

Abstract
Transcriptome profiles derived from the site of human disease have led to the identification of genes that contribute to pathogenesis, yet the complex mixture of cell types in these lesions has been an obstacle for defining specific mechanisms. Leprosy provides an outstanding model to study host defense and pathogenesis in a human infectious disease, given its clinical spectrum, which interrelates with the host immunologic and pathologic responses. Here, we investigated gene expression profiles derived from skin lesions for each clinical subtype of leprosy, analyzing gene coexpression modules by cell-type deconvolution. In lesions from tuberculoid leprosy patients, those with the self-limited form of the disease, dendritic cells were linked with MMP12 as part of a tissue remodeling network that contributes to granuloma formation. In lesions from lepromatous leprosy patients, those with disseminated disease, macrophages were linked with a gene network that programs phagocytosis. In erythema nodosum leprosum, neutrophil and endothelial cell gene networks were identified as part of the vasculitis that results in tissue injury. The present integrated computational approach provides a systems approach toward identifying cell-defined functional networks that contribute to host defense and immunopathology at the site of human infectious disease.
An integrated computational analysis provides a systems approach towards identifying cell-specific functional networks that contribute to host defense and immunopathology in leprosy.
Introduction
The simultaneous detection of the expression patterns for tens of thousands of genes in patient samples can provide valuable insights into disease pathogenesis. In infectious disease, analysis of gene expression profiles has provided an understanding of host immune responses with the potential to improve diagnosis, develop vaccines, and design new therapies (1–3). Leprosy offers an attractive model for investigating immune pathways of host defense and disease pathogenesis (4), as the disease forms a spectrum in which clinical manifestations correlate with the type of immune response. At one end of the spectrum, tuberculoid leprosy (T-lep) is characterized by few skin lesions containing few bacilli, i.e., paucibacillary (PB), and is typically self-limited, indicating that the immune response is containing the infection. At the opposite end of the spectrum, lepromatous leprosy (L-lep), patients have many lesions containing numerous bacilli, i.e., multibacillary (MB), and, if untreated, this form of the disease is progressive, indicating that the immune response is ineffective.
The various forms of leprosy enable the study of disease complications, as the disease spectrum is not static and patients can develop various types of inflammatory and pathologic reactions, either spontaneously or during therapy. These reactions, “reversal reactions” (RR, also known as “type 1 reactions”) (5) and erythema nodosum leprosum (ENL, also known as a “type 2 reaction”) (6), provide a window into the dynamic immune events associated with mechanisms of immunoregulation and immune-mediated tissue injury in human disease. RR is generally considered to represent a naturally occurring delayed-type hypersensitivity response to Mycobacterium leprae antigens; RR is associated with clearance of bacilli from lesions, enhanced cell-mediated immunity against the pathogen resulting in inflammation, and an upgrade from the lepromatous pole toward the tuberculoid pole (7–11). RR is characterized as an inflammatory reaction characterized by erythema, edema, and induration of existing lesions, with associated neuritis (5, 12). ENL is thought to be mediated by immune complex deposition (13) but also involves IL-1 secretion, which can trigger neutrophil recruitment (14). ENL lesions are characterized as subcutaneous erythematous nodules, with systemic symptoms such as fever, arthritis, and neuritis (12). Because leprosy is a disease localized to skin, analysis of the gene expression dynamics in biopsy specimens from leprosy skin lesions has provided new insights into human immune responses at the site of infection that are often not possible to study in other conditions, such as tuberculosis and some autoimmune diseases.
Previously, investigation of the gene expression profiles from leprosy lesions has been limited to the comparison of specific subgroups or the grouping of all leprosy patients in comparison to 15 other infectious, inflammatory, and neoplastic skin diseases (15). Here, we investigated the specific genes and pathways associated with immune cell types that contribute to host defense and tissue injury at the site of infection in the different subtypes of leprosy. The approach we used was motivated by the observation that these lesions differ in the relative abundance and states of specific cell types. Therefore, to characterize the gene expression differences between disease subtypes and reactions, it is essential to leverage existing cell-type and inflammatory signatures and overlay these with well-characterized immune pathways. We synthesize these observations using multipartite networks that reveal the interconnectivity among individual genes, pathways, and cell types.
Results
Gene expression profiles derived from leprosy lesions.
Lesional biopsy samples (n = 29) from patients with the following leprosy subtypes were obtained for determination of gene expression profiles and analysis of gene subtype profiles: L-lep (n = 6), T-lep (n = 10), RR (n = 7), and ENL (n = 6) (Tables 1 and 2 and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.88843DS1). Although obtained simultaneously with the ENL profiles, the T-lep, L-lep, and RR gene expression profiles were previously published (15). All T-lep and L-lep lesions were obtained before the onset of chemotherapy. Gene expression profiles were derived via Affymetrix HG U133 Plus 2.0 microarrays and normalized using frozen robust multiarray average (fRMA), as previously described (16).
Table 1. Clinical characteristics of leprosy patients.

Table 2. Clinical characteristics of leprosy patients with reaction.

Principal component analysis (PCA) of the gene expression profiles demonstrated separation of the leprosy samples into three groups enriched for L-lep, ENL, and T-lep/RR (Figure 1A). The T-lep and RR forms of the disease clustered together, which prevented separation of the two forms by clustering, in line with previous results (16). Although the gene expression profiles of these subforms of leprosy overlapped, the RR samples showed a greater heterogeneity, as indicated by the size of the ellipsoid that represents the 95% confidence interval. The heterogeneity of the RR samples may relate to differences in the severity and/or duration of the reactional state or intrinsic host ability to respond, as was also observed for RR blood samples (17). It is noteworthy that the histologic differentiation of T-lep and RR forms is often difficult (18, 19). In contrast, the ENL samples formed a separate group that was equally dissimilar to both the L-lep and T-lep/RR clusters. RR7 was located near the ENL samples.
Figure 1. Differential distribution of leprosy subtypes.
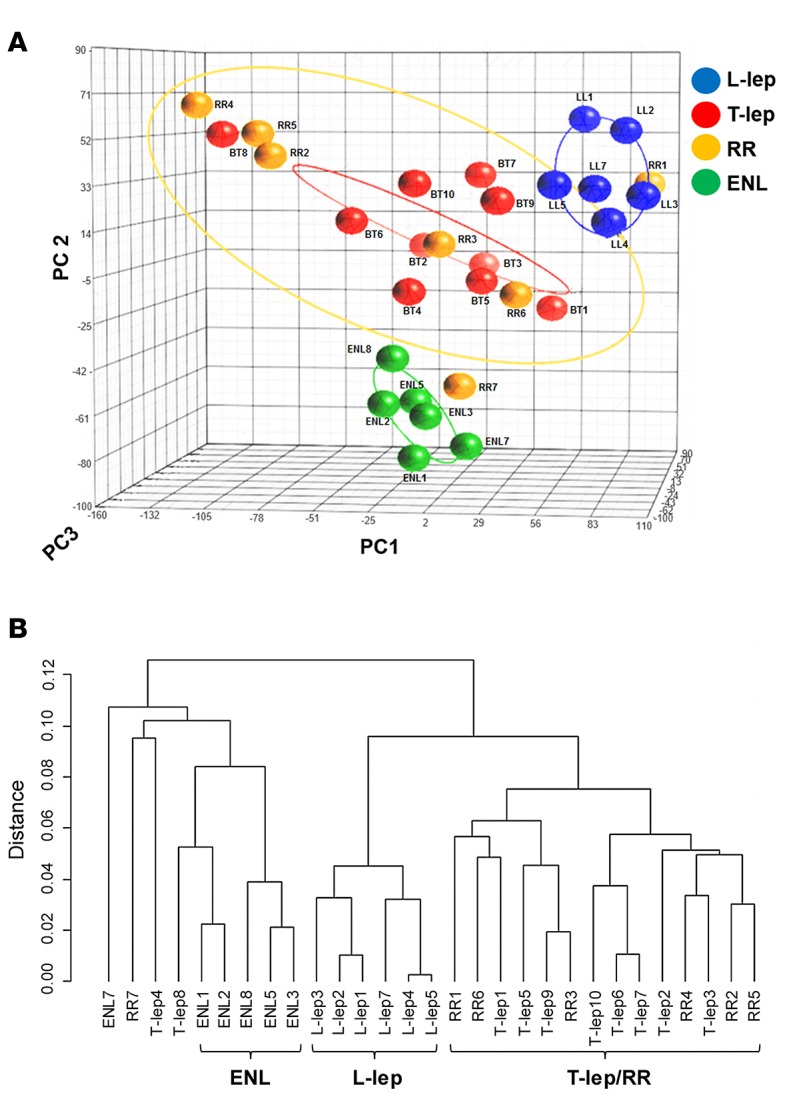
(A) Unsupervised principal component analysis (PCA) of 29 leprosy biopsy specimens. Frozen RMA–normalized and filtered gene expression profiles from 29 leprosy biopsy specimens were characterized using PCA and displayed, with each profile displayed as a colored sphere and individual identification. Ellipsoids represent the 95% confidence interval for sample distribution. Total 3-dimensional PCA mapping represents 56% of variance (PC1 = 29.5%; PC2 = 16.1%, and PC3 = 10.3%). (B) Unsupervised hierarchical clustering of leprosy biopsy specimens. Gene expression profiles from A were clustered using average Pearson correlation and displayed in a tree, with each terminal leaf representing a biopsy sample. ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy; RR, reversal reaction; T-lep, tuberculoid leprosy.
Hierarchical clustering using average linkage again showed three distinct subtype groups of L-lep, ENL, and T-lep/RR (Figure 1B). There were a few exceptions to the overall clustering pattern; three non-ENL samples (2 T-lep, 1 RR) clustered with an ENL sample on a branch adjacent to the other ENL samples, although the relative distance of these three samples to the ENL group was great, consistent with the distant branch points in the dendrogram. Furthermore, of these three possible outliers, only one, RR7, was located in the near vicinity of the ENL cluster by PCA analysis. Moreover, the PCA analysis used a subset of genes used for hierarchal clustering, filtered by a coefficient of variance of ≥ 1.0. There was no clinical information to suggest that the three potential outliers were distinct.
Proportional median signatures.
In order to identify genes that were highly expressed in one subtype relative to all others, we calculated proportional median values for all filtered probe sets in every subtype (Supplemental Table 1). Briefly, the proportional median is a measure for comparing three or more conditions (15), and it is calculated for each probe set in each disease by dividing the median expression of that probe in that disease by the median expression of that same probe across the disease subtypes. Thus, ranking probes by their proportional median measures the relative expression in one subtype compared with all others.
Cell-type deconvolution by specific immune signatures.
In order to be able to connect specific genes that comprise the leprosy subtype signatures with immune cell types, a deconvolution analysis was performed. Cell-type–specific signatures were previously developed by Swindell et al. (20, 21) by comparing relative gene expression fold changes for a set of publicly available microarray data, which was selected as being representative of specific cell types. Briefly, the 50 genes most enriched for each cell type were identified using moderated t tests and fold changes (20, 21). Scores for each cell-type signature were assessed in disease subtype gene expression profiles by computing log intensities of the genes in that subtype relative to all others and then calculating an arithmetic mean of the log expression gene specificity to a certain cell type. Scores were then grouped by hierarchical clustering according to Euclidian distance (Figure 2A). The signatures for CD3+, CD4+, and CD8+ T cells were prominent in T-lep and RR lesions, albeit not significantly enriched compared with the other leprosy subtypes. The DC signature was strongest in T-lep lesions, consistent with the role of DC in activating T cell responses as part of cell-mediated immunity in T-lep patients. A keratinocyte signature was found to be enriched in T-lep lesions (Z score = 2.2). These data correlate with classical histopathological observations that T cells are abundant in T-lep and RR lesions (4, 22) and that keratinocytes in T-lep lesions express high levels of MHC class II, a sign of immune activation (23). Deconvolution of the L-lep gene expression profile identified a significant signature score for macrophages (Z score = 3.2) and a prominent enrichment for CD138+ plasma cells, both of which are indeed prominent in lesions (24), consistent with the elevated antibody levels in L-lep patients. Finally, ENL lesions were characterized by gene signatures for vascular and lymphatic endothelial cells as well as neutrophils, consistent with the presence histologically of a neutrophilic vasculitis. The strong keratinocyte signature in ENL lesions (Z score = 1.9) is also consistent with their robust expression of MHC class II in the epidermis overlying ENL lesions (23).
Figure 2. Cell-type–specific enrichment (deconvolution).

(A) Cell-type–specific deconvolution of all leprosy clinical subtypes. For each of 19 immune cell–type–specific signatures, signature enrichment scores were calculated using average gene expression for each leprosy subtype and normalized to Z scores. Each Z score represents the enrichment for a particular immune cell–type signature in the gene expression profile of one leprosy subtype relative to the other subtypes. Enrichment profiles for each condition were clustered using Euclidean distance and displayed in a heatmap, for which columns correspond to leprosy subtypes and rows correspond to cell types. Each individual square corresponds to the enrichment for one immune cell type in a specific leprosy subtype, with darker squares indicating higher enrichment. (B) Cell-type deconvolution of the proportional median lists for all leprosy subtypes. The gene count represents the number of genes in the proportional median list that overlapped with the specific cell-type list. ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy; RR, reversal reaction; T-lep, tuberculoid leprosy.
A similar analysis of the proportional median gene lists for each subtype identified enrichment for specific cell types and genes of significance to immune pathogenesis (Figure 2B). In the T-lep proportional median list, DC were enriched (6 of 50 genes, including MMP12, CD1B, and CD1C, P = 0.002). Macrophages were enriched in L-lep lesions, including the genes associated with the M2 macrophages in L-lep lesions (25) (14 of 50 genes, including MARCO and MSR1, P = 5 × 10–11), as were CD138+ plasma cells (5 of 50 genes, including IGHD and TNFSFR17, P = 0.01). The top proportional median gene in L-lep lesions was CHRNA1, the cholinergic receptor nicotinic α 1 subunit. Although this gene was not connected to the cell-type signatures or any of the pathways analyses, it has been shown to contribute to macrophage-induced inflammation in atherosclerosis (26, 27), in addition to its established function at the neuromuscular junction (28). The role of CHRNA1 in microbial pathogenesis is unknown. In ENL lesions, a fibroblast gene signature was found to be enriched (6 of 50 genes, including MMP3, P = 0.001).
Cell-type deconvolution of disease-specific gene modules by weighted gene correlation network analysis.
In order to identify genes and functional pathways associated with leprosy subtypes, we performed weighted gene correlation network analysis (WGCNA) on the filtered leprosy gene expression profiles (29, 30). WGCNA is a clustering approach that identifies the typical pattern of each cluster (the eigengene). The WGCNA algorithm uses correlations to group genes into modules, similar to traditional clustering analysis; unlike other approaches, it raises each correlation to a power, thus lending more weight to stronger, more reliable correlations. We then correlated these patterns with binary vectors that encode each subtype (i.e., a sample is assigned a 1 if it is part of the subtype and 0 if not). Therefore, a significant module-subtype correlation implies that samples from one specific subtype have higher expression than those of other subtypes. WGCNA analysis identified 17 modules of coexpressed genes (Figure 3, y axis). To determine which modules were associated with each disease subtype, we performed module eigengene correlation of the samples clustering within the main subtype branch by coding traits as a binary matrix of zeros and ones: each sample had a value of “1” for its corresponding subtype and a value of “0” for all other subtypes (30).
Figure 3. Identification of clinical subtype gene modules in leprosy patients.

Correlation of WGCNA modules to leprosy subtypes was calculated by correlating module eigengene expression to a binary matrix representation of sample membership for each particular subtype. Module eigengenes, as well as the corresponding number of genes in each module, are labeled on the y axis, and leprosy subtypes are labeled on the x axis. Each square in the heatmap is colored according to correlation, with P values for correlation given in parentheses. ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy; RR, reversal reaction; T-lep, tuberculoid leprosy.
The “purple” module was significantly correlated to T-lep (correlation = 0.41; P = 0.03). The “tan” module was significantly correlated to RR (correlation = 0.43, P = 0.02). Four modules, “gray60,” “brown,” “salmon,” and “cyan” were significantly correlated to L-lep (correlation = 0.7, 0.85, 0.55, and 0.45; P = 2 × 10–5, 5 × 10–9, 0.002, and 0.01, respectively). Five modules, “magenta,” “black,” “blue,” “red,” and “turquoise,” were significantly correlated to ENL (correlation = 0.38, 0.75, 0.47, 0.48, and 0.82; P = 0.04, 2 × 10–6, 0.01, 0.009 and 5 × 10–8, respectively).
The WGCNA modules were next evaluated by cell-type deconvolution to identify the specific immune cells that contribute to specific genes and the correlated networks (Figure 4). The “purple” T-lep module overlappedkeratinocytes (5 of 50 genes) and DC (2 of 50 genes, including MMP12 and CCL22). The “tan” module associated with RR did not significantly overlap with a specific cell-type signature. For the L-lep modules, the “brown” module overlapped with macrophages (28 of 50 genes, including MSR1, CHIT1, ATP6V0D2, MARCO, and TREM2, P = 2.7 × 10–8), monocytes (8 of 50 genes, including TLR5, TLR7, NLRP3, CD14, CD33, and DENND1A), and B cells (7 of 50 genes, including CD22), with “gray60” overlapped with CD138+ plasma cells (6 of 50 genes, including TNFRSF17, MZB1, and IGHD, P = 3.7 × 10–4). The “brown” module also overlapped with the DC signature. For the ENL modules, the “black” module overlapped with the cell-type signatures for vascular and lymphatic endothelial cells (5 of 50 and 4 of 50 genes, respectively) as well as fibroblasts (11 of 50 genes, P = 2.7 × 10–2). In addition, the “turquoise” module overlapped with many other cell-type signatures, including vascular endothelial cells (5 of 50 genes, P = 2.0 × 10–3) and lymphatic endothelial cells (9 of 50 genes), fibroblasts (8 of 50 genes), and neutrophils (3 of 50 genes). Although not statistically significant, this overlap for neutrophils was greater than that any other module, consistent with the characteristic presence of neutrophils in ENL lesions. Overall, there was high concordance for the cell types identified by deconvolution of the subtype-specific proportional median signatures and the corresponding subtype-specific WGCNA modules. These results suggest that the clustering pattern of expression profiles across disease subtypes is dictated in part by the cell-type–specific expression of genes, with genes highly expressed in certain cell types concentrated within particular modules.
Figure 4. Integration of WGCNA gene modules with cell-type–specific gene signatures.

For each of the 17 modules of related genes derived from WGCNA analysis, enrichment for cell-type–specific gene signatures for 18 cell types with immune or structural functions were calculated and displayed in a heatmap of Z scores. Z scores were calculated from log2 fold change enrichment scores, using average gene expression for each leprosy subtype for each cell type. Cell-type names are provided in rows, and WGCNA module names from Figure 2 are provided in the column. Modules that were significantly associated with one leprosy subtype are labeled. ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy; RR, reversal reaction; T-lep, tuberculoid leprosy.
Integrative analysis reveals gene pathways associated with leprosy subtypes.
To define gene networks for each disease subtype, we used WGCNA to identify coexpression relationships and integrated the information from functional pathways and cell-type deconvolution analysis with an analysis of the subtype-defined proportional median genes. In L-lep lesions, integration of the functional pathway analysis of the “brown” and “gray60” modules as well as the top 250 proportional median genes identified pathways relating to macrophage function, including “phagosomes,” “endocytosis,” and “endosomes” as well as “lipid binding,” which by the deconvolution analysis was connected with the macrophage and monocyte signatures. In addition, a number of immunoglobulin genes were connected to “innate immunity” and “receptor signaling/activity,” which, in turn, connected with the plasma cell and B cell signatures in the deconvolution analysis (Figure 5A).
Figure 5. Specific networks for clinical subtypes of leprosy (L-lep and ENL).

Gene ontology, KEGG pathway, and Reactome analysis were performed with ClueGO (Cytoscape software) for the most relevant WGCNA modules and the top 250 genes of the proportional median (PM) list for each leprosy clinical subtype. Genes selected from gene sets composed of either PM lists or WGCNA modules were overlapped with the top 250 genes in the specific cell-type signatures related with (A) L-lep and (B) ENL. Connections were visualized by Gephi software. Blue circles represent genes, red circles denote immune functions, yellow circles show cell type, and gray lines represent connections between genes and immune functions and/or cell types. ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy.
We noted in the gene set containing the “black” and “turquoise” modules as well as the ENL proportional median list, a collection of genes encoding collagen polypeptides associated with extracellular matrix organization. Gene pathways for “angiogenesis” and “vasculature development” were prominent, reflecting the vascular and lymphatic endothelial cell as well as the fibroblast signatures detected by cell-type deconvolution. The “cell migration,” “chemotaxis,” and “cell motility” pathways were related to the neutrophil gene signature identified by cell-type deconvolution (Figure 5B).
To identify genes differentially associated with RR, we overlapped the top 250 proportional median genes with “tan” module gene set. Functional analysis identified pathways related to nerve biology, including “response to IFN-γ,” which included CYP27B1, part of the vitamin D antimicrobial pathway, which was also identified in the proportional median genes in T-lep lesions (Figure 6A). In RR, gene pathways for “neurogenesis,” “neurotrophin signaling pathway,” “axon guidance,” and “glial cell differentiation” were detected. Gene pathway analysis further identified immune networks related to cell death and cell survival, including “apoptosis,” “response to stress,” and “autophagy.” The intersection of gene pathways involved in nerve biology and immune-mediated cell death and cell survival reflects the process of immune-mediated nerve injury in RR.
Figure 6. Specific networks for clinical subtypes of leprosy (T-lep and RR).

Gene ontology, KEGG pathway, and Reactome analysis were performed with ClueGO (Cytoscape software) for the most relevant WGCNA modules and the top 250 genes of the proportional median (PM) list for each leprosy clinical subtype. Genes selected from gene sets composed of either PM lists or WGCNA modules were overlapped with the top 250 genes in the specific cell-type signatures related with (A) RR and (B) T-lep. Connections were visualized by Gephi software. Blue circles represent genes, red circles denote immune functions, yellow circles show cell type, and gray lines represent connections between genes and immune functions and/or cell types. (A) RR function analysis shows genes (blue circles) connected with immune and neural biology (red) functions. RR, reversal reaction; T-lep, tuberculoid leprosy.
The gene pathways that are involved in T-lep were examined by integrating the WGCNA “purple” module, associated with T-lep, alongside the top 250 proportional median genes for T-lep (Figure 6B). The top proportional median gene in T-lep was matrix metalloproteinase-12 (MMP12), which was connected in both comparisons to the “response to wounding,” “chemotaxis,” “cell proliferation,” and “tissue morphogenesis” pathways. MMP12 was also connected to DC by the deconvolution analysis.
Gene expression and tissue immunolabeling of leprosy skin lesions show higher expression of MMP12 in T-lep versus L-lep.
MMP12 was the gene with the highest proportional median value in the T-lep lesions. As a result, we evaluated MMP12 mRNA expression in our leprosy subtype gene expression profiles as well as additional samples collected at a different time. These samples comprised 13 previously published ENL (n = 6) and L-lep (n = 7) samples (14) and 9 newly obtained L-lep (n = 3), RR (n = 3), and T-lep (n = 3) gene expression profiles derived from deidentified leprosy skin biopsy specimens, for a total of 51 gene expression profiles. MMP12 mRNA expression was significantly higher in the T-lep lesions in comparison to that in the L-lep and ENL lesions (P < 0.01 for each, Figure 7). The expression of MMP12 mRNA in RR was variable and statistically indistinguishable from the other samples, albeit somewhat lower than in T-lep lesions, with strong expression in only 4 of 10 RR samples as compared with 9 of 13 T-lep samples. MMP-12 protein was detected by immunohistochemistry and was also more highly expressed in T-lep and RR lesions, as compared with L-lep or ENL lesions (Figure 8). MMP-12 protein expression was strong throughout the granuloma but was also detected in cells with elongated nuclei, presumably fibroblasts, in the connective tissue (Figure 8A). Quantification of MMP-12 expression in skin lesions by automated image analysis showed that T-lep lesions have significantly higher expression of MMP-12 compared with L-lep and ENL samples (P < 0.001 and P < 0.01, respectively). RR lesions were characterized by a lower expression of MMP-12 compared with T-lep lesions (not significant) but a significantly higher expression level when compared with L-lep lesions (P < 0.05) (Figure 8B).
Figure 7. MMP12 regulation in leprosy.
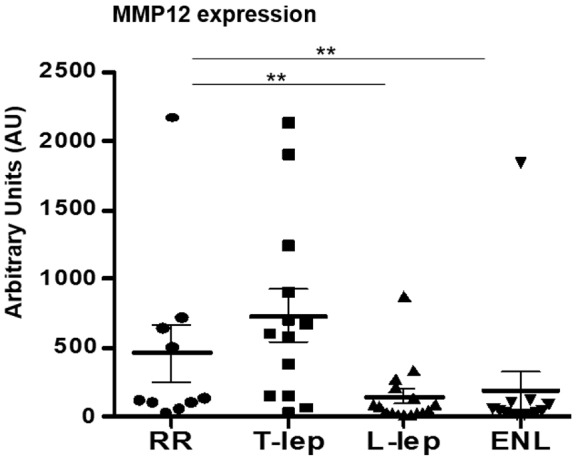
Distribution of MMP12 mRNA by microarray analysis of skin lesions shown in arbitrary units (AU). The graph shows the total number of samples per clinical subtype (mean and SEM). Data represent mean ± SEM, RR = 10 (457.2 ± 207.2 AU), T-lep = 13 (732.7 ± 186.3 AU), L-lep = 16 (143.2 ± 54.41 AU), and ENL = 12 (186.3 ± 138.6 AU). Significance was determined by Kruskal-Wallis test (nonparametric ANOVA style test) using GraphPad Prism software and post-hoc analysis (Dunn’s multiple comparison test). **P < 0.01. ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy; RR, reversal reaction; T-lep, tuberculoid leprosy.
Figure 8. MMP-12 expression in leprosy lesions.

MMP-12 protein expression was evaluated in leprosy lesions (T-lep, L-lep, RR, and ENL). (A) CD3 expression was used as a marker for T cells, and IgG1 was used as a negative control; 1 representative labeled section is shown of at least 5 (each obtained from a different patient). Scale bar: 40 μm. Original magnification: ×100. (B) Automated image analysis of MMP12 protein expression. Each dot represents the percentage of MMP-12–stained area (diaminobenzidine [DAB]) per nuclear area for each individual photomicrograph (n = 5 for each group). Data represent mean ± SEM, T-lep (77.5 ± 10.7), L-lep (17.8 ± 8), RR (54.76 ± 10.2), and ENL 13 (32.14 ± 10.2). One-way ANOVA analysis was performed (P = 0.0004) using GraphPad Prism software, and post-hoc analysis (Bonferroni test) is indicated (*P < 0.05, **P < 0.01, ***P < 0.001). ENL, erythema nodosum leprosum; L-lep, lepromatous leprosy; RR, reversal reaction; T-lep, tuberculoid leprosy.
Discussion
The disease spectrum of leprosy provides an extraordinary model to investigate the relationship between the immune response at the site of infection and the clinical presentation and in particular to understand the gene networks that contribute to host defense versus immunopathology. Here, integration of cell-type deconvolution with the functional pathways associated with the proportional median lists and the coexpression gene networks for each disease subtype revealed new and distinct immune mechanisms at the site of disease. For example, it was unanticipated that this approach would identify an integrated network involving DC linked to MMP12 as part of tissue remodeling and inflammation functional programs in T‑lep lesions, in which the infection is contained. In the other forms of leprosy, gene networks that are known to contribute to host defense and tissue injury were identified, including those pertinent to IFN-γ responses, nerve injury, and vasculitis. The present integrated computational approach provides a systems approach toward identifying cell-defined functional networks at the site of disease.
In T-lep lesions, the top proportional median gene was MMP12, an elastase, suggesting a key role for tissue remodeling in the host defense response to M. leprae. We demonstrated that the MMP-12 protein was more highly expressed in T-lep and RR lesions versus L-lep and ENL lesions, as is most evident throughout the granulomatous infiltrate. MMP12 and its coexpressed genes were linked to “response to wounding,” “chemotaxis,” “cell proliferation,” and “tissue morphogenesis” pathways. In DC, MMP-12 is induced by a combination of inflammatory cytokines, including TNF-α, IL-1β, IL-6 and GM-CSF (31). MMP-12 is also known to be expressed in monocytes, macrophages, and fibroblasts. Like other matrix metalloproteinases, MMP-12 is involved in the breakdown of the extracellular matrix. Although the major substrate for MMP-12 is elastin, MMP-12 is able to degrade a broad spectrum of substrates, such as type IV collagen, fibronectin, laminin, vitronectin, proteoglycans, chondroitin sulfate, α-1-antitrypsin, plasminogen, and myelin basic protein, which is related to nerve damage in the disease (32). MMP-12 contributes to cell migration (33) of DC and leukocytes (31, 34). The ability of MMP-12 to cleave off IFN-α receptor 2–binding sites (35) blocks the action of type I IFN, which in previous work we showed blocks antimicrobial responses in leprosy (16). The presence of MMP-12 in T-lep and RR granulomas, as well as other granulomatous diseases such as sarcoidosis (36), suggests that MMP-12 is part of a tissue remodeling and inflammation gene network that contributes to granuloma formation and/or maintenance. The induction of MMP-12 in RR lesions indicates the dynamic upregulation of this protein during the clinical reduction in the bacillary load and gain in immunity.
The identification of MMP-12 as a contributor to the local immune response in mycobacterial infection was unexpected, since previous studies have focused on the role of a different metalloproteinase family member, MMP-9. In addition to MMP-12, MMP-9 was also present in the top T-lep proportional median genes, more highly expressed in T-lep, RR, and L-lep versus ENL. Previously, MMP-9 protein was found to be more highly expressed in T-lep versus L-lep lesions (37), including in Schwann cells (38), and could be induced by M. leprae in cultured Schwann cells (39). In Mycobacterium marinum infection of zebrafish, studied as a model for human tuberculosis, MMP-9 has been shown to be required for granuloma formation (40). MMP-9 and MMP-12 cooperate in tissue remodeling in the lung (41) and may coregulate, as MMP-12 upregulated MMP-9 expression in the presence of IL-1β (42). In the gene networks in T-lep lesions, MMP9 and MMP12 were connected by the common gene ontology terms: “chemotaxis” and “tissue remodeling”. As part of this network, other genes involved in “chemotaxis” and “tissue remodeling” included IRF6, ADIPOQ, OVOL1, and FOXQ1 (43–48). Another prominent gene, CYP27B1, a member of the cytochrome p450 family, is a critical part of the vitamin D antimicrobial pathway to kill mycobacteria (16, 25, 49, 50). Together, these networks link chemotaxis, tissue remodeling, and antimicrobial activity as part of the effective host response to limit mycobacterial infection.
Our previous studies failed to identify specific gene networks associated with RR lesions (16). However, here, we analyzed the gene expression profiles in RR patient lesions by integrating the top proportional median genes and the WGCNA “tan” module, which was most highly correlated with RR. Gene networks related to cell death and nerve biology were identified. Genes associated with neuronal death and subsequent regeneration (LIFR, BCL11A, AATK, BCL2L11, GSK3A, PLCG1) suggest an ongoing cycle of axon degradation and regeneration. Specifically, these gene signatures are indicative of neurotrophin signaling through Trk and p75 receptors, reflecting the essentiality of this pathway for vertebrate neuronal maintenance (51). In addition, EGFR (also known as ERBB1) and ERBB3, found in these lesions, are part of the ErbB family, which has been shown to be critical for Schwann cell differentiation and survival (52). Genes involved in cell death included TNFSF10 (also know as TRAIL), CXCR3, CCL19, and IDO, which regulate apoptosis (53–56), consistent with the prominent apoptosis present in RR lesions in comparison to nonreactional patients (57, 58). We previously demonstrated that M. leprae ligands induce apoptosis of Schwann cells through TLR2, providing one mechanism by which activation of the innate immune response contributes to nerve injury in leprosy (59). The immune pathway “response to IFN-γ” was associated with RR lesions, including the genes that contribute to antimicrobial activity, including CCL19, CYP27B1, and GBP5. Together, those pathways provide insight into clinical presentation of RR, characterized as sudden episodes of inflammation, edema, and neuritis, associated with severe peripheral nerve impairment and disabilities associated with enhanced immunity against M. leprae (60–62).
In L-lep lesions, gene networks linked to macrophages and B cells were defined that clearly relate to the clinical presentation of these patients. Networks of genes involved in “endocytosis,” “lysosome,” and “phagosome” were identified and associated with macrophages, possibly reflecting the presence of IL-10–derived CD163+ M2 macrophages, which are programmed to be highly phagocytic (16, 25). A lipid-binding pathway was present in L-lep lesions, which is consistent with the observation that host-derived lipids accumulate in macrophages in L-lep lesions (63). Genes involved in humoral immunity, including IGAH1, IGHM, and IGJ genes, which encode the protein components forming IgA and IgM multimers, were prominent in L-lep lesions, consistent with the previous identification of B cell genes in L-lep lesions (24, 64). These Ig genes were connected to B cells and plasma cells, which are present at a greater frequency in L-lep lesions versus T-lep lesions (24, 65). We previously found that IgA and IgM mRNA and protein were more strongly expressed in L-lep lesions versus T-lep lesions, with IgM colocalizing with CD138+ plasma cells (24). These data support the dogma that humoral immune responses found in the L-lep disease do not contribute to effective host defense against M. leprae infection but could contribute to disease pathology.
ENL occurs in patients toward the L-lep part of the disease spectrum, presenting as a neutrophilic vasculitis and panniculitis. Previously, we had identified the gene pathways involved in neutrophil recruitment in ENL lesions (14). Here, we defined CXCL1, CXCL5, and CCR2 as associated with a neutrophil signature and linked with a “chemotaxis” gene network in ENL lesions. In addition, the present data defined “angiogenesis” and “vascular development” gene networks in ENL lesions, consistent with the histological prominence of new blood vessel formation in ENL (66). In particular, these analyses identified genes like ROBO4, NOTCH1, HEY1, SEMA5A, and WNT5A, which are indicative of active angiogenesis. The ENL signature identified here was consistent with vascular remodeling events, as per the high number of matrix proteins, and the maturation and organization of pericyte-endothelial interactions. The high frequency of matrix proteins that are either integral components of the basement membrane (COL4A1, COL8A1, LAMA5) or frequently associated with blood vessels indicates that this tissue is actively engaged in either repairing and/or supporting the establishment of a newly formed vascular tree. Genes associated with late responses to inflammatory events were also represented (IL6, CXCL12) as well as a subgroup of genes that is required for the active engagement of endothelial-pericyte/smooth muscle cell interactions (PDGFA, PDGFRB, NOTCH3, CYR61), also indicative of vascular maturation. The clinical response of ENL patients to the drug thalidomide has been related to its ability to inhibit angiogenesis (67).
In this work, we found that two key measures of disease-specific gene expression profiles, the proportional median and the WGCNA module, could be analyzed and integrated to identify disease mechanisms. By cell-type deconvolution of the proportional median list and disease-related WGCNA module, along with gene ontology analysis, it was possible to link cell types with functional pathways to identify the gene networks that contribute to the clinical manifestations of disease as well as pathophysiologic disease mechanisms. Using this approach, we indicated that DC are linked to MMP12 and a tissue remodeling network that contributes to host defense at the site of infection in leprosy. We also identified several pathways involved in pathogenesis and tissue injury, such as neutrophil and endothelial cell gene networks that contribute to chemotaxis and angiogenesis, respectively, in ENL. Delineation of cell-related functional networks can enhance our understanding not only of leprosy but of other diseases with a complex immunological component and offer greater possibilities than previous work in identifying targets for host-directed therapy in human infectious diseases.
Methods
Microarray normalization and clustering.
Skin lesional biopsy specimens were obtained from patients with various multidrug therapy treatment duration (Tables 1 and 2) and included the following leprosy subtypes: L-lep (n = 6), T-lep (n = 10), RR (n = 7), and ENL (n = 6). Although obtained simultaneously with the ENL profiles, the T-lep, L-lep, and RR gene expression profiles were previously published (15). All L-lep samples were classified according to the Ridley-Jopling criteria as lepromatous (LL), and most of T-lep patients were classified as borderline tuberculoid (BT); only one of them was classified as borderline (BB) (Table 1). For RR, 4 patients were classified as borderline lepromatous (BL) patients at the time of diagnosis, 2 patients were classified as BT, and 2 patients classified as BB or BT/BB. Most of the RR occurred during chemotherapy. In most of the RR patients, the bacillary index was positive, except for 2 patients with bacillary index of 0.5 (Table 2). All ENL patients had received multidrug therapy and were classified at the time of diagnosis as LL, 3 patients developed ENL after completion of multidrug therapy, and 3 developed ENL during multidrug therapy (Table 2). All skin biopsy specimens from patients with one of the reactional states were biopsied at the site of reaction, at the time of diagnosis of RR or ENL, and before start of additional chemotherapy. Some of the RR and ENL patients presented in reaction before chemotherapy, and others developed their reactional state during chemotherapy. mRNA was extracted from lesions, and gene expression profiles were derived via Affymetrix HG U133 Plus 2.0 microarrays (Affymetrix) and normalized using fRMA, as previously described in Bleharski et al. (64). Test samples were obtained from publicity available data on NCBI GEO (GSE17763 for 10 T-lep, 6 LL, and 7 RR samples and GSE8489 for ENL samples). Additional data for validation of MMP-12 gene expression were obtained from publicly available data on NCBI GEO (GSE16844; L-lep: n = 7; and ENL: n = 6) (14) (validation set “V1”) and a second batch of skin lesional biopsy specimens (L-lep: n = 3; T-lep: n = 3; RR: n = 3) (validation set “V2”); both sets were derived via Affymetrix HG U133 Plus 2.0 microarrays. All data were normalized using fRMA to minimize between-batch variation (68). Samples were deposited in GEO under numbers GSE17763 and GSE16844
Normalized data were filtered at a mean intensity of at least 150 in any one subtype. Filtered data were used in the proportional median signature, PCA, clustering, and WGCNA analyses. For the PCA analysis, genes were further filtered using a coefficient of variance of ≥ 1.0 and PCA was performed using Partek v6.4. Hierarchical clustering of genes with a mean intensity of ≥ 150 was performed using average correlation distances and the “amap” package in R.
Proportional median signatures.
The proportional median metric was defined as the median intensity of a probe set within one leprosy subtype divided by the median intensity of the same probe set across all samples. Proportional median values were calculated for each subtype and ranked according to highest proportional median as previously described (15).
Cell-type–specific signature enrichment.
Cell-specific enrichment was calculated using an adapted methodology from Swindell et al. (21). Using 50 gene signatures for each cell type, signature scores were calculated for each subtype based on the mean score of the log-transformed, mean-centered values. The scores for each signature were then clustered based on average Euclidian distance.
WGCNA.
WGCNA was performed on the filtered leprosy subtype gene expression profiles (“wgcna” package in R). Automatic network construction was carried out with a power of 14 and a minimum module size of 50. For each module, networks were constructed using the topological overlap matrix. The top 50 probes from each network were selected by filtering using kME (intramodular connectivity) and converted to gene names before displaying. Networks were built using VisANT (69). Module correlation to leprosy subtypes was calculated by computing the correlation of each module eigengene to a binary matrix of traits, which corresponded to individual subtypes (30). Correlation and significance calculations, as well as heatmap display, were calculated using built-in functions from the “wgcna” R package. P values for overlap of modules with cell-type–specific signatures were calculated using the hypergeometric distribution and were corrected using a Bonferroni adjustment (n = 16).
Functional analysis.
Gene ontology, KEGG pathway, and Reactome analysis were performed with Cytoscape (ClueGO application) with Bonferroni correction for proportional median signatures, cell-specific signatures, and WGCNA modules. Gephi software was used to visualization of functional networks.
Tissue immunostaining.
For each leprosy subtype, 5 leprosy skin biopsies (from 5 different patients) were prepared, from which 1 representative sample was selected. Frozen tissue sections were blocked with normal horse serum before incubation with MMP-12 (clone 4D, R&D Systems, MAB917), CD3 (clone SK7, BD Bioscience, 340851) monoclonal antibodies, and isotype control (clone MOPC 21, Sigma-Aldrich, M5284) for 60 minutes, followed by incubation with biotinylated horse anti-mouse IgG (Vector Laboratories, B-2000) for 30 minutes (70). Slides were counterstained with hematoxylin and mounted in crystal mounting medium (Biomeda) and were visualized using the ABC Elite system (Vector Laboratories). Skin sections were examined using a Leica microscope (Leica 2500). Expression of MMP-12 in photomicrographs was quantified using ImmunoRatio, an automated image analysis application, blind to the diagnosis, which calculates the percentage of diaminobenzidine-stained nuclear area per total area (71). This application was developed based on ImageJ quantification, and it is part of the ImageJ plug-ins.
Statistics.
Statistical methods for specific analyses are detailed in paragraphs above. In brief, microarrays were normalized with the fRMA method (“frma” R package) (63). Microarray clustering was performed using hierarchical clustering with average Euclidian distance, and microarray module-finding analysis was carried out by weighted gene coexpression analysis (“wgcna” R package) (23). P values for module overlap with cell-type–specific signatures were calculated using a Bonferroni-corrected hypergeometric distribution. For MMP12 gene expression and MMP-12 protein expression, multiple comparison tests were used for statistical analysis. They were selected based on normal distribution analysis; tests and P values are listed in the figure legends. P values of less than 0.05 were considered significant.
Study approval.
Ethical approval for the use of human leprosy biopsy specimens was obtained from the UCLA Institutional Review Board. Written informed consent was obtained from all patients included in the study.
Author contributions
MSI, RMBT, DP, CAM, AG, THMO, BRB, MP, and RLM wrote the manuscript. MSI, RMBT, MA, MN, WRS, THMO, MP, and RLM designed the experiments and computational analyses. RMBT, ENS, THR, MTO, and MLIA obtained the leprosy biopsy samples. MSI, RMBT, PRA, DL, MA, WRS, MP, and RLM performed the computational analyses. RMBT and DP conducted the experiments.
Supplementary Material
Acknowledgments
This study was supported by the Order of Malta-Grants-for-Leprosy-Research, the Heiser Program for Research in Leprosy in The New York Community Trust (P13-000392), the NIH (AR063020, AI022553, AR040312, and AI04786), the Q.M. Gastmann-Wichers Foundation, and the Netherlands Leprosy Relief Foundation/Turing Foundation. MN is supported by the National Institute for Health Research Biomedical Research Centre at University College London Hospital.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(15):e88843. doi:10.1172/jci.insight.88843.
Contributor Information
Megan S. Inkeles, Email: myso@g.ucla.edu.
Delila Pouldar, Email: dpouldar@mednet.ucla.edu.
Priscila R. Andrade, Email: PRibeiroAndrade@mednet.ucla.edu.
Cressida A. Madigan, Email: cmadigan@g.ucla.edu.
David Lopez, Email: davidlopez@ucla.edu.
Mike Ambrose, Email: mikeambrose@berkeley.edu.
Mahdad Noursadeghi, Email: m.noursadeghi@ucl.ac.uk.
M. Luisa Iruela-Arispe, Email: arispe@mcdb.ucla.edu.
Tom H.M. Ottenhoff, Email: t.h.m.ottenhoff@lumc.nl.
Annemieke Geluk, Email: A.Geluk@lumc.nl.
Barry R. Bloom, Email: bbloom@hsph.harvard.edu.
Matteo Pellegrini, Email: matteop@mcdb.ucla.edu.
Robert L. Modlin, Email: rmodlin@mednet.ucla.edu.
References
- 1.Zak DE, Tam VC, Aderem A. Systems-level analysis of innate immunity. Annu Rev Immunol. 2014;32:547–577. doi: 10.1146/annurev-immunol-032713-120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joosten SA, et al. Identification of biomarkers for tuberculosis disease using a novel dual-color RT-MLPA assay. Genes Immun. 2012;13(1):71–82. doi: 10.1038/gene.2011.64. [DOI] [PubMed] [Google Scholar]
- 3.Kaforou M, et al. Detection of tuberculosis in HIV-infected and -uninfected African adults using whole blood RNA expression signatures: a case-control study. PLoS Med. 2013;10(10):e88843. doi: 10.1371/journal.pmed.1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr Other Mycobact Dis. 1966;34(3):255–273. [PubMed] [Google Scholar]
- 5.Andrade PR, et al. Type 1 reaction in leprosy: a model for a better understanding of tissue immunity under an immunopathological condition. Expert Rev Clin Immunol. 2015;11(3):391–407. doi: 10.1586/1744666X.2015.1012501. [DOI] [PubMed] [Google Scholar]
- 6.Modlin RL, Bakke AC, Vaccaro SA, Horwitz DA, Taylor CR, Rea TH. Tissue and blood T-lymphocyte subpopulations in erythema nodosum leprosum. Arch Dermatol. 1985;121(2):216–219. [PubMed] [Google Scholar]
- 7.Crawford CL. Mechanisms of reaction in leprosy. Lancet. 1972;2(7783):925–926. doi: 10.1016/s0140-6736(72)92563-9. [DOI] [PubMed] [Google Scholar]
- 8.Godal T, Myrvang B, Samuel DR, Ross WF, Lofgren M. Mechanism of “reactions” in borderline tuberculoid (BT) leprosy. A preliminary report. Acta Pathol Microbiol Scand Suppl. 1973;236(0):45–53. [PubMed] [Google Scholar]
- 9.Barnetson RS, Bjune G, Pearson JM, Kronvall G. Cell mediated and humoral immunity in “reversal reactions”. Int J Lepr Other Mycobact Dis. 1976;44(1-2):267–274. [PubMed] [Google Scholar]
- 10.Bjune G, Barnetson RS, Ridley DS, Kronvall G. Lymphocyte transformation test in leprosy; correlation of the response with inflammation of lesions. Clin Exp Immunol. 1976;25(1):85–94. [PMC free article] [PubMed] [Google Scholar]
- 11.Rea TH, Taylor CR. Serum and tissue lysozyme in leprosy. Infect Immun. 1977;18(3):847–856. doi: 10.1128/iai.18.3.847-856.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19(2):338–381. doi: 10.1128/CMR.19.2.338-381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjorvatn B, Barnetson RS, Kronvall G, Zubler RH, Lambert PH. Immune complexes and complement hypercatabolism in patients with leprosy. Clin Exp Immunol. 1976;26(3):388–396. [PMC free article] [PubMed] [Google Scholar]
- 14.Lee DJ, et al. Integrated pathways for neutrophil recruitment and inflammation in leprosy. J Infect Dis. 2010;201(4):558–569. doi: 10.1086/650318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inkeles MS, et al. Comparison of molecular signatures from multiple skin diseases identifies mechanisms of immunopathogenesis. J Invest Dermatol. 2015;135(1):151–159. doi: 10.1038/jid.2014.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teles RM, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339(6126):1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khadge S, et al. Longitudinal immune profiles in type 1 leprosy reactions in Bangladesh, Brazil, Ethiopia and Nepal. BMC Infect Dis. 2015;15:e88843. doi: 10.1186/s12879-015-1128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lockwood DN, Lucas SB, Desikan KV, Ebenezer G, Suneetha S, Nicholls P. The histological diagnosis of leprosy type 1 reactions: identification of key variables and an analysis of the process of histological diagnosis. J Clin Pathol. 2008;61(5):595–600. doi: 10.1136/jcp.2007.053389. [DOI] [PubMed] [Google Scholar]
- 19.Lockwood DN, et al. Comparing the clinical and histological diagnosis of leprosy and leprosy reactions in the INFIR cohort of Indian patients with multibacillary leprosy. PLoS Negl Trop Dis. 2012;6(6):e88843. doi: 10.1371/journal.pntd.0001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swindell WR, et al. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS ONE. 2012;7(3):e88843. doi: 10.1371/journal.pone.0034594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swindell WR, Johnston A, Voorhees JJ, Elder JT, Gudjonsson JE. Dissecting the psoriasis transcriptome: inflammatory- and cytokine-driven gene expression in lesions from 163 patients. BMC Genomics. 2013;14:e88843. doi: 10.1186/1471-2164-14-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridley DS. Histological classification and the immunological spectrum of leprosy. Bull World Health Organ. 1974;51(5):451–465. [PMC free article] [PubMed] [Google Scholar]
- 23.Rea TH, Shen JY, Modlin RL. Epidermal keratinocyte Ia expression, Langerhans cell hyperplasia and lymphocytic infiltration in skin lesions of leprosy. Clin Exp Immunol. 1986;65(2):253–259. [PMC free article] [PubMed] [Google Scholar]
- 24.Ochoa MT, et al. A role for interleukin-5 in promoting increased immunoglobulin M at the site of disease in leprosy. Immunology. 2010;131(3):405–414. doi: 10.1111/j.1365-2567.2010.03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montoya D, et al. Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host Microbe. 2009;6(4):343–353. doi: 10.1016/j.chom.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang G, et al. Nicotinic acetylcholine receptor α1 promotes calpain-1 activation and macrophage inflammation in hypercholesterolemic nephropathy. Lab Invest. 2011;91(1):106–123. doi: 10.1038/labinvest.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang G, et al. In vivo knockdown of nicotinic acetylcholine receptor α1 diminishes aortic atherosclerosis. Atherosclerosis. 2011;215(1):34–42. doi: 10.1016/j.atherosclerosis.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89(1):73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:e88843. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montoya D, et al. IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci Transl Med. 2014;6(250):e88843. doi: 10.1126/scitranslmed.3009546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kis-Toth K, Bacskai I, Gogolak P, Mazlo A, Szatmari I, Rajnavolgyi E. Monocyte-derived dendritic cell subpopulations use different types of matrix metalloproteinases inhibited by GM6001. Immunobiology. 2013;218(11):1361–1369. doi: 10.1016/j.imbio.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 32.Gronski TJ, et al. Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J Biol Chem. 1997;272(18):12189–12194. doi: 10.1074/jbc.272.18.12189. [DOI] [PubMed] [Google Scholar]
- 33.Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A. 1996;93(9):3942–3946. doi: 10.1073/pnas.93.9.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leppert D, Waubant E, Galardy R, Bunnett NW, Hauser SL. T cell gelatinases mediate basement membrane transmigration in vitro. J Immunol. 1995;154(9):4379–4389. [PubMed] [Google Scholar]
- 35.Marchant DJ, et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat Med. 2014;20(5):493–502. doi: 10.1038/nm.3508. [DOI] [PubMed] [Google Scholar]
- 36.Vaalamo M, Kariniemi AL, Shapiro SD, Saarialho-Kere U. Enhanced expression of human metalloelastase (MMP-12) in cutaneous granulomas and macrophage migration. J Invest Dermatol. 1999;112(4):499–505. doi: 10.1046/j.1523-1747.1999.00547.x. [DOI] [PubMed] [Google Scholar]
- 37.Teles RM, et al. High matrix metalloproteinase production correlates with immune activation and leukocyte migration in leprosy reactional lesions. Infect Immun. 2010;78(3):1012–1021. doi: 10.1128/IAI.00896-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teles RM, et al. Expression of metalloproteinases (MMP-2, MMP-9, and TACE) and TNF-alpha in the nerves of leprosy patients. J Peripher Nerv Syst. 2007;12(3):195–204. doi: 10.1111/j.1529-8027.2007.00139.x. [DOI] [PubMed] [Google Scholar]
- 39.Oliveira AL, et al. Schwann cells producing matrix metalloproteinases under Mycobacterium leprae stimulation may play a role in the outcome of leprous neuropathy. J Neuropathol Exp Neurol. 2010;69(1):27–39. doi: 10.1097/NEN.0b013e3181c6515c. [DOI] [PubMed] [Google Scholar]
- 40.Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. 2010;327(5964):466–469. doi: 10.1126/science.1179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toumpanakis D, et al. Inspiratory resistive breathing induces MMP-9 and MMP-12 expression in the lung. Am J Physiol Lung Cell Mol Physiol. 2015;308(7):L683–L692. doi: 10.1152/ajplung.00133.2014. [DOI] [PubMed] [Google Scholar]
- 42.Oh H, Yang S, Park M, Chun JS. Matrix metalloproteinase (MMP)-12 regulates MMP-9 expression in interleukin-1beta-treated articular chondrocytes. J Cell Biochem. 2008;105(6):1443–1450. doi: 10.1002/jcb.21963. [DOI] [PubMed] [Google Scholar]
- 43.Biggs LC, et al. Interferon regulatory factor 6 regulates keratinocyte migration. J Cell Sci. 2014;127(Pt 13):2840–2848. doi: 10.1242/jcs.139246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shibata S, et al. Adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signaling pathway. J Immunol. 2012;189(6):3231–3241. doi: 10.4049/jimmunol.1101739. [DOI] [PubMed] [Google Scholar]
- 45.Nair M, Teng A, Bilanchone V, Agrawal A, Li B, Dai X. Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J Cell Biol. 2006;173(2):253–264. doi: 10.1083/jcb.200508196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roca H, et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE. 2013;8(10):e88843. doi: 10.1371/journal.pone.0076773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, et al. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res. 2011;71(4):1292–1301. doi: 10.1158/0008-5472.CAN-10-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan DM, Feng XS, Qi PW, Chen YW. Forkhead factor FOXQ1 promotes TGF-β1 expression and induces epithelial-mesenchymal transition. Mol Cell Biochem. 2014;397(1-2):179–186. doi: 10.1007/s11010-014-2185-1. [DOI] [PubMed] [Google Scholar]
- 49.Liu PT, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 50.Fabri M, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3(104):e88843. doi: 10.1126/scitranslmed.3003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 52.Wagner B, Natarajan A, Grünaug S, Kroismayr R, Wagner EF, Sibilia M. Neuronal survival depends on EGFR signaling in cortical but not midbrain astrocytes. EMBO J. 2006;25(4):752–762. doi: 10.1038/sj.emboj.7600988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiley SR, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 54.Billottet C, Quemener C, Bikfalvi A. CXCR3, a double-edged sword in tumor progression and angiogenesis. Biochim Biophys Acta. 2013;1836(2):287–295. doi: 10.1016/j.bbcan.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 55.Liu C, et al. Gamma-ray irradiation impairs dendritic cell migration to CCL19 by down-regulation of CCR7 and induction of cell apoptosis. Int J Biol Sci. 2011;7(2):168–179. doi: 10.7150/ijbs.7.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fallarino F, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9(10):1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 57.Ajith C, et al. Study of apoptosis in skin lesions of leprosy in relation to treatment and lepra reactions. Int J Lepr Other Mycobact Dis. 2005;73(4):269–276. [PubMed] [Google Scholar]
- 58.Patnaik N, Agarwal S, Sharma S, Sharma S, Pandhi D. Evaluation of apoptosis in skin biopsies of patients of borderline leprosy and lepra type 1 reaction. Indian J Dermatol. 2015;60(1):60–65. doi: 10.4103/0019-5154.147795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oliveira RB, et al. Expression of Toll-like receptor 2 on human Schwann cells: a mechanism of nerve damage in leprosy. Infect Immun. 2003;71(3):1427–1433. doi: 10.1128/IAI.71.3.1427-1433.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scollard DM, Smith T, Bhoopat L, Theetranont C, Rangdaeng S, Morens DM. Epidemiologic characteristics of leprosy reactions. Int J Lepr Other Mycobact Dis. 1994;62(4):559–567. [PubMed] [Google Scholar]
- 61.Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25(2):165–172. doi: 10.1016/j.clindermatol.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 62.Lockwood DN, et al. Cytokine and protein markers of leprosy reactions in skin and nerves: baseline results for the North Indian INFIR cohort. PLoS Negl Trop Dis. 2011;5(12):e88843. doi: 10.1371/journal.pntd.0001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cruz D, et al. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J Clin Invest. 2008;118(8):2917–2928. doi: 10.1172/JCI34189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bleharski JR, et al. Use of genetic profiling in leprosy to discriminate clinical forms of the disease. Science. 2003;301(5639):1527–1530. doi: 10.1126/science.1087785. [DOI] [PubMed] [Google Scholar]
- 65.Iyer AM, et al. Leprosy-specific B-cells within cellular infiltrates in active leprosy lesions. Hum Pathol. 2007;38(7):1065–1073. doi: 10.1016/j.humpath.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 66.Soares CT, et al. Angiogenesis and lymphangiogenesis in the spectrum of leprosy and its reactional forms. PLoS ONE. 2013;8(9):e88843. doi: 10.1371/journal.pone.0074651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91(9):4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11(2):242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu Z, Mellor J, Wu J, DeLisi C. VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004;5:e88843. doi: 10.1186/1471-2105-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ochoa MT, Loncaric A, Krutzik SR, Becker TC, Modlin RL. “Dermal dendritic cells” comprise two distinct populations: CD1+ dendritic cells and CD209+ macrophages. J Invest Dermatol. 2008;128(9):2225–2231. doi: 10.1038/jid.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tuominen VJ, Ruotoistenmäki S, Viitanen A, Jumppanen M, Isola J. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 2010;12(4):e88843. doi: 10.1186/bcr2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.