

Abstract
Antibody‐drug conjugates (ADCs) are a rapidly growing therapeutic platform for the treatment of cancer. ADCs consist of a cytotoxic small molecule drug linked to an antibody to provide targeted delivery of the cytotoxic agent to the tumor. Understanding the pharmacokinetics (PK) and pharmacodynamics (PD) of ADCs is crucial in their design to optimize dose and regimen, to maximize efficacy and to minimize toxicity in patients. Significant progress has been made in recent years in this area, however, many fundamental questions still remain. This review discusses factors to consider while assessing the disposition of ADCs, and the unique challenges associated with these therapeutics. Current tools that are available and strategies to enable appropriate assessment are also discussed. © 2015 Genentech Inc. Biopharmaceutics & Drug Disposition Published by John Wiley & Sons Ltd.
Keywords: antibody drug conjugate, pharmacokinetics, preclinical, biotherapeutics, cancer
Introduction
Antibody‐drug conjugates (ADCs) are a rapidly growing biotherapeutic platform for the treatment of cancer that provides targeted delivery of a cytotoxic drug to the tumor cell 1, 2, 3. Two ADCs have been approved recently by the FDA: Kadcyla® (ado‐trastuzumab emtansine) for the treatment of HER2 positive metastatic breast cancer, and Adcetris® (brentuximab vedotin) for the treatment of Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Several more ADCs are at various stages of development from the preclinical to clinical pipeline 4, 5. Three main components of an ADC are (i) a cytotoxic drug, (ii) a monoclonal antibody and (iii) a linker that attaches these two components together. Some of the desirable features of an ADC include a monoclonal antibody that is targeted to a tumor specific antigen (or expressed at higher levels on the tumor vs. normal cells), and is internalized upon binding to the target, a highly potent cytotoxic drug that is stable at physiological pH, and a linker that is stable in circulation but releases the active drug in the cell upon internalization of the antigen–antibody complex. Types of currently used cytotoxic drugs, linkers and conjugation chemistries are summarized in Table 1.
Table 1.
Types of currently used ADC components
| Component | Type | Desired characteristics |
|---|---|---|
| Payloads | Microtubule inhibitors | Potent cytotoxic amenable to linking |
| • Auristatins (MMAE, MMAF) | ||
| • Maytansinoids (DM1, DM4) | ||
| DNA‐damaging agents | ||
| • Calicheamicin | ||
| • Ducaramycins | ||
| • Pyrrolobenzodiazepines (PBD) | ||
| RNA polymerase (alpha‐amanitin) | ||
| Linkers | Cleavable | Stable in circulation but released in tumor |
| • Protease cleavable e.g. valine‐citruline | ||
| • Acid labile e.g. hydrazone | ||
| • Disulfide linkers e.g. SPDB, SPP | ||
| Non‐cleavable (e.g. MCC) | ||
| Conjugation chemistry | Via lysine residues | Homogenous mixture, stable |
| Via cysteines derived from reduced interchain disulfides | ||
| Site specific conjugation | ||
| • Engineered cysteines | ||
| • Unnatural amino acids | ||
| • Enzymatic conjugation |
Considerable efforts are ongoing to understand the optimal characteristics needed for an antibody‐drug conjugate in order to maximize its efficacy, while minimizing its toxicity. Several aspects of an ADC can influence its pharmacokinetics (PK) (i.e. distribution, metabolism/catabolism, excretion) as well as its pharmacodynamics (PD). These include the antibody component, type of linker, site of conjugation, drug to antibody ratio (DAR) and the properties of the cytotoxic agent including its mechanism of action, potential for efflux and metabolite profile 6, 7, 8, 9, 10, 11. The aspects of the antibody component that could impact PK are similar to that of the naked antibody, and include antigen binding, target mediated drug disposition, tissue distribution, immunogenicity, binding to FcRn, effector functions, aggregation, etc.
The stability of the linker which is dependent on the type of linker, as well as the site and type of conjugation, can impact ADC clearance. The type of linker (cleavable vs. non‐cleavable) can also impact the metabolite/catabolite profile of an ADC, which could lead to differences in its activity. Another important aspect that impacts PK is the drug to antibody ratio of the ADC that can be manipulated by different conjugation chemistry 10, 11. Two main types of cytotoxic agents used with the current ADCs are microtubule inhibitors (auristatins, maytansinoids) and DNA‐damaging agents. The different mechanisms of action of these cytotoxic drugs can have an effect on the PK drivers of efficacy and toxicity of the ADC. Gaining a mechanistic understanding of the pharmacokinetics (PK) and disposition of ADCs can help us to take advantage of the opportunity to increase the safety window of an ADC. However, this complex molecule offers multiple challenges to assess its PK. While there has been a lot of progress in understanding the PK of ADCs, many questions still remain. This review discusses key PK findings for different types of ADCs in development, challenges associated with PK characterization, and the strategies and technologies being used to address these issues.
Mechanism of Antibody‐Drug Conjugate disposition
The pharmacokinetics of an antibody drug conjugate is driven primarily by its antibody component rather than its small molecule component, and is characterized by its slow clearance, long half‐life and limited tissue distribution 12. The proposed mechanism of ADC disposition in vivo is shown in Figure 1 1, 6. The ADC binds to its target on tumor cells and is internalized via receptor‐mediated endocytosis. Upon internalization of the ADC–antigen complex, it goes from the endosome to the lysosome, where the cytotoxic drug is released from the ADC in these intracellular compartments and causes cell death. The process of cytotoxic drug release from an ADC can occur via deconjugation, where the linker is cleaved to release the drug, or via catabolism where the entire ADC is degraded by proteolysis, thereby releasing the drug 1, 6. Depending on the stability of the linker, deconjugation and release of the cytotoxic drug can take place in the systemic circulation (unstable linker) and/or inside the cell (stable linker). The site of drug release has a huge impact on its toxicity profile and the main rationale for an ADC therapeutic is to have the drug released in the target tumor cell and not in the systemic circulation. However, toxicity could also result from drug release in normal cells following either target‐mediated uptake in normal cells (if the target is expressed on normal cells) and/or non‐specific uptake (target‐independent uptake) of ADCs via pinocytosis 6. Hence it is critical to understand ADC disposition and its relationship to efficacy and toxicity.
Figure 1.
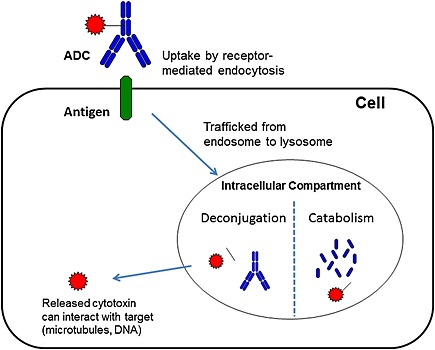
Proposed mechanism of ADC disposition. Upon binding to its target antigen on tumor cell, the ADC is internalized via receptor‐mediated endocytosis and trafficked from the endosome to the lysosome. The cytotoxic drug is released from the ADC by either deconjugation or catabolism in these intracellular compartments. The released cytotoxic drug then binds to its target and causes cell death
The key aspects that need to be investigated to characterize the pharmacokinetics of an ADC include:
Deconjugation: suitable linker stability to deliver the ADC to the target but sufficient lability to release the active drug once internalized.
In vivo exposure, preclinally in the efficacy and toxicity species, as well as clinically in patients: the choice of analytes to be measured is important and is discussed in more detail in the section below.
Tissue distribution: assess target and non‐target tissues that the ADC can distribute to, and the accumulation in those tissues.
Catabolite/metabolite profile: assess what is released, their activity, profile in various tissues and major routes of elimination.
Other important assessments include drug efflux, drug–drug interactions, exposure–response analysis, immunogenicity and the effect of organ impairment on PK: these assessments can help to adjust the dose in patients to maintain an appropriate exposure.
In addition, assessing the impact of conjugation on the PK, i.e. site of conjugation, type of conjugation, linker chemistry, as well as the physicochemical properties of the cytotoxic, can further the understanding of structure–activity relationships and help to improve ADC design.
Challenges in Pharmacokinetics Assessment of ADCs
Due to the complex structure of ADCs, the characterization of PK and the types of mechanistic studies are different from that used for small molecules. The pharmacokinetics of ADC is more similar to its large molecule component, i.e. the naked antibody in terms of target‐mediated clearance, FcRn recycling, Fc gamma interactions and immunogenicity. Antibody‐drug conjugates also have limited distribution into tissues, similar to naked antibodies, and it is important to assess tissue distribution to explore the relationship between tissue concentrations and activity. Some of the challenges in characterizing the PK of ADCs are discussed below.
Limited in vitro assays
Unlike small molecules that have validated in vitro systems to assess ADME (absorption, distribution, metabolism and excretion), the currently available systems are not easily applicable or relevant to large molecule therapeutics, such as naked antibodies and ADCs 13. Information on the small molecule component can be obtained using in vitro systems and include assessments of its permeability, enzymes involved in its metabolism and whether it is a substrate/inhibitor of transporters such as P‐glycoprotein. However, for the ADC molecule as a whole, the most commonly used in vitro system is a plasma stability assay to assess linker stability. Limited information on catabolite profiles could be obtained in vitro using target expressing tumor cells where the ADC can be taken up by the cells. For other organs such as liver, lung etc., where the uptake of the ADCs into the cells in the in vitro setting may be limited due to a lack of target expression, it is difficult to obtain comprehensive catabolite profiles that would be similar to the in vivo situation. To better assess distribution, accumulation and catabolism of ADCs in these organs, in vivo studies may be more appropriate.
Species differences
Similar to naked antibodies, the pharmacokinetics of ADCs could also be species dependent, i.e. different in animals vs. humans 14. This can stem from both a specific target mediated clearance process due to possible differences in antigen binding, as well as a non‐specific clearance process such as differences in FcRn binding 15. In addition to known challenges associated with naked antibody, ADCs have some additional considerations such as linker stability and deconjugation processes that may differ between species. It is important to investigate any species differences in pharmacokinetics and the mechanisms of deconjugation and catabolism to ensure translatability of pharmacokinetics data from efficacy and toxicity in animal species to predict exposure in humans and to estimate the therapeutic index. Linker stability across species can be assessed in in vitro plasma stability studies as well as in vivo by comparing pharmacokinetics profiles of different analytes that are described below.
Heterogeneity
Additional complexity is introduced due to conjugation of the small molecule component via a linker resulting in a heterogeneous mixture of different molecular species. The distinct structures of these various molecular species are due to different drug to antibody ratios, i.e. having a varying number of drugs on each antibody, as well as different attachment locations on the antibody, depending on the conjugation chemistry utilized 11. For example, Kadcyla® which is conjugated via lysine residues, and Adcetris® which is conjugated via cysteines derived from reduced internal disulfides, both have DARs ranging from 0 to 8. The DAR of an ADC can influence its stability, solubility, antigen binding, clearance and biodistribution, resulting in different DAR species having distinct pharmacokinetics behaviors 10, 11, 16, 17. Studies have shown that ADCs with higher drug loads were cleared faster than those with lower drug loads 10, 17. In addition, the tolerability of higher DAR ADCs was also lower than the lower DAR ADCs. It is possible that this increase in clearance with the higher DAR ADCs could result in the rapid delivery of the ADC to normal organs, leading to toxicity and hence less tolerability. Recent advances in site‐specific conjugation have made it possible to control some of the heterogeneity by allowing synthesis of ADCs with lower DARs ranging from 0 to 2 11. However, additional heterogeneity can arise in vivo when the ADC undergoes biotransformation via deconjugation and/or catabolism resulting in multiple species with varying DARs as well as various fragments and adducts.
Bioanalytical challenges
The presence of these multiple molecular species, including the different DAR species, as well as unconjugated drug and antibody, gives rise to several questions on the critical analytes to measure in order to appropriately characterize ADC PK and elucidate exposure–response relationships where different species could have varying contributions to the efficacy and/or toxicity. The dynamically changing nature of the mixture in vivo including changing DARs and various catabolites/adducts, etc. due to biotransformation, adds to the complexity of this analysis and makes developing quantitative assays very challenging as different DAR species could behave differently in the assays. Strategies and methods for the bioanalysis of ADCs are still evolving, although significant progress has been made in recent years 18.
Tools to Assess Pharmacokinetics of ADCs
Bioanalytical methods
There have been great advances in the types of analytical methods to measure the different components of the ADCs such as the total antibody, conjugated and unconjugated drug, DAR distribution, catabolites and metabolites in various matrices such as plasma, bile, tissues from in vitro or in vivo studies depending on the different stages of drug development and type of information desired 18, 19. Commonly used methods are enzyme‐linked immunosorbent assays (ELISAs) and more recently liquid chromatography‐mass spectrometry assays (LC‐MS). Some of the analytes used to assess pharmacokinetics include total antibody (Tab: measures both conjugated and naked antibody), conjugated antibody (measures antibody that has at least one drug attached to it), antibody‐conjugated drug (measures any drug still conjugated to the antibody) and unconjugated drug (measures drug that is not associated with the antibody) 18, 19. Typical PK profiles of these analytes are shown in Figure 2. The information from the profiles of these various analytes can provide insights into linker stability, target mediated drug disposition, etc. and can be used to assess exposure–response relationships. For example, the comparison of the Tab and conjugated antibody analytes allows for an assessment of linker stability, where the greater the separation the greater the instability. Additional bioanalytical assays using LC‐MS/MS methods have been developed to assess DAR distribution and to provide information into the products of biotransformation pathways such as maleimide exchange, adduct formation and the formation of various other catabolites/metabolites 20, 21, 22. Strategies for appropriate analytes to measure for PK evaluation are discussed in depth in several recent bioanalytical reviews 18, 19.
Figure 2.
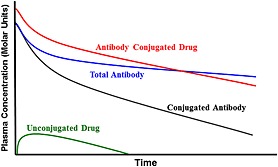
Typical pharmacokinetic profiles of commonly measured ADC analytes. Total antibody which measures both conjugated and naked antibody; conjugated antibody which measures antibody that has at least one drug attached to it; antibody‐conjugated drug which measures any drug still conjugated to the antibody; unconjugated drug which measures drug that is not associated with the antibody
Biodistribution methods
Investigation of biodistribution of ADCs to various tissues, their accumulation and their catabolite/metabolite profiles are important to obtain a mechanistic understanding of the ADME of ADCs. It is critical to understand the distribution of ADCs to tumor vs. normal tissues, especially when normal tissues could have low levels of target expression, since that could possibly lead to toxicity in those tissues 23. One commonly used technique to assess ADC biodistribution is the use of radiolabeled ADCs (labeling either the antibody or small molecule component) with tissue cut and count techniques where the tissues are harvested at various timepoints and radioactivity is measured 23, 24, 25, 26. Commonly used radioactive probes include 125I‐labeled antibody which reflects tissue uptake kinetics and 111In‐DOTA labeled antibody, a residualizing probe that is charged and highly polar, causing it to accumulate in cells if the labeled ADC is internalized. A radiolabel (3H or 14C) can also be applied on the small molecule component of the ADC 24. Comparison of the ADC biodistribution with that of the unconjugated antibody or unconjugated drug can be conducted to assess the impact of conjugation on the antibody biodistribution. Other imaging technologies that can be used in animals as well as in clinical studies include single photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging 26, 27.
Pharmacokinetics/pharmacodynamics modeling
Linking the ADC PK to its response is useful in understanding how exposure can impact safety and efficacy. This analysis can be used to optimize dose and dose regimens to maximize efficacy while minimizing toxicity. Several PK/PD models have been proposed for ADCs ranging from simplified compartment models to multi‐scale mechanistic models 28, 29, 30, 31. Challenges for developing PK/PD models for ADCs include (i) multiple analytes for ADCs with their distinct physiochemical properties impacting distribution/elimination, (ii) dynamic nature of the ADC with constantly changing DARs in vivo, (iii) multiple elimination pathways such as deconjugation and catabolism in addition to the target‐mediated drug disposition for the antibody component, and (iv) possible immunogenicity that can lead to anti‐therapeutic antibody (ATA) formation. As more in vitro and in vivo data become available, both preclinically and clinically, these models can be expanded and help in furthering our mechanistic understanding of ADCs. These analyses can also guide the choice of critical analytes to measure in larger clinical studies.
PK Characteristics of Selected ADCs
Most of the ADCs in the clinical pipeline have tubulin binding agents such as maytansinoids and auristatins, or DNA damaging agents as their cytotoxic components. The different mechanism of action of these cytotoxins can impact their PK drivers of efficacy and safety. Different types of linkers, drug loads and conjugation chemistry have been used with these payloads. The pharmacokinetics characteristics of selected ADCs in each of these three categories are described in more detail below. A recent report showed the clinical PK comparison of selected ADCs from Phase 1 studies using standardized methods across all studies 32.
Pharmacokinetics of maytansinoid ADCs
Maytansinoid payloads that are commonly used are DM1 and DM4. The most advanced of the maytansinoid ADC is the FDA approved Kadcyla® (ado‐trastuzumab emtansine), where DM1 (payload) is conjugated to trastuzumab (anti‐Her2 antibody, IgG1 isotype) via a non‐cleavable thioether linker (MCC) using lysine conjugation chemistry. The average DAR is 3.5, however, the product is heterogeneous having multiple species with DARs ranging from 0 to 8. Kadcyla® showed linear pharmacokinetics in mice and rats (non‐binding species) and non‐linear pharmacokinetics in cynomolgus monkeys and humans (both binding species) where the clearance decreased with an increase in dose, possibly due to saturation of the target at the higher doses 33, 34, 35. In the Phase 1 study in patients with HER2‐positive metastatic breast cancer 35, at a dose of 3.6 mg/kg given intravenously (i.v.) every 3 weeks, the PK parameters of the ADC were as follows: CL of 12.9 ml/day/kg, half‐life of 3.5 days and volume of distribution at steady state (V ss) of 60 ml/kg. Biodistribution studies conducted in rats showed non‐specific distribution to highly perfused organs without accumulation in any organs 33. Similar catabolites were found in rat and human plasma from in vivo studies and included trastuzumab, DM1 and linker containing catabolites, MCC‐DM1 and Lys‐MCC‐DM1 33. Similar to Kadcyla®, other maytansinoid ADCs using lysine conjugation such as SAR3419 (anti‐CD19 IgG1 antibody conjugated to DM4 via SPDB disulfide linker) and IMGN901 (anti‐CD56 IgG1 antibody conjugated to DM1 via SPP disulfide linker) showed similar tissue distribution to the unconjugated antibody 7, 33.
For maytansinoid‐ADCs with cleavable disulfide linkers conjugated to an anti‐Her2 antibody, the ADC clearance in mice was found to be dependent on the degree of steric hindrance of the disulfide linkage, with the least hindered linker (SPDP‐DM1) having a higher clearance than the most hindered linker (SSNP‐DM4) 36. In the same study, clearance of the non‐cleavable MCC linker was comparable to that of the most hindered disulfide linker (i.e. most stable linker, SSNP‐DM4). The cleavable disulfide linkers also produced multiple catabolites, some with increased cell permeability which led to bystander effects, whereby a cytotoxic agent released in one cell diffuses to neighboring cells and exerts its effect 9, 37. This showed that the linker could impact the type of catabolites formed and in turn impact the activity of the ADC.
Pharmacokinetics of auristatin ADCs
Auristatin payloads that have been used for ADCs include MMAE and MMAF. The most advanced auristatin ADC is the FDA approved Adcetris® (brentuximab vedotin), where MMAE (payload) is conjugated to an anti‐CD30 antibody cAC10 (IgG1 isotype) via a cleavable linker (MC‐VC‐PAB) using maleimide chemistry to cysteines derived from the reduction of interchain disulfides. This has an average DAR of 4 with a heterogeneous mixture consisting of multiple DAR species ranging from 0 to 8. The pharmacokinetics of Adcetris® in a Phase 1 study was dose proportional in the narrow dose range tested, and the PK parameters at the maximum tolerated dose of 1.8 mg/kg given i.v. every 3 weeks were as follows: CL of 25.1 ml/day/kg, half‐life of 4.43 days and V ss of 117 ml/kg 32, 38.
Studies in mice and rats 10, 17 using either MMAE or MMAF MC‐VC‐PAB linked ADCs have shown that ADCs with higher drug loads (DARs of 6 and 8) showed higher clearance and shorter half‐lives than ADCs with lower drug loads (DARs of 2 and 4). Higher DAR auristatin ADCs were also less tolerated than lower DAR ADCs 17. Use of site specific conjugation with engineered cysteines (such as Genentech's THIOMAB™ technology among others) can produce more homogeneous ADC mixtures and eliminate higher DAR species 11. The conjugation site can impact the stability and PK of the ADC, depending on the solvent accessibility and local charge at the site 39, 40.
Tissue distribution studies with auristatin ADCs in rodents showed similar distribution profiles of the ADC to the unconjugated antibody with a trend towards slightly increased hepatic uptake 24, 25.
Pharmacokinetics of DNA damaging ADCs
The DNA damaging agents as payloads used in ADCs include calicheamicin, pyrolobenzodiazepines, etc. These agents are very cytotoxic with different mechanisms of action than the tubulin binding agents and hence have a different spectrum of toxicity 41. The more advanced calechemicin ADCs are Mylotarg® (gemtuzumab ozogamicin), which was approved by the FDA in 2000 and withdrawn in 2010 over concerns of clinical benefit, and CMC544 (inotuzumab ozogamicin). Both of these ADCs have calicheamicin conjugated to an IgG4 isotype antibody (anti‐CD33 for Mylotarg® and anti‐CD22 for CMC544) via an acid labile hydrazone linker. These two calicheamicin ADCs showed higher clearance values compared with the other ADCs, which could be a combination of various factors including the antibody, target, indication, linker, etc. 32. In addition, their MTDs were lower, possibly reflecting the potent cytotoxicity of calicheamicin. In a Phase 1 study, Mylotarg® at a dose of 0.22 mg/kg i.v. had a CL of 90.8 ml/day/kg, half‐life of 3 days and V ss of 300 ml/kg 32, 42. CMC544 showed non‐linear PK in a Phase 1 study and its PK parameters at a dose of 0.045 mg/kg i.v. were CL of 91.2 ml/day/kg, a very short half‐life of 0.71 days and V ss of 89.1 ml/kg 32. The biodistribution of a calechaemicin ADC, CMD‐193 (anti‐CD174 antibody conjugated to calicheamicin via an acid labile hydrazone linker) was evaluated in a Phase 1 study and compared with a previous study using the parent unconjugated antibody, hu3S193 27. The ADC showed faster clearance, increased hepatic uptake and much lower tumor uptake than the parent antibody, showing that conjugation can have an impact on PK and biodistribution. ADCs with other DNA damaging agents such as pyrolobenzodiapines are still early in the pipeline with limited data available 43.
Summary
Information on the pharmacokinetics and biodistribution for ADCs using different linkers, payloads and conjugation chemistry is increasing as more molecules are evaluated both preclinically and then advance into the clinic. In addition, the field is rapidly moving ahead with new designs in conjugation chemistry, novel linkers, as well as novel payloads. While progress has been made in gaining knowledge on ADC pharmacokinetics and pharmacodynamics, as more data become available, there should be increased understanding of the mechanistic aspects of ADC disposition and how it relates to efficacy and safety. Improvements in current tools as well as new advancements in technologies for bioanalysis, PK/PD models and in vitro models, will help to answer fundamental questions on the disposition of ADCs and help to improve ADC design as well as optimize dose and dose regimen for improved patient benefit. Translation of PK/PD for ADCs from in vitro systems as well as from animals to humans, are key focus areas that will require our sustained efforts. The strategies and PK considerations for ADCs will continue to evolve as assessment tools and technologies advance and more data become available on diverse types of ADCs.
Acknowledgements and Disclosures
All authors are current or former employees of Genentech, a member of the Roche Group, and hold financial interest in Hoffman‐La Roche.
Kamath, A. V. , and Iyer, S. (2016) Challenges and advances in the assessment of the disposition of antibody‐drug conjugates. Biopharm. Drug Dispos., 37: 66–74. doi: 10.1002/bdd.1957.
References
- 1. Sievers EL, Senter PD. Antibody‐drug conjugates in cancer therapy. Annu Rev Med 2013; 64: 15–29. [DOI] [PubMed] [Google Scholar]
- 2. Carter PJ, Senter PD. Antibody‐drug conjugates for cancer therapy. Cancer J 2008; 14(3): 154–169. [DOI] [PubMed] [Google Scholar]
- 3. Lambert JM. Drug‐conjugated antibodies for the treatment of cancer. Br J Clin Pharmacol 2012; 76(2): 248–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mullard A. Maturing antibody‐drug conjugate pipeline hits 30. Nature Rev Drug Discov 2013; 12: 329–332. [DOI] [PubMed] [Google Scholar]
- 5. Beck A, Reichert JM. Antibody‐drug conjugates: present and future mAbs 2014; 6: 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin K, Tibbitts J. Pharmacokinetic considerations for antibody drug conjugates. Pharm Res 2012; 29: 2354–2366. [DOI] [PubMed] [Google Scholar]
- 7. Erickson HK, Lambert JM. ADME of antibody‐maytansinoid conjugates. AAPS J 2012; 14(4): 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flygare JA, Pillow TH, Aristoff P. Antibody–drug conjugates for the treatment of cancer. Chem Biol Drug Des 2013; 81: 113–121. [DOI] [PubMed] [Google Scholar]
- 9. Erickson HK, Lewis Phillips GD, Leipold DD, et al. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther 2012; 11(5): 1133–1142. [DOI] [PubMed] [Google Scholar]
- 10. Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10: 7063–7070. [DOI] [PubMed] [Google Scholar]
- 11. Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site‐specific antibody drug conjugates for cancer therapy mAbs 2014; 6(1): 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng R, Jin F, Prabhu S, Iyer S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamics considerations for drug development? Expert Opin Drug Metab Toxicol 2012; 8(2): 141–160. [DOI] [PubMed] [Google Scholar]
- 13. Lin JH. Pharmacokinetics of biotech drugs: peptides, proteins and monoclonal antibodies. Curr Drug Metabol 2009; 10: 661–691. [DOI] [PubMed] [Google Scholar]
- 14. Xin Y, Bai S, Damico‐Beyer LA, et al. Anti‐neuropilin‐1 (MNRP1685A): unexpected pharmacokinetic differences across species, from preclinical models to humans. Pharm Res 2012; 29(9): 2512–2521. [DOI] [PubMed] [Google Scholar]
- 15. Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross‐species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem 2010; 285: 4826–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adem YT1, Schwarz KA, Duenas E, Patapoff TW, Galush WJ, Esue O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug Chem 2014; 25(4): 656–664. [DOI] [PubMed] [Google Scholar]
- 17. Leipold DD, Jumbe N, Duggar D, et al Trastuzumab‐Mc‐vc‐PAB‐MMAF: the effects of the drug:antibody ratio (DAR) on efficacy, toxicity and pharmacokinetics. AACR Annual Meeting 2007, Abstract #1551.
- 18. Kaur S, Xu K, Saad OM, Dere FC, Carrasco‐Triguero M. Bioanalytical assay strategies for the development of antibody‐drug conjugate biotherapeutics. Bioanalysis 2013; 5(2): 201–226. [DOI] [PubMed] [Google Scholar]
- 19. Gorovits B, Alley SC, Bilic S, et al. Bioanalysis of antibody‐drug conjugates: American Association of Pharmaceutical Scientists Antibody‐Drug Conjugate Working Group position paper. Bioanalysis 2013; 5(9): 997–1006. [DOI] [PubMed] [Google Scholar]
- 20. Xu K, Liu L, Saad OM, et al. Characterization of intact antibody–drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography‐mass spectrometry. Anal Biochem 2011; 412: 56–66. [DOI] [PubMed] [Google Scholar]
- 21. Xu K, Liu L, Dere R, et al. Characterization of the drug‐to‐antibody ratio distribution for antibody‐drug conjugates in plasma/serum. Bioanalysis 2013; 5(9): 1057–1071. [DOI] [PubMed] [Google Scholar]
- 22. Hengel SM, Sanderson R, Valliere‐Douglass J, Nicholas N, Leiske C, Alley SC. Measurement of in vivo drug load distribution of cysteine‐linked antibody‐drug conjugates using microscale liquid chromatography mass spectrometry. Anal Chem 2014; 86(7): 3420–3425. [DOI] [PubMed] [Google Scholar]
- 23. Boswell CA, Mundo EE, Zhang C, et al. Differential effects of predosing on tumor and tissue uptake of an 111In‐labeled anti‐TENB2 antibody‐drug conjugate. J Nucl Med 2012; 53(9): 1454–1461. [DOI] [PubMed] [Google Scholar]
- 24. Alley SC, Zhang X, Okeley NM, et al. The pharmacologic basis for antibody–auristatin conjugate activity. J Pharmacol Exp Ther 2009; 330(3): 932–938. [DOI] [PubMed] [Google Scholar]
- 25. Boswell CA, Mundo EE, Zhang C, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti‐STEAP1 antibody‐drug conjugates in rats. Bioconjug Chem 2011; 22(10): 1994–2004. [DOI] [PubMed] [Google Scholar]
- 26. Boswell CA, Bumbaca D, Fielder PJ, Khawli LA. Compartmental tissue distribution of antibody therapeutics: experimental approaches and interpretations. AAPS J 2012; 14(3): 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herbertson RA, Tebbutt NC, Lee FT, et al. Phase I biodistribution and pharmacokinetic study of Lewis Y‐targeting immunoconjugate CMD‐193 in patients with advanced epithelial cancers. Clin Cancer Res 2009; 15(21): 6709–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bender B, Leipold DD, Xu K, Shen BQ, Tibbitts J, Friberg LE. A mechanistic pharmacokinetic model elucidating the disposition of trastuzumab emtansine (T‐DM1), an antibody‐drug conjugate (ADC) for treatment of metastatic breast cancer. AAPS J 2014; 16(5): 994–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu D, Girish S, Gao Y, et al. Population pharmacokinetics of trastuzumab emtansine (T‐DM1), a HER2‐targeted antibody‐drug conjugate, in patients with HER2‐positive metastatic breast cancer: clinical implications of the effect of covariates. Cancer Chemother Pharmacol 2014; 74(2): 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model and its approximations for antibody‐drug conjugates. J Pharmacokinet Pharmacodyn 2014; 41(1): 35–47. [DOI] [PubMed] [Google Scholar]
- 31. Shah DK, Haddish‐Berhane N, Betts A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab‐vedotin. J Pharmacokinet Pharmacodyn 2012; 39(6): 643–659. [DOI] [PubMed] [Google Scholar]
- 32. Deslandes A. Comparative clinical pharmacokinetics of antibody‐drug conjugates in first‐in‐human Phase 1 studies. MAbs 2014; 6(4): 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen BQ, Bumbaca D, Saad O, et al. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T‐DM1): an emphasis on preclinical and clinical catabolism. Curr Drug Metab 2012; 13(7): 901–910. [DOI] [PubMed] [Google Scholar]
- 34. Poon KA, Flagella K, Beyer J, et al. Preclinical safety profile of trastuzumab emtansine (T‐DM1): mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol Appl Pharmacol 2013; 273(2): 298–313. [DOI] [PubMed] [Google Scholar]
- 35. Krop IE, Beeram M, Modi S, et al. Phase I study of trastuzumab‐DM1, an HER2 antibody‐drug conjugate, given every 3 weeks to patients with HER2‐positive metastatic breast cancer. J Clin Oncol 2010; 28(16): 2698–2704. [DOI] [PubMed] [Google Scholar]
- 36. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody‐cytotoxic drug conjugate. Cancer Res 2008; 68(22): 9280–9290. [DOI] [PubMed] [Google Scholar]
- 37. Sun X, Widdison W, Mayo M, et al. Design of antibody‐maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug Chem 2011; 22(4): 728–735. [DOI] [PubMed] [Google Scholar]
- 38. Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN‐35) for relapsed CD30‐positive lymphomas. N Engl J Med 2010; 363(19): 1812–1821. [DOI] [PubMed] [Google Scholar]
- 39. Shen BQ, Xu K, Liu L, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody‐drug conjugates. Nat Biotechnol 2012; 30: 184–189. [DOI] [PubMed] [Google Scholar]
- 40. Strop P1, Liu SH, Dorywalska M, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 2013; 20(2): 161–167. [DOI] [PubMed] [Google Scholar]
- 41. Leal M, Sapra P, Hurvitz SA, et al. Antibody‐drug conjugates: an emerging modality for the treatment of cancer. Ann NY Acad Sci 2014; 1321(1): 41–54. [DOI] [PubMed] [Google Scholar]
- 42. Dowell JA, Korth‐Bradley J, Liu H, King SP, Berger MS. Pharmacokinetics of gemtuzumab ozogamicin, an antibody‐targeted chemotherapy agent for the treatment of patients with acute myeloid leukemia in first relapse. J Clin Pharmacol 2001; 41: 1206–1214. [DOI] [PubMed] [Google Scholar]
- 43. Kung Sutherland MS, Walter RB, Jeffrey SC, et al. SGN‐CD33A: a novel CD33‐targeting antibody‐drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug‐resistant AML. Blood 2013; 122(8): 1455–1463. [DOI] [PubMed] [Google Scholar]