

Abstract
Background
Neurodegenerative diseases are a heterogeneous group of disorders characterized by loss of neurons and are commonly associated with a genetic mutation.
Hypothesis/Objectives
To characterize the clinical and histopathological features of a novel degenerative neurological disease affecting the brain of young adult Nova Scotia Duck Tolling Retrievers (NSDTRs).
Animals
Nine, young adult, related NSDTRs were evaluated for neurological dysfunction and rapid eye movement sleep behavior disorder.
Methods
Case series review.
Results
Clinical signs of neurological dysfunction began between 2 months and 5 years of age and were progressive in nature. They were characterized by episodes of marked movements during sleep, increased anxiety, noise phobia, and gait abnormalities. Magnetic resonance imaging documented symmetrical, progressively increasing, T2‐weighted image intensity, predominantly within the caudate nuclei, consistent with necrosis secondary to gray matter degeneration. Abnormalities were not detected on clinicopathological analysis of blood and cerebrospinal fluid, infectious disease screening or urine metabolite screening in most cases. Postmortem examination of brain tissue identified symmetrical malacia of the caudate nuclei and axonal dystrophy within the brainstem and spinal cord. Genealogical analysis supports an autosomal recessive mode of inheritance.
Conclusions and Clinical Importance
A degenerative encephalopathy was identified in young adult NSDTRs consistent with a hereditary disease. The prognosis is guarded due to the progressive nature of the disease, which is minimally responsive to empirical treatment.
Keywords: Axonopathy, Basal nuclei, Behavioral changes, Familial
Abbreviations
- NSDTR
Nova Scotia Duck Tolling Retriever
- SRMA
steroid‐responsive meningitis‐arteritis
- MRI
magnetic resonance imaging
- CSF
cerebrospinal fluid
- GFAP
glial fibrillary acidic protein‐positive
- NAD
neuroaxonal dystrophy
- REM
rapid eye movement
Neurodegenerative diseases occur in both humans and animals.1, 2 They form a large heterogeneous group of diseases characterized by loss of neurons leading to distinct involvement of functional systems, mostly caused by a genetic anomaly.3, 4, 5 Clinical signs are often progressive, possibly delayed in onset, and reflect the area(s) of the central nervous system most affected. The end result of these diseases might be encephalomalacia, which is a nonspecific finding characterized by a hyperintense signal on T2‐weighted magnetic resonance imaging (MRI) as a result of fluid accumulation. Although rapid eye movement (REM) sleep behavior disorders are a common finding in human neurodegenerative diseases such as Parkinson's disease, multiple systems atrophy, and dementia with Lewy bodies,6, 7 sleep disorders have not been associated with neurodegenerative diseases in dogs.8, 9, 10
Neurologists in the United Kingdom, Canada, and Sweden have recently recognized the emergence of a novel degenerative encephalopathy in the Nova Scotia Duck Tolling Retriever (NSDTR), presenting with progressive neurological dysfunction including REM sleep behavior disorder, not previously reported in dogs. The purpose of this study was to characterize the phenotype and potential etiology of this condition to provide the basis for elimination of the disease in the NSDTR population.
Materials and Methods
From 2013 to 2014, 9 purebred NSDTR located in United Kingdom, Canada, and Sweden were investigated for slowly progressive neurological dysfunction. Clinical and historical information regarding these dogs were obtained by medical record review and consultation with owners and referring veterinarians. Complete blood counts, routine serum biochemistry profiles, brain MRI, cerebrospinal fluid (CSF) analysis, infectious disease screening (serology or PCR for Toxoplasma gondii, Neospora caninum, and canine distemper virus), and urine metabolic screening were performed in various animals at the discretion of the neurologist conducting each case work‐up and reviewed where available.
After euthanasia due to progressive clinical signs, postmortem examination was performed in 3 dogs, and light microscopy histopathological studies of brain, muscles, and other tissues were performed on collected samples. The brain of a 15‐year‐old, neutered male NSDTR that was euthanized as a result of choroid plexus carcinoma and lacked microscopic lesions seen in affected dogs was also examined as a control. The familial relationship between this dog and affected dogs is unknown. Tissues were fixed by immersion in neutral buffered formalin, embedded in paraffin, and sectioned at 5 μm thickness. Sections were stained with hematoxylin and eosin (H&E), Bielschowsky silver stain for cell processes, Perl's stain for iron, and luxol fast blue stain for myelin. Immunohistochemical staining was performed on selected sections to detect the following factors: neurofilament proteins in axons; neuronal calcium‐binding proteins calbindin, calretinin, and parvalbumin in selected populations of neurons; NeuN in neurons; and glial fibrillary acidic protein (GFAP) in astroglia (for methods used with immunohistochemical reagents see Table S1).11, 12, 13
To determine the genealogical relationships among affected dogs, data were collected either from pedigree information provided by the dog owners or accessed through public canine registries.1, 2
Results
Case Histories/Clinical Findings/Response to Treatment
These data are available for individual dogs in Table S2.
Clinical signs were reported in dogs 2 months to 5 years of age, were progressive in nature, and were similar in all cases. The most consistent clinical sign was a REM sleep behavior disorder characterized by frequent episodes of marked involuntary movements during sleep (see Video S1). These episodes of involuntary movements were both extreme and progressive over time, in contrast to the sleep movements of clinically normal dogs. The observed episodes were violent and owners of affected dogs reported that the dogs had fallen from their bed and owners were awakened by the agitation of the dog. The duration of episodes typically ranged from a few seconds to 15–30 minutes, although some owners reported occasional episodes lasting longer than 1 hour and occurred up to 10 times in a sleep period. The dogs made running movements, sometimes lifting their heads, and frequently vocalized. Some owners also reported that the dog would be difficult to awaken from sleep.
The dogs learned basic obedience, but had difficulty with advanced training. Affected dogs had increased anxiety and sensitivity to sensory stimuli, which sometimes manifested as noise phobia or obsessive‐compulsive behaviors such as tail chasing, chasing any moving object, or repeatedly investigating their food bowl. With progression of the disease 4 dogs began having aggressive interactions (#4,5,7,9), or lacked self‐control, with other dogs and sometimes their owners. Five owners reported abnormal swimming motion (#4,5,7–9; the dog adopting a vertical position in water—see Video S2), whereas others felt this was part of normal learning to swim and others were never witnessed swimming. The owners also reported reduced tail movement and gait abnormalities. Two dogs were reported to have been urinary and fecally incontinent (#2,9). To date, 5 dogs (#1,4,7–9) have been euthanized due to progressive debilitating behavior anomalies.
Abnormalities were not detected on general physical examination in 7 dogs (#3–9). One dog (#2) had a heart murmur, although abnormalities were not detected on subsequent electrocardiography and echocardiography. One dog (#1) was small in stature and also had a prior history of reactive lymphadenopathy of unknown etiology. One dog (#3) had a prior history of steroid‐responsive meningitis‐arteritis (SRMA) and had relapse of clinical signs (pyrexia, neck pain, and lethargy) concomitant with the appearance of the first movement disturbances during sleep.
Neurologically, the majority of dogs were mentally alert. Affected dogs showed varying degrees of a generalized ataxia and tetraparesis, with mild hypermetria in the thoracic limbs. A fine head tremor was reported in 1 dog (#1). Mild conscious proprioceptive deficits were occasionally noted. The tendon reflexes were brisk, and crossed extensor reflexes were recorded in 2 dogs (#1,2). Abnormalities were more exaggerated in the pelvic limbs in comparison with the thoracic limbs in all but 1 dog (#3). In 1 dog, neurological deficits were lateralized (#5), while, in the remainder, they were symmetrical. Of the cranial nerves, mild menace deficits were occasionally reported, as was abnormal vision and strabismus (#2,3,5). One dog (#1) acutely developed keratoconjunctivitis sicca, suspected to be neurogenic in origin, with bilateral corneal ulceration, in the absence of an underlying cause, and that was poorly responsive to conventional treatment for non‐neurogenic keratoconjunctivitis sicca.
One dog had an equivocal response to anti‐epileptic medication (levetiracetam, imepitoin). Response to corticosteroids was positive in 1 dog (#2) and resulted in a progression of behavioral signs in (#3).
Clinical Pathology
Hematology and serum biochemistry results were available for 6 affected dogs (#1,2,3,5,8,9). Other than 1 dog (#3) with a mild, mature neutrophilia (16.10 × 103/μL, reference interval 2.95–11.64 × 103/μL) and increased C‐reactive protein (121 mg/L, reference interval 0–35 mg/L) that were attributed to concurrent SRMA, none of the results were considered remarkable. Abnormalities were not detected on additional tests, performed at the discretion of the attending clinician: bile acid stimulation test (#1), serum cobalamin (#1), serum thiamine (#2,3), cisternal CSF analysis (#1,2,4,8), urine organic metabolites (#1,2,5), ACTH stimulation test (#1), T. gondii and N. caninum serology (#1,2) and PCR of CSF (#1), and canine distemper virus PCR of CSF (#1,2).
Magnetic Resonance Imaging Findings
Magnetic resonance imaging of the brain was performed in 4 dogs (#1,2,3,8). The most striking lesions were bilateral symmetrical, rounded, T2‐weighted hyperintensities, which were hypointense on FLAIR, within the caudate nucleus, particularly rostrally (Fig 1). These lesions intensity did not change on T1‐weighted images after IV injection of gadolinium contrast agent and compared to precontrast injection such that no hyperintensity was noted. In addition, moderately hyperintense lesions forming bands within the brainstem running from ventrolateral to dorsomedial and within the lateral cuneate nuclei, caudal cerebellar peduncles, and spinal tract of the trigeminal nuclei were also noted on T2‐weighted images (Fig 2). In 1 dog, follow‐up MRI performed at intervals of 7 and 17 months after the initial presentation documented mild progression of these changes (Fig 1E–G).
Figure 1.
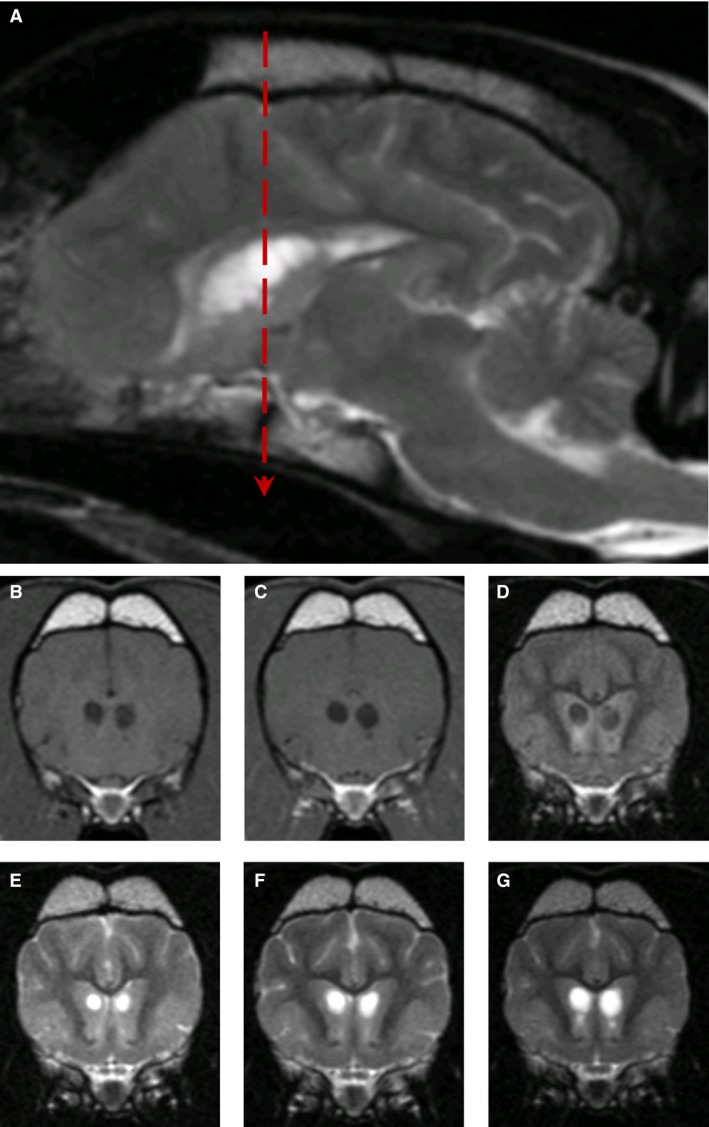
MR images from Dog #1. T2‐weighted image (A), parasagittal to the right of midline at the level of the caudate nuclei lesions, showing well‐defined hyperintense lesions at age 47 months. Transverse T1‐weighted (B), T1‐weighted postcontrast (C), and FLAIR (D) images at the level of caudate nuclei (indicated by red dashed line on parasagittal view) at age 47 months. Transverse T2‐weighted images at the level of caudate nuclei obtained at 30 (E), 37 (F), and 47 months (G), demonstrating progressive cavitation.
Figure 2.
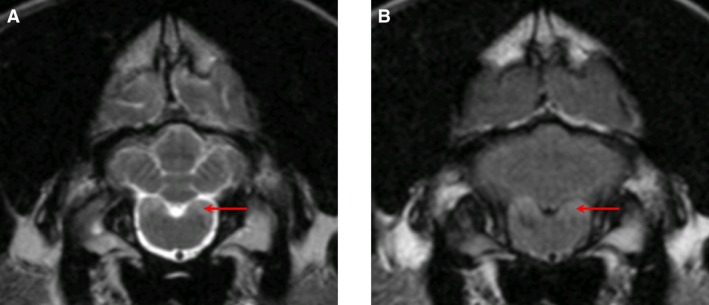
MR images from Dog #2. T2‐weighted (A) and FLAIR (B) transverse images at the level of the lateral cuneate nuclei showing a hyperintense signal (red arrow).
Postmortem Examination
Postmortem examination was performed in 3 dogs (#1,8,9). Transverse sectioning of the brain revealed bilaterally symmetrical cavities in the head of the caudate nucleus, corresponding to the changes seen on MRI, in all dogs examined (Fig 3). Abnormalities were not detected during the remainder of the postmortem examinations.
Figure 3.
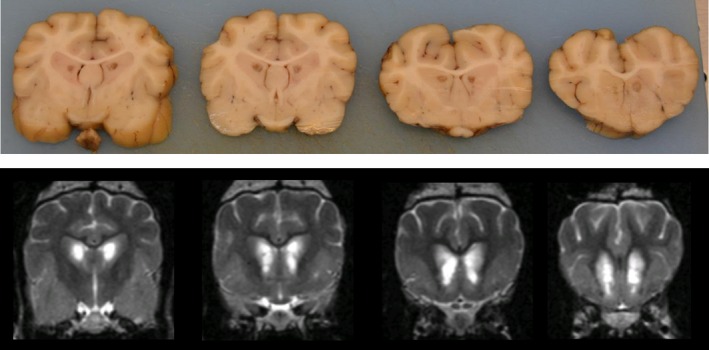
Fixed brain tissue sections (upper series; Dog #9) along transverse T2‐weighted MR images (lower series; Dog #2) moving rostrally from the level of the pituitary gland, illustrating the homogeneity of the disease among cases. Symmetrical cavitatory lesions are visualized in portions of the caudate nuclei, corresponding to the hyperintense lesions seen on T2‐weighted magnetic resonance imaging.
Histopathological Distribution and General Characteristics of Brain Lesions
All affected dogs had symmetrical cavitatory lesions at necropsy that affected portions of the caudate nucleus, as seen grossly (Fig 3). The centers of these cavitations were unstained, and gitter cells were generally uncommon, except near tissue margins. The irregular periphery of the cavitations was crossed by slender strands of neuropil containing thin‐walled, often patent, blood vessels ensheathed by glia (Fig 4). Irregular spongiform change loosened the neuropil around the cavity, with coincident astrocytosis. Neurons remained in unaffected portions of the nuclei. One dog had a tiny area of iron pigment peripheral to the cavity, but the other 2 dogs and the unaffected NSDTRs were negative for iron. Cavitation did not affect the brain of the unaffected dog.
Figure 4.
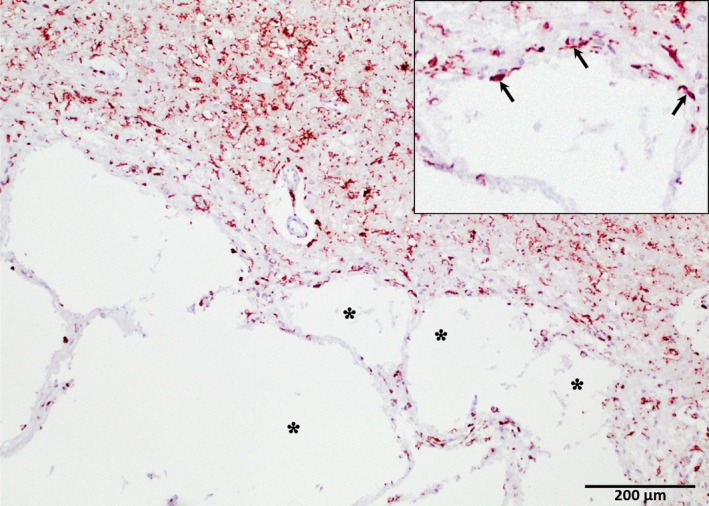
Cavitation of the caudate nucleus from one of the affected dogs. The tissue intersecting with the edges of the cavity (asterisks) are irregular, with astrocyte‐rich parenchymal strands crossing the margin and peripheral astrocytosis (inset, arrows; under ×2 greater magnification) (glial fibrillary acidic protein‐positive immunohistochemistry; hematoxylin counterstain). Original magnification 40×.
In addition to caudate cavitations, all dogs had prominent swollen, eosinophilic neurites of various sizes observed with H&E staining (Fig 5A, B), which were most obvious in the brainstem, cerebellum, and medulla, but which occurred throughout the neuraxis, including the spinal cord of the 1 dog examined. These were present in the internal granular and molecular layers of the cerebellum and in white matter of cerebellar folia. However, they were overall most numerous in the cerebellar roof nuclei, superior olives, trapezoid, cuneate, and gracilis nuclei of the medulla and inferior olives (Fig 5A). In cross section, these swellings measured up to 50 μm diameter. Longitudinal profiles of swollen axons were apparent in associated white tracts. Droplet‐like swollen neurites were found at low density in the thalamus, hippocampus, striate nuclei, cochlear nuclei, and colliculi (Fig 5B).
Figure 5.
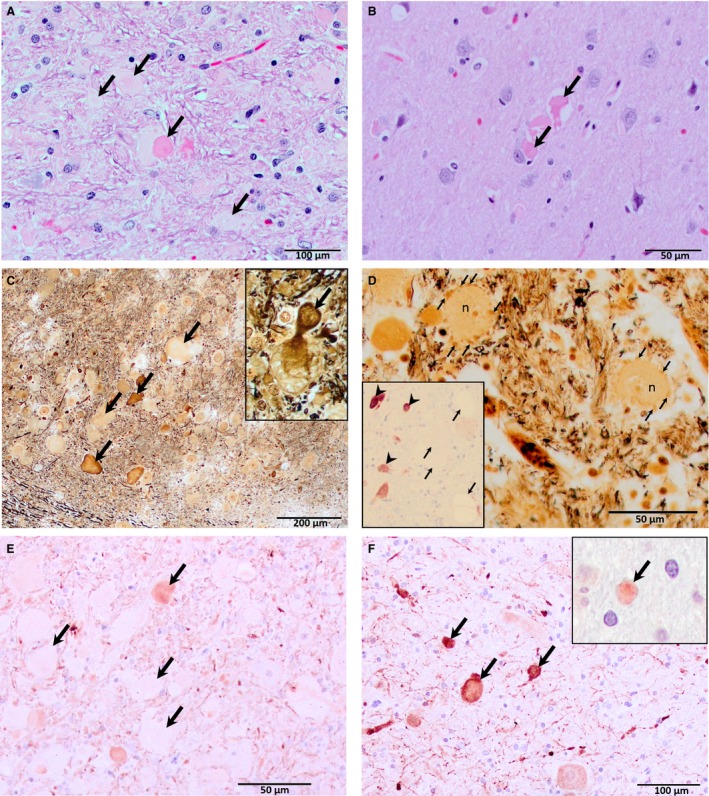
H&E (A and B), Bielschowsky (C and D), phosphorylated neurofilament (E), and calretinin (F) staining showing distended neurites in the brainstem (trapezoid nucleus, A, C, D, and E; inferior olive, F) and the cerebral cortex (B). Swellings up to 50 μm in diameter occur in the brainstem (A, C, and E; arrows) and medulla (F; arrows), with irregular shapes and variable intensity characteristic of H&E and Bielschowsky staining, whereas swellings in the forebrain (B; arrows) were more uniformly and intensely stained. Rarely, there is continuity between a cell body and swollen neurites (inset, C; arrow), identifying them as axons. In another area of the trapezoid nucleus small swollen neurites form haloes (D, arrows) around neuron cell bodies (n). NeuN immunohistochemistry (inset, D; hematoxylin counterstain) revealed viable neurons (arrow heads) within affected brain nuclei, whereas, as expected, axonal spheroids (arrows) remained unstained. Immunohistochemical staining for axonal neurofilaments vary in intensity (E; arrows), whereas those detecting calcium‐binding proteins are more consistent (F and inset, F, calbindin staining of a single neurite, cerebellar molecular layer; arrows). Original magnifications 200× (A and F); 400× (B, D and E); 100× (C).
Immunohistochemistry for Neurons and Their Projections
Bielschowsky and phosphorylated neurofilament stains were used to identify nerve fibers and NeuN to identify neuronal cell bodies. Calcium‐binding protein stains were used because they stain both the soma and cell processes. Swollen neurites had 2 relatively distinct morphologies. Smaller homogeneous deeply eosinophilic, droplet‐like swellings were more common in the forebrain and did not stain with Bielschowsky stains. Larger swellings had rounded to somewhat irregular, lumpy shapes and stained heterogeneously pale to intensely eosinophilic with H&E and variably with Bielschowsky stains (Fig 5A, C). In a few areas, rosettes of small, rounded neurites were clustered around neurons, forming a halo around the soma (Fig 5D). Rarely, continuity could be observed between axonal swellings and a nucleated nerve cell body (Fig 5C, inset). NeuN immunohistochemistry of specimens containing numerous swollen axons revealed the continued presence of at least some nerve cell bodies (Fig 5D, inset) despite the neurite/axonal changes. The larger dilations in the hindbrain were often irregularly positive for expression of phosphorylated neurofilaments, which identifies axons with relative specificity (Fig 5E). Phosphorylated neurofilament staining was not found in nerve cell bodies, as expected in normal brain. Phosphorylated neurofilaments were not detected in the smaller swellings in the forebrain. Calcium‐binding proteins were utilized to examine the morphology of several populations of neurons in the cerebellum and brainstem (Fig 5F). Staining of these soluble proteins was more homogeneous than staining of neurofilaments using immunohistochemistry or the Bielschowsky method. They also verified the presence of swollen neurites in white matter and nuclei, as both the soma and cell processes stain with these reagents. The calcium‐binding proteins immunohistochemistry appeared normal.
Immunohistochemistry for Astrocytes
Astrocytes were examined with immunohistochemistry using GFAP staining. Degenerative changes in neurites were frequently associated with profound enlargement of astrocytes in affected brainstem areas. Astrocytes often had gemistocytic morphology and greatly enlarged nuclei and were intimately associated with neurons and swollen axons (Fig 6).
Figure 6.
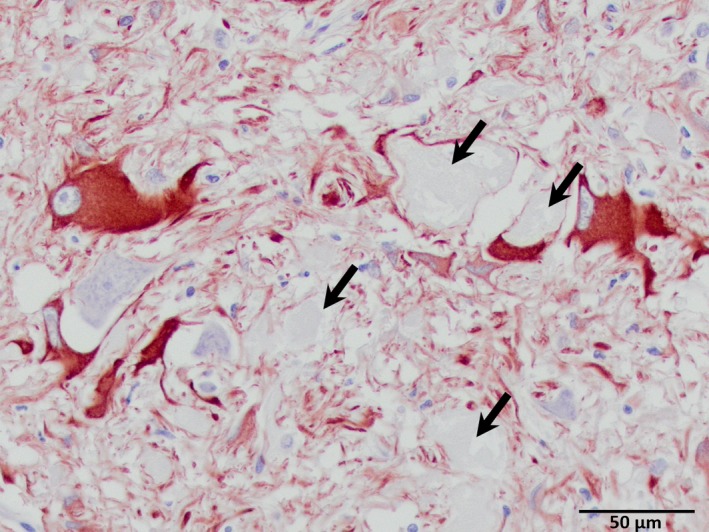
Astrocytosis associated with swollen axons and their neurons. Arrows denote nearby swollen axons (glial fibrillary acidic protein‐positive; hematoxylin counterstain). Original magnification 400×.
Histopathology of Other Organs/Parts of the Nervous System
Abnormalities were not detected on histopathological examination of muscle, spinal cord, peripheral nerves, liver, and kidney tissue in all dogs but 1 (#1). This dog showed changes consistent with mild, chronic hepatitis and a mild myopathy characterized by patchy lymphocytic infiltration and mild variation in myocyte diameter.
Pedigree Analysis
Affected dogs were offspring to unaffected parents (Fig 7). There was no significant difference between numbers of affected males and females (males 6, females 3 known affected; c.f. 26 males and 20 females total in affected litters; χ2, P = .49) and 28–43% of the dogs in the litters were reported to be affected. Common ancestors on both maternal and paternal lines were frequent, with all affected dogs having descended from 5 founder members (data not shown).
Figure 7.
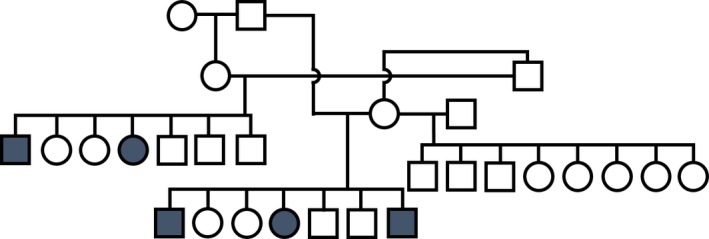
Pedigree for a cluster of the affected dogs (blue squares/circles).
Discussion
We report here a novel, progressive encephalopathy of the NSDTR presenting with REM sleep behavior disorder, abnormal behavior, and ataxia. MRI of affected individuals is characterized by symmetrical T2W hyperintensities of the rostral caudate nuclei, corresponding to malacia with neurite swelling on histopathology. The marked involuntary movements during sleep have the characteristics of a REM sleep behavior disorder (also called REM without atonia)14 although polysomnographic recordings were not performed to confirm that the episodes occurred during the REM phase of sleep. Confirmed or suspected REM sleep behavior disorders occur in dogs, a minority of which are associated with concurrent central nervous system disorders (seizures, ataxia, and anxiety disorders).8, 9, 10 MRI in 2 affected dogs was normal.
Experimental models of caudate nucleus ablation in cats, as well as instances of spontaneous damage to the caudate nucleus, result in ataxia, apathy, obsessive‐compulsive behavior, cognitive deficits, and hyperactivity;15 much of which were reported in affected dogs described within this study. However, REM sleep behavior disorder is not a feature of caudate nucleus destruction in cats, and in contrast, hyposomnia was present.15 In humans, REM sleep behavior anomalies are often the initial clinical sign in extrapyramidal diseases such as Parkinson's disease, multiple system atrophy, and dementia with Lewy bodies.7 Again, however, neither degeneration of the substantia nigra or cerebellum, nor Lewy bodies were observed in the NSDTR, and the pathomechanism of the REM sleep behavior disorder in this condition remains unknown.
Differential diagnoses for the bilaterally symmetrical lesions in the basal nuclei (comprising the caudate nucleus and putamen) included the following: toxic (e.g. carbon monoxide, methanol), metabolic, infectious, immune‐mediated, vascular, and neurodegenerative diseases (including multiple systems atrophies).16, 17, 18 The chronic, progressive nature of the disease, and its worldwide distribution made toxic insult highly unlikely. No evidence of other inborn errors of metabolism, acquired metabolic disease, nutritional deficiencies, or infectious disease were found on clinical pathology and review of clinical history (including the observation of simultaneous development of disease on different continents). However, rare hereditary malabsorptive process or vitamin metabolism disorder19 could not be excluded. The absence of inflammatory infiltrates in the brain tissue made an immune‐mediated destruction or an infectious etiology unlikely. The virtual absence of hemosiderin and gitter cells in affected NSDTR reduces the likelihood of an acute vascular etiology. The occurrence of nearly identical clinical signs, MRI appearance, and histopathological findings in related dogs of the same breed supported a hereditary neurodegenerative disorder.
Hereditary neurodegenerative disorders characterized by bilateral necrosis of gray matter, including the basal nuclei, occur in dogs. Leigh‐like syndrome, or polioencephalomyelopathy, occurs as an autosomal recessive disease in Australian cattle dogs, American Staffordshire bull terriers, Alaskan huskies, and Yorkshire terriers.20, 21, 22, 23 In contrast to the affected NSDTR described in this study, affected dogs present with a combination of seizures, and progressive dysphagia and spastic tetraparesis. As in Leigh disease of humans, a mitochondrial disorder had been suspected; however, a recent study identified a mutation in the thiamine transporter 2 gene (SLC19A3) associated with Leigh‐like syndrome in the Alaskan husky.24 Progressive cavitation of the caudate nuclei occurs in Kerry Blue terriers and Chinese Crested dogs with multisystem neurodegeneration.25, 26 However, these changes are preceded by loss of cerebellar Purkinje cells, with associated cerebellar ataxia, and are followed by neuronal depletion in the substantia nigra. Necrosis of the basal nuclei also occurred in a young Shi Tzu with severe neurological signs and a disorder of fatty acid metabolism.27
Degenerative encephalopathies with necrosis of the basal nuclei also occur in noncanine species.28, 29 Well characterized human diseases include Huntington's disease (autosomal dominant, adult onset), chorea‐acanthocytosis (autosomal recessive; adult onset), infantile bilateral striatal necrosis (autosomal recessive, neonatal onset), and progressive supranuclear palsy (adult onset).29, 30, 31, 32 However, the heterogeneity of clinical signs, age of onset, rate of progression, and underlying pathological mechanism of these different neurodegenerative diseases reflect how nonspecific basal nuclei malacia is as a finding.
In addition to the grossly evident cavitation of the caudate nuclei, locally severe swelling of axons was present in all NSDTR examined with histopathology. Although 2 types of swollen neurites were identified with routine histopathology, we further used immunohistochemistry to visualize both neuronal projections and cell bodies. It confirmed the presence of 2 types of neurite swelling (e.g., with the Bielschowsky and phosphorylated neurofilament stains, some swollen neurites were pale and some were strongly stained) and also helped to classify the neurites as axons due to their relationship with neuronal cell bodies. The NeuN stain also allowed identifying viable neurons despite the extensive axonal lesions. Finally, calcium‐binding proteins, widely expressed through the central nervous system, were stained to further assess whether there was depletion of these molecules which could cause impairment of motor coordination for example. However, abnormalities were not detected using these staining techniques, although we did not quantify stained material against controls. The extent of astrogliosis associated with abnormal axons, in the absence of concurrent inflammatory infiltration, and the presence of caudate cavitations are supportive of this being a chronic the disease process, and it is surprising that the degree of dysfunction was not more marked in these dogs at the time of euthanasia. The axonal swellings are consistent with a neuroaxonal dystrophy (NAD), which could cause the ataxia observed. NAD is characterized by localized swellings (spheroids) and axonal atrophy, but is a nonspecific finding and might occur as a progressive hereditary disease or develop secondary to insults such as vitamin E deficiency, hydrocarbon exposure, and as an age‐related change.33 Ultrastructural studies often show compartmentalization in axonal swellings with separation of neurofilaments and organelles, perhaps accounting for the variable staining pattern. Fetal‐onset and juvenile‐onset NADs, resembling human infantile NAD, occur in Papillons, Jack Russell terriers, Border Collies, Rottweilers, and a laboratory colony of crossbreed dogs.34, 35, 36, 37, 38 How the axonal changes relate to the dramatic caudate necrosis is unclear as this has not been described in the reports of NAD. A lysosomal storage disease cannot be ruled out, due to the progressive nature of the disease and the presence of PAS‐positive material in the medulla of affected dogs; however, the marked malacia is not a feature typical of lysosomal storage disease. There was no evidence of vacuolation in non‐nervous tissue examined, and the medullary changes were more consistent with axonal dystrophy.
Due to a small number of founder dogs, the NSDTR has a high inbreeding coefficient (0.26; where 0.25 would equate to the genetic equivalent of a dog produced from a son to mother mating) and low effective population size;39 however, in a study of Norwegian NSDTR estimating genetic diversity using standard canine panels of microsatellite markers, the level of genetic variation is found to be only slightly lower than that of many other dog breeds.40 Inbreeding does not cause hereditary diseases as such, but makes the expression of recessive traits more likely. Inspection of pedigrees described herein supported an autosomal recessive mode of inheritance of the degenerative encephalopathy. Nine affected dogs were investigated in this study, and some dogs did not have an exhaustive work‐up to exclude all potential causes of a degenerative encephalopathy. Thus, additional studies would be needed to completely rule out other causes of the condition or confirm a hereditary disease by identifying a variant that cosegregates with the phenotype in a larger population. As this disease has not been previously described in animals, this could either represent a recent de novo mutation within the breed or a failure to associate the subtle MRI changes with the clinical signs. Identification of the genetic abnormality associated with this disease could be used to determine the frequency of carriers within the population. Identification of carriers would permit breeders to use wise breeding strategies to avoid affected dogs in the future while maintaining desirable traits and genetic diversity in the breed.
Supporting information
Table S1. Methods and reagents used for immunohistochemical staining of sections.
Table S2. Summaries of case signalment, reported clinical signs (general and neurological), physical examination findings (general and neurological), clinical tests performed, advanced imaging findings, response to treatment, and outcome.
Video S1. Home video recording of affected NSDTR demonstrating marked involuntary movements during sleep.
Video S2. Home video of affected NSDTR demonstrating abnormal vertical position adopted during swimming.
Acknowledgments
The authors thank Drs. Carolina Duque and Andrea Finnen for collecting samples, Candace Kassel for preparation of histopathology tissues, and Karen Clifford for help with preparing histopathology illustrations.
Grant support: This study was supported in part by the Swedish Toller Club and the Norwegian Retriever Club.
Conflict of Interest Declaration: Authors disclose no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
Where the work was performed: Clinical cases were examined, and postmortem examinations were performed in Canada, UK, and Sweden. Additional case evaluation and histopathology was performed in Missouri, USA.
Meeting at which data were presented: This work was presented in part at the ECVN congress held in Amsterdam, the Netherlands, in September 2015.
Footnotes
References
- 1. Martin JB. Molecular basis of the neurodegenerative disorders. N Engl J Med 1999;340:1970–1980. [DOI] [PubMed] [Google Scholar]
- 2. Siso S, Hanzlicek D, Fluehmann G, et al. Neurodegenerative diseases in domestic animals: A comparative review. Vet J 2006;171:20–38. [DOI] [PubMed] [Google Scholar]
- 3. O'Brien DP, Leeb T. DNA testing in neurologic diseases. J Vet Intern Med 2014;28:1186–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kovacs GG. Molecular pathological classification of neurodegenerative diseases: Turning towards precision medicine. Int J Mol Sci 2016;17:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carrell RW, Lomas DA. Conformational disease. Lancet 1997;350:134–138. [DOI] [PubMed] [Google Scholar]
- 6. Howell MJ, Schenck CH. Rapid eye movement sleep behavior disorder and neurodegenerative disease. JAMA Neurol 2015;72:707–712. [DOI] [PubMed] [Google Scholar]
- 7. Iranzo A, Santamaria J, Tolosa E. The clinical and pathophysiological relevance of REM sleep behavior disorder in neurodegenerative diseases. Sleep Med Rev 2009;13:385–401. [DOI] [PubMed] [Google Scholar]
- 8. Bush WW, Barr CS, Stecker MM, et al. Diagnosis of rapid eye movement sleep disorder with electroencephalography and treatment with tricyclic antidepressants in a dog. J Am Anim Hosp Assoc 2004;40:495–500. [DOI] [PubMed] [Google Scholar]
- 9. Hendricks JC, Lager A, O'Brien D, et al. Movement‐disorders during sleep in cats and dogs. J Am Vet Med Assoc 1989;194:686–689. [PubMed] [Google Scholar]
- 10. Schubert TA, Chidester RM, Chrisman CL. Clinical characteristics, management and long‐term outcome of suspected rapid eye movement sleep behaviour disorder in 14 dogs. J Small Anim Pract 2011;52:93–100. [DOI] [PubMed] [Google Scholar]
- 11. Siso S, Tort S, Aparici C, et al. Abnormal neuronal expression of the calcium‐binding proteins, parvalbumin and calbindin D‐28k, in aged dogs. J Comp Pathol 2003;128:9–14. [DOI] [PubMed] [Google Scholar]
- 12. Bastianelli E. Distribution of calcium‐binding proteins in the cerebellum. Cerebellum 2003;2:242–262. [DOI] [PubMed] [Google Scholar]
- 13. Shea TB, Beermann ML. Evidence that the monoclonal antibodies SMI‐31 and SMI‐34 recognize different phosphorylation‐dependent epitopes of the murine high molecular mass neurofilament subunit. J Neuroimmunol 1993;44:117–121. [DOI] [PubMed] [Google Scholar]
- 14. Hendricks JC, Morrison AR. Normal and abnormal sleep in mammals. J Am Vet Med Assoc 1981;178:121–126. [PubMed] [Google Scholar]
- 15. Villablanca J. Why do we have a caudate nucleus? Acta Neurobiol Exp 2010;70:95–105. [DOI] [PubMed] [Google Scholar]
- 16. Hegde AN, Mohan S, Lath N, et al. Differential diagnosis for bilateral abnormalities of the basal ganglia and thalamus. Radiographics 2011;31:5–30. [DOI] [PubMed] [Google Scholar]
- 17. Vandevelde M, Higgins RJ, Oevermann A. Degenerative diseases In: Vandevelde M, Higgins RJ, Oevermann A, eds. Veterinary Neuropathology: Essentials of Theory and Practice. Chichester, West Sussex, UK: Wiley‐Blackwell; 2012:157–189. [Google Scholar]
- 18. Singh M, Thompson M, Sullivan N, et al. Thiamine deficiency in dogs due to the feeding of sulphite preserved meat. Aust Vet J 2005;83:412–417. [DOI] [PubMed] [Google Scholar]
- 19. Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden‐Spatz syndrome) and pantothenate kinase‐associated neurodegeneration. Mov Disord 2004;19:36–42. [DOI] [PubMed] [Google Scholar]
- 20. Collins D, Angles JM, Christodoulou J, et al. Severe subacute necrotizing encephalopathy (Leigh‐like syndrome) in American Staffordshire Bull Terrier Dogs. J Comp Pathol 2013;148:345–353. [DOI] [PubMed] [Google Scholar]
- 21. Baiker K, Hofmann S, Fischer A, et al. Leigh‐like subacute necrotising encephalopathy in Yorkshire Terriers: Neuropathological characterisation, respiratory chain activities and mitochondrial DNA. Acta Neuropathol 2009;118:697–709. [DOI] [PubMed] [Google Scholar]
- 22. Brenner O, de Lahunta A, Summers BA, et al. Hereditary polioencephalomyelopathy of the Australian cattle dog. Acta Neuropathol 1997;94:54–66. [DOI] [PubMed] [Google Scholar]
- 23. Brenner O, Wakshlag JJ, Summers BA, et al. Alaskan Husky encephalopathy—A canine neurodegenerative disorder resembling subacute necrotizing encephalomyelopathy (Leigh syndrome). Acta Neuropathol 2000;100:50–62. [DOI] [PubMed] [Google Scholar]
- 24. Vernau KM, Runstadler JA, Brown EA, et al. Genome‐wide association analysis identifies a mutation in the thiamine transporter 2 (SLC19A3) gene associated with Alaskan Husky encephalopathy. PLoS ONE 2013;8:e57195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montgomery DL, Storts RW. Hereditary striatonigral and cerebello‐olivary degeneration of the Kerry blue terrier. I. Gross and light microscopic central nervous system lesions. Vet Pathol 1983;20:143–159. [DOI] [PubMed] [Google Scholar]
- 26. O'Brien DP, Johnson GS, Schnabel RD, et al. Genetic mapping of canine multiple system degeneration and ectodermal dysplasia loci. J Hered 2005;96:727–734. [DOI] [PubMed] [Google Scholar]
- 27. Biegen VR, McCue JP, Donovan TA, et al. Metabolic encephalopathy and lipid storage myopathy associated with a presumptive mitochondrial fatty acid oxidation defect in a dog. Front Vet Sci 2015;2:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fravel V, Van Bonn W, Dennison S, et al. Bilaterally symmetrical lesions of the caudate nucleus in a northern fur seal pup (Callorhinus ursinus). Vet Rec Case Rep 2015;3:e000176. [Google Scholar]
- 29. Walker FO. Huntington's disease. Lancet 2007;369:218–228. [DOI] [PubMed] [Google Scholar]
- 30. Sokolov E, Schneider SA, Bain PG. Chorea‐acanthocytosis. Pract Neurol 2012;12:40–43. [DOI] [PubMed] [Google Scholar]
- 31. Basel‐Vanagaite L, Muncher L, Straussberg R, et al. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol 2006;60:214–222. [DOI] [PubMed] [Google Scholar]
- 32. Williams DR, Lees AJ. Progressive supranuclear palsy: Clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009;8:270–279. [DOI] [PubMed] [Google Scholar]
- 33. Carpenter S, Soares H, Brandao O, et al. A novel type of familial proximal axonal dystrophy: Three cases and a review of the axonal dystrophies. Eur J Paediatr Neurol 2012;16:292–300. [DOI] [PubMed] [Google Scholar]
- 34. Fyfe JC, Al‐Tamimi RA, Castellani RJ, et al. Inherited neuroaxonal dystrophy in dogs causing lethal, fetal‐onset motor system dysfunction and cerebellar hypoplasia. J Comp Neurol 2010;518:3771–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chrisman CL, Cork LC, Gamble DA. Neuroaxonal dystrophy of Rottweiler dogs. J Am Vet Med Assoc 1984;184:464–467. [PubMed] [Google Scholar]
- 36. Nibe K, Nakayama H, Uchida K. Immunohistochemical features of dystrophic axons in Papillon dogs with neuroaxonal dystrophy. Vet Pathol 2009;46:474–483. [DOI] [PubMed] [Google Scholar]
- 37. Sacre BJ, Cummings JF, De Lahunta A. Neuroaxonal dystrophy in a Jack Russell terrier pup resembling human infantile neuroaxonal dystrophy. Cornell Vet 1993;83:133–142. [PubMed] [Google Scholar]
- 38. Clark RG, Hartley WJ, Burgess GS, et al. Suspected inherited cerebellar neuroaxonal dystrophy in collie sheep dogs. N Zeal Vet J 1982;30:102–103. [DOI] [PubMed] [Google Scholar]
- 39. Maki K. Population structure and genetic diversity of worldwide Nova Scotia Duck Tolling Retriever and Lancashire Heeler dog populations. J Anim Breed Genet 2010;127:318–326. [DOI] [PubMed] [Google Scholar]
- 40. Melis C, Borg AA, Espelien IS, et al. Low neutral genetic variability in a specialist puffin hunter: The Norwegian Lundehund. Anim Genet 2013;44:348–351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Methods and reagents used for immunohistochemical staining of sections.
Table S2. Summaries of case signalment, reported clinical signs (general and neurological), physical examination findings (general and neurological), clinical tests performed, advanced imaging findings, response to treatment, and outcome.
Video S1. Home video recording of affected NSDTR demonstrating marked involuntary movements during sleep.
Video S2. Home video of affected NSDTR demonstrating abnormal vertical position adopted during swimming.