions and have a compatible sequence (Topham et al., 1993). Fifth, the side chains are modeled based on their intrinsic conformational preferences and on the conformation of the equivalent side chains in the template structures (Sutcliffe et al., 1987). Finally, the stereochemistry of the model is improved either by a restrained energy minimization or a molecular dynamics refinement. The accuracy of a model can be somewhat increased when more than one template structure is used to construct the framework and when the templates are averaged into the framework using weights corresponding to their sequence similarities to the target sequence (Srinivasan and Blundell, 1993). Possible future improvements of modeling by rigid-body assembly include incorporation of rigid body shifts, such as the relative shifts in the packing of a helices and β-sheets (Nagarajaram et al., 1999). Three other programs that implement this method are 3D-JIGSAW (Bates et al., 2001), RosettaCM (Song et al., 2013), and SWISS-MODEL (Schwede et al., 2003).
Modeling by segment matching or coordinate reconstruction
The basis of modeling by coordinate reconstruction is the finding that most hexapeptide segments of protein structure can be clustered into only 100 structurally different classes (Jones and Thirup, 1986; Claessens et al., 1989; Unger et al., 1989; Levitt, 1992; Bystroff and Baker, 1998). Thus, comparative models can be constructed by using a subset of atomic positions from template structures as guiding positions to identify and assemble short, all-atom segments that fit these guiding positions. The guiding positions usually correspond to the Cα atoms of the segments that are conserved in the alignment between the template structure and the target sequence. The all-atom segments that fit the guiding positions can be obtained either by scanning all known protein structures, including those that are not related to the sequence being modeled (Claessens et al., 1989; Holm and Sander, 1991), or by a conformational search restrained by an energy function (Bruccoleri and Karplus, 1987; van Gelder et al., 1994). This method can construct both main-chain and side-chain atoms, and can also model unaligned regions (gaps). It is implemented in the program SegMod (Levitt, 1992). Even some side-chain modeling methods (Chinea et al., 1995) and the class of loop-construction methods based on finding suitable fragments in the database of known structures (Jones and Thirup, 1986) can be seen as segment-matching or coordinate-reconstruction methods.
Modeling by satisfaction of spatial restraints
The methods in this class begin by generating many constraints or restraints on the structure of the target sequence, using its alignment to related protein structures as a guide. The procedure is conceptually similar to that used in determination of protein structures from NMR-derived restraints. The restraints are generally obtained by assuming that the corresponding distances between aligned residues in the template and the target structures are similar. These homology-derived restraints are usually supplemented by stereochemical restraints on bond lengths, bond angles, dihedral angles, and nonbonded atom-atom contacts that are obtained from a molecular mechanics force field. The model is then derived by minimizing the violations of all the restraints. This optimization can be achieved either by distance geometry or real-space optimization. For example, an elegant distance geometry approach constructs all-atom models from lower and upper bounds on distances and dihedral angles (Havel and Snow, 1991).
Comparative protein structure modeling by MODELLER
MODELLER, the authors’ own program for comparative modeling, belongs to this group of methods (Sali and Blundell, 1993; Sali and Overington, 1994; Fiser et al., 2000; Fiser et al., 2002). MODELLER implements comparative protein structure modeling by satisfaction of spatial restraints. The program was designed to use as many different types of information about the target sequence as possible.
Homology-derived restraints
In the first step of model building, distance and dihedral angle restraints on the target sequence are derived from its alignment with template 3-D structures. The form of these restraints was obtained from a statistical analysis of the relationships between similar protein structures. The analysis relied on a database of 105 family alignments that included 416 proteins of known 3-D structure (Sali and Overington, 1994). By scanning the database of alignments, tables quantifying various correlations were obtained, such as the correlations between two equivalent Cα-Cα distances, or between equivalent main-chain dihedral angles from two related proteins (Sali and Blundell, 1993). These relationships are expressed as conditional probability density functions (pdf’s), and can be used directly as spatial restraints. For example, probabilities for different values of the main-chain dihedral angles are calculated from the type of residue considered, from main-chain conformation of an equivalent residue, and from sequence similarity between the two proteins. Another example is the pdf for a certain Cα-Cα distance given equivalent distances in two related protein structures. An important feature of the method is that the form of spatial restraints was obtained empirically, from a database of protein structure alignments.
Stereochemical restraints
In the second step, the spatial restraints and the CHARMM22 force field terms enforcing proper stereochemistry (MacKerell et al., 1998) are combined into an objective function. The general form of the objective function is similar to that in molecular dynamics programs, such as CHARMM22 (MacKerell et al., 1998). The objective function depends on the Cartesian coordinates of ~10,000 atoms (3-D points) that form the modeled molecules. For a 10,000-atom system, there can be on the order of 200,000 restraints. The functional form of each term is simple; it includes a quadratic function, harmonic lower and upper bounds, cosine, a weighted sum of a few Gaussian functions, Coulomb law, Lennard-Jones potential, and cubic splines. The geometric features presently include a distance, an angle, a dihedral angle, a pair of dihedral angles between two, three, four, and eight atoms, respectively, the shortest distance in the set of distances, solvent accessibility, and atom density expressed as the number of atoms around the central atom. Some restraints can be used to restrain pseudo-atoms, e.g., the gravity center of several atoms.
Optimization of the objective function
Finally, the model is obtained by optimizing the objective function in Cartesian space. The optimization is carried out by the use of the variable target function method (Braun and Go, 1985), employing methods of conjugate gradients and molecular dynamics with simulated annealing (Clore et al., 1986). Several slightly different models can be calculated by varying the initial structure, and the variability among these models can be used to estimate the lower bound on the errors in the corresponding regions of the fold.
Restraints derived from experimental data
Because modeling by satisfaction of spatial restraints can use many different types of information about the target sequence, it is perhaps the most promising of all comparative modeling techniques. One of the strengths of modeling by satisfaction of spatial restraints is that restraints derived from a number of different sources can easily be added to the homology-derived restraints. For example, restraints could be provided by rules for secondary-structure packing (Cohen et al., 1989), analyses of hydrophobicity (Aszodi and Taylor, 1994) and correlated mutations (Taylor et al., 1994), empirical potentials of mean force (Sippl, 1990), nuclear magnetic resonance (NMR) experiments (Sutcliffe et al., 1992), cross-linking experiments, fluorescence spectroscopy, image reconstruction in electron microscopy, site-directed mutagenesis (Boissel et al., 1993), and intuition, among other sources. Especially in difficult cases, a comparative model could be improved by making it consistent with available experimental data and/or with more general knowledge about protein structure.
Relative accuracy, flexibility, and automation
Accuracies of the various model-building methods are relatively similar when used optimally (Marti-Renom et al., 2002). Other factors such as template selection and alignment accuracy usually have a larger impact on the model accuracy, especially for models based on low sequence identity to the templates. However, it is important that a modeling method allow a degree of flexibility and automation to obtain better models more easily and rapidly. For example, a method should allow for an easy recalculation of a model when a change is made in the alignment. It should also be straightforward enough to calculate models based on several templates, and should provide tools for incorporation of prior knowledge about the target (e.g., cross-linking restraints, predicted secondary structure) and allow ab initio modeling of insertions (e.g., loops), which can be crucial for annotation of function.
Loop modeling
Loop modeling is an especially important aspect of comparative modeling in the range from 30% to 50% sequence identity. In this range of overall similarity, loops among the homologs vary while the core regions are still relatively conserved and accurately aligned. Loops often play an important role in defining the functional specificity of a given protein, forming the active and binding sites. Loop modeling can be seen as a mini protein-folding problem, because the correct conformation of a given segment of a polypeptide chain has to be calculated mainly from the sequence of the segment itself. However, loops are generally too short to provide sufficient information about their local fold. Even identical decapeptides in different proteins do not always have the same conformation (Kabsch and Sander, 1984; Mezei, 1998). Some additional restraints are provided by the core anchor regions that span the loop and by the structure of the rest of the protein that cradles the loop. Although many loop-modeling methods have been described, it is still challenging to correctly and confidently model loops longer than ~10 to 12 residues (Fiser et al., 2000; Jacobson et al., 2004; Zhu et al., 2006).
There are two main classes of loop-modeling methods: (i) database search approaches that scan a database of all known protein structures to find segments fitting the anchor core regions (Jones and Thirup, 1986; Chothia and Lesk, 1987); (ii) conformational search approaches that rely on optimizing a scoring function (Moult and James, 1986; Bruccoleri and Karplus, 1987; Shenkin et al., 1987). There are also methods that combine these two approaches (van Vlijmen and Karplus, 1997; Deane and Blundell, 2001).
Loop modeling by database search
The database search approach to loop modeling is accurate and efficient when a database of specific loops is created to address the modeling of the same class of loops, such as β-hairpins (Sibanda et al., 1989), or loops on a specific fold, such as the hypervariable regions in the immunoglobulin fold (Chothia and Lesk, 1987; Chothia et al., 1989). There are attempts to classify loop conformations into more general categories, thus extending the applicability of the database search approach (Ring et al., 1992; Oliva et al., 1997; Rufino et al., 1997; Fernandez-Fuentes and Fiser, 2006). However, the database methods are limited because the number of possible conformations increases exponentially with the length of a loop, and until the late 1990s only loops up to 7 residues long could be modeled using the database of known protein structures (Fidelis et al., 1994; Lessel and Schomburg, 1994). However, the growth of the PDB in recent years has largely eliminated this problem (Fernandez-Fuentes and Fiser, 2006).
Loop modeling by conformational search
There are many such methods, exploiting different protein representations, objective functions, and optimization or enumeration algorithms. The search algorithms include the minimum perturbation method (Fine et al., 1986), dihedral angle search through a rotamer library (Zhu et al., 2006; Sellers et al., 2008), molecular dynamics simulations (Bruccoleri and Karplus, 1990; van Vlijmen and Karplus, 1997), genetic algorithms (Ring et al., 1993), Monte Carlo and simulated annealing (Higo et al., 1992; Collura et al., 1993; Abagyan and Totrov, 1994), multiple copy simultaneous search (Zheng et al., 1993), self-consistent field optimization (Koehl and Delarue, 1995), robotics-inspired kinematic closure (Mandell et al., 2009), and enumeration based on graph theory (Samudrala and Moult, 1998). The accuracy of loop predictions can be further improved by clustering the sampled loop conformations and partially accounting for the entropic contribution to the free energy (Xiang et al., 2002). Another way to improve the accuracy of loop predictions is to consider the solvent effects. Improvements in implicit solvation models, such as the Generalized Born solvation model, motivated their use in loop modeling. The solvent contribution to the free energy can be added to the scoring function for optimization, or it can be used to rank the sampled loop conformations after they are generated with a scoring function that does not include the solvent terms (Fiser et al., 2000; Felts et al., 2002; de Bakker et al., 2003; DePristo et al., 2003).
Loop modeling in MODELLER
The loop-modeling module in MODELLER implements the optimization-based approach (Fiser et al., 2000; Fiser and Sali, 2003b). The main reasons for choosing this implementation are the generality and conceptual simplicity of scoring function minimization. Loop prediction by optimization is applicable to simultaneous modeling of several loops and loops interacting with ligands, which is not straightforward with the database-search approaches. Loop optimization in MODELLER relies on conjugate gradients and molecular dynamics with simulated annealing. The pseudo energy function is a sum of many terms, including some terms from the CHARMM22 molecular mechanics force field (MacKerell et al., 1998), spatial restraints based on distributions of distances (Sippl, 1990; Melo et al., 2002), and dihedral angles in known protein structures. The method was tested on a large number of loops of known structure, both in native and near-native environments (Fiser et al., 2000).
Comparative model building by iterative alignment, model building, and model assessment
Comparative or homology protein structure modeling is severely limited by errors in the alignment of a modeled sequence with related proteins of known three-dimensional structure. To ameliorate this problem, one can use an iterative method that optimizes both the alignment and the model implied by it (Sanchez and Sali, 1997a; Miwa et al., 1999). This task can be achieved by a genetic algorithm protocol that starts with a set of initial alignments and then iterates through realignment, model building, and model assessment to optimize a model assessment score (John and Sali, 2003). During this iterative process: (1) new alignments are constructed by the application of a number of genetic algorithm operators, such as alignment mutations and cross-overs; (2) comparative models corresponding to these alignments are built by satisfaction of spatial restraints, as implemented in the program MODELLER; and (3) the models are assessed by a composite score, partly depending on an atomic statistical potential (Melo et al., 2002). When testing the procedure on a very difficult set of 19 modeling targets sharing only 4% to 27% sequence identity with their template structures, the average final alignment accuracy increased from 37% to 45% relative to the initial alignment (the alignment accuracy was measured as the percentage of positions in the tested alignment that were identical to the reference structure-based alignment). Correspondingly, the average model accuracy increased from 43% to 54% (the model accuracy was measured as the percentage of the Cα atoms of the model that were within 5 Å of the corresponding Cα atoms in the superimposed native structure).
Errors in comparative models
As the similarity between the target and the templates decreases, the errors in the model increase. Errors in comparative models can be divided into five categories (Sanchez and Sali, 1997a, 1997b; Fig. 5.6.14), as follows:
Figure 5.6.14.
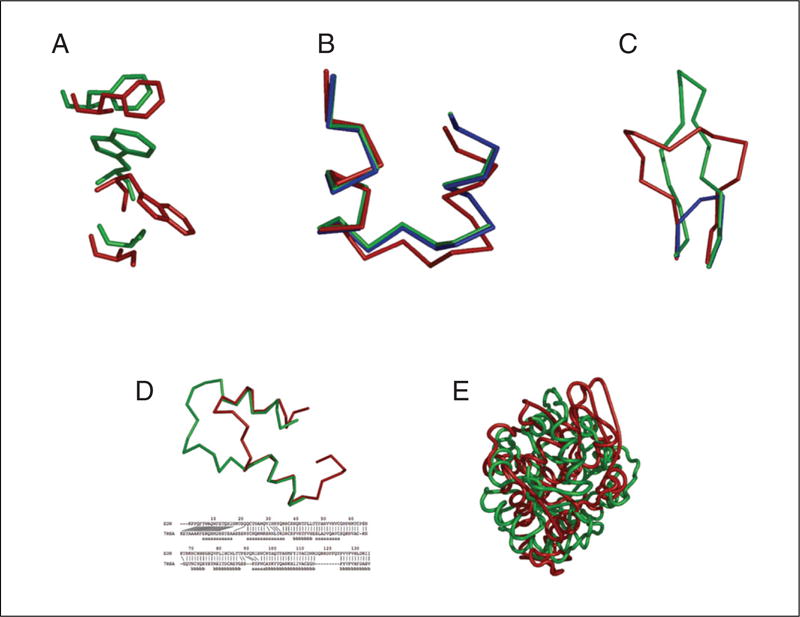
Typical errors in comparative modeling. (A) Errors in side chain packing. The Trp 109 residue in the crystal structure of mouse cellular retinoic acid binding protein I (red) is compared with its model (green). (B) Distortions and shifts in correctly aligned regions. A region in the crystal structure of mouse cellular retinoic acid binding protein I (red) is compared with its model (green) and with the template fatty acid binding protein (blue). (C) Errors in regions without a template. The Cα trace of the 112-117 loop is shown for the X-ray structure of human eosinophil neurotoxin (red), its model (green), and the template ribonuclease A structure (residues 111-117; blue). (D) Errors due to misalignments. The N-terminal region in the crystal structure of human eosinophil neurotoxin (red) is compared with its model (green). The corresponding region of the alignment with the template ribonuclease A is shown. The red lines show correct equivalences, that is, residues whose Cα atoms are within 5 Å of each other in the optimal least-squares superposition of the two X-ray structures. The “a” characters in the bottom line indicate helical residues and “b” characters, the residues in sheets. (E) Errors due to an incorrect template. The X-ray structure of α-trichosanthin (red) is compared with its model (green) that was calculated using indole-3-glycerophosphate synthase as the template.
Errors in side-chain packing (Fig. 5.6.14A)
As the sequences diverge, the packing of side-chains in the protein core changes. Sometimes even the conformation of identical side chains is not conserved, a pitfall for many comparative modeling methods. Side-chain errors are critical if they occur in regions that are involved in protein function, such as active sites and ligand-binding sites.
Distortions and shifts in correctly aligned regions (Fig. 5.6.14B)
As a consequence of sequence divergence, the main-chain conformation changes, even if the overall fold remains the same. Therefore, it is possible that in some correctly aligned segments of a model, the template is locally different (>3 Å) from the target, resulting in errors in that region. The structural differences are sometimes not due to differences in sequence, but are a consequence of artifacts in structure determination or structure determination in different environments (e.g., packing of subunits in a crystal). The simultaneous use of several templates can minimize this kind of error (Srinivasan and Blundell, 1993; Sanchez and Sali, 1997a, 1997b).
Errors in regions without a template (Fig. 5.6.14C)
Segments of the target sequence that have no equivalent region in the template structure (i.e., insertions or loops) are the most difficult regions to model. If the insertion is relatively short, <9 residues long, some methods can correctly predict the conformation of the backbone (van Vlijmen and Karplus, 1997; Fiser et al., 2000; Jacobson et al., 2004). Conditions for successful prediction are the correct alignment and an accurately modeled environment surrounding the insertion.
Errors due to misalignments (Fig. 5.6.14D)
The largest single source of errors in comparative modeling is misalignments, especially when the target-template sequence identity decreases below 30%. However, alignment errors can be minimized in two ways. First, it is usually possible to use a large number of sequences to construct a multiple alignment, even if most of these sequences do not have known structures. Multiple alignments are generally more reliable than pairwise alignments (Barton and Sternberg, 1987; Taylor et al., 1994). The second way of improving the alignment is to iteratively modify those regions in the alignment that correspond to predicted errors in the model (Sanchez and Sali, 1997a, 1997b; John and Sali, 2003).
Incorrect templates (Fig. 5.6.14E)
This is a potential problem when distantly related proteins are used as templates (i.e., < 25% sequence identity). Distinguishing between a model based on an incorrect template and a model based on an incorrect alignment with a correct template is difficult. In both cases, the evaluation methods will predict an unreliable model. The conservation of the key functional or structural residues in the target sequence increases the confidence in a given fold assignment.
Predicting the model accuracy
The accuracy of the predicted model determines the information that can be extracted from it. Thus, estimating the accuracy of a model in the absence of the known structure is essential for interpreting it.
Initial assessment of the fold
As discussed earlier, a model calculated using a template structure that shares more than 30% sequence identity is indicative of an overall accurate structure. However, when the sequence identity is lower, the first aspect of model evaluation is to confirm whether or not a correct template was used for modeling. It is often the case, when operating in this regime, that the fold-assignment step produces only false positives. A further complication is that at such low similarities the alignment generally contains many errors, making it difficult to distinguish between an incorrect template on one hand and an incorrect alignment with a correct template on the other hand. There are several methods that use 3-D profiles and statistical potentials (Sippl, 1990; Luthy et al., 1992; Melo et al., 2002) to assess the compatibility between the sequence and modeled structure by evaluating the environment of each residue in a model with respect to the expected environment as found in native high-resolution experimental structures. These methods can be used to assess whether or not the correct template was used for the modeling. They include VERIFY3D (Luthy et al., 1992), Prosa, 2003 (Sippl, 1993; Wiederstein and Sippl, 2007), HARMONY (Topham et al., 1994), ANOLEA (Melo and Feytmans, 1998), DFIRE (Zhou and Zhou, 2002), DOPE (Shen and Sali, 2006), SOAP (Dong et al., 2013), QMEAN local (Benkert et al., 2011), and TSVMod (Eramian et al., 2008).
Even when the model is based on alignments that have >30% sequence identity, other factors, including the environment, can strongly influence the accuracy of a model. For instance, some calcium-binding proteins undergo large conformational changes when bound to calcium. If a calcium-free template is used to model the calcium-bound state of the target, it is likely that the model will be incorrect irrespective of the target-template similarity or accuracy of the template structure (Pawlowski et al., 1996).
Evaluations of self-consistency
The model should also be subjected to evaluations of self-consistency to ensure that it satisfies the restraints used to calculate it. Additionally, the stereochemistry of the model (e.g., bond-lengths, bond-angles, backbone torsion angles, and nonbonded contacts) may be evaluated using programs such as PROCHECK (Laskowski et al., 1993) and WHATCHECK (Hooft et al., 1996). Although errors in stereochemistry are rare and less informative than errors detected by statistical potentials, a cluster of stereochemical errors may indicate that there are larger errors (e.g., alignment errors) in that region.
Applications
To maximize the impact of comparative models, the modeling process used to generate them should be clearly described, for example by publishing or identifying a protocol similar to that described here. This protocol should also include assessment using the best computational tools currently available, such as the model assessment methods covered in this text, and those cataloged at the Protein Model Portal (http://proteinmodelportal.org; Arnold et al., 2009; Haas et al., 2013). Finally, the study should both rationalize existing experimental data and make testable predictions. For example, comparative models can be helpful in designing mutants to test hypotheses about the protein’s function (Wu et al., 1999; Vernal et al., 2002); in identifying active and binding sites (Sheng et al., 1996); in searching for, designing, and improving ligand binding strength for a given binding site (Ring et al., 1993; Li et al., 1996; Selzer et al., 1997; Enyedy et al., 2001; Que et al., 2002); modeling substrate specificity (Xu et al., 1996); in predicting antigenic epitopes (Sali and Blundell, 1993); in simulating protein-protein docking (Vakser, 1995); in inferring function from calculated electrostatic potential around the protein (Matsumoto et al., 1995); in facilitating molecular replacement in X-ray structure determination (Howell et al., 1992); in refining models based on NMR constraints (Modi et al., 1996); in testing and improving a sequence-structure alignment (Wolf et al., 1998); in annotating single nucleotide polymorphisms (Mirkovic et al., 2004; Karchin et al., 2005); in structural characterization of large complexes by docking to low-resolution cryo-electron density maps (Spahn et al., 2001; Gao et al., 2003); and in rationalizing known experimental observations.
Fortunately, a 3-D model does not have to be absolutely perfect to be helpful in biology, as demonstrated by the applications listed above. The type of a question that can be addressed with a particular model does depend on its accuracy (Fig. 5.6.15).
Figure 5.6.15.
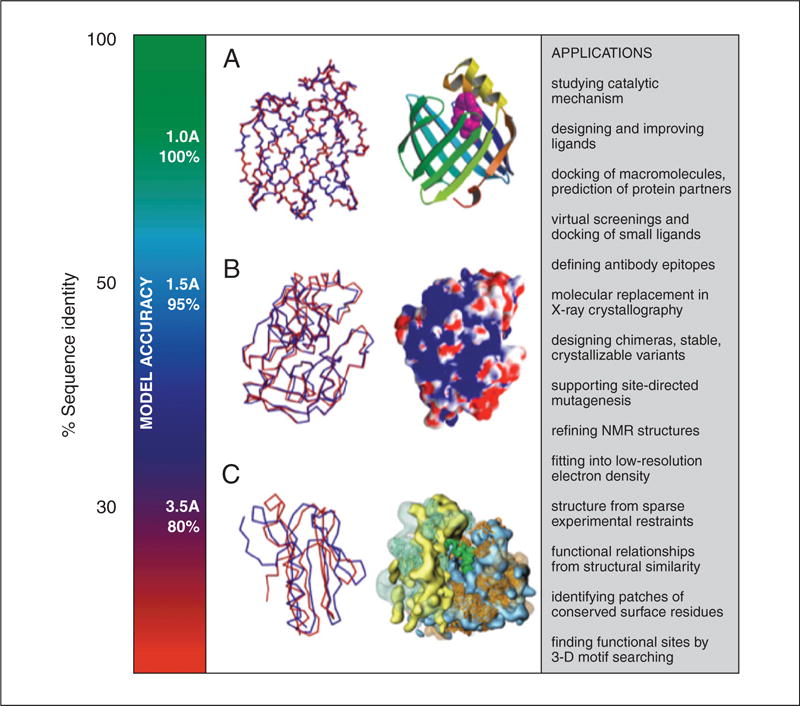
Accuracy and application of protein structure models. The vertical axis indicates the different ranges of applicability of comparative protein structure modeling, the corresponding accuracy of protein structure models, and their sample applications. (A) The docosahexaenoic fatty acid ligand (violet) was docked into a high accuracy comparative model of brain lipid-binding protein (right), modeled based on its 62% sequence identity to the crystallographic structure of adipocyte lipid-binding protein (PDB code 1adl). A number of fatty acids were ranked for their affinity to brain lipid-binding protein consistently with site-directed mutagenesis and affinity chromatography experiments (Xu et al., 1996), even though the ligand specificity profile of this protein is different from that of the template structure. Typical overall accuracy of a comparative model in this range of sequence similarity is indicated by a comparison of a model for adipocyte fatty acid binding protein with its actual structure (left). (B) A putative proteoglycan binding patch was identified on a medium-accuracy comparative model of mouse mast cell protease 7 (right), modeled based on its 39% sequence identity to the crystallographic structure of bovine pancreatic trypsin (2ptn) that does not bind proteoglycans. The prediction was confirmed by site-directed mutagenesis and heparin-affinity chromatography experiments (Matsumoto et al., 1995). Typical accuracy of a comparative model in this range of sequence similarity is indicated by a comparison of a trypsin model with the actual structure. (C) A molecular model of the whole yeast ribosome (right) was calculated by fitting atomic rRNA and protein models into the electron density of the 80S ribosomal particle, obtained by electron microscopy at 15 Å resolution (Spahn et al., 2001). Most of the models for 40 out of the 75 ribosomal proteins were based on template structures that were approximately 30% sequentially identical. Typical accuracy of a comparative model in this range of sequence similarity is indicated by a comparison of a model for a domain in L2 protein from B. stearothermophilus with the actual structure (1rl2).
At the low end of the accuracy spectrum, there are models that are based on less than 25% sequence identity and that sometimes have less than 50% of their Cα atoms within 3.5 Å of their correct positions. However, such models may still have the correct fold, and even knowing only the fold of a protein may sometimes be sufficient to predict its approximate biochemical function. Models in this low range of accuracy, combined with model evaluation, can be used for confirming or rejecting a match between remotely related proteins (Sanchez and Sali, 1997a, 1998).
In the middle of the accuracy spectrum are the models based on approximately 35% sequence identity, corresponding to 85% of the Cα atoms modeled within 3.5 Å of their correct positions. Fortunately, the active and binding sites are frequently more conserved than the rest of the fold, and are thus modeled more accurately (Sanchez and Sali, 1998). In general, medium-resolution models frequently allow a refinement of the functional prediction based on sequence alone, because ligand binding is most directly determined by the structure of the binding site rather than its sequence. It is frequently possible to correctly predict important features of the target protein that do not occur in the template structure. For example, the location of a binding site can be predicted from clusters of charged residues (Matsumoto et al., 1995), and the size of a ligand may be predicted from the volume of the binding-site cleft (Xu et al., 1996). Medium-resolution models can also be used to construct site-directed mutants with altered or destroyed binding capacity, which in turn could test hypotheses about the sequence-structure-function relationships. Other problems that can be addressed with medium-resolution comparative models include designing proteins that have compact structures, without long tails, loops, and exposed hydrophobic residues, for better crystallization, or designing proteins with added disulfide bonds for extra stability.
The high end of the accuracy spectrum corresponds to models based on 50% sequence identity or more. The average accuracy of these models approaches that of low-resolution X-ray structures (3 Å resolution) or medium-resolution NMR structures (10 distance restraints per residue; Sanchez and Sali, 1997b). The alignments on which these models are based generally contain almost no errors. Models with such high accuracy have been shown to be useful even for refining crystallographic structures by the method of molecular replacement (Howell et al., 1992; Baker and Sali, 2001; Jones, 2001; Claude et al., 2004; Schwarzenbacher et al., 2004).
A good comparative modeling study tends to include novel experimental data, used in model derivation, assessment, and/or interpretation. In some instances, however, only models, rationalizations, and/or predictions, even without new experimental data, can be impactful, accelerating the cycle of science, consisting of data generation, interpretation, modeling, and hypothesizing.
Conclusion
Over the past few years, there has been a gradual increase in both the accuracy of comparative models and the fraction of protein sequences that can be modeled with useful accuracy (Marti-Renom et al., 2000; Baker and Sali, 2001; Pieper et al., 2014). The magnitude of errors in fold assignment, alignment, and the modeling of side-chains and loops have decreased considerably. These improvements are a consequence both of better techniques and a larger number of known protein sequences and structures. Nevertheless, all the errors remain significant and demand future methodological improvements. In addition, there is a great need for more accurate modeling of distortions and rigid-body shifts, as well as detection of errors in a given protein structure model. Error detection is useful both for refinement and interpretation of the models.
Acknowledgments
The authors wish to express gratitude to all members of their research group. This review is partially based on the authors’ previous reviews (Marti-Renom et al., 2000; Eswar et al., 2003; Fiser and Sali, 2003a; also see the previous versions of this unit at http://onlinelibrary.wiley.com/doi/10.1002/0471140864.ps0209s50/pdf. They wish to acknowledge funding from Sandler Family Supporting Foundation, NIH R01 GM54762, P01 GM71790, P01 1AI091575, and U54 GM62529, as well as hardware gifts from IBM and Intel.
Literature Cited
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A, Howorth D, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006;34:W604–W608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Arnold K, Kiefer F, Kopp J, Battey JN, Podvinec M, Westbrook JD, Berman HM, Bordoli L, Schwede T. The protein model portal. J Struct Funct Genomics. 2009;10:1–8. doi: 10.1007/s10969-008-9048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aszodi A, Taylor WR. Secondary structure formation in model polypeptide chains. Protein Eng. 1994;7:633–644. doi: 10.1093/protein/7.5.633. [DOI] [PubMed] [Google Scholar]
- Attwood TK, Coletta A, Muirhead G, Pavlopoulou A, Philippou PB, Popov I, Roma-Mateo C, Theodosiou A, Mitchell AL. The PRINTS database: A fine-grained protein sequence annotation and analysis resource–its status in 2012. Database (Oxford) 2012;2012:bas019. doi: 10.1093/database/bas019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Barton GJ, Sternberg MJ. A strategy for the rapid multiple alignment of protein sequences. Confidence levels from tertiary structure comparisons. J Mol Biol. 1987;198:327–337. doi: 10.1016/0022-2836(87)90316-0. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates PA, Kelley LA, MacCallum RM, Sternberg MJ. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins. 2001;(Suppl 5):39–46. doi: 10.1002/prot.1168. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell TL, Sibanda BL, Sternberg MJ, Thornton JM. Knowledge-based prediction of protein structures and the design of novel molecules. Nature. 1987;326:347–352. doi: 10.1038/326347a0. [DOI] [PubMed] [Google Scholar]
- Boissel JP, Lee WR, Presnell SR, Cohen FE, Bunn HF. Erythropoietin structure-function relationships. Mutant proteins that test a model of tertiary structure. J Biol Chem. 1993;268:15983–15993. [PubMed] [Google Scholar]
- Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Braun W, Go N. Calculation of protein conformations by proton-proton distance constraints. A new efficient algorithm. J Mol Biol. 1985;186:611–626. doi: 10.1016/0022-2836(85)90134-2. [DOI] [PubMed] [Google Scholar]
- Brenner SE, Chothia C, Hubbard TJ. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc Natl Acad Sci USA. 1998;95:6073–6078. doi: 10.1073/pnas.95.11.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Babbitt PC. Inference of functional properties from large-scale analysis of enzyme superfamilies. J Biol Chem. 2012;287:35–42. doi: 10.1074/jbc.R111.283408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne WJ, North AC, Phillips DC, Brew K, Vanaman TC, Hill RL. A possible three-dimensional structure of bovine alpha-lactalbumin based on that of hen’s egg-white lysozyme. J Mol Biol. 1969;42:65–86. doi: 10.1016/0022-2836(69)90487-2. [DOI] [PubMed] [Google Scholar]
- Bru C, Courcelle E, Carrere S, Beausse Y, Dalmar S, Kahn D. The ProDom database of protein domain families: More emphasis on 3D. Nucleic Acids Res. 2005;33:D212–D215. doi: 10.1093/nar/gki034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruccoleri RE, Karplus M. Prediction of the folding of short polypeptide segments by uniform conformational sampling. Biopolymers. 1987;26:137–168. doi: 10.1002/bip.360260114. [DOI] [PubMed] [Google Scholar]
- Bruccoleri RE, Karplus M. Conformational sampling using high-temperature molecular dynamics. Biopolymers. 1990;29:1847–1862. doi: 10.1002/bip.360291415. [DOI] [PubMed] [Google Scholar]
- Bystroff C, Baker D. Prediction of local structure in proteins using a library of sequence-structure motifs. J Mol Biol. 1998;281:565–577. doi: 10.1006/jmbi.1998.1943. [DOI] [PubMed] [Google Scholar]
- Chinea G, Padron G, Hooft RW, Sander C, Vriend G. The use of position-specific rotamers in model building by homology. Proteins. 1995;23:415–421. doi: 10.1002/prot.340230315. [DOI] [PubMed] [Google Scholar]
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, Colman PM, Spinelli S, Alzari PM, Poljak RJ. Conformations of immunoglobulin hypervariable regions. Nature. 1989;342:877–883. doi: 10.1038/342877a0. [DOI] [PubMed] [Google Scholar]
- Claessens M, Van Cutsem E, Lasters I, Wodak S. Modelling the polypeptide backbone with ‘spare parts’ from known protein structures. Protein Eng. 1989;2:335–345. doi: 10.1093/protein/2.5.335. [DOI] [PubMed] [Google Scholar]
- Claude JB, Suhre K, Notredame C, Claverie JM, Abergel C. CaspR: A web server for automated molecular replacement using homology modelling. Nucleic Acids Res. 2004;32:W606–W609. doi: 10.1093/nar/gkh400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Brunger AT, Karplus M, Gronenborn AM. Application of molecular dynamics with interproton distance restraints to three-dimensional protein structure determination. A model study of crambin. J Mol Biol. 1986;191:523–551. doi: 10.1016/0022-2836(86)90146-4. [DOI] [PubMed] [Google Scholar]
- Cohen FE, Gregoret L, Presnell SR, Kuntz ID. Protein structure predictions: New theoretical approaches. Prog Clin Biol Res. 1989;289:75–85. [PubMed] [Google Scholar]
- Collura V, Higo J, Garnier J. Modeling of protein loops by simulated annealing. Protein Sci. 1993;2:1502–1510. doi: 10.1002/pro.5560020915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker PI, DePristo MA, Burke DF, Blundell TL. Ab initio construction of polypeptide fragments: Accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force field with the Generalized Born solvation model. Proteins. 2003;51:21–40. doi: 10.1002/prot.10235. [DOI] [PubMed] [Google Scholar]
- Deane CM, Blundell TL. CODA: A combined algorithm for predicting the structurally variable regions of protein models. Protein Sci. 2001;10:599–612. doi: 10.1110/ps.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, de Bakker PI, Lovell SC, Blundell TL. Ab initio construction of polypeptide fragments: Efficient generation of accurate, representative ensembles. Proteins. 2003;51:41–55. doi: 10.1002/prot.10285. [DOI] [PubMed] [Google Scholar]
- Dietmann S, Park J, Notredame C, Heger A, Lappe M, Holm L. A fully automatic evolutionary classification of protein folds: Dali Domain Dictionary version 3. Nucleic Acids Res. 2001;29:55–57. doi: 10.1093/nar/29.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong GQ, Fan H, Schneidman-Duhovny D, Webb B, Sali A. Optimized atomic statistical potentials: Assessment of protein interfaces and loops. Bioinformatics. 2013;29:3158–3166. doi: 10.1093/bioinformatics/btt560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror O, Benyamini H, Nussinov R, Wolfson H. MASS: Multiple structural alignment by secondary structures. Bioinformatics. 2003;19(Suppl 1):i95–i104. doi: 10.1093/bioinformatics/btg1012. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Sjolander K. A comparison of scoring functions for protein sequence profile alignment. Bioinformatics. 2004;20:1301–1308. doi: 10.1093/bioinformatics/bth090. [DOI] [PubMed] [Google Scholar]
- Enyedy IJ, Lee SL, Kuo AH, Dickson RB, Lin CY, Wang S. Structure-based approach for the discovery of bis-benzamidines as novel inhibitors of matriptase. J Med Chem. 2001;44:1349–1355. doi: 10.1021/jm000395x. [DOI] [PubMed] [Google Scholar]
- Eramian D, Eswar N, Shen M, Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17:1881–1893. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B, Sali A. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003;31:3375–3380. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP—Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. doi: 10.1111/j.1096-0031.1989.tb00562.x. [DOI] [Google Scholar]
- Felts AK, Gallicchio E, Wallqvist A, Levy RM. Distinguishing native conformations of proteins from decoys with an effective free energy estimator based on the OPLS all-atom force field and the Surface Generalized Born solvent model. Proteins. 2002;48:404–422. doi: 10.1002/prot.10171. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fuentes N, Fiser A. Saturating representation of loop conformational fragments in structure databanks. BMC Struct Biol. 2006;6:15. doi: 10.1186/1472-6807-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Fuentes N, Rai BK, Madrid-Aliste CJ, Fajardo JE, Fiser A. Comparative protein structure modeling by combining multiple templates and optimizing sequence-to-structure alignments. Bioinformatics. 2007;23:2558–2565. doi: 10.1093/bioinformatics/btm377. [DOI] [PubMed] [Google Scholar]
- Fidelis K, Stern PS, Bacon D, Moult J. Comparison of systematic search and database methods for constructing segments of protein structure. Protein Eng. 1994;7:953–960. doi: 10.1093/protein/7.8.953. [DOI] [PubMed] [Google Scholar]
- Fine RM, Wang H, Shenkin PS, Yarmush DL, Levinthal C. Predicting antibody hypervariable loop conformations. II: Minimization and molecular dynamics studies of MCPC603 from many randomly generated loop conformations. Proteins. 1986;1:342–362. doi: 10.1002/prot.340010408. [DOI] [PubMed] [Google Scholar]
- Fiser A. Protein structure modeling in the proteomics era. Expert Rev Proteomics. 2004;1:97–110. doi: 10.1586/14789450.1.1.97. [DOI] [PubMed] [Google Scholar]
- Fiser A, Sali A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003a;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- Fiser A, Sali A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics. 2003b;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- Fiser A, Do RKG, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, Feig M, Brooks CL, Sali A. Evolution and physics in comparative protein structure modeling. Acc Chem Res. 2002;35:413–421. doi: 10.1021/ar010061h. [DOI] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Garcia-Giron C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kahari AK, Keenan S, Komorowska M, Kulesha E, Longden I, Maurel T, McLaren WM, Muffato M, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ritchie GR, Ruffier M, Schuster M, Sheppard D, Sobral D, Taylor K, Thormann A, Trevanion S, White S, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I, Harrow J, Herrero J, Hubbard TJ, Johnson N, Kinsella R, Parker A, Spudich G, Yates A, Zadissa A, Searle SM. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HX, Sengupta J, Valle M, Korostelev A, Eswar N, Stagg SM, Van Roey P, Agrawal RK, Harvey SC, Sali A, Chapman MS, Frank J. Study of the structural dynamics of the E-coli 70S ribosome using real-space refinement. Cell. 2003;113:789–801. doi: 10.1016/S0092-8674(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Godzik A. Fold recognition methods. Methods Biochem Anal. 2003;44:525–546. doi: 10.1002/0471721204.ch26. [DOI] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- Greer J. Comparative model-building of the mammalian serine proteases. J Mol Biol. 1981;153:1027–1042. doi: 10.1016/0022-2836(81)90465-4. [DOI] [PubMed] [Google Scholar]
- Gribskov M, McLachlan AD, Eisenberg D. Profile analysis: Detection of distantly related proteins. Proc Natl Acad Sci USA. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerler A, Knapp EW. Novel protein folds and their nonsequential structural analogs. Protein Sci. 2008;17:1374–1382. doi: 10.1110/ps.035469.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J, Roth S, Arnold K, Kiefer F, Schmidt T, Bordoli L, Schwede T. The Protein Model Portal–a comprehensive resource for protein structure and model information. Database (Oxford) 2013;2013:bat031. doi: 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel TF, Snow ME. A new method for building protein conformations from sequence alignments with homologues of known structure. J Mol Biol. 1991;217:1–7. doi: 10.1016/0022-2836(91)90603-4. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Position-based sequence weights. J Mol Biol. 1994;243:574–578. doi: 10.1016/0022-2836(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Higo J, Collura V, Garnier J. Development of an extended simulated annealing method: Application to the modeling of complementary determining regions of immunoglobulins. Biopolymers. 1992;32:33–43. doi: 10.1002/bip.360320106. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Database algorithm for generating protein backbone and side-chain co-ordinates from a C alpha trace application to model building and detection of co-ordinate errors. J Mol Biol. 1991;218:183–194. doi: 10.1016/0022-2836(91)90883-8. [DOI] [PubMed] [Google Scholar]
- Holm L, Kääriäinen S, Wilton C, Plewczynski D. Using Dali for structural comparison of proteins. Curr Protoc Bioinform. 2006;14:5.5.1–5.5.24. doi: 10.1002/0471250953.bi0505s14. [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Howell PL, Almo SC, Parsons MR, Hajdu J, Petsko GA. Structure determination of turkey egg-white lysozyme using Laue diffraction data. Acta Crystallogr., B. 1992;48:200–207. doi: 10.1107/S0108768191012466. [DOI] [PubMed] [Google Scholar]
- Huang H, Hu ZZ, Arighi CN, Wu CH. Integration of bioinformatics resources for functional analysis of gene expression and proteomic data. Front Biosci. 2007;12:5071–5088. doi: 10.2741/2449. [DOI] [PubMed] [Google Scholar]
- Huang CC, Novak WR, Babbitt PC, Jewett AI, Ferrin TE, Klein TE. Integrated tools for structural and sequence alignment and analysis. Pac Symp Biocomput. 2000:230–241. doi: 10.1142/9789814447331_0022. [DOI] [PubMed] [Google Scholar]
- Hulo N, Bairoch A, Bulliard V, Cerutti L, De Castro E, Langendijk-Genevaux PS, Pagni M, Sigrist CJ. The PROSITE database. Nucleic Acids Res. 2006;34:D227–D230. doi: 10.1093/nar/gkj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. FFAS03: A server for profile–profile sequence alignments. Nucleic Acids Res. 2005;33:W284–W288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Jones DT. Evaluating the potential of using fold-recognition models for molecular replacement. Acta Crystallogr D Biol Crystallogr. 2001;57:1428–1434. doi: 10.1107/S0907444901013403. [DOI] [PubMed] [Google Scholar]
- Jones TA, Thirup S. Using known substructures in protein model building and crystallography. EMBO J. 1986;5:819–822. doi: 10.1002/j.1460-2075.1986.tb04287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. On the use of sequence homologies to predict protein structure: Identical pentapeptides can have completely different conformations. Proc Natl Acad Sci USA. 1984;81:1075–1078. doi: 10.1073/pnas.81.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan W, Littlejohn TG. Swiss-PDB Viewer (Deep View) Brief Bioinformatics. 2001;2:195–197. doi: 10.1093/bib/2.2.195. [DOI] [PubMed] [Google Scholar]
- Karchin R, Cline M, Mandel-Gutfreund Y, Karplus K. Hidden Markov models that use predicted local structure for fold recognition: Alphabets of backbone geometry. Proteins. 2003;51:504–514. doi: 10.1002/prot.10369. [DOI] [PubMed] [Google Scholar]
- Karchin R, Diekhans M, Kelly L, Thomas DJ, Pieper U, Eswar N, Haussler D, Sali A. LS-SNP: Large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- Karplus K. SAM-T08, HMM-based protein structure prediction. Nucleic Acids Res. 2009;37:W492–W497. doi: 10.1093/nar/gkp403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Karplus K, Karchin R, Draper J, Casper J, Mandel-Gutfreund Y, Diekhans M, Hughey R. Combining local-structure, fold-recognition, and new fold methods for protein structure prediction. Proteins. 2003;53(Suppl 6):491–496. doi: 10.1002/prot.10540. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJ. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- Khafizov K, Staritzbichler R, Stamm M, Forrest LR. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry. 2010;49:10702–10713. doi: 10.1021/bi101256x. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl P, Delarue M. A self consistent mean field approach to simultaneous gap closure and side-chain positioning in homology modelling. Nat Struct Biol. 1995;2:163–170. doi: 10.1038/nsb0295-163. [DOI] [PubMed] [Google Scholar]
- Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: A multiple structural alignment algorithm. Proteins. 2006;64:559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK-a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Lessel U, Schomburg D. Similarities between protein 3-D structures. Protein Eng. 1994;7:1175–1187. doi: 10.1093/protein/7.10.1175. [DOI] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M. Accurate modeling of protein conformation by automatic segment matching. J Mol Biol. 1992;226:507–533. doi: 10.1016/0022-2836(92)90964-L. [DOI] [PubMed] [Google Scholar]
- Li R, Chen X, Gong B, Selzer PM, Li Z, Davidson E, Kurzban G, Miller RE, Nuzum EO, McKerrow JH, Fletterick RJ, Gillmor SA, Craik CS, Kuntz ID, Cohen FE, Kenyon GL. Structure-based design of parasitic protease inhibitors. Bioorg Med Chem. 1996;4:1421–1427. doi: 10.1016/0968-0896(96)00136-8. [DOI] [PubMed] [Google Scholar]
- Lin J, Qian J, Greenbaum D, Bertone P, Das R, Echols N, Senes A, Stenger B, Gerstein M. GeneCensus: Genome comparisons in terms of metabolic pathway activity and protein family sharing. Nucleic Acids Res. 2002;30:4574–4582. doi: 10.1093/nar/gkf555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E, Elofsson A. Identification of related proteins on family, superfamily and fold level. J Mol Biol. 2000;295:613–625. doi: 10.1006/jmbi.1999.3377. [DOI] [PubMed] [Google Scholar]
- Lupyan D, Leo-Macias A, Ortiz AR. A new progressive-iterative algorithm for multiple structure alignment. Bioinformatics. 2005;21:3255–3263. doi: 10.1093/bioinformatics/bti527. [DOI] [PubMed] [Google Scholar]
- Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Madhusudhan MS, Marti-Renom MA, Sanchez R, Sali A. Variable gap penalty for protein sequence-structure alignment. Protein Eng Des Sel. 2006;19:129–133. doi: 10.1093/protein/gzj005. [DOI] [PubMed] [Google Scholar]
- Madhusudhan MS, Webb BM, Marti-Renom MA, Eswar N, Sali A. Alignment of multiple protein structures based on sequence and structure features. Protein Eng Des Sel. 2009;22:569–574. doi: 10.1093/protein/gzp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods. 2009;6:551–552. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom MA, Ilyin VA, Sali A. DBAli: A database of protein structure alignments. Bioinformatics. 2001;17:746–747. doi: 10.1093/bioinformatics/17.8.746. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Madhusudhan MS, Sali A. Alignment of protein sequences by their profiles. Protein Sci. 2004;13:1071–1087. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom MA, Madhusudhan MS, Fiser A, Rost B, Sali A. Reliability of assessment of protein structure prediction methods. Structure. 2002;10:435–440. doi: 10.1016/S0969-2126(02)00731-1. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Matsumoto R, Sali A, Ghildyal N, Karplus M, Stevens RL. Packaging of proteases and proteoglycans in the granules of mast cells and other hematopoietic cells. A cluster of histidines on mouse mast cell protease 7 regulates its binding to heparin serglycin proteoglycans. J Biol Chem. 1995;270:19524–19531. doi: 10.1074/jbc.270.33.19524. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Jones DT. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277:1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- Melo F, Sali A. Fold assessment for comparative protein structure modeling. Protein Sci. 2007;16:2412–2426. doi: 10.1110/ps.072895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo F, Sanchez R, Sali A. Statistical potentials for fold assessment. Protein Sci. 2002;11:430–448. doi: 10.1002/pro.110430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezei M. Chameleon sequences in the PDB. Protein Eng. 1998;11:411–414. doi: 10.1093/protein/11.6.411. [DOI] [PubMed] [Google Scholar]
- Mirkovic N, Marti-Renom MA, Weber BL, Sali A, Monteiro AN. Structure-based assessment of missense mutations in human BRCA1: Implications for breast and ovarian cancer predisposition. Cancer Res. 2004;64:3790–3797. doi: 10.1158/0008-5472.CAN-03-3009. [DOI] [PubMed] [Google Scholar]
- Misura KM, Baker D. Progress and challenges in high-resolution refinement of protein structure models. Proteins. 2005;59:15–29. doi: 10.1002/prot.20376. [DOI] [PubMed] [Google Scholar]
- Misura KM, Chivian D, Rohl CA, Kim DE, Baker D. Physically realistic homology models built with ROSETTA can be more accurate than their templates. Proc Natl Acad Sci USA. 2006;103:5361–5366. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa JM, Ibanez-Tallon I, Crabtree GW, Sanchez R, Sali A, Role LW, Heintz N. lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron. 1999;23:105–114. doi: 10.1016/S0896-6273(00)80757-6. [DOI] [PubMed] [Google Scholar]
- Modi S, Paine MJ, Sutcliffe MJ, Lian LY, Primrose WU, Wolf CR, Roberts GC. A model for human cytochrome P450 2D6 based on homology modeling and NMR studies of substrate binding. Biochemistry. 1996;35:4540–4550. doi: 10.1021/bi952742o. [DOI] [PubMed] [Google Scholar]
- Moult J. A decade of CASP: Progress, bottlenecks and prognosis in protein structure prediction. Curr Opin Struct Biol. 2005;15:285–289. doi: 10.1016/j.sbi.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Moult J, James MN. An algorithm for determining the conformation of polypeptide segments in proteins by systematic search. Proteins. 1986;1:146–163. doi: 10.1002/prot.340010207. [DOI] [PubMed] [Google Scholar]
- Moult J, Fidelis K, Zemla A, Hubbard T. Critical assessment of methods of protein structure prediction (CASP)-round V. Proteins. 2003;53(Suppl 6):334–339. doi: 10.1002/prot.10556. [DOI] [PubMed] [Google Scholar]
- Moult J, Fidelis K, Kryshtafovych A, Rost B, Tramontano A. Critical assessment of methods of protein structure prediction—Round VIII. Proteins. 2009;77(Suppl 9):1–4. doi: 10.1002/prot.22589. [DOI] [PubMed] [Google Scholar]
- Nagarajaram HA, Reddy BV, Blundell TL. Analysis and prediction of inter-strand packing distances between beta-sheets of globular proteins. Protein Eng. 1999;12:1055–1062. doi: 10.1093/protein/12.12.1055. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Notredame C. Computing multiple sequence/structure alignments with the T-coffee package. Curr Protoc Bioinformatics. 2010;29:3.8.1–3.8.25. doi: 10.1002/0471250953.bi0308s04. 3.8. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Ohlson T, Wallner B, Elofsson A. Profile-profile methods provide improved fold-recognition: A study of different profile-profile alignment methods. Proteins. 2004;57:188–197. doi: 10.1002/prot.20184. [DOI] [PubMed] [Google Scholar]
- Oliva B, Bates PA, Querol E, Aviles FX, Sternberg MJ. An automated classification of the structure of protein loops. J Mol Biol. 1997;266:814–830. doi: 10.1006/jmbi.1996.0819. [DOI] [PubMed] [Google Scholar]
- Ortiz AR, Strauss CEM, Olmea O. MAMMOTH (matching molecular models obtained from theory): An automated method for model comparison. Protein Sci. 2002;11:2606–2621. doi: 10.1110/ps.0215902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko AR. Finding weak similarities between proteins by sequence profile comparison. Nucleic Acids Res. 2003;31:683–689. doi: 10.1093/nar/gkg154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J Mol Biol. 1998;284:1201–1210. doi: 10.1006/jmbi.1998.2221. [DOI] [PubMed] [Google Scholar]
- Pawlowski K, Bierzynski A, Godzik A. Structural diversity in a family of homologous proteins. J Mol Biol. 1996;258:349–366. doi: 10.1006/jmbi.1996.0255. [DOI] [PubMed] [Google Scholar]
- Pearl F, Todd A, Sillitoe I, Dibley M, Redfern O, Lewis T, Bennett C, Marsden R, Grant A, Lee D, Akpor A, Maibaum M, Harrison A, Dallman T, Reeves G, Diboun I, Addou S, Lise S, Johnston C, Sillero A, Thornton J, Orengo C. The CATH Domain Structure Database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucleic Acids Res. 2005;33:D247–D251. doi: 10.1093/nar/gki024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR. Using the FASTA program to search protein and DNA sequence databases. Methods Mol Biol. 1994;24:307–331. doi: 10.1385/0-89603-246-9:307. [DOI] [PubMed] [Google Scholar]
- Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- Pegg SC, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Leveraging enzyme structure-function relationships for functional inference and experimental design: The structure-function linkage database. Biochemistry. 2006;45:2545–2555. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- Pei J, Kim BH, Grishin NV. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrey D, Honig B. Protein structure prediction: Inroads to biology. Mol Cell. 2005;20:811–819. doi: 10.1016/j.molcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Pieper U, Webb B, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, Khuri N, Spill Y, Weinkam P, Hammel M, Tainer J, Nilges M, Sali A. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2014;42:336–346. doi: 10.1093/nar/gkt1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjolander K, Ferrin TE, Burley SK, Sali A. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2011;39:465–474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski S. Searching databases of conserved sequence regions by aligning protein multiple-alignments. Nucleic Acids Res. 1996;24:3836–3845. doi: 10.1093/nar/24.19.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prlic A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE. Pre-calculated protein structure alignments at the RCSB PDB website. Bioinformatics. 2010;26:2983–2985. doi: 10.1093/bioinformatics/btq572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que X, Brinen LS, Perkins P, Herdman S, Hirata K, Torian BE, Rubin H, McKerrow JH, Reed SL. Cysteine proteinases from distinct cellular compartments are recruited to phagocytic vesicles by Entamoeba histolytica. Mol Biochem Parasitol. 2002;119:23–32. doi: 10.1016/S0166-6851(01)00387-5. [DOI] [PubMed] [Google Scholar]
- Ray A, Lindahl E, Wallner B. Improved model quality assessment using ProQ2. BMC Bioinformatics. 2012;13:224. doi: 10.1186/1471-2105-13-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Hauser A, Soding J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- Ring CS, Kneller DG, Langridge R, Cohen FE. Taxonomy and conformational analysis of loops in proteins. J Mol Biol. 1992;224:685–699. doi: 10.1016/0022-2836(92)90553-V. [DOI] [PubMed] [Google Scholar]
- Ring CS, Sun E, McKerrow JH, Lee GK, Rosenthal PJ, Kuntz ID, Cohen FE. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc Natl Acad Sci U S A. 1993;90:3583–3587. doi: 10.1073/pnas.90.8.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche DB, Buenavista MT, Tetchner SJ, McGuffin LJ. The IntFOLD server: An integrated web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction. Nucleic Acids Res. 2011;39:W171–176. doi: 10.1093/nar/gkr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufino SD, Donate LE, Canard LH, Blundell TL. Predicting the conformational class of short and medium size loops connecting regular secondary structures: Application to comparative modelling. J Mol Biol. 1997;267:352–367. doi: 10.1006/jmbi.1996.0851. [DOI] [PubMed] [Google Scholar]
- Rychlewski L, Zhang B, Godzik A. Fold and function predictions for Mycoplasma genitalium proteins. Fold Des. 1998;3:229–238. doi: 10.1016/S1359-0278(98)00034-0. [DOI] [PubMed] [Google Scholar]
- Sadreyev R, Grishin N. COMPASS: A tool for comparison of multiple protein alignments with assessment of statistical significance. J Mol Biol. 2003;326:317–336. doi: 10.1016/S0022-2836(02)01371-2. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sali A, Overington JP. Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci. 1994;3:1582–1596. doi: 10.1002/pro.5560030923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudrala R, Moult J. A graph-theoretic algorithm for comparative modeling of protein structure. J Mol Biol. 1998;279:287–302. doi: 10.1006/jmbi.1998.1689. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Sali A. Advances in comparative protein-structure modelling. Curr Opin Struct Biol. 1997a;7:206–214. doi: 10.1016/S0959-440X(97)80027-9. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Sali A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins. 1997b;(Suppl 1):50–58. doi: 10.1002/(sici)1097-0134(1997)1+<50::aid-prot8>3.3.co;2-w. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Sali A. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc Natl Acad Sci USA. 1998;95:13597–13602. doi: 10.1073/pnas.95.23.13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saqi MA, Russell RB, Sternberg MJ. Misleading local sequence alignments: Implications for comparative protein modelling. Protein Eng. 1998;11:627–630. doi: 10.1093/protein/11.8.627. [DOI] [PubMed] [Google Scholar]
- Sauder JM, Arthur JW, Dunbrack RL., Jr Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins. 2000;40:6–22. doi: 10.1002/(SICI)1097-0134(20000701)40:1%3c6::AID-PROT30%3e3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Sali A. FoXS: A web server for rapid computation and fitting of SAXS Profiles. Nucleic Acids Res. 2010;38:541–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L. The importance of alignment accuracy for molecular replacement. Acta Crystallogr D Biol Crystallogr. 2004;60:1229–1236. doi: 10.1107/S0907444904010145. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers BD, Zhu K, Zhao S, Friesner RA, Jacobson MP. Toward better refinement of comparative models: Predicting loops in inexact environments. Proteins. 2008;72:959–971. doi: 10.1002/prot.21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer PM, Chen X, Chan VJ, Cheng M, Kenyon GL, Kuntz ID, Sakanari JA, Cohen FE, McKerrow JH. Leishmania major: Molecular modeling of cysteine proteases and prediction of new nonpeptide inhibitors. Exp Parasitol. 1997;87:212–221. doi: 10.1006/expr.1997.4220. [DOI] [PubMed] [Google Scholar]
- Shatsky M, Nussinov R, Wolfson HJ. A method for simultaneous alignment of multiple protein structures. Proteins. 2004;56:143–156. doi: 10.1002/prot.10628. [DOI] [PubMed] [Google Scholar]
- Shatsky M, Nussinov R, Wolfson HJ. Optimization of multiple-sequence alignment based on multiple-structure alignment. Proteins. 2006;62:209–217. doi: 10.1002/prot.20665. [DOI] [PubMed] [Google Scholar]
- Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y, Sali A, Herzog H, Lahnstein J, Krilis SA. Site-directed mutagenesis of recombinant human beta 2-glycoprotein I identifies a cluster of lysine residues that are critical for phospholipid binding and anti-cardiolipin antibody activity. J Immunol. 1996;157:3744–3751. [PubMed] [Google Scholar]
- Shenkin PS, Yarmush DL, Fine RM, Wang HJ, Levinthal C. Predicting antibody hypervariable loop conformation. I Ensembles of random conformations for ringlike structures. Biopolymers. 1987;26:2053–2085. doi: 10.1002/bip.360261207. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Sibanda BL, Blundell TL, Thornton JM. Conformation of beta-hairpins in protein structures. A systematic classification with applications to modelling by homology, electron density fitting and protein engineering. J Mol Biol. 1989;206:759–777. doi: 10.1016/0022-2836(89)90583-4. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990;213:859–883. doi: 10.1016/S0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. Knowledge-based potentials for proteins. Curr Opin Struct Biol. 1995;5:229–235. doi: 10.1016/0959-440X(95)80081-6. [DOI] [PubMed] [Google Scholar]
- Skolnick J, Kihara D. Defrosting the frozen approximation: PROSPECTOR–a new approach to threading. Proteins. 2001;42:319–331. doi: 10.1002/1097-0134(20010215)42:3%3c319::AID-PROT30%3e3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Dimaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-Resolution Comparative Modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA-ribosome and subunit-subunit interactions. Cell. 2001;107:373–386. doi: 10.1016/S0092-8674(01)00539-6. [DOI] [PubMed] [Google Scholar]
- Srinivasan N, Blundell TL. An evaluation of the performance of an automated procedure for comparative modelling of protein tertiary structure. Protein Eng. 1993;6:501–512. doi: 10.1093/protein/6.5.501. [DOI] [PubMed] [Google Scholar]
- Sutcliffe MJ, Dobson CM, Oswald RE. Solution structure of neuronal bungarotoxin determined by two-dimensional NMR spectroscopy: Calculation of tertiary structure using systematic homologous model building, dynamical simulated annealing, and restrained molecular dynamics. Biochemistry. 1992;31:2962–2970. doi: 10.1021/bi00126a017. [DOI] [PubMed] [Google Scholar]
- Sutcliffe MJ, Haneef I, Carney D, Blundell TL. Knowledge based modelling of homologous proteins, Part I: Three-dimensional frameworks derived from the simultaneous superposition of multiple structures. Protein Eng. 1987;1:377–384. doi: 10.1093/protein/1.5.377. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Flores TP, Orengo CA. Multiple protein structure alignment. Protein Sci. 1994;3:1858–1870. doi: 10.1002/pro.5560031025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham CM, Srinivasan N, Thorpe CJ, Overington JP, Kalsheker NA. Comparative modelling of major house dust mite allergen Der p I: Structure validation using an extended environmental amino acid propensity table. Protein Eng. 1994;7:869–894. doi: 10.1093/protein/7.7.869. [DOI] [PubMed] [Google Scholar]
- Topham CM, McLeod A, Eisenmenger F, Overington JP, Johnson MS, Blundell TL. Fragment ranking in modelling of protein structure. Conformationally constrained environmental amino acid substitution tables. J Mol Biol. 1993;229:194–220. doi: 10.1006/jmbi.1993.1018. [DOI] [PubMed] [Google Scholar]
- Unger R, Harel D, Wherland S, Sussman JL. A 3D building blocks approach to analyzing and predicting structure of proteins. Proteins. 1989;5:355–373. doi: 10.1002/prot.340050410. [DOI] [PubMed] [Google Scholar]
- Vakser IA. Protein docking for low-resolution structures. Protein Eng. 1995;8:371–377. doi: 10.1093/protein/8.4.371. [DOI] [PubMed] [Google Scholar]
- van Gelder CW, Leusen FJ, Leunissen JA, Noordik JH. A molecular dynamics approach for the generation of complete protein structures from limited coordinate data. Proteins. 1994;18:174–185. doi: 10.1002/prot.340180209. [DOI] [PubMed] [Google Scholar]
- van Vlijmen HW, Karplus M. PDB-based protein loop prediction: Parameters for selection and methods for optimization. J Mol Biol. 1997;267:975–1001. doi: 10.1006/jmbi.1996.0857. [DOI] [PubMed] [Google Scholar]
- Vernal J, Fiser A, Sali A, Muller M, Cazzulo JJ, Nowicki C. Probing the specificity of a trypanosomal aromatic alpha-hydroxy acid dehydrogenase by site-directed mutagenesis. Biochem Biophys Res Commun. 2002;293:633–639. doi: 10.1016/S0006-291X(02)00270-X. [DOI] [PubMed] [Google Scholar]
- von Ohsen N, Sommer I, Zimmer R. Profile-profile alignment: A powerful tool for protein structure prediction. Pac Symp Biocomput. 2003:252–263. [PubMed] [Google Scholar]
- Wang G, Dunbrack RL., Jr Scoring profile-to-profile sequence alignments. Protein Sci. 2004;13:1612–1626. doi: 10.1110/ps.03601504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Canutescu AA, Dunbrack RL., Jr SCWRL and MolIDE: Computer programs for side-chain conformation prediction and homology modeling. Nat Protoc. 2008;3:1832–1847. doi: 10.1038/nprot.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinkam P, Pons J, Sali A. Structure-based model of allostery predicts coupling between distant sites. Proc Natl Acad Sci USA. 2012;109:4875–4880. doi: 10.1073/pnas.1116274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley SK. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell. 1998;94:439–449. doi: 10.1016/S0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhang Y. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Fiser A, ter Kuile B, Sali A, Muller M. Convergent evolution of Trichomonas vaginalis lactate dehydrogenase from malate dehydrogenase. Proc Natl Acad Sci U S A. 1999;96:6285–6290. doi: 10.1073/pnas.96.11.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Soto CS, Honig B. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc Natl Acad Sci USA. 2002;99:7432–7437. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LZ, Sanchez R, Sali A, Heintz N. Ligand specificity of brain lipid-binding protein. J Biol Chem. 1996;271:24711–24719. doi: 10.1074/jbc.271.40.24711. [DOI] [PubMed] [Google Scholar]
- Xu J, Li M, Kim D, Xu Y. RAPTOR: Optimal protein threading by linear programming. J Bioinform Comput Biol. 2003;1:95–117. doi: 10.1142/S0219720003000186. [DOI] [PubMed] [Google Scholar]
- Yona G, Levitt M. Within the twilight zone: A sensitive profile-profile comparison tool based on information theory. J Mol Biol. 2002;315:1257–1275. doi: 10.1006/jmbi.2001.5293. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Rosenfeld R, Vajda S, DeLisi C. Determining protein loop conformation using scaling-relaxation techniques. Protein Sci. 1993;2:1242–1248. doi: 10.1002/pro.5560020806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005;58:321–328. doi: 10.1002/prot.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Pincus DL, Zhao S, Friesner RA. Long loop prediction using the protein local optimization program. Proteins. 2006;65:438–452. doi: 10.1002/prot.21040. [DOI] [PubMed] [Google Scholar]
Internet Resources
- http://salilab.org/modeller/; Webb B, Madhusudhan MS, Shen M-Y, Dong GQ, Marti-Renom MA, Eswar N, Alber F, Topf M, Oliva B, Fiser A, Sanchez R, Yerkovich B, Badretdinov A, Melo F, Overington JP, Feyfant E, Sali A. MODELLER, A Protein Structure Modeling Program, Release 9.15 2015 [Google Scholar]
- http://salilab.org/modbase/; Pieper U, Webb B, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, Khuri N, Spill YG, Weinkam P, Hammel M, Tainer JA, Nilges M, Sali A. MODBASE, a database of annotated comparative protein structure models. 2015 doi: 10.1093/nar/gkt1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://salilab.org/modeval/; Eramian D, Shen M-Y, Melo F, Pieper U, Webb B, Eswar N, Sanchez R, Sali A. ModEval, a web server for evaluating protein structure models 2015 [Google Scholar]
- http://salilab.org/modloop/; Fiser A, Do R, Webb B, Pieper U, Sali A. ModLoop, a web server for modeling of loops in protein structures 2003 [Google Scholar]
- http://salilab.org/allosmod/; Weinkam P, Pons J, Webb B, Pieper U, Tjioe E, Sali A. AllosMod, a web server to set up and run simulations based on modeled energy landscapes 2012 [Google Scholar]
- http://salilab.org/foxs/; Schneidman-Duhovny D, Hammel M, Tainer J, Sali A. FoXS, a web server for fast SAXS profile computation with Debye formula 2013 [Google Scholar]