

Abstract
Proprotein convertase subtilisin kexin 9 (PCSK9) is a key regulator of low-density lipoprotein receptor levels and LDL-cholesterol levels. Loss-of-function mutations in PCSK9 gene are associated with hypocholesterolaemia and protection against cardiovascular disease, identifying PCSK9 inhibition as a valid therapeutic approach to manage hypercholesterolaemia and related diseases. Although PCSK9 is expressed mainly in the liver, it is present also in other tissues and organs with specific functions, raising the question of whether a pharmacological inhibition of PCSK9 to treat hypercholesterolaemia and associated cardiovascular diseases might be helpful or deleterious in non-hepatic tissues. For example, PCSK9 is expressed in the vascular wall, in the kidneys, and in the brain, where it was proposed to play a role in development, neurocognitive process, and neuronal apoptosis. A link between PCSK9 and immunity was also proposed as both sepsis and viral infections are differentially affected in the presence or absence of PCSK9. Despite the increasing number of observations, the debate on the exact roles of PCSK9 in extrahepatic tissues is still ongoing, and as very effective drugs that inhibit PCSK9 have become available to the clinician, a better understanding of the biological roles of PCSK9 is warranted.
Keywords: PCSK9, LDL, APO B, Monoclonal antibodies, LDLR
1. Introduction
Proprotein convertase subtilisin kexin 9 (PCSK9) is a serine protease that plays a key role in the regulation of hepatic low-density lipoprotein receptor (LDLR) function. In fact, circulating PCSK9 binds LDLR, targeting it to lysosomal degradation within cells,1 which leads to reduced LDLR expression on the cell membrane, decreased LDL catabolism, and increased plasma levels of LDL-cholesterol (LDL-C). Loss-of-function (LOF) mutations in the gene coding for PCSK9 result in lower levels of LDL-C and protection against cardiovascular disease,2–4 whereas the opposite is observed with gain-of-function (GOF) mutations for PCSK9.5 These findings set the stage for investigating PCSK9 inhibition as a way to reduce plasma LDL-C levels. The development and therapeutic availability of monoclonal antibodies (mAbs) against circulating PCSK9, which effectively reduce LDL-C up to 70%, is the result of this approach.5–7 In this context, however, it is worth noting that such mAbs can block only extracellular circulating PCSK9. The physiopathological relevance of intracellular PCSK9, if any, requires better understanding not only for controlling the LDLR levels8 but also for other, yet poorly investigated, activities.
Although PCSK9 is mainly produced in the liver, it is expressed also in other tissues, including kidney, pancreas, and brain,9 raising legitimate questions regarding extrahepatic effects of PCSK9 and unwanted effects of pharmacological inhibition. The aim of this review is to discuss critically the effects of PCSK9 on circulating lipoproteins as well as on extrahepatic tissues thus providing the biological bases for monitoring additional consequences of PCSK9 inhibition independent of LDL-C lowering.
2. PCSK9 and apoB-containing lipoproteins
There is a bidirectional connection between apoB-containing lipoproteins and PCSK9: on one hand PCSK9, by enhancing LDLR degradation, increases LDL-C levels; on the other hand, apoB-containing lipoproteins act also as bulk plasma carriers of PCSK9.10 Indeed, PCSK9-LDL complexes have been observed initially in in vitro systems or in plasma from transgenic mice expressing human PCSK9.11,12 Later this was also confirmed in human plasma,13,10 where up to 40% of total PCSK9 appears to be associated with LDL,10,14 with a Kd of 160–320 nM at neutral pH.10,15 Although a large percentage of PCSK9 is bound to LDL, only one in 500–1000 apoB-containing lipoproteins carries a single PCSK9 molecule.14 This interaction makes it likely that the correlation between these two molecules in plasma16,17 is the result of a complex series of events. For example, it is not known where and how the association of PCSK9 with LDL takes place. Interestingly, although PCSK9 binds to apoB within the hepatocyte,11 it does not associate with the apoB-containing VLDL secreted by hepatocytes;10–13 therefore, the association of PCSK9 with LDL occurs in plasma and requires VLDL catabolism and perhaps apoB conformational changes or surface exposure of specific, yet to be identified, lipid structures hidden in the much larger VLDL or exposed during lipolysis.
The potential clinical relevance of PCSK9 association with LDL was first demonstrated in patients undergoing lipoprotein apheresis treatment,14,18–20 a dialysis procedure that acutely removes apoB-containing lipoproteins from plasma. Apheresis using dextran sulfate cellulose beads columns or heparin column reduces plasma PCSK9 levels by more than 50%, along with the known 70–80% reduction in LDL.14,19–21 The loss of PCSK9 during apheresis is mainly due to the removal of LDL-bound PCSK9, but also to the loss of some free PCSK9. One could speculate that PCSK9 removal contributes to the overall effects of regular apheresis on plasma LDL levels.
In addition, it should be considered that at least two forms of PCSK9 exist in plasma, the intact protein (62 kDa) and the furin-cleaved form (55 kDa), which is less active,22–24 but still maintains a significant residual ability to remove surface LDLR.25 The observation that, in plasma, most of LDL-bound PCSK9 is in the intact form, whereas the furin-cleaved form is mostly found not associated with apoB-containing lipoproteins suggests that LDL-bound PCSK9 might represent the active form of PCSK9 or at least contains the site for interaction with LDL13,10,22 (Figure 1).
Figure 1.
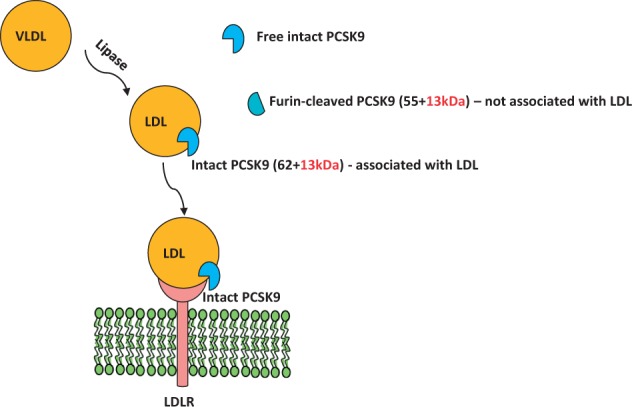
Plasma PCSK9. The intact form of PCSK9 (62 + 13 kDa) in plasma is found predominantly on LDL but not on VLDL. The furin-cleaved form of PCSK9 (55 + 13 kDa) in plasma is found predominantly as apoB-free.
In vitro, LDL inhibits PCSK9-mediated LDLR degradation,10 suggesting that LDL can compete for the binding of intact PCSK9 to the LDLR at the cell surface. However, as most of the intact PCSK9 in vivo is already associated with LDL,14,19 the relevance of these in vitro findings remains unclear. Therefore, one might speculate that LDLR turnover may be mostly controlled by the few LDL particles (one every 500/1000) carrying PCSK9, an intriguing sequitur to the old knowledge that LDLR recycles a few hundred times before finding its demise. Currently available enzyme-linked immunosorbent assays only measure total PCSK9 levels in plasma, without distinguishing between intact and furin-cleaved forms, or between LDL-bound and apoB-free forms. Thus, new methods to quantify plasma PCSK9 forms that serve as surrogate markers for PCSK9 function are needed, as data showing that genetically determined low levels of PCSK9 are associated with low LDL-C levels and reduced risk of CHD,2 whereas total plasma PCSK9 levels only in some studies but not others independently predicted the incidence of CVD events.26–29
3. PCSK9 and lipoprotein synthesis
It is becoming clear that the pro-atherosclerotic action of PCSK9 is not exclusively due to the effect on plasma LDL clearance. Population studies have shown a correlation between plasma PCSK9 and fasting TG levels in both genders30–34 although not in obese people.35 In line with this, subjects carrying the PCSK9 S127R Gain Of Function (GOF) mutation present increased levels of LDL, VLDL, and remnants.36 Therapies that inhibit PCSK9 significantly reduce plasma TG levels,37,38 although not as much as statins.39
Both secretion and degradation of hepatic TG-rich lipoproteins are negatively regulated by LDLR,40 and thus PCSK9-induced degradation of LDLR can result in hypertriglyceridaemia through multiple mechanisms. PCSK9 is believed to play a role in the secretion of hepatic apoB-containing lipoproteins from the liver; indeed, acute adenoviral PCSK9 overexpression in fasted mice causes hypertriglyceridaemia due to dramatically increased hepatic production of VLDL-triglyceride and apoB.41 In addition, chronic hepatic PCSK9 overexpression stimulates hepatic VLDL production;11 transgenic expression of human PCSK9 in mice increases hepatic lipogenesis in an apoE and LDLR-dependent manner;42 and changes in hepatic VLDL production parallel those of PCSK9 production in hepatocytes.43 Finally, in mice the absence of PCSK9 results in a reduced post-prandial TG peak.44
The mechanisms by which PCSK9 impacts hepatic TG-rich lipoproteins secretion are poorly understood. PCSK9 overexpression in mice modulates hepatic TG metabolism through regulation of the SREBP1c pathway, with both transcriptional and post-transcriptional regulation of FAS, and through the up-regulated synthesis of non-sterol receptor element (SRE) regulated genes such as apoB, apoE, DGAT1, and microsomal triglyceride transfer protein (MTTP),42 an ER apoB chaperone critical for the transfer of lipids to apoB.
The intestine is the other major source of TG-rich lipoproteins, accounting for a significant portion of plasma lipids in the postprandial state (transported in chylomicrons and chylomicron remnants), with apoB48 as their signature apolipoprotein.45 PCSK9 expression in the intestine is quantitatively second only to hepatic expression,9 and studies have shown that absence of PCSK9 reduces apoB48 secretion and protects mice from postprandial hypertriglyceridaemia.44,46 In addition, in vivo and in vitro studies have shown that PCSK9 increases assembly and secretion of intestinal TG-rich lipoproteins through mechanisms encompassing the transcriptional (increased FAS, SCD, and DGAT2 expression) and post-transcriptional (increased MTTP activity and decreased apoB degradation) mechanisms, and resulting in lipid accumulation in enterocytes.47 PCSK9 also affects intestinal cholesterol absorption, as the addition of exogenous PCSK9 to cultured enterocytes reduced the expression of NPC1L1, a key protein in intestinal cholesterol absorption.48 As it was shown that LDLR directly influences apoB stability by promoting autophagic degradation in hepatocytes and thus reducing the rate of lipoprotein secretion,49 it is expected that lower LDLR levels in enterocytes48 will have a similar effect. A summary of mechanisms of PCSK9 induction of the synthesis of apoB-containing lipoproteins is shown in Figure 2.
Figure 2.

PCSK9 and apoB-containing lipoproteins metabolism. PCSK9 regulates surface LDLR levels via targeting of both proteins to lysosomal degradation (1). The main consequence of the decreased PCSK9 interaction with the LDLR is the increase in the intracellular cholesterol pool which promotes: (2) a reduction of the activity of the SRE-dependent pathway and of intracellular cholesterol synthesis, (3) a reduction in the expression of non-SRE genes involved in lipogenesis, (4) a reduction of apoB lipidation, and (5) a reduction of apoB/apoE particle uptake and degradation.
Although the exact cellular mechanisms explaining PCSK9 stimulation of TG-rich lipoproteins in enterocytes and hepatocytes are not fully understood, available evidence extends the established notion that apoB is regulated exclusively at the post-transcriptional level50 and supports the hypothesis for a role of PCSK9 inhibition on cellular lipid trafficking. To date, clinical studies with anti PCSK9 antibodies were not designed to address the changes in plasma TG levels in hypertriglyceridaemic cohorts; moreover, the antibodies do not directly affect intracellular PCSK9 thus halting from appreciating a direct effect on cellular PCSK9 function.
4. PCSK9, glucose metabolism, and obesity
In addition to the LDLR, PCSK9 targets also the VLDLR and ApoER251,52 which are intimately involved in the hydrolysis of triglyceride-rich lipoproteins, a critical step for fat utilization in the heart and in the muscles or for fat storage in adipose tissue. Pcsk9−/− mice fed on a chow diet present adipocyte hypertrophy and increased in vivo fatty acid uptake,51 which results in increased visceral adiposity. Of note, this phenotype is independent of LDLR expression, as it is also observed in Pcsk9−/−/Ldlr−/− mice. Even more importantly, PCSK9 deficiency was associated with increased VLDLR expression in fat depots51 and also with increased cluster of differentiation (CD) 36 expression.53 PCSK9-mediated CD36 degradation was proposed to limit fatty acid uptake and triglyceride accumulation in tissues, such as the liver.53 In parallel, studies using transgenic animals with the specific inactivation of PCSK9 in the liver further showed that VLDLR expression is regulated by circulating PCSK9. These data suggest the possibility that by targeting VLDLr and/or CD36, PCSK9 could be involved also in the modulation of TG metabolism.
Indeed, a lower postprandial response was observed in Pcsk9−/− mice, with a reduced triglyceride peak after olive oil gavage, caused by both reduced intestinal apoB secretion in the lymph and increased clearance of plasma chylomicrons, an effect which was not dependent only on LDLR.44 Additionally, tumour necrosis factor alpha (TNF-α), which is produced during low chronic inflammatory conditions such as those observed in obesity,54 was shown to induce PCSK9 expression in hepatocytes through the involvement of SREBP-1 and HNF-1 alpha,55 via SOCS3 (Suppressor of Cytokine Signalling 3), a contributor to insulin resistance. These findings further confirm how the inhibition of PCSK9 will decrease plasma TG levels by increasing catabolism of TG-rich lipoproteins, but as a consequence support the possibility that it might also increase peripheral fat accumulation.
The link between PCSK9 and metabolic dysfunction extends beyond VLDLR modulation, as PCSK9 is expressed also in the pancreas (Figure 3), mainly in δ cells.56 Pcsk9−/− mice present an altered morphology of pancreatic islets,57 although it is not clear whether this is associated with impaired insulin secretion. Indeed, two conflicting reports in animal models were recently published: one showing a neutral effect of PCSK9 on insulin secretion and glucose tolerance,56 and the other showing that Pcsk9−/− mice are hypoinsulinaemic, hyperglycaemic, and intolerant to glucose.57 In the latter study, pancreatic islets from Pcsk9−/− mice presented with signs of malformation, apoptosis and inflammation. Differences in the experimental design, including the age of the mice (8–10 weeks56 or 4–5 months57) and the strains (with different genetic background) used in these studies may explain such discrepancies. Also in humans, data from carriers of PCSK9 LOF mutations are conflicting, with two reports indicating no pancreatic dysfunction58 or increased incidence of diabetes59 with another linking PCSK9 LOF to an increased incidence of diabetes.60 The possibility that a long-term cholesterol reduction results in metabolic changes, which favour glucose utilization and therefore prompt an increase in plasma glucose levels, cannot be excluded and this is indeed a favoured hypothesis for the effect of statins on incident diabetes.61,62 **As the LDLR-cholesterol axis has been suggested to play a key role in regulating beta cell function and insulin secretion,63 the possibility that PCSK9 plays a physiological role also in the pancreas cannot be excluded.
Figure 3.

Expression and function of PCSK9. PCSK9 is mainly expressed in the liver, where it is involved in the binding and degradation of LDLR. PCSK9 is expressed also in other tissues and organs, where it plays additional functions.
5. PCSK9 and vascular tissues
PCSK9 is expressed in many tissues, including liver, small intestine, kidney, and pancreas9,56 (Figure 3). Recent evidence shows that PCSK9 expression is also observed in cells of the artery wall, including endothelium cell (EC), smooth muscle cell (SMC), and macrophages,64–66 with local effects that can impact vascular homeostasis and atherosclerosis.
Low shear stress up-regulates PCSK9 expression in vascular EC and SMC, and in these cells is coupled with reactive oxygen species (ROS) production, with phosphorylated NF-κB playing a bridging role in LOX-1-mediated PCSK9 expression.67 In turn, PCSK9 derived from SMC cleaves LDLR at the surface of arterial macrophages64 a process that may promote LDL accumulation in the artery wall and oxidized LDL (OxLDL) formation; this could trigger a feed-forward loop generating additional intracellular ROS that result in further LDL modification and their binding to LOX-1.67
Whether PCSK9 is expressed by macrophages is currently a subject of discussion. Murine J774A macrophages do not express PCSK964,66 whereas murine peritoneal macrophages have been reported to secrete functional PCSK9 by Giunzioni et al.,66 while by another group to not express PCSK9, but to respond to PCSK9 expressed by nearby tissues.64 Interestingly, the real-time polymerase chain reaction (PCR) primers used to detect PCSK9 expression in hepatocytes do not detect gene expression in mouse peritoneal macrophages,64,66 whereas semi-quantitative PCR methods66 show PCSK9 expression in macrophages, though at a much lower level compared with hepatocytes. Experiments with PCSK9-expressing bone marrow cells transplanted into PCSK9 knockout (KO) mice show that macrophage-derived PCSK9 is secreted and reaches the circulation, with <1% of total plasma PCSK9 being macrophage derived.66 Further, PCSK9 secreted by macrophages and/or SMC is functionally active and reduces LDLR expression in macrophages with a possible local paracrine and autocrine effect on LDLR degradation in atheroma cells.64,66
The role of macrophage LDLR on lipid accumulation in the artery wall and atherosclerotic lesion development has been established in C57BL6 mice with macrophages expressing or lacking LDLR.68 Animals receiving LDLR−/− bone marrow developed significantly smaller lesions than did animals receiving normal bone marrow, indicating that macrophage LDLR influences the rate of foam cell formation in the artery wall and, consequently, atherosclerosis progression.68,69 It can be speculated, therefore, that reduction of the LDLR number by PCSK9 in macrophages results in a reduced rate of foam cell formation in the artery wall and less atherosclerosis. However, in apoE−/− mice, bone marrow transplantation of leucocytes overexpressing human PCSK9, although resulting in reduced surface LDLR levels on macrophages, did not translate in increased atherosclerotic lesion size,66 thus limiting the impact of macrophage-derived PCSK9 in this context.
More recently, the direct effect of local PCSK9 on the atheroma was studied in the absence of systemic lipid changes.42,66 Local expression of PCSK9 from macrophages affected neither plasma lipid levels nor atherosclerotic lesion size, but PCSK9 accumulated in the artery wall and induced infiltration of Ly6chi monocytes into the atherosclerotic lesion.66 Ly6Chi monocytosis is a hallmark of tissue inflammation,70 as Ly6Chi monocytes are precursors of macrophages with a pro-inflammatory phenotype. Interestingly, the accumulation of PCSK9 in the artery wall, and its effect on Ly6Chi monocyte recruitment into the artery wall were completely abolished when macrophages did not express LDLR.66 The direct involvement of PCSK9 in the formation of atherosclerotic plaque has been shown by a study that evaluated the effect of the anti-PCSK9 mAb alirocumab in a mouse model of atherosclerosis.71 This study showed that alirocumab alone or in combination with atorvastatin significantly reduced plasma lipids by reducing LDLR degradation; this resulted in a reduced atherosclerosis development and the improvement of plaque morphology, with a decrease of macrophage and necrotic content and increased smooth muscle cells and collagen content.71 These findings suggest the possibility that anti-PCSK9 mAb may prevent the formation of new lesions.
Changes in PCSK9 plasma levels may also affect macrophage function, as PCSK9 deficiency was associated with a reduction in OxLDL-induced cytokine expression by macrophages.72 Moreover, transgenic overexpression of PCSK9 promotes lesion inflammation in the vascular wall of mice on apoE−/− but not LDLR−/− background. Several additional findings support a possible role for PCSK9 in the inflammatory processes: (i) PCSK9 levels correlate with white blood cell count in patients with stable coronary artery disease;73 (ii) the pro-inflammatory cytokine interleukin (IL)-1 β disrupts the cholesterol-mediated feedback regulation of LDLR in SMC, thus causing massive uptake of native LDL and foam cell transformation through mechanisms involving mTOR activation both in vitro and in apoE−/− mice,74,75 suggesting a potential role for proteins affecting the LDLR activity such as PCSK9; (iii) macrophage-derived PCSK9 promotes inflammatory macrophage differentiation in vitro and increases monocyte infiltration in the artery wall in apoE−/− mice,66 and anti-PCSK9 treatment reduces monocyte recruitment in apoE3Leiden/cholesteryl ester transfer protein (CETP) mice;71 (iv) lack of PCSK9 protects against septic shock induced by LPS administration, an effect not seen in LDLR−/− mice and in FH patients without functional LDLR;76 (v) PCSK9 gene expression is regulated by HNF-1α, an acute-phase response transcriptional controller, which suggests that PCSK9 expression takes part in the inflammatory process underlying the pathogenesis of coronary heart disease;77 and (vi) PCSK9 expression is also regulated by the pro-inflammatory cytokine TNF-α, in a SOCS3-dependent manner, suggesting a direct link between inflammation and the regulation of lipid metabolism via PCSK9.55
Systemic and local expression of PCSK9 (by either SMC or macrophages) affects macrophage LDLR levels,64,66 and thus the absence of LDLR may mitigate the negative effect of PCSK9 on inflammation. This mechanism explains, at least in part, the anti-atherogenic effect of LDLR absence in macrophages68 and the possible effect of PCSK9 on lesion inflammation. Moreover, it was suggested that PCSK9 interacts with other members of the LDLR family, such as LRP1 and leads to its degradation in many cell types,78 including macrophages.66 As macrophage LRP1 deficiency in mice has been associated with increased atherosclerosis, it is also possible that PCSK9 interaction with LRP1 in the plaque enhances the local inflammatory response.79 Potential pathways involved in PCSK9-mediated inflammation are depicted in Figure 4.
Figure 4.

Pathways involved in PCSK9-mediated inflammation. Intracellular PCSK9, including the internalized one which escapes lysosomal degradation could eventually undergoes re-secretion but exerts cytoplasmic effects that might regulate the expression of genes controlling inflammation (1). PCSK9 could also target LRP-1 which is involved in the activation of JAK/STAT and ERK pathways (2).
6. PCSK9 and the kidney
The kidney is another organ producing PCSK9 to a significant level (Figure 3),9 and in this site the epithelial Na+ channel (ENaC) was identified as a PCSK9 target.80 ENaC plays a critical role in the re-absorption of Na+ from urine across epithelium, and defects in ENaC regulation are responsible for most of the known genetic forms of hypertension.80 PCSK9 interacts with ENaC and reduces its cell surface expression mainly by increasing its degradation in the biosynthetic pathway thus reducing the amount of ENaC available for exocytosis.80 Thus, reductions in PCSK9 might result in increased Na+ renal absorption and increased risk of hypertension. However, PCSK9 deficiency neither increases ENaC expression nor alters blood pressure in mouse models of hypertension,81 a finding in line with the observation that subjects carrying LOF mutations of PCSK9 do not exhibit increased prevalence of hypertension compared with non-carriers.2 Furthermore, in studies with anti-PCSK9 mAbs such as evolocumab or alirocumab, no effect on blood pressure has been reported to date.38,82–87
Patients with impaired renal function exhibit altered lipid metabolism and dyslipidaemia,88 which may in turn contribute to the worsening of renal function and to the development of cardiovascular complications.89,90 Of note, in rats with experimental chronic renal failure, serum PCSK9 levels are significantly increased and liver LDLR decreased compared with pair-fed and controls; higher levels of total cholesterol and LDL-C were also observed, which positively correlated with circulating PCSK9 and negatively with levels of LDLR.91
Several studies have also evaluated the correlation between circulating levels of PCSK9 and renal disease. PCSK9 plasma levels are elevated in subjects with proteinuria compared with matched healthy individuals, and the extent of PCSK9 level increase is proportional to the degree of proteinuria and is not affected by antiproteinuric therapy.92 Within the proteinuric group, statin treatment is associated with higher PCSK9 levels, a finding in agreement with the known effect of statins to increase both LDLR and PCSK9 expressions.93
Patients with severe glomerular proteinuria, also referred to as nephrotic syndrome, exhibit elevated levels of total and LDL-C, mainly due to an acquired LDLR deficiency, which results in a reduced removal of LDL particles from the circulation.94 These patients also have higher PCSK9 levels compared with matched healthy subjects, with a significant direct correlation between plasma PCSK9 and total cholesterol and LDL-C concentrations.95 This finding suggests that increased PCSK9 levels can cause LDLR deficiency, which in turn regulates plasma PCSK9 levels in a reciprocal regulation loop,13 thus contributing to the onset of hypercholesterolaemia in these patients. The mechanisms underpinning these effects are not clear but indicate a possible benefit of PCSK9 inhibition in these patients.
Similarly, plasma PCSK9 levels are higher in patients with chronickidneydisease (CKD) compared with healthy subjects;96 among these patients, those not taking statins showed an inverse correlation between PCSK9 levels and estimated glomerular filtration rate and a direct correlation with total cholesterol and LDL-C.96 In addition, PCSK9 plasma levels were positively correlated with TG and negatively with HDL-C.96 Similar results were reported in another study showing that subjects with CKD on haemodialysis have lower PCSK9 levels when compared with healthy subjects, that the subgroup taking statins have increased PCSK9 levels compared with the subgroup not taking statins, and that there is a positive correlation between PCSK9 levels and TG levels.97
The mechanisms responsible for the increase of PCSK9 levels in patients with CKD are still not known; however, in a model of experimental renal failure, a reduced liver clearance of PCSK9 from the circulation was reported.98 In addition, the progression of CKD may also reduce renal LDLR-mediated clearance of PCSK9, thus contributing to the increase of PCSK9 levels. Other mechanisms contribute to the increased PCSK9 levels in CKD patients, as PCSK9 expression is up-regulated by the chronic pro-inflammatory status present in CKD patients,99 and this mechanism might contribute to induce PCSK9 expression and increase its plasma levels. Altogether, these observations support the hypothesis for a PCSK9 role in the development of dyslipidaemia in renal pathologies, and suggest that a study on the benefit of PCSK9 inhibition should be performed in CKD patients.
7. PCSK9 and infections
Metabolism and levels of lipids and lipoproteins are dramatically altered during infections and therefore systemic factors modulating lipid metabolism, such as PCSK9, may play a role under these conditions.
7.1 Bacterial infections/sepsis
Lipoproteins and their receptors play a key role during sepsis as they favour the hepatic clearance of endotoxins.100–102 Following infection, lipid microbial moieties, which are often present in the microbial cell wall, do not circulate in the free form but are quickly bound to specific proteins, including the bactericidal permeability increasing protein and the microbial LPS-binding protein100–102 which are mainly present on HDL and support a role for this class of lipoproteins in immunity.100–102 Of note, proteins that have been critically associated with lipoprotein metabolism such as CETP and phospholipid transfer protein (PLTP) also bind lipid pathogens and therefore their deficiency, in animal models, was shown to be associated with decreased survival following sepsis.103 These findings support the concept that the clearance of cholesterol and that of pathogen lipids share common pathways, likely due to their incorporation with and transfer among lipoproteins.104,105 Factors improving LDL clearance in sepsis could therefore reduce endotoxaemia and improve survival. Indeed increased LDLR expression was associated with protection from severe sepsis106 and, more importantly, the PCSK9–LDLR interaction was found to play a key role in this context (Figure 5). Indeed, Pcsk9−/−-deficient mice displayed blunted cardiovascular and systemic responses to LPS treatment76 and administration of PCSK9-blocking antibodies increased survival and blunted the inflammatory response after sepsis in wild-type mice.76 This effect was shown to be critically dependent on LDLR, as PCSK9 inhibition in LDLR KO mice did not result in additional protection.76
Figure 5.

PCSK9 and infection. (A) PCSK9 reduces LDL uptake thus reducing LPS clearance and increasing inflammatory response during sepsis. (B) PCSK9 reduces LDLR thus resulting in reduced LDL-associated HCV uptake and decreased viral infection.
Plasma PCSK9 levels significantly increase during sepsis, and this results in a reduced clearance of bacterial endotoxin via the LDLR pathway;107 moreover, PCSK9 plasma levels correlate with the onset of multiple organ failure.107 The critical role for PCSK9 in human sepsis has been confirmed by the observation that patients carrying PCSK9 LOF mutations displayed increased survival compared with patients without LOF,76 whereas, on the contrary, patients with a GOF genetic variant showed a reduced survival.76 When carriers of PCSK9 LOF mutations were exposed to LPS, they showed a reduced inflammatory response as determined by changes in plasma IL-6 levels.76 Also in humans, the PCSK9-dependent effect was the direct consequence of the modulation of the LDLR and indeed carriers of a polymorphism within the LDLR gene altering the ability of PCSK9 to bind the EGF-A domain were insensitive to any protective effect related to PCSK9 LOF.76 These findings suggest a critical role for PCSK9 in limiting clearance of lipid pathogens, and support the hypothesis that PCSK9 inhibition represents a novel strategy for sepsis treatment.
7.2 Viral infections
Hepatitis C virus (HCV) is a major cause of chronic hepatitis that may lead to cirrhosis and hepatocellular carcinoma; several surface proteins have been proposed to have a role in HCV entry within cells, and a major role was proposed for LDLR and tetraspanin CD81 (Figure 5).108 In the circulation, HCV associates with LDL and VLDL, and may use the LDLR route to enter hepatic cells.108 In vitro studies showed that up-regulation of LDLR facilitates HCV infection in vitro and that the transfection of LDLR in cells not expressing this receptor, and thus unable to bind HCV, significantly increased the binding of HCV.108 However, some contrasting observations regarding the role of LDLR in HCV entry have been reported; in fact, it has been proposed that LDLR might play a relevant role in HCV replication but does not seem to have a major role in the productive HCV entry.109 In vitro HCV infection increased LDLR expression both at the protein level and at the transcriptional level (SREBP), thus resulting in an increased uptake of LDL, and the livers from patients with chronic hepatitis C exhibit increased expression of LDLR compared with the livers from non-HCV donors.110 The down-regulation of LDLR activity resulted in a reduced HCV infectivity109 probably due to a reduced uptake of cholesterol and other lipids that can affect HCV replication, suggesting that LDLR may be involved in viral processes downstream of entry, such as intracellular accumulation of lipids regulating HCV virus particle assembly and secretion rates. PCSK9 targets and reduces cell surface expression of both LDLR and CD81, the latter causing reduced susceptibility of cells to infection by HCV.111 Although PCSK9 mRNA expression was not affected, PCSK9 protein expression was reduced, likely due to increased targeting to proteasomal degradation.110 In Pcsk9−/− mice, livers exhibited a higher level of CD81 compared with wild-type animals, and a similar observations were reported in the double KO Pcsk9−/−/Ldlr−/−, confirming the LDLR-independency of the effect on CD81.111 In line with these observations, incubation with PCSK9 resulted in the inability of HCV to infect cells.111 Data obtained with GOF mutants of PCSK9 showed that although D129N and D374H mutants increased LDLR but not CD81 degradation compared with wild type, the GOF D374Y decreased both LDLR and CD81 expression, indicating a crucial role of the residue in this position.112 On the contrary, some LOF mutations on the catalytic domain of PCSK9 result in the inability to bind and degrade either LDLR and CD81 or only LDLR.112
Altogether these observations suggest that, at least in vitro, PCSK9 has the potential to protect against HCV and thus, the correlation between PCSK9 inhibition and the incidence of HCV infection needs to be carefully monitored in phase 3 clinical trials as well as in post-marketing studies.
7.3 Parasitic infections
Although cholesterol is the main sterol in parasites, they do not have the ability to synthesize it and thus their cholesterol is derived from cellular and circulating sources in the host through specific lipoprotein receptor-like proteins.113 Parasitic infection is usually associated with reduced plasma cholesterol levels and altered lipoprotein composition113 in the host. Furthermore, similar to viruses, parasites may utilize the host LDLR pathway for cell invasion.114,115
These observations suggest that parasitic infections might interfere with the axis LDL-C/LDLR and that, due to the competition for cholesterol, genetic variants that may represent an advantage for the host, the parasite, or both may have evolved.
Out of the three major nonsense single-nucleotide polymorphisms (SNPs) of PCSK9—R46L, Y142X, and C679X—the first is more frequent among white subjects, whereas the other two nonsense mutations are more frequent among African Americans, and are associated with a 40% reduction in LDL-C and a 88% reduction in CHD rates.2 It was proposed that the high frequency of these mutations might be protective against parasitic infections.113 In this context, PCSK9 LOF SNPs may have been naturally selected due to the dual protection of the host, i.e. a more efficient removal of circulating lipoproteins and the inhibition of parasite proliferation and activity.113 If this hypothesis is correct, then these two SNPs should be particularly frequent in areas where parasitic infections are common; of note, the C679X variant was identified in 3.7% of a women population attending two prenatal clinics in Zimbabwe58 but no data on the incidence of parasitic infections are available.
8. PCSK9 and the brain
PCSK9 was initially discovered as a protein expressed in the brain (NARC-1);9 its role in the brain, however, is to date controversial, and indeed, either a pro-apoptotic effect or a protective role in the development of the nervous system has been proposed. PCSK9 has been detected also in the cerebrospinal fluid, although to a level which is approximately 50–60 times lower than that detected in the serum.116
8.1 Role of PCSK9 in neuronal development
PCSK9 is highly expressed in cells that present an elevated proliferative index, and these include also embryonic brain telencephalon neurons,9 which, when transfected to overexpress PCSK9, present a higher recruitment rate of undifferentiated neural progenitor cells.9 The relevance of PCSK9 in this process was observed also in pluripotent mouse P19 embryonal carcinoma cells: although naïve P19 cells exhibited a very low level of PCSK9, after neuroectodermal induction by retinoic acid PCSK9 expression increased and reached a peak after 2 days, suggesting that early PCSK9 increased expression may be required to modulate cell differentiation.117 This is in agreement with the observation of a transient increased expression of PCSK9 in the telencephalon and cerebellum during the gestational period in rodents. This was not seen in adulthood, with the exception of the rostral extension of the olfactory peduncle (RE-OP).9 PCSK9 and LDLR are co-expressed in the telencephalon and cerebellum during active neurogenesis, and in RE-OP of adult animals.118 During brain development, LDLR is decreased following the increased expression of PCSK9 in wild-type mice, whereas LDLR protein levels are higher in telencephalon and cerebellum of Pcsk9−/− mice compared with wild-type animals. LDLR expression is not affected in RE-OP and olfactory bulb of adult Pcks9−/− mice, suggesting that in these brain areas PCSK9 does not promote LDLR degradation.118 In agreement with these observations, apolipoprotein E, the main apolipoprotein in brain and a ligand for LDLR, is significantly reduced during brain development in Pcsk9−/− mice, due to an increased expression of LDLR, but not in adulthood RE-OP or olfactory bulb, where the expression is similar to that of wild-type mice.118 In addition Pcsk9−/− mice are viable46 and do not exhibit relevant alterations in the cerebellum, hippocampus, or cortex,119 in agreement with the observation that humans carrying complete LOF mutations of PCSK9 show no major neurological defects.58,120 In zebrafish, which has a pattern of expression of PCSK9 similar to that observed in mice,117 the embryonic knockdown of PCSK9, however, results in defective neurogenesis, disorganization of cerebellar neurons, and embryonic death at 96 h after fertilization.117 This last finding suggests a possible different role for PCSK9 in the development of central nervous system (CNS) in mammals and fishes.
Following experimental transient ischaemic stroke in mice, PCSK9 mRNA levels increase in the lesioned area 24–72 h after reperfusion, and return to basal levels after 1 week.118 Of note, PCSK9 expression was not observed in the infarct and penumbra areas (suggesting that it may not play a role in cell death) but rather in the area where neurogenesis takes place.118 In spite of this specific up-regulation, Pcsk9−/− and wild-type mice presented a similar level of cell proliferation thus perhaps limiting the role for PCSK9 in de novo neurogenesis.118 After the ischaemic stroke, LDLR expression was decreased in the lesioned area of the brain, but in Pcsk9−/− mice the reduction in LDLR levels was significantly attenuated;118 nevertheless, apoE levels increased similarly in both groups in lesion areas compared with the non-lesion side.118 This suggests that the higher levels of LDLR observed in Pcsk9−/− mice after transient ischaemic stroke do not enhance the degradation of cerebral apoE. These findings indicate that the inhibition of PCSK9 should not interfere with brain development or impact brain recovery after an ischaemic stroke, in agreement with the observations that subjects carrying LOF variants of PCSK9 are healthy.3,120 Although not conclusive, these data have a clinical relevance; in fact, although under physiological conditions antibodies do not cross the blood–brain barrier (BBB), some pathological conditions, however, may increase its permeability. As example, after the onset of acute ischaemic stroke, the BBB is rapidly disrupted and this perturbation persists for days.121 Other pathological conditions which might result in increased permeability of BBB include diabetes mellitus (due to microvascular anomalies in the brain leading to BBB dysfunction),122 acute and chronic cerebrovascular diseases123 and, in some cases, after cardiac surgery.124
Finally, as the two PCSK9 targets, VLDLR and ApoER2,52 play a critical role also in neuronal migration125 and when both are defective the cerebellar development is arrested,126 it remains to be addressed whether PCSK9 inhibition in this context will be beneficial.
8.2 Role of PCSK9 in neuronal apoptosis
Neuronal apoptosis is a crucial process during normal CNS development, but it can also have a pathological connotation in the aetiology of neurodegenerative disease in adult brain, as it can induce the loss of neuronal network integrity.127 PCSK9 was identified as one of several genes whose expression was up-regulated during apoptosis induced by withdrawal of potassium and serum in cultured cerebellar granule neurons (CGN).128 Accordingly, the transient overexpression of recombinant NARC1/PCSK9 in CGN is pro-apoptotic and only partially sensitive to caspase inhibitors, thus defining both a caspase-dependent and a caspase-independent component of PCSK9 pro-apoptotic effect.129 Human gliomas are malignant tumours of the CNS characterized by aggressive proliferation and expansion into surrounding brain tissue. Recently, it has been shown that PCSK9 regulates apoptosis in a model of human neuroglioma; indeed, silencing of PCSK9 by siRNA inhibited cell proliferation and increased the pro-apoptotic to anti-apoptotic protein ratio.130 On the contrary, PCSK9 overexpression promoted cell proliferation,130 suggesting an anti-apoptotic effects in these cells. It could be hypothesized that PCSK9 inhibition might represent a possible therapeutic strategy to treat neuroglioma. The data available, however, are scarce and in vivo data are still lacking.
PCSK9 RNA interference (RNAi) also resulted in the inhibition of caspase-3 cleavage and phosphorylation of c-Jun in potassium-deprived CGN, thus resulting in an increased cell survival.131 CGN endogenously express ApoER2 and VLDLR; a decrease in their expression is observed after potassium deprivation131 and is associated with a reduction of cell survival (Figure 6). This effect was proposed to be the consequence of PCSK9 up-regulation; however, studies with PCSK9 RNAi have shown a significant increase mainly in ApoER2 but not in VLDLR levels131 thus prompting the need of additional studies to clarify this issue. More importantly, PCSK9 RNAi was not effective in an ApoER2 knockdown model, further suggesting a critical role for ApoER2 in these experimental conditions.131 Data obtained in vitro were not fully recapitulated in vivo; in fact, although PCSK9 can bind LDLR, VLDLR and ApoER2 in a cell-free assay, changes in PCSK9 expression do not modify the expression of these receptors in the adult mouse brain. Furthermore, although liver LDLR levels were changed in Pcsk9−/− or PCSK9 overexpressing mice compared with wild-type mice, no alteration was observed in LDLR levels in the brain of these animals.132 These contrasting results may be explained assuming that PCSK9 could have a cell/tissue-specific function, or by the observation that, being PCSK9 level in the adult mouse brain much lower than in the liver, it could not modulate receptor levels.
Figure 6.
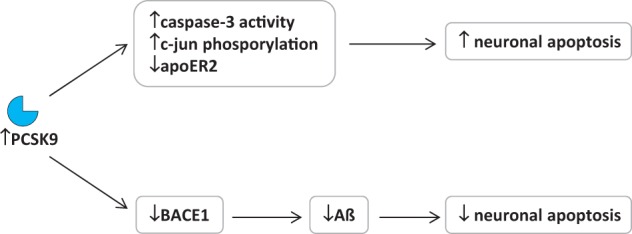
PCSK9 and the brain. Role of PCSK9 in neuronal apoptosis and AD progression.
8.3 PCSK9 and neurocognitive dysfunction
Theoretically, cholesterol-lowering might induce effects on brain; this organ, in fact, has a high content of cholesterol, an essential component for neuronal development as well as for brain function133 and thus the reduction of plasma cholesterol levels to a large extent might negatively impact brain function. In this context, statins have received attention for their potential to impair neurocognitive function, but the reported symptoms were generally of poor grade, reversible upon statin discontinuation and not associated with fixed or progressive neurodegenerative disease such as Alzheimer’s disease (AD).134 On the contrary, hypercholesterolaemia is considered an important risk factor for AD and other neurodegenerative diseases and lipid-lowering therapies can attenuate the risk of such pathological conditions.133
Whether these considerations could be extended also to other drugs reducing LDL-C levels, including PCSK9 inhibitors, is still a matter of debate. It has to be acknowledged that low levels of plasma cholesterol such as those due to LOF variants in PCSK9 gene have not been associated with cognitive impairment.58,120,135,136
Is it possible that PCSK9 affects neurological function independent of lipoprotein receptors modulation? Neurodegenerative processes leading to the progressive loss of neural tissues characterize neurological diseases, such as Parkinson’s disease and AD. In particular the excessive production and accumulation of amyloid ß peptide (Aß) in the brain is a hallmark of AD, thus resulting in the formation of lesions and neurofibrillary tangles; in addition, ‘pathological’ neuronal apoptosis represents a process relevant in the pathogenesis of this disease, which can be induced by a large number of factors.137 BACE1 [ß-site amyloid precursor protein (APP)-cleaving enzyme 1] is a membrane protein that catalyses the rate limiting step in the amyloidogenic metabolism of APP that results in the generation of Aß.138 BACE1 is transiently acetylated on seven different lysine residues in the lumen of the endoplasmic reticulum followed by deacetylation in the lumen of the Golgi apparatus after full maturation;139 this transient acetylation is required for the nascent protein to leave the ER and proceed towards the secretory pathway, whereas non-acetylated protein is retained and degraded.139 PCSK9 was shown to contribute to the disposal of this non-acetylated BACE1 form.140 In vitro, PCSK9 overexpression results in the reduction of endogenous BACE1 levels (Figure 6), with a decrease of both immature and mature forms, whereas the down-regulation of PCSK9 by siRNA completely normalized the levels of BACE1.140 When cells were transfected with either the wild type or a mutant form of BACE1, silencing of PCSK9 increased the levels of wild type and loss-of-acetylation mutants of BACE1 but not those of gain-of-acetylation mutant.140 In addition, Pcsk9−/− mice showed higher levels of BACE1 and Aß in the neocortex.140 These findings suggest that in the brain PCSK9 regulates the metabolism of BACE1 and the rate of Aß production. However, similarly to the results described for ApoER2 and VLDLR, another work reported a lack of effect of PCSK9 overexpression or deletion on the levels of BACE1 in the mouse brain, with a concomitant lack of effect also on Aß levels,132 and the authors concluded that PCSK9 has no effect on these receptors and enzyme in vivo. Therefore, the role of PCSK9 on brain biology remains controversial.
In randomized clinical trials with PCSK9 mAb, neurocognitive adverse events were uncommon and PCSK9 mAb treatment was not associated with increased incidence of neurocognitive adverse events in most studies.37,82,83,85,141–143 However, two trials reported, although not statistically significant, an increase of neurocognitive adverse events in the group treated with PCSK9 mAb.87,144 A key aspect might be represented by the criteria used to define the neurocognitive disorders (often self-reported), which consist of delirium (including confusion), cognitive and attention disorders and disturbances, dementia and amnestic disorders, disturbances in thinking and perception, mental impairment disorders. On the other hand, the data from the recent meta-analyses are not unequivocal.145,146 In fact, Lipinski et al.145 reported a significantly increased incidence of neurocognitive adverse events with PCSK9 inhibitors (odds ratio = 2.34, P = 0.02); however, this meta-analysis included both OSLER trials (with OSLER-2 largely contributing to the results) and their parent studies, thus resulting in the inclusion of the same patients twice. Thus, when re-analysed based on this consideration without OSLER-2, PCSK9 treatment did not result associate with increased neurocognitive adverse events (P = 0.48).146 A further reassurance of a lack of adverse neurocognitive events following therapy with PCSK9 mAbs comes from a comprehensive analysis of 28 trials showing that both alirocumab and evolocumab are associated with rate of adverse neurocognitive events similar to that of controls (0.7% with placebo vs. 0.8% with alirocumab; 1.0% with ezetimibe vs. 0.9% with alirocumab; 0.3% any control vs. 0.1% evolocumab; 0.2% with standard of care vs. 0.6% with evolocumab and standard of care).147,148 To date, an ongoing trial (EBBINGHAUS: Evaluating PCSK9 Binding Antibody Influence on Cognitive Health in High Cardiovascular Risk Subjects, NCT02207634) is evaluating change over time in neurocognitive testing in patients receiving statins in association with evolocumab or placebo and has as estimated completion date February 2018.
Of note, no correlation was found between neurocognitive events and the degree of LDL-C reduction obtained,144 suggesting that very low levels of LDL-C (<25 mg/dL) cannot directly elicit neurocognitive dysfunctions. This observation is in agreement with the fact that lipoproteins carrying cholesterol cannot cross the BBB and thus brain cholesterol is synthesized de novo and is independent of circulating levels of cholesterol. Although, as described above, mAbs to PCSK9 do not cross the BBB due to their size, some concerns may raise also from the observation that neurological diseases such as AD and multiple sclerosis are associated with BBB disruption, an event that would increase the permeability of substances normally excluded from the brain149 such as the antibodies. Altogether these observations suggest the need for longer follow-up together with a more rigorous assessment of neurocognitive adverse events to define a possible role of PCSK9 inhibition in increasing the risk of neurocognitive disorders.
In summary, the role for PCSK9 in the brain is still largely debated and most of the controversial results stem from in vitro cell systems and selective manipulations in animal models, whereas data in humans are still inconclusive.
9. Conclusions
Since the discovery of its physiological role, PCSK9 has become the focus for the development of therapeutic approaches to hypercholesterolaemia. The impetus of research and the magnitude of effects achieved in the area of plasma lipids and lipoproteins, mainly by the use of mAbs, have somehow limited investigations on the intracellular or other possible physiologic roles of PCSK9. In this review, we have addressed the areas of research that in our opinion require serious scrutiny to better understand the biological role of this intriguing protein, an information which is critical and may add or detract value from different approaches to modulate PCSK9 activity.
Some key points in our opinion require further investigation:
Investigating the role of PCSK9 in modulating lipid and lipoprotein metabolism especially in the area of lipoprotein assembly in both the intestines and liver, and the binding of PCSK9 to specific circulating lipoproteins, might help in explaining some of the effects observed in relation to atherosclerosis, for example modulating post-prandial lipaemia, or decreasing Lp(a) plasma levels.
A better knowledge of PCSK9 biology will also offer the opportunity to unravel other possible physiological roles lipoproteins may play that were in place thousands of years ago such as, perhaps, protection from infections. This role is of potential value under some circumstances such as acute infections. Of note, the fact that different PCSK9 polymorphisms give rise to a selective effect on CD81 and viral infection as compared to the interaction with the LDLR raises the question as to whether we can design molecules not affecting CD81 but controlling only the LDLR. This knowledge might help to mitigate the consequences of bacterial or even viral infections. At the same time, the control of the outcomes following infections in patients being treated with PCSK9 inhibitors should be carefully investigated.
Another intriguing observation refers to the effects of PCSK9 on the distribution of adipose tissue and glucose metabolism. Are we somehow modulating the metabolism of adipocytes via modulation of VLDL receptors? Is the effect of PCSK9 KO in mice relevant to man as per the pancreatic function? To date the clinical trials appear reassuring; however we should bear in mind that some of these correlations may be linked to the intracellular presence of PCSK9, not targeted by mAbs; the fact that humans homozygotes for LOF mutations do not recapitulate the observations in mice may not be taken as the final proof.
Last but not least the possible role for PCSK9 in the nervous system. The preliminary report of neurocognitive effects following PCSK9 inhibition with mAbs has raised a number of questions. The role of PCSK9 in modulating apoptosis in the CNS in mice certainly suggests that a better understanding of this function is requested. A clinical trial currently being performed with mAbs will provide the final answer.
A final point relates to the fact that systemic inhibition of PCSK9 in the circulation, as it occurs with the mAbs now available for therapy, may not have the same effect as inhibiting also intracellular levels of PCSK9 as it occurs with other approaches (such as siRNA). This calls for a word of caution when other approaches are sought for inhibiting PCSK9 that will affect intracellular levels of the protein. Some unexpected findings may occur.
In summary although we are already in the clinic to inhibit the function of PCSK9 for controlling plasma lipids, we are far from fully understanding the physiological role of this protein: a number of experimental and some clinical data suggest that PCSK9 may not serve only the role of controlling plasma cholesterol. The recognition of LOF and GOF mutations as well as some well-designed mechanistic studies might offer a key to unravel these mechanisms in humans.
Conflict of interest: Receipt of grants/research supports: Pfizer, Sanofi, Regeneron, Merck, Mediolanum SigmaTau, Menarini, Kowa, Recordati, Eli Lilly. Receipt of honoraria or consultation fees: Pfizer, Merck, Sanofi, Aegerion, Amgen, Genzyme, ISIS. Participation in a company sponsored speaker’s bureau: Merck, Sanofi, Regeneron, Pfizer, AstraZeneca, Amgen, Sigma Tau, Recordati, Aegerion, ISIS, Kowa.
Funding
A.L.C. was supported by H2020 Grant REPROGRAM and CARIPLO (2012-0549 and 2015-0524); G.D.N. was supported by grants from Telethon Foundation (GGP13002), Ministero della Salute WFR GR-2011-02346974; and H.T. and S.F. were partially supported by the National Institutes of Health (National Heart, Lung, and Blood Institute) through grant R01-HL106845.
References
- 1.Lagace TA. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Curr Opin Lipidol 2014;25:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264–1272. [DOI] [PubMed] [Google Scholar]
- 3.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005;37:161–165. [DOI] [PubMed] [Google Scholar]
- 4.Kathiresan S. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. N Engl J Med 2008;358:2299–2300. [DOI] [PubMed] [Google Scholar]
- 5.Norata GD, Tibolla G, Catapano AL. PCSK9 inhibition for the treatment of hypercholesterolemia: promises and emerging challenges. Vascul Pharmacol 2014;62:103–111. [DOI] [PubMed] [Google Scholar]
- 6.Tibolla G, Norata GD, Artali R, Meneghetti F, Catapano AL. Proprotein convertase subtilisin/kexin type 9 (PCSK9): from structure-function relation to therapeutic inhibition. Nutr Metab Cardiovascular diseases: NMCD 2011;21:835–843. [DOI] [PubMed] [Google Scholar]
- 7.Norata GD, Ballantyne CM, Catapano AL. New therapeutic principles in dyslipidaemia: focus on LDL and Lp(a) lowering drugs. Eur Heart J 2013;34:1783–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, Parker R, Prat A, Seidah NG. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem 2009;284:28856–28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 2003;100:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem 2013;288:8279–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng BB. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol 2012;32:1585–1595. [DOI] [PubMed] [Google Scholar]
- 12.Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, Fazio S. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry 2008;47:1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tavori H, Fan D, Blakemore JL, Yancey PG, Ding L, Linton MF, Fazio S. Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-density lipoprotein receptor: evidence for a reciprocal regulation. Circulation 2013;127:2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavori H, Giunzioni I, Linton MF, Fazio S. Loss of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) after lipoprotein apheresis. Circ Res 2013;113:1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem 2015;290:11649–11662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raal F, Panz V, Immelman A, Pilcher G. Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J Am Heart Assoc 2013;2:e000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK. The PCSK9 decade. J Lipid Res 2012;53:2515–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Julius U, Milton M, Stoellner D, Rader D, Gordon B, Polk D, Waldmann E, Parhofer KG, Moriarty PM. Effects of lipoprotein apheresis on PCSK9 levels. Atheroscler Suppl 2015;18:180–186. [DOI] [PubMed] [Google Scholar]
- 19.Hori M, Ishihara M, Yuasa Y, Makino H, Yanagi K, Tamanaha T, Kishimoto I, Kujiraoka T, Hattori H, Harada-Shiba M. Removal of plasma mature and furin-cleaved proprotein convertase subtilisin/kexin 9 by low-density lipoprotein-apheresis in familial hypercholesterolemia: development and application of a new assay for PCSK9. J Clin Endocrinol Metab 2015;100:E41–49. [DOI] [PubMed] [Google Scholar]
- 20.Duell PB DG, Seidah NG, Davignon J. Clearance of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) by LDL apheresis. Circ Res 2014. 115:e3–4. [DOI] [PubMed] [Google Scholar]
- 21.Drouin-Chartier JP, Tremblay AJ, Bergeron J, Pelletier M, Laflamme N, Lamarche B, Couture P. Comparison of two low-density lipoprotein apheresis systems in patients with homozygous familial hypercholesterolemia. J Clin Apher 2015. [DOI] [PubMed] [Google Scholar]
- 22.Han B, Eacho PI, Knierman MD, Troutt JS, Konrad RJ, Yu X, Schroeder KM. Isolation and characterization of the circulating truncated form of PCSK9. J Lipid Res 2014;55:1505–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Essalmani R, Susan-Resiga D, Chamberland A, Abifadel M, Creemers JW, Boileau C, Seidah NG, Prat A. In vivo evidence that furin from hepatocytes inactivates PCSK9. J Biol Chem 2011;286:4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem 2006;281:30561–30572. [DOI] [PubMed] [Google Scholar]
- 25.Lipari MT, Li W, Moran P, Kong-Beltran M, Sai T, Lai J, Lin SJ, Kolumam G, Zavala-Solorio J, Izrael-Tomasevic A, Arnott D, Wang J, Peterson AS, Kirchhofer D. Furin-cleaved proprotein convertase subtilisin/kexin type 9 (PCSK9) is active and modulates low density lipoprotein receptor and serum cholesterol levels. J Biol Chem 2012;287:43482–43491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridker PM, Rifai N, Bradwin G, Rose L. Plasma proprotein convertase subtilisin/kexin type 9 levels and the risk of first cardiovascular events. Eur Heart J 2015;37:554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li JJ, Li S, Zhang Y, Xu RX, Guo YL, Zhu CG, Wu NQ, Qing P, Gao Y, Sun J, Liu G, Dong Q. Proprotein convertase subtilisin/kexin type 9, C-reactive protein, coronary severity, and outcomes in patients with stable coronary artery disease: a prospective observational cohort study. Medicine 2015;94:e2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Werner C, Hoffmann MM, Winkler K, Bohm M, Laufs U. Risk prediction with proprotein convertase subtilisin/kexin type 9 (PCSK9) in patients with stable coronary disease on statin treatment. Vascul Pharmacol 2014;62:94–102. [DOI] [PubMed] [Google Scholar]
- 29.Zhu YM, Anderson TJ, Sikdar K, Fung M, McQueen MJ, Lonn EM, Verma S. Association of proprotein convertase subtilisin/kexin type 9 (PCSK9) with cardiovascular risk in primary prevention. Arterioscler Thromb Vasc Biol 2015;35:2254–2259. [DOI] [PubMed] [Google Scholar]
- 30.Cariou B, Langhi C, Le BM, Bortolotti M, Le KA, Theytaz F, Le MC, Guyomarc'h-Delasalle B, Zair Y, Kreis R, Boesch C, Krempf M, Tappy L, Costet P. Plasma PCSK9 concentrations during an oral fat load and after short term high-fat, high-fat high-protein and high-fructose diets. Nutr Metab 2013;10:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab 2009;94:2537–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baass A, Dubuc G, Tremblay M, Delvin EE, O'Loughlin J, Levy E, Davignon J, Lambert M. Plasma PCSK9 is associated with age, sex, and multiple metabolic markers in a population-based sample of children and adolescents. Clin Chem 2009;55:1637–1645. [DOI] [PubMed] [Google Scholar]
- 33.Janis MT, Tarasov K, Ta HX, Suoniemi M, Ekroos K, Hurme R, Lehtimaki T, Paiva H, Kleber ME, Marz W, Prat A, Seidah NG, Laaksonen R. Beyond LDL-C lowering: distinct molecular sphingolipids are good indicators of proprotein convertase subtilisin/kexin type 9 (PCSK9) deficiency. Atherosclerosis 2013;228:380–385. [DOI] [PubMed] [Google Scholar]
- 34.Kwakernaak AJ, Lambert G, Dullaart RP. Plasma proprotein convertase subtilisin-kexin type 9 is predominantly related to intermediate density lipoproteins. Clin Biochem 2014;47:679–682. [DOI] [PubMed] [Google Scholar]
- 35.Sullivan S, Fabbrini E, Horton JD, Korenblat K, Patterson BW, Klein S. Lack of a relationship between plasma PCSK9 concentrations and hepatic lipoprotein kinetics in obese people. Transl Res 2011;158:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouguerram K, Chetiveaux M, Zair Y, Costet P, Abifadel M, Varret M, Boileau C, Magot T, Krempf M. Apolipoprotein B100 metabolism in autosomal-dominant hypercholesterolemia related to mutations in PCSK9. Arterioscler Thromb Vasc Biol 2004;24:1448–1453. [DOI] [PubMed] [Google Scholar]
- 37.Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol 2014;63:2531–2540. [DOI] [PubMed] [Google Scholar]
- 38.Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, Burgess L, Ceska R, Roth E, Koren MJ, Ballantyne CM, Monsalvo ML, Tsirtsonis K, Kim JB, Scott R, Wasserman SM, Stein EA. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014;370:1809–1819. [DOI] [PubMed] [Google Scholar]
- 39.Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A, Watts GF. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J 2011;32:1345–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blasiole DA, Oler AT, Attie AD. Regulation of ApoB secretion by the low density lipoprotein receptor requires exit from the endoplasmic reticulum and interaction with ApoE or ApoB. J Biol Chem 2008;283:11374–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costet P, Cariou B, Lambert G, Lalanne F, Lardeux B, Jarnoux AL, Grefhorst A, Staels B, Krempf M. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J Biol Chem 2006;281:6211–6218. [DOI] [PubMed] [Google Scholar]
- 42.Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, Devay RM, Liang H, Rashid S, Linton MF, Fazio S. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res 2016;110:268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sniderman AD, Qi Y, Ma CI, Wang RH, Naples M, Baker C, Zhang J, Adeli K, Kiss RS. Hepatic cholesterol homeostasis: is the low-density lipoprotein pathway a regulatory or a shunt pathway? Arterioscler Thromb Vasc Biol 2013;33:2481–2490. [DOI] [PubMed] [Google Scholar]
- 44.Le May C, Kourimate S, Langhi C, Chetiveaux M, Jarry A, Comera C, Collet X, Kuipers F, Krempf M, Cariou B, Costet P. Proprotein convertase subtilisin kexin type 9 null mice are protected from postprandial triglyceridemia. Arterioscler Thromb Vasc Biol 2009;29:684–690. [DOI] [PubMed] [Google Scholar]
- 45.Havel RJ. Triglyceride-rich lipoproteins and plasma lipid transport. Arterioscler Thromb Vasc Biol 2010;30:9–19. [DOI] [PubMed] [Google Scholar]
- 46.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A 2005;102:5374–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, Fazio S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation 2014;130:431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levy E, Ben Djoudi Ouadda A, Spahis S, Sane AT, Garofalo C, Grenier E, Emonnot L, Yara S, Couture P, Beaulieu JF, Menard D, Seidah NG, Elchebly M. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis 2013;227:297–306. [DOI] [PubMed] [Google Scholar]
- 49.Gillian-Daniel DL, Bates PW, Tebon A, Attie AD. Endoplasmic reticulum localization of the low density lipoprotein receptor mediates presecretory degradation of apolipoprotein B. Proc Natl Acad Sci U S A 2002;99:4337–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olofsson SO, Boren J. Apolipoprotein B secretory regulation by degradation. Arterioscler Thromb Vasc Biol 2012;32:1334–1338. [DOI] [PubMed] [Google Scholar]
- 51.Roubtsova A, Munkonda MN, Awan Z, Marcinkiewicz J, Chamberland A, Lazure C, Cianflone K, Seidah NG, Prat A. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler Thromb Vasc Biol 2011;31:785–791. [DOI] [PubMed] [Google Scholar]
- 52.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 2008;283:2363–2372. [DOI] [PubMed] [Google Scholar]
- 53.Demers A, Samami S, Lauzier B, Des Rosiers C, Sock ET, Ong H, Mayer G. PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler Thromb Vasc Biol 2015;35:2517–2525. [DOI] [PubMed] [Google Scholar]
- 54.Norata GD, Caligiuri G, Chavakis T, Matarese G, Netea MG, Nicoletti A, O'Neill LA, Marelli-Berg FM. The cellular and molecular basis of translational immunometabolism. Immunity 2015;43:421–434. [DOI] [PubMed] [Google Scholar]
- 55.Ruscica M, Ricci C, Macchi C, Magni P, Cristofani R, Liu J, Corsini A, Ferri N. Suppressor of cytokine signaling-3 (SOCS-3) induces proprotein convertase subtilisin kexin type 9 (PCSK9) expression in hepatic HepG2 cell line. J Biol Chem 2016;291:3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langhi C, Le May C, Gmyr V, Vandewalle B, Kerr-Conte J, Krempf M, Pattou F, Costet P, Cariou B. PCSK9 is expressed in pancreatic delta-cells and does not alter insulin secretion. Biochem Biophys Res Commun 2009;390:1288–1293. [DOI] [PubMed] [Google Scholar]
- 57.Mbikay M, Sirois F, Mayne J, Wang GS, Chen A, Dewpura T, Prat A, Seidah NG, Chretien M, Scott FW. PCSK9-deficient mice exhibit impaired glucose tolerance and pancreatic islet abnormalities. FEBS Lett 2010;584:701–706. [DOI] [PubMed] [Google Scholar]
- 58.Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007;193:445–448. [DOI] [PubMed] [Google Scholar]
- 59.Bonnefond A, Yengo L, Le May C, Fumeron F, Marre M, Balkau B, Charpentier G, Franc S, Froguel P, Cariou B. The loss-of-function PCSK9 p.R46L genetic variant does not alter glucose homeostasis. Diabetologia 2015;58:2051–2055. [DOI] [PubMed] [Google Scholar]
- 60.Saavedra YG, Dufour R, Baass A. Familial hypercholesterolemia: PCSK9 InsLEU genetic variant and prediabetes/diabetes risk. J Clin Lipidol 2015;9:786–793 e781. [DOI] [PubMed] [Google Scholar]
- 61.Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012;380:565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corrao G, Ibrahim B, Nicotra F, Soranna D, Merlino L, Catapano AL, Tragni E, Casula M, Grassi G, Mancia G. Statins and the risk of diabetes: evidence from a large population-based cohort study. Diabetes Care 2014;37:2225–2232. [DOI] [PubMed] [Google Scholar]
- 63.Fryirs M, Barter PJ, Rye KA. Cholesterol metabolism and pancreatic beta-cell function. Curr Opin Lipidol 2009;20:159–164. [DOI] [PubMed] [Google Scholar]
- 64.Ferri N, Tibolla G, Pirillo A, Cipollone F, Mezzetti A, Pacia S, Corsini A, Catapano AL. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis 2012;220:381–386. [DOI] [PubMed] [Google Scholar]
- 65.Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS, Liu LS. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase9-caspase3 pathway. Mol Cell Biochem 2012;359:347–358. [DOI] [PubMed] [Google Scholar]
- 66.Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, DeVay RM, Hong L, Fan D, Predazzi IM, Rashid S, Linton MF, Fazio S. Local effects of human PCSK9 on the atherosclerotic lesion. J Pathol 2016;238:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, Wang Y, Mehta JL. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal 2015;22:760–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Linton MF, Babaev VR, Gleaves LA, Fazio S. A direct role for the macrophage low density lipoprotein receptor in atherosclerotic lesion formation. J Biol Chem 1999;274:19204–19210. [DOI] [PubMed] [Google Scholar]
- 69.Herijgers N, Van Eck M, Groot PH, Hoogerbrugge PM, Van Berkel TJ. Low density lipoprotein receptor of macrophages facilitates atherosclerotic lesion formation in C57Bl/6 mice. Arterioscler Thromb Vasc Biol 2000;20:1961–1967. [DOI] [PubMed] [Google Scholar]
- 70.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuhnast S, van der Hoorn JW, Pieterman EJ, van den Hoek AM, Sasiela WJ, Gusarova V, Peyman A, Schafer HL, Schwahn U, Jukema JW, Princen HM. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res 2014;55:2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, Pan L, Jiang Z, Liu L. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-kappaB activation in THP-1-derived macrophages. Int J Mol Med 2012;30:931–938. [DOI] [PubMed] [Google Scholar]
- 73.Li S, Guo YL, Xu RX, Zhang Y, Zhu CG, Sun J, Qing P, Wu NQ, Jiang LX, Li JJ. Association of plasma PCSK9 levels with white blood cell count and its subsets in patients with stable coronary artery disease. Atherosclerosis 2014;234:441–445. [DOI] [PubMed] [Google Scholar]
- 74.Ruan XZ, Moorhead JF, Tao JL, Ma KL, Wheeler DC, Powis SH, Varghese Z. Mechanisms of dysregulation of low-density lipoprotein receptor expression in vascular smooth muscle cells by inflammatory cytokines. Arterioscler Thromb Vasc Biol 2006;26:1150–1155. [DOI] [PubMed] [Google Scholar]
- 75.Ma KL, Liu J, Wang CX, Ni J, Zhang Y, Wu Y, Lv LL, Ruan XZ, Liu BC. Activation of mTOR modulates SREBP-2 to induce foam cell formation through increased retinoblastoma protein phosphorylation. Cardiovasc Res 2013;100:450–460. [DOI] [PubMed] [Google Scholar]
- 76.Walley KR, Thain KR, Russell JA, Reilly MP, Meyer NJ, Ferguson JF, Christie JD, Nakada TA, Fjell CD, Thair SA, Cirstea MS, Boyd JH. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med 2014;6:258ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Armendariz AD, Krauss RM. Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol 2009;20:106–111. [DOI] [PubMed] [Google Scholar]
- 78.Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 2013;8:e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res 2007;100:670–677. [DOI] [PubMed] [Google Scholar]
- 80.Sharotri V, Collier DM, Olson DR, Zhou R, Snyder PM. Regulation of epithelial sodium channel trafficking by proprotein convertase subtilisin/kexin type 9 (PCSK9). J Biol Chem 2012;287:19266–19274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Berger JM, Vaillant N, Le May C, Calderon C, Bregeon J, Prieur X, Hadchouel J, Loirand G, Cariou B. PCSK9-deficiency does not alter blood pressure and sodium balance in mouse models of hypertension. Atherosclerosis 2015;239:252–259. [DOI] [PubMed] [Google Scholar]
- 82.Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R, Chaudhari U, Colhoun HM. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J 2015;36:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, Blom D, Civeira F, Krempf M, Lorenzato C, Zhao J, Pordy R, Baccara-Dinet MT, Gipe DA, Geiger MJ, Farnier M. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015;36:2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bays H, Gaudet D, Weiss R, Ruiz JL, Watts GF, Gouni-Berthold I, Robinson J, Zhao J, Hanotin C, Donahue S. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J Clin Endocrinol Metab 2015;100:3140–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farnier M, Jones P, Severance R, Averna M, Steinhagen-Thiessen E, Colhoun HM, Du Y, Hanotin C, Donahue S. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: the ODYSSEY OPTIONS II randomized trial. Atherosclerosis 2016;244:138–146. [DOI] [PubMed] [Google Scholar]
- 86.Moriarty PM, Thompson PD, Cannon CP, Guyton JR, Bergeron J, Zieve F, Bruckert E, Jacobson TA, Kopecky SL, Baccara-Dinet MT, Du L, Pordy R, Gipe D. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol 2015;9:758–769. [DOI] [PubMed] [Google Scholar]
- 87.Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015;372:1489–1499. [DOI] [PubMed] [Google Scholar]
- 88.Pandya V, Rao A, Chaudhary K. Lipid abnormalities in kidney disease and management strategies. World J Nephrol 2015;4:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baragetti A, Norata GD, Sarcina C, Rastelli F, Grigore L, Garlaschelli K, Uboldi P, Baragetti I, Pozzi C, Catapano AL. High density lipoprotein cholesterol levels are an independent predictor of the progression of chronic kidney disease. J Intern Med 2013;274:252–262. [DOI] [PubMed] [Google Scholar]
- 90.Balla S, Nusair MB, Alpert MA. Risk factors for atherosclerosis in patients with chronic kidney disease: recognition and management. Curr Opin Pharmacol 2013;13:192–199. [DOI] [PubMed] [Google Scholar]
- 91.Sucajtys-Szulc E, Szolkiewicz M, Swierczynski J, Rutkowski B. Up-regulation of liver Pcsk9 gene expression as a possible cause of hypercholesterolemia in experimental chronic renal failure. Mol Cell Biochem 2015;411:281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kwakernaak AJ, Lambert G, Slagman MC, Waanders F, Laverman GD, Petrides F, Dikkeschei BD, Navis G, Dullaart RP. Proprotein convertase subtilisin-kexin type 9 is elevated in proteinuric subjects: relationship with lipoprotein response to antiproteinuric treatment. Atherosclerosis 2013;226:459–465. [DOI] [PubMed] [Google Scholar]
- 93.Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, Lahaye SA, Mbikay M, Ooi TC, Chretien M. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis 2008;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vaziri ND. Molecular mechanisms of lipid disorders in nephrotic syndrome. Kidney Int 2003;63:1964–1976. [DOI] [PubMed] [Google Scholar]
- 95.Jin K, Park BS, Kim YW, Vaziri ND. Plasma PCSK9 in nephrotic syndrome and in peritoneal dialysis: a cross-sectional study. Am J Kidney Dis 2014;63:584–589. [DOI] [PubMed] [Google Scholar]
- 96.Konarzewski M, Szolkiewicz M, Sucajtys-Szulc E, Blaszak J, Lizakowski S, Swierczynski J, Rutkowski B. Elevated circulating PCSK-9 concentration in renal failure patients is corrected by renal replacement therapy. Am J Nephrol 2014;40:157–163. [DOI] [PubMed] [Google Scholar]
- 97.Abujrad H, Mayne J, Ruzicka M, Cousins M, Raymond A, Cheesman J, Taljaard M, Sorisky A, Burns K, Ooi TC. Chronic kidney disease on hemodialysis is associated with decreased serum PCSK9 levels. Atherosclerosis 2014;233:123–129. [DOI] [PubMed] [Google Scholar]
- 98.Chmielewski M, Sucajtys-Szulc E, Kossowska E, Swierczynski J, Rutkowski B, Boguslawski W. Increased gene expression of liver SREBP-2 in experimental chronic renal failure. Atherosclerosis 2007;191:326–332. [DOI] [PubMed] [Google Scholar]
- 99.Libetta C, Sepe V, Esposito P, Galli F, Dal Canton A. Oxidative stress and inflammation: Implications in uremia and hemodialysis. Clin Biochem 2011;44:1189–1198. [DOI] [PubMed] [Google Scholar]
- 100.Pirillo A, Catapano AL, Norata GD. HDL in infectious diseases and sepsis. Handb Exp Pharmacol 2015;224:483–508. [DOI] [PubMed] [Google Scholar]
- 101.Catapano AL, Pirillo A, Bonacina F, Norata GD. HDL in innate and adaptive immunity. Cardiovasc Res 2014;103:372–383. [DOI] [PubMed] [Google Scholar]
- 102.Norata GD, Pirillo A, Ammirati E, Catapano AL. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis 2011;220:11–21. [DOI] [PubMed] [Google Scholar]
- 103.Azzam KM, Fessler MB. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol Metab 2012;23:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Levels JH, Marquart JA, Abraham PR, van den Ende AE, Molhuizen HO, van Deventer SJ, Meijers JC. Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect Immun 2005;73:2321–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hailman E, Albers JJ, Wolfbauer G, Tu AY, Wright SD. Neutralization and transfer of lipopolysaccharide by phospholipid transfer protein. J Biol Chem 1996;271:12172–12178. [DOI] [PubMed] [Google Scholar]
- 106.dos Santos C, Marshall JC. Bridging lipid metabolism and innate host defense. Sci Transl Med 2014;6:258fs241. [DOI] [PubMed] [Google Scholar]
- 107.Boyd JH, Fjell CD, Russell JA, Sirounis D, Cirstea MS, Walley KR. Increased plasma PCSK9 levels are associated with reduced endotoxin clearance and the development of acute organ failures during sepsis. J Innate Immun 2016;8:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Diedrich G. How does hepatitis C virus enter cells? FEBS J 2006;273:3871–3885. [DOI] [PubMed] [Google Scholar]
- 109.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Terce F, Duverlie G, Rouille Y, Dubuisson J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012;55:998–1007. [DOI] [PubMed] [Google Scholar]
- 110.Syed GH, Tang H, Khan M, Hassanein T, Liu J, Siddiqui A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J Virol 2014;88:2519–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Labonte P, Begley S, Guevin C, Asselin MC, Nassoury N, Mayer G, Prat A, Seidah NG. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 2009;50:17–24. [DOI] [PubMed] [Google Scholar]
- 112.Le QT, Blanchet M, Seidah NG, Labonte P. Plasma membrane tetraspanin CD81 complexes with proprotein convertase subtilisin/kexin type 9 (PCSK9) and low density lipoprotein receptor (LDLR), and its levels are reduced by PCSK9. J Biol Chem 2015;290:23385–23400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mbikay M, Mayne J, Seidah NG, Chretien M. Of PCSK9, cholesterol homeostasis and parasitic infections: possible survival benefits of loss-of-function PCSK9 genetic polymorphisms. Med Hypotheses 2007;69:1010–1017. [DOI] [PubMed] [Google Scholar]
- 114.Nagajyothi F, Weiss LM, Silver DL, Desruisseaux MS, Scherer PE, Herz J, Tanowitz HB. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl Trop Dis 2011;5:e953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Portugal LR, Fernandes LR, Pietra Pedroso VS, Santiago HC, Gazzinelli RT, Alvarez-Leite JI. Influence of low-density lipoprotein (LDL) receptor on lipid composition, inflammation and parasitism during Toxoplasma gondii infection. Microbes Infect 2008;10:276–284. [DOI] [PubMed] [Google Scholar]
- 116.Chen YQ, Troutt JS, Konrad RJ. PCSK9 is present in human cerebrospinal fluid and is maintained at remarkably constant concentrations throughout the course of the day. Lipids 2014;49:445–455. [DOI] [PubMed] [Google Scholar]
- 117.Poirier S, Prat A, Marcinkiewicz E, Paquin J, Chitramuthu BP, Baranowski D, Cadieux B, Bennett HP, Seidah NG. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem 2006;98:838–850. [DOI] [PubMed] [Google Scholar]
- 118.Rousselet E, Marcinkiewicz J, Kriz J, Zhou A, Hatten ME, Prat A, Seidah NG. PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J Lipid Res 2011;52:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seidah NG, Mayer G, Zaid A, Rousselet E, Nassoury N, Poirier S, Essalmani R, Prat A. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol 2008;40:1111–1125. [DOI] [PubMed] [Google Scholar]
- 120.Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, Cohen JC, Hobbs HH. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006;79:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008;32:200–219. [DOI] [PubMed] [Google Scholar]
- 122.Yu X, Xu X, Jackson A, Sun J, Huang P, Mao Y, Chen Z, Lou M, Jiang Q, Zhang M. Blood brain barrier disruption in diabetic stroke related to unfavorable outcome. Cerebrovasc Dis 2016;42:49–56. [DOI] [PubMed] [Google Scholar]
- 123.Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011;42:3323–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Merino JG, Latour LL, Tso A, Lee KY, Kang DW, Davis LA, Lazar RM, Horvath KA, Corso PJ, Warach S. Blood-brain barrier disruption after cardiac surgery. AJNR Am J Neuroradiol 2013;34:518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sharaf A, Bock HH, Spittau B, Bouche E, Krieglstein K. ApoER2 and VLDLr are required for mediating reelin signalling pathway for normal migration and positioning of mesencephalic dopaminergic neurons. PLoS One 2013;8:e71091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999;97:689–701. [DOI] [PubMed] [Google Scholar]
- 127.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature 2000;407:802–809. [DOI] [PubMed] [Google Scholar]
- 128.Chiang LW, Grenier JM, Ettwiller L, Jenkins LP, Ficenec D, Martin J, Jin F, DiStefano PS, Wood A. An orchestrated gene expression component of neuronal programmed cell death revealed by cDNA array analysis. Proc Natl Acad Sci U S A 2001;98:2814–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bingham B, Shen R, Kotnis S, Lo CF, Ozenberger BA, Ghosh N, Kennedy JD, Jacobsen JS, Grenier JM, DiStefano PS, Chiang LW, Wood A. Proapoptotic effects of NARC 1 (= PCSK9), the gene encoding a novel serine proteinase. Cytometry A 2006;69:1123–1131. [DOI] [PubMed] [Google Scholar]
- 130.Piao MX, Bai JW, Zhang PF, Zhang YZ. PCSK9 regulates apoptosis in human neuroglioma u251 cells via mitochondrial signaling pathways. Int J Clin Exp Pathol 2015;8:2787–2794. [PMC free article] [PubMed] [Google Scholar]
- 131.Kysenius K, Muggalla P, Matlik K, Arumae U, Huttunen HJ. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cell Mol Life Sci 2012;69:1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu M, Wu G, Baysarowich J, Kavana M, Addona GH, Bierilo KK, Mudgett JS, Pavlovic G, Sitlani A, Renger JJ, Hubbard BK, Fisher TS, Zerbinatti CV. PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J Lipid Res 2010;51:2611–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xue-Shan Z, Juan P, Qi W, Zhong R, Li-Hong P, Zhi-Han T, Zhi-Sheng J, Gui-Xue W, Lu-Shan L. Imbalanced cholesterol metabolism in Alzheimer's disease. Clin Chim Acta 2016;456:107–114. [DOI] [PubMed] [Google Scholar]
- 134.Patil S, Chan C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci Lett 2005;384:288–293. [DOI] [PubMed] [Google Scholar]
- 135.Postmus I, Trompet S, de Craen AJ, Buckley BM, Ford I, Stott DJ, Sattar N, Slagboom PE, Westendorp RG, Jukema JW. PCSK9 SNP rs11591147 is associated with low cholesterol levels but not with cognitive performance or noncardiovascular clinical events in an elderly population. J Lipid Res 2013;54:561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cariou B, Ouguerram K, Zair Y, Guerois R, Langhi C, Kourimate S, Benoit I, Le May C, Gayet C, Belabbas K, Dufernez F, Chetiveaux M, Tarugi P, Krempf M, Benlian P, Costet P. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol 2009;29:2191–2197. [DOI] [PubMed] [Google Scholar]
- 137.Obulesu M, Lakshmi MJ. Apoptosis in Alzheimer's disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res 2014;39:2301–2312. [DOI] [PubMed] [Google Scholar]
- 138.Hunt CE, Turner AJ. Cell biology, regulation and inhibition of beta-secretase (BACE-1). FEBS J 2009;276:1845–1859. [DOI] [PubMed] [Google Scholar]
- 139.Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J 2007;407:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep 2008;9:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D, Somaratne R, Legg JC, Nelson P, Scott R, Wasserman SM, Weiss R. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014;311:1870–1882. [DOI] [PubMed] [Google Scholar]
- 142.Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, Bruckert E, Cho L, Dent R, Knusel B, Xue A, Scott R, Wasserman SM, Rocco M. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol 2014;63:2541–2548. [DOI] [PubMed] [Google Scholar]
- 143.Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:341–350. [DOI] [PubMed] [Google Scholar]
- 144.Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015;372:1500–1509. [DOI] [PubMed] [Google Scholar]
- 145.Lipinski MJ, Benedetto U, Escarcega RO, Biondi-Zoccai G, Lhermusier T, Baker NC, Torguson R, Brewer HB, Jr, Waksman R. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J 2016;37:536–545. [DOI] [PubMed] [Google Scholar]
- 146.Kolodziejczak M, Navarese EP. Role of PCSK9 antibodies in cardiovascular disease: Critical considerations of mortality and neurocognitive findings from the current literature. Atherosclerosis 2016;247:189–192. [DOI] [PubMed] [Google Scholar]
- 147.Food and Drug Administration Briefing Document. The Endocrinologic and Metabolic Drugs Advisory Committee Meeting. Repatha (evolocumab) injections. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM450072.pdf. (10 June 2015, date last accessed).
- 148.Food and Drug Administration Briefing Document, The Endocrinologic and Metabolic Drugs Advisory Committee Meeting. Praluent (alirocumab) injections. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM449865.pdf. (10 June 2015, date last accessed).
- 149.Patel JP, Frey BN. Disruption in the blood-brain barrier: the missing link between brain and body inflammation in bipolar disorder? Neural Plast 2015;2015:708306. [DOI] [PMC free article] [PubMed] [Google Scholar]