

Abstract
Genome-wide association studies of asthma have identified genetic variants in the IL1RL1 gene, but the molecular mechanisms conferring risk are unknown. IL1RL1 encodes the ST2 receptor (ST2L) for IL-33 and an inhibitory decoy receptor (sST2). IL-33 promotes type 2 inflammation, which is present in some but not all asthmatics. We find that two single nucleotide polymorphisms (SNPs) in IL1RL1 — rs1420101 and rs11685480 — are strongly associated with plasma sST2 levels, though neither is an expression quantitative trait locus (eQTL) in whole blood. Rather, rs1420101 and rs11685480 mark eQTLs in airway epithelial cells and distal lung parenchyma, respectively. We find that the genetically determined plasma sST2 reservoir, derived from the lung, neutralizes IL-33 activity, and these eQTL SNPs additively increase the risk of airway type 2 inflammation among asthmatics. These risk variants define a population of asthmatics at risk of IL-33–driven type 2 inflammation.
The molecular mechanism underlying IL1RL1 genetic asthma risk variants and their relationship to type 2 inflammation in the airway are demonstrated.
Introduction
Advances in molecular phenotyping reveal heterogeneity within the asthmatic population (1–3). The most consistent endotype to emerge is characterized by excessive airway type 2 inflammation (type 2-high asthma) (2–6). IL-33 signaling through the ST2 receptor (encoded by the IL1RL1 gene) is an important driver of type 2 immune responses (7, 8). Genetic variation in both the IL33 and IL1RL1 genes is strongly associated with asthma in genome-wide association study (GWAS) meta-analyses of European and North American populations (9, 10). The molecular mechanism and the influence of these risk variants on airway type 2 inflammation is unknown.
Two major transcript variants are produced from the IL1RL1 gene (11). Transcript variant 1 is a long transcript that encodes the membrane-bound ST2 receptor (ST2L) for IL-33, while transcript variant 2 is short and encodes a soluble decoy receptor (sST2) for IL-33. ST2 transcripts can be expressed from two spatially distinct promoters. The distal promoter can drive either ST2L or sST2 expression, while the proximal promoter directs expression of sST2 only. Two studies indicate that single nucleotide polymorphisms (SNPs), some overlapping with the asthma GWAS variants, are strongly associated with sST2 plasma protein levels (12, 13), while a single study demonstrates that SNPs in IL1RL1 are associated with ST2 gene expression in lung parenchymal tissue (14). Together, these studies suggest that SNPs in the IL1RL1 gene act as expression quantitative trait loci (eQTL) in the lung.
These observations raise several important questions with regard to the consequences of genetic control by IL1RL1 variants of ST2 transcript expression in the asthmatic airway: (i) what is the relationship between sST2 plasma levels and ST2 transcript expression in the lung, (ii) do one or more genetic variants drive sST2 expression in the lung, (iii) do genetically determined sST2 plasma levels contribute to asthma disease risk, and (iv) are IL1RL1 eQTL variants associated with the type 2 inflammatory endotype of asthma? To answer these questions, we explored the role of IL1RL1 genetics in determining sST2 plasma levels, blood and lung sST2 transcript expression, and airway type 2 inflammation in a large cohort of asthmatics and controls (Figure 1 and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/jci.insight.87871DS1). IL1RL1 genetic variant expression effects were studied in different lung cell types using a large dataset of cultured primary human airway epithelial cells and a publicly available expression database of distal lung parenchymal tissue (Figure 1). Finally, we evaluated the relationship between IL1RL1 eQTL variants that mark distinct lung cell–specific expression and risk for the type 2-high asthma endotype (Figure 1).
Figure 1. Biospecimens used to examine genetic regulation of the IL1RL1 locus.
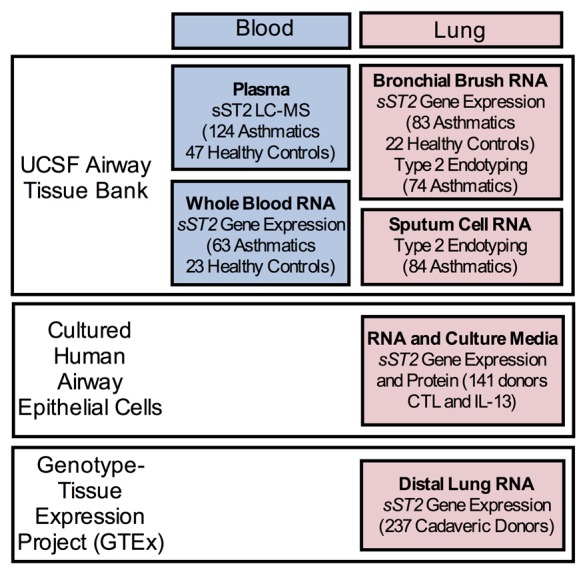
We explored protein and RNA expression genetics in both blood and lung tissue. We employed a clinical cohort of asthmatic and healthy control subjects, recruited at UCSF, to examine plasma protein sST2 levels using liquid chromatography mass-spectroscopy (LC-MS) and whole blood sST2 gene expression. We used a bank of cultured airway epithelial cells (both unstimulated [CTL] and stimulated with L-13) and an NIH-sponsored distal lung organ tissue gene expression repository (Genotype and Tissue Expression Project [GTEx], Broad Institute) to determine sST2 expression genetics in 2 lung tissue types: airway epithelium and distal lung tissue, respectively. Finally, we used a cohort of asthmatic subjects (UCSF) with airway measures of type 2 inflammation (i.e., endotyping) either by bronchial brush gene expression signatures or sputum cell cytokine expression profiles, to examine IL1RL1 genetic influence on asthma endotype.
Results
IL1RL1 asthma risk variants are associated with sST2 plasma protein levels but not whole blood sST2 gene expression levels.
To examine genetic influences on plasma sST2 protein levels, we selected 5 candidate protein quantitative trait loci (pQTL) SNPs at the IL1RL1 locus (Figure 2A and Supplemental Figure 1A). Based on 1,000 genomes data in the CEU population (Utah residents with ancestry from northern and western Europe), these SNPs tag 96 variants distributed across the locus. Minor allele frequencies and linkage disequilibrium maps for our study population can be found in Supplemental Figure 1, A and B. These variants are eQTL candidates based on reported genetic associations with asthma, allergic diseases, eosinophilia, sST2 plasma protein levels, and in vitro sST2 expression (9, 10, 12, 13, 15–18) (Supplemental Figure 1A). Among 90 self-identified white non-Hispanic asthmatic and healthy control subjects, we found strong associations between 3 of the SNPs, and liquid chromatography mass spectroscopy (LC-MS) determined sST2 plasma protein levels (Figure 2B). These 3 associations were also observed in the full mixed-race cohort of 171 asthmatic and healthy control subjects (Figure 2B). The direction of effect we observed for these variants (Supplemental Figure 2, A and B) is consistent with prior reports from a pediatric Dutch cohort and from the Framingham Offspring Cohort, which also found that rs1420101 and rs11685480 were among the most significant associations with circulating sST2 levels (12, 13). We hypothesized that these variants mediate their effect on protein levels through regulation of gene expression. To test this, we measured sST2 transcript expression levels in whole blood RNA from 63 asthmatics and 23 healthy control subjects (Figure 2B). None of the SNPs tested were associated with expression of sST2 transcript in whole blood RNA. We found no difference in either sST2 plasma protein levels or whole blood sST2 transcript expression in asthmatics compared with healthy control subjects (Supplemental Figure 3, B and C).
Figure 2. Single nucleotide polymorphisms (SNPs) in the IL1RL1 gene influence plasma protein sST2 levels but not whole blood cell sST2 expression.

(A) Schematic showing the IL1RL1 gene transcript structure highlighting 5 candidate expression quantitative trait loci (eQTL) SNPs. Blue exons are specific to the ST2L transcript, green exons are specific to the sST2 transcript, and red exons are shared by both transcripts. The distal promoter can produce both the long ST2L and the short sST2 transcript, while the proximal can produce only the short sST2 transcript. (B) sST2 plasma protein levels (red) as measured by liquid chromatography-mass spectroscopy (LC-MS) are associated with 3 SNPs in the IL1RL1 gene: rs11685480, rs1420101, and rs1921622 in white non-Hispanic subjects only (WNH) and among the entire cohort (All). The tested allele in all cases is associated with lower-circulating sST2 levels. None of the tested SNPs was an eQTL for sST2 expression in whole blood RNA (blue). SNP regression coefficient and the corresponding 95% CIs are plotted with P values listed below. Additive linear regression models were used. Nonitalicized P values indicate the WNH, while the italicized P values shown for the entire cohort (All).
rs11685480 and rs1420101 mark cell type–specific lung eQTL.
The strong genetic association of IL1RL1 variants with sST2 plasma protein but not blood gene expression measurements suggests that other tissues may be a source of circulating sST2 protein. Given the importance of the airway epithelium in asthma, we examined sST2 transcript expression and protein secretion in cultured primary airway epithelial cells from 141 donors. These airway epithelial cells were harvested from cadaver tracheas and cultured at air-liquid interface to produce muco-ciliary epithelium that was examined in the unstimulated condition and following stimulation with the type 2 cytokine IL-13. We found the A allele of the rs1420101 SNP, which was associated with lower plasma sST2 protein levels, was also associated with lower sST2 gene expression and sST2 protein secretion both in the unstimulated and the IL-13–stimulated culture conditions (Figure 3, A and B, and Supplemental Figure 2C). In contrast, the other blood pQTLs, rs11685480 and rs1921622, were not associated with sST2 transcript or sST2 protein levels (Figure 3, A and B, and Supplemental Figure 2D). We thus considered the possibility that the rs11685480 SNP influences gene expression in another lung cell type. We tested the association between the blood pQTL SNPs and sST2 expression in distal lung tissue using data from 237 white non-Hispanic donors generated by the Genotype-Tissue Expression Project (GTEx) consortium. Notably, whole lung specimens are largely composed of alveolar epithelial and microvascular endothelial cells, with few airway epithelial cells. In contrast to airway epithelial cells, we found that both rs11685480 and rs1420101 SNPs were strongly associated with distal lung expression of sST2 (Figure 3C). Again, the same allele of both of these SNPs was associated with lower sST2 gene expression (Supplemental Figure 2, E and F).
Figure 3. Differences in airway epithelial cell and distal lung tissue expression genetics is driven by differential promoter usage.

In cultured airway epithelial cells, rs1420101 (blue) — but not rs11685480 (red) or rs1921622 (black) — is associated with sST2 gene expression (n = 127) (A) and sST2 secreted protein levels (n = 141) (B) in both the unstimulated and IL-13–stimulated conditions. White non-Hispanic subjects are indicated with a circle, while the entire cohort is indicated with a square. SNP regression coefficient and the corresponding 95% CIs are plotted. Additive linear regression models were used. *P < 0.05, **P < 0.01. (C) In contrast, both rs1420101 (blue) and rs11685480 (red) are associated with sST2 gene expression in distal lung parenchymal tissue in GTEx cadaveric donors (n = 237). SNP regression coefficient and the corresponding 95% CIs are plotted with P value listed below. Additive linear regression models were used. (D) Schema showing that sST2 transcripts can be derived from either the IL1RL1 distal or proximal promoters. sST2 transcripts derived from the distal promoter will contain exon 1a, while those derived from the proximal promoter will contain exon 1b. Primers were designed to specifically amplify exon 1a and exon 1b, as indicated by black arrowheads. (E) Cultured human airway epithelial cells (n = 6) and cultured human lung endothelial cells (n = 3) use the proximal promoter, while human alveolar epithelial cells (n = 7) use both promoters. *P < 0.05, ***P < 0.001 using 2-tailed paired t test.
The differences in eQTL status between the rs1420101 and rs11685480 SNPs by lung cell type suggest these SNPs mark independent, cell type–specific molecular effects. Given that sST2 can be expressed from either or both the distal and proximal promoters (Figure 3D) (18), differential promoter usage by cell type could explain this effect. Supporting this, the rs11685480 SNP is located in the distal IL1RL1 promoter and has previously been shown to regulate distal promoter sST2 transcript expression (18). We investigated promoter usage in airway epithelial cells, alveolar epithelial, and lung microvascular endothelial cells using quantitative PCR (qPCR) assays to amplify exon 1a or exon 1b of ST2 transcripts (Figure 3D). Exon 1a is exclusively found in transcripts derived from the distal IL1RL1 promoter, while exon 1b is found in transcripts derived from the proximal promoter. We find that airway epithelial cells and lung microvascular endothelial cells largely use the proximal promoter, while alveolar epithelial cells use the proximal and distal promoters equally (Figure 3E). Our results identifying rs11685480 as an eQTL for distal lung tissue but not airway epithelial cells are consistent with rs11685480 effects on distal promoter–driven expression in alveolar but not airway epithelial cells. Likewise, our results identifying rs1420101 as an eQTL in both the airway epithelium and distal lung tissue suggest that the expression effect is mediated through the proximal promoter that is used in both cell types.
Plasma sST2 levels are correlated with whole blood EC50 for IL-33.
To determine the contribution of plasma sST2 levels to IL-33 signaling, we examined IL-33–stimulated PBMCs and whole blood cells from 4 healthy subjects. This paired design allowed us to examine the effect of plasma proteins on IL-33 signaling. We find the half maximal effective concentration (EC50) for IL-33 across 4 donors showed little variability in PBMCs, while the EC50 in whole blood is markedly higher and considerably more variable, suggesting the presence of an IL-33 inhibitor in whole blood (Figure 4, A and B). Soluble ST2 directly inhibits IL-33 signaling in both mouse models and in vitro cultures (19). Consistent with this, we find that plasma sST2 levels are positively correlated with whole blood cell EC50 to IL-33 (Figure 4C), suggesting that soluble ST2 in circulating plasma is a functional inhibitor of IL-33 signaling in whole blood.
Figure 4. Plasma sST2 levels are correlated with the EC50 dose of IL-33 for whole blood cell activation.

(A) Paired peripheral blood mononuclear cells (PBMC) and whole blood cells from human donors (n = 4; each color represents a unique subject) were stimulated with IL-12 and a biologically active variant of IL-33 with mutated cysteine residues. (B) Compared with PBMC, IL-33 signaling is more variable and markedly reduced in whole blood, suggesting the presence of an IL-33 inhibitor (n = 4). Statistical analysis performed using 2-tailed paired t test. (C) The level of plasma sST2 is significantly and positively associated with the half maximal effective concentration (EC50) for IL-33 in whole blood cells (n = 20). Simple linear regression with P value listed.
Airway epithelial sST2 expression is associated with type 2 inflammation and is directly regulated by IL-13.
In order to understand the significance of a genetically determined deficiency of sST2 in promoting airway inflammation in asthma, we examined sST2 expression in bronchial brush RNA from 22 healthy and 83 asthmatic subjects. In previous studies, the degree of airway type 2 inflammation was measured in these asthmatics using a gene expression signature consistent with IL-13 activation of the epithelium (2, 20). A single metric derived from the qPCR-based expression of POSTN, CLCA1, and SERPINB2 in airway epithelial cells was used to classify asthmatics as type 2-high or -low. We find that sST2 gene expression is increased in asthmatics compared with healthy controls (Supplemental Figure 3D); however, this effect is present only in the subset with evidence of airway type 2 inflammation (type 2-high asthma) (Figure 5A). These findings suggest that sST2 expression is regulated by type 2 cytokines in airway epithelial cells. To test this directly, we examined sST2 gene expression and protein secretion in cultured human airway epithelial cells from 141 donors in unstimulated and IL-13–stimulated conditions (21). IL-13 markedly increased sST2 gene expression and secreted sST2 protein in these cells (Figure 5, B and C). We did not find significant associations between IL1RL1 SNPs and in vivo bronchial brush sST2 gene expression (Supplemental Table 3), which may be due to confounding mediated by differences in type 2 inflammation across the subjects. This finding contrasts with the eQTL and pQTL effects of rs1420101 revealed in the unstimulated and defined exposure to IL-13 in the in vitro airway epithelial cultures (Figure 3, A and B). Thus, there appears to be dual regulation of airway epithelial sST2 gene expression in vivo. The genetic effect of rs1420101 acts though the proximal IL1RL1 promoter to lower sST2 gene expression. At the same time, the type 2 cytokine IL-13 dramatically upregulates sST2 expression by airway epithelial cells. Both in vivo and in vitro, sST2 acts as a negative regulator of type 2 inflammation via direct inhibition of IL-33 signaling (19, 22–25). Our results suggest that this negative regulation is most prominent in asthmatics with excessive type 2 inflammation and lead us to predict that IL1RL1 lung eQTL SNPs, which are associated with decreased sST2 expression, will increase the risk of type 2-high asthma endotype.
Figure 5. sST2 expression is increased in the airway in the subset of asthmatics with type 2 inflammation and is regulated by IL-13.
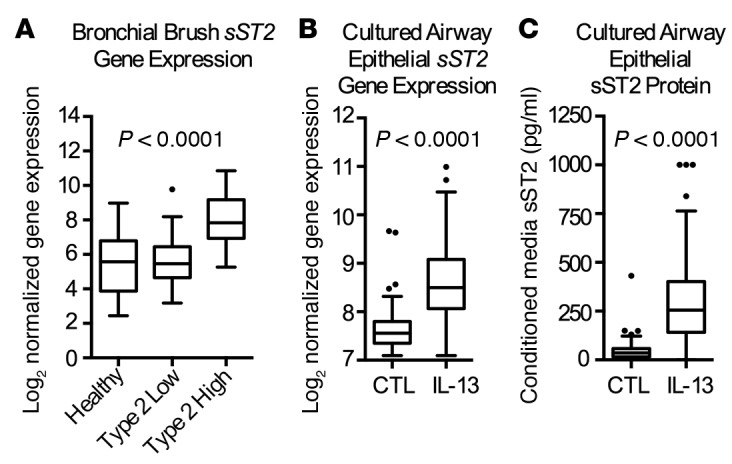
(A) In bronchial brush RNA from asthmatics (n = 83) and healthy controls (n = 22), sST2 gene expression is increased only in the subset that demonstrates evidence of excessive type 2 airway inflammation (type 2-high). P value reflects ordinary 1-way ANOVA. In cultured human airway epithelial cells, we find that variance stabilizing transformation–normalized (VST-normalized) sST2 gene expression (n = 127) (B) and secreted protein (n = 141) (C) are strongly increased from baseline levels (CTL) following IL-13 stimulation. P values reflects 2-tailed paired t tests. Data presented as Tukey box plots with box containing the first and third quartile, line at median, and whiskers at 1.5 interquartile range.
rs11685480 and rs1420101 eQTL variants are associated with the type 2-high asthma endotype.
To determine if IL1RL1 eQTL/pQTL SNPs are associated with airway type 2 inflammation in asthma, we examined 150 asthmatics with airway type 2 endotyping data (Supplemental Table 2). Type 2 endotype (high vs. low) was determined in our prior studies by the presence of airway gene expression signatures consistent with type 2 inflammation in either bronchial brush or sputum cell RNA (1, 3). Type 2-high asthmatics demonstrate markedly increased blood eosinophil counts, serum IgE measures, and a fraction of exhaled nitric oxide (FeNO), as well as lower prebronchodilator forced expiratory volume in 1 second (FEV1) percent predicted compared with type 2-low asthmatics (Supplemental Table 2). In our genetic analysis, we find that both eQTL/pQTL SNPs, rs11685480 and rs1420101, are associated with increased risk of the type 2-high endotype of asthma (odds ratio [OR] 2.32, P = 0.002; OR 1.74, P = 0.032, respectively) (Figure 6, A and B). We observed this association despite the fact that inhaled steroid use can reduce airway type 2 inflammation (20), an effect that could result in subject misclassification and a bias toward a null result. Consistent with the function of sST2 as an inhibitor of IL-33 and type 2 inflammation, the allele for both SNPs that is associated with lower sST2 gene and sST2 protein expression is associated with increased risk of the type 2-high endotype. Based on our promoter usage data suggesting an independent effect for the rs11685480 and rs1420101 SNPs on sST2 expression, we considered if carrying risk alleles from both SNPs would be associated with further decreases in sST2 plasma levels. We found a strong negative relationship between the number of rs11685480/rs1420101 risk alleles carried and circulating sST2 plasma protein levels (Figure 6C). Consistent with our model, the risk estimate for type 2-high asthma in the combined 3–4 allele–carrying group versus the 0–2 allele–carrying group was higher than the risk estimate for either SNP alone (OR 2.85, P = 0.02) (Figure 6C). These results provide strong evidence that reduced sST2 expression is a risk factor for the type 2-high endotype of asthma.
Figure 6. IL1RL1 eQTL single nucleotide polymorphisms (SNPs) are associated with risk of type 2 airway inflammation.

(A) Both eQTL SNPs rs1420101 and rs11685480 are associated with risk of type 2 airway inflammation in asthma. Alleles associated with lower plasma sST2 and lower lung sST2 expression are also associated with increased risk of type 2-high asthma endotype. SNP odds ratios (OR) represent results of logistic regression analysis. (B) The increasing number of risk alleles for rs1420101 and rs11685480 demonstrates a step-wise decrease in circulating sST2 plasma levels (red, white non-Hispanic; blue, entire cohort [All]). P values from parametric ANOVA trend test across increasing number of risk alleles. Data presented as Tukey box plots with box containing the first and third quartile, line at median, and whiskers at 1.5 interquartile range. (C) The presence of 3–4 rs1420101/rs11685480 risk alleles is associated with increased risk of type 2 airway inflammation in asthma compared with the presence of 0–2 risk alleles, with an OR of 2.85, which is greater than either risk genotype alone.
Discussion
In the last decade, both clinical and translational research has revealed heterogeneity in the pathological mechanisms underlying asthma. The most clinically important disease endotype described to date is characterized by excessive airway type 2 inflammation and affects about half of asthmatics (2, 26). Using genetically modified mice, many of the molecular mechanisms of type 2 inflammation have been discovered (27); however, little is known regarding the genetic predisposition to this endotype in human asthma. In this study, we demonstrate that asthma risk variants in the IL1RL1 gene specifically increase the risk of type 2-high disease. Moreover, we show that these variants confer risk by reducing plasma and airway levels of sST2, an inhibitor of IL-33 signaling and type 2 inflammation. Together, our findings define a population of asthmatics that is at risk for IL-33–driven type 2 inflammation.
To date, 7 candidate gene studies and 4 GWAS studies — including European and North American GWAS meta-analysis studies involving 26,000 and 17,000 subjects, respectively — have implicated genetic variation at the IL1RL1/IL18R1 locus in the development of asthma (9, 10, 12, 15–17, 28–32). Studies showing a relationship between several IL1RL1 GWAS variants and plasma sST2 protein levels suggest that these variants function as eQTLs (12, 13, 18). Most notably, a GWAS for sST2 plasma levels in the Framingham Offspring Cohort found strong evidence for genetic regulation of circulating protein (13). The most strongly associated SNP in this study was rs950880, which is in complete linkage disequilibrium with rs1420101 and within the haplotype we implicated as a blood pQTL. The rs1420101 SNP was also associated with plasma sST2 levels in a large pediatric Dutch cohort (PIAMA) (12). We find that rs1420101 is an eQTL in airway epithelium and distal lung parenchyma, tissues that use the proximal IL1RL1 promoter. These results are consistent with the effect of the rs1420101 SNP being mediated through this promoter. Analyzing CEU data from the 1,000 genomes project, we find 12 other variants in strong linkage disequilibrium (r2 > 0.8) with the rs1420101 variant. While rs1420101 and rs950880 are located in the first and fifth introns, respectively, 3 variants within this block are located in the proximal promoter. Molecular genetic studies of these variants in airway epithelial cells will be needed to determine if the eQTL effect of the rs1420101 block is mediated through the proximal promoter and to further narrow the causal variant within this SNP block.
One of the SNPs most strongly associated with soluble ST2 levels in the above referenced Framingham cohort, rs953934, is a tag for the rs11685480 (CEU r2 = 0.98) SNP that we have implicated as a blood pQTL and eQTL driving expression through the distal IL1RL1 promoter in the lung parenchyma, specifically within alveolar epithelial cells (13). Moreover, the rs11685424 SNP, which is in complete LD with rs11685480 SNP in the CEU population, is strongly associated with sST2 expression in parenchymal lung tissue measured by microarray (14), consistent with the distal lung tissue expression data presented here. Notably, rs11685480 resides within a distal promoter haplotype that is associated with atopic dermatitis and modulates ST2 expression in a human mast cell line (18). Our observation that both the distal and proximal promoters are used by alveolar epithelial cells presents the possibility that ST2L is also expressed and affected by this eQTL. However, we note that ST2L is weakly expressed in the GTEx lung tissue data versus sST2 (median RPKM expression levels sST2 = 56.4 vs. ST2L = 0.83). Taken together, these data strongly suggest that rs11685480 alone or in combination with other SNPs alters sST2 expression through the distal promoter in alveolar epithelial cells and thereby contributes to circulating sST2 levels.
Our experimental findings support the presence of lung cell type–specific eQTLs tagged by SNPs rs1420101 and rs11685480. We did find a significant association between the rs1921622 SNP and circulating levels of sST2 protein, though the association was less significant than for rs1420101 or rs11685480. Moreover, we found no association with this SNP and airway epithelial cell sST2 gene expression and only a weak association with distal lung tissue sST2 expression. We hypothesize that the rs1921622 SNP effect may be explained by linkage disequilibrium with the rs1421010 SNP or that this SNP may function as an eQTL in another untested cell type. Further molecular studies will be needed to delineate this mechanism. While our results support that the identified circulating sST2 pQTLs are the result of these SNPs functioning as eQTLs in lung cells but not blood cells, we acknowledge that eQTL effects of these SNPs in specific blood cell types could be missed by our mixed blood cell type analysis. Additionally, these SNPs could mediate posttranslational changes to sST2 protein and thus contribute to the observed pQTL.
Our data suggest that the rs11685480 and rs1420101 SNPs or variation in LD with these SNPs mark independent eQTL loci that contribute to asthma pathogenesis by reducing circulating and lung sST2 levels to promote type 2 airway inflammation. This is consistent with several well-powered genetic epidemiology studies of asthma and allergic phenotypes (12, 15). Specifically, the A allele of rs1420101 SNP is strongly associated with increased risk of asthma in a comparison of 44,890 controls versus 7,996 European asthmatics (15). Stratification of asthmatics in that cohort indicates that the association is strongest among atopic asthmatics, consistent with our finding that the rs1420101 A allele is associated with the type 2-high asthma endotype. The rs1420101 variant is also one of the 5 genome-wide significant hits from a GWAS of blood eosinophil levels conducted in over 20,000 European and 5,000 Asian patients (15). In that study, the A allele of rs1420101 is associated with higher levels of both eosinophils and IgE. Although the rs11685480 SNP has not been screened in GWAS studies, the allele of this SNP that is associated with lower sST2 levels is also associated with type 2-high disease, matching the rs1420101 findings. Moreover, carrying multiple risk alleles for rs1420101 and rs1685480 is associated in further reductions in plasma sST2 levels and an even higher risk of type-2 high asthma, suggesting that these 2 loci function independently.
Our genetic mechanism is strongly supported by mouse models demonstrating that IL-33 promotes type 2 inflammation and is inhibited by sST2 (19). We find that this inflammatory pathway is altered in the airway of asthmatics with excessive type 2 inflammation. In these subjects, sST2 is markedly upregulated, an effect that is mediated by the type 2 cytokine IL-13. Genetic variation that reduces local airway sST2 production and disrupts a finely tuned balance between IL-33–driven type 2 cytokine production and the IL-13–induced negative feedback loop may be the primary driver of the risk we observe. However, leakage of plasma proteins into the airways during inflammation may allow a reservoir of biologically active sST2 to restrain type 2 inflammation. In this way, the genetically determined level of plasma sST2 protein, derived from the lung, would influence risk of type 2 inflammation in asthma.
Our translational study of in vivo and in vitro lung tissue genetics allows us to develop a model linking the strong IL1RL1 genetic epidemiology to basic immunology and to demonstrate a key role for IL-33 in asthmatic type 2 inflammation (Supplemental Figure 4) (8, 33–35). This multicellular model suggests that local airway sST2 levels, as well as circulating plasma sST2 levels, contribute to neutralization of IL-33 in the tissue. A genetically determined deficiency in sST2 in the airway (driven by the proximal promoter), as well as a deficiency in sST2 in the plasma (driven though both the proximal and distal promoters), act to increase IL-33 signaling and promote airway type 2 inflammation. These genetic variants define an asthmatic population that is at increased risk for IL-33–driven type 2 inflammation and that may benefit from targeted blockade of the IL-33/ST2 signaling axis.
Methods
UCSF Airway Tissue Bank (UCSF ATB)
We studied human subjects who had biospecimens stored in the UCSF ATB. We included subjects who had available DNA, as well as any of the following: plasma, whole blood RNA, bronchial brush RNA, or induced sputum cell RNA (Figure 1 and Supplemental Table 1). These samples were collected in research studies at UCSF from 2000–2012 and stored in the UCSF ATB. Subject characterization and biospecimen collections were performed according to standardized and uniform protocols. These biospecimens have been used in other reported studies from our group (1–3, 20, 36–39). Asthma was defined as physician diagnosis and confirmed by measures of airway hyperresponsiveness or reversible airway obstruction. Healthy control subjects were nonsmokers with no history of lung disease and no evidence of airway hyperresponsiveness.
Measurement of sST2 protein
sST2 was measured by ELISA (Presage ST2 Assay, Critical Diagnostics) per the manufacturer’s instructions and by immunocapture LC-MS (IC LC-MS). The IC LC-MS was performed by first incubating plasma overnight with biotinylated anti-ST2 (BAF523, R&D Systems), followed by bead capture, acid elution, and tryptic digestion. Peptides unique to ST2 were quantitated against a peptide control and standard curve with ST2-Fc. Plasma measurements of sST2 made with the Critical Diagnostics assay correlated strongly with the measurements made by IC LC-MS (Supplemental Figure 3A).
Cultured human airway epithelial cells
Airway epithelial cells were harvested from the tracheas of 141 organ donors obtained via the California Transplant Donor Network (21). DNA was extracted from cells following harvest using DNeasy Blood and Tissue Kit (Qiagen). Harvested cells were also expanded for one passage only in basal epithelial growth media (Lonza) and then plated to 12-mm diameter transwells. Cells were grown for 21–23 days in 2% Ultroser G (Pall Corporation) in DMEM/F12 media and then stimulated with media alone or IL-13 (10 ng/ml) (R&D Systems) every 24 hours for 2 days. RNA was extracted using RNeasy kits (Qiagen). Basal conditioned media was collected and analyzed by ELISA for sST2 protein (sST2 Duoset, R&D Systems).
Lung microvascular endothelial cell culture
Human lung microvascular endothelial cells (HMVEC-L) were purchased from Lonza and grown according to the distributor’s instructions in EGM-2MV Bullet Kit Medium (Lonza) for one passage. Cells were harvested at confluence, and RNA was extracted as described above.
Lung alveolar epithelial cells
Alveolar type II epithelial cells were isolated from deidentified organ donors whose lungs were not suitable for transplantation and donated for medical research through the National Disease Research Interchange and the International Institute for the Advancement of Medicine. The type II cells were dissociated from the lung by treatment with elastase, and they were purified by discontinuous density gradient centrifugation and positively selected by binding to magnetic beads through CD326 (EP-CAM) ( Miltenyi Biotec) (40, 41).
Measurement of gene expression
Whole blood cell (PAXGENE) RNA and bronchial brush RNA expression levels of sST2 were measured by qPCR. Briefly, 20 ng of RNA was reverse transcribed using the SuperScript VILO cDNA Synthesis Kit and Master Mix (Invitrogen). Bronchial brush cDNA (4 ng) was PCR amplified in multiplex for housekeeping genes PPIA, RPL13A, and EEF1A1 and sST2; 4 ng of PAXGENE cDNA was PCR amplified for housekeeping genes GAPDH, PPIA, and RPL37A and sST2. Nested taqman primers and probes were then used in a standard 40-cycle taqman reaction using the preamplified PCR product and Taqman Universal Master Mix (Invitrogen). Primer and probe sequences can be found in Supplemental Table 4. The gene expression levels of sST2 in cultured airway epithelial cells (control and IL-13 stimulated) were determined from whole transcriptome sequencing. Specifically, Kapa Biosystems mRNA-seq library kits (catalog KK8421) were used to generate sequencing libraries with barcodes and adapters compatible with the Ion Torrent Proton sequencing instrument (Thermo Fisher Scientific). Sequencing was conducted on an Ion Torrent Proton sequencer using P1 chips. Sequencing reads for each sample from different sequencing runs were consolidated and downsampled to the same number of reads within each control and IL-13–treated pair, with a total number of reads within 8 million to 12 million reads per sample. Downsampled reads from each sample were pseudoaligned and quantified using Kallisto (version 0.42.3) (42) and the human transcriptome sequence index generated from the iGenomes transcript annotation file (downloaded from Illumina’s FTP site on 01/15/2013). Estimated transcript counts from the Kallisto abundance files were rounded to the nearest integer and merged to form a count table for all 254 paired samples. Variance stabilizing transformation (VST) (DESeq2) was performed with an experimental design labeling samples based on IL-13 and control stimulation (blind=FALSE) (43). The NM_003856 transcript was used for expression of sST2. For the distal lung eQTL analysis, we used data generated by the GTEx consortium downloaded from database of Genotypes and Phenotypes (dbGap; http://www.ncbi.nlm.nih.gov/gap) (dbGap Study Accession: phs000424.v6.p1). We used raw counts generated by FluxCapacitor for the 320 GTEx_MidPoint_RNAseq samples (phe0000006.v2) from lung tissue, marked as best suited for eQTL analysis. We then selected 242 individuals with reported white race and available GTEx_MidPoint_Imputation genotypes (phg000520.v2). We then performed PCA analysis and identified 5 subjects who did not cluster with the remaining white-race individuals and those were removed from subsequent analyses, resulting in a total of 237 individuals with complete genotype and expression data. We performed variance stabilizing transformation on GTEx transcript expression. Transcript quantitation for GTEx was performed with the ensembl GTF; therefore, sST2 expression for this cohort was represented by the ENST00000311734.2 transcript. The ENST00000311734.2 transcript is equivalent to the RefSeq NM_003856 transcript, differing only in the 5′) end of the first exon.
qPCR for IL1RL1 exon 1a and 1b was performed on primary cultured airway epithelial cell RNA, lung microvascular endothelial cell RNA, and alveolar epithelial cell RNA. RNA was reverse transcribed as described above. Taqman primers and probes were then used in a standard 40-cycle taqman reaction using 2 μg of unamplified cDNA and Taqman Universal Master Mix (Invitrogen) to measure exon 1a, exon 1b, and housekeeping genes PPIA, RPL13A, and EEF1A1. Primer and probe sequences are indicated in Supplemental Table 4.
Blood Cell Cultures
Human heparinized whole blood or mononuclear cells (PBMCs) were primed with IL-12 for 2 hours followed by the addition of IL-33 Cys-Ser variant (var) for 15–20 hours. IL-33 var was expressed and purified from E. coli by expression of a variant of IL-33, amino acids 112–270, with all 4 cysteines replaced by serine. The activity of IL-33 var is more stable in tissue culture than WT IL-33 (44). IFNγ was measured in the supernatant by mesoscale discovery immunoassays. EC50 of IL-33 was calculated following log transformation of the IFNγ levels. The type 1–stimulating conditions (IL-12 and IL-33) were used in these whole blood and PBMC assays rather than type 2–stimulating conditions (IL-33 and TSLP) because we find very few circulating type 2 cytokine–producing cells in the blood of healthy volunteers, resulting in very little type 2 cytokine response.
Airway type 2 classification
Data for the classification of asthmatics as type 2-high or -low was available from our prior studies (3, 20). In 76 subjects, the classification was made using a 3-gene expression signature of IL-13–responsive epithelial cell genes from bronchial brush RNA (1). The 3 genes are POSTN, SERPINB2, and CLCA1, and their individual expression values can be combined into a single 3-gene mean metric to determine the presence of type 2 inflammation in the airway epithelium, as previously described (20). In 84 subjects, the classification was made using gene expression for type 2 cytokines in induced sputum cells (3). The three type 2 cytokine genes are IL4, IL5, and IL13, and their individual expression values are combined into a similar 3-gene metric to determine the presence of type 2 inflammation in the airway lumen, as previously described (20). Ten subjects had measures of both bronchial brush and induced sputum type 2 inflammation. Subjects were classified as type 2-high if either the bronchial brush or the induced sputum cell standardized measurement exceeded 2 standard deviations of the mean value for healthy controls (3, 20).
Genotyping
UCSF ATB samples were genotyped for IL1RL1 SNPs rs1041973, rs1420101, rs10192157, and rs1921622 by Taqman genotyping assays. We used LNA Primetime probes and primers (IDT DNA) to genotype the rs11685480 SNP. Taqman and LNA reactions were performed with 5–10 ng of DNA according to manufacturer’s recommendations using either an ABI 7900HT or Quantstudio 6 Flex real-time thermocyclers (Invitrogen). Genotypes of the rs1420101 and rs1921622 SNPs for the donor cultured airway epithelial cells were extracted from Illumina Infinium Genome-wide genotyping array data. The rs11685480 SNP was genotyped in donor cultured airway epithelial cells with the assay and method described above. Genotype data for the rs11685480, rs1420101, and rs1921622 IL1RL1 SNPs in the GTEx lung donors was obtained from National Human Genome Research Institute (NHGRI) GTEx data release phs000424.v6.p1.
Statistics
UCSF ATB genetic analyses.
Protein and gene expression data was transformed where appropriate to more closely approximate a normal distribution for genetic analysis. Specifically, normalized blood paxgene expression data was log2 transformed; normalized bronchial brush expression data was log2 transformed and then square root transformed. Blood sST2 protein levels were natural log (ln) transformed. Genetic association between IL1RL1 SNPs and gene and protein expression levels was tested by linear regression models coding the SNPs in an additive fashion (e.g., 0 = GG, 1 = AG, 2 = AA). UCSF ATB analyses of only white non-Hispanic subjects included asthma status, inhaled steroid use, age, and sex as covariates. The analysis of all subjects included the formerly stated covariates with the addition of 5 dichotomous variables for race/ethnicity based on self-identification, namely (i) white non-Hispanic, (ii) black non-Hispanic, (iii) Asian non-Hispanic and Native Hawaiian and Other Pacific Islanders, (iv) Hispanic any race, (v) and other, which included subjects reporting as mixed race non-Hispanic, white with no ethnicity, or non-Hispanic with American Indian/Alaska native race.
Cultured human airway epithelial cells and GTEx lung genetic analyses.
VST normalized gene expression values, from airway epithelial cell cultures, were used to perform probabilistic estimation of expression residuals (PEER) normalization (45) and to generate PEER residuals corrected for plink (version 1.07) (46) determined sex, genome-wide genetic data determined genetic ancestry principle components (pc1, pc2, and pc3), and 64 hidden PEER factors (corresponding to ~25% of sample size). Similarly, GTEx lung VST-normalized gene expression data was used to perform PEER normalization and to generate PEER residuals corrected for age, sex, genome-wide genetic data–determined genetic ancestry principle components (pc1 and pc2), and 59 hidden PEER factors (corresponding to ~25% of sample size) for the sST2 transcript. Both the GTEx and cultured airway epithelial cell sST2 gene expression PEER residuals were analyzed for association with IL1RL1 SNPs by linear regression analysis using an additive genetic model. The airway epithelial bank IL-13–stimulated sST2 peer residuals were ln transformed for analysis. The untransformed control and square root–transformed IL-13–stimulated sST2 protein levels were also tested for association with IL1RL1 SNPs by linear regression analysis using an additive genetic model including sex and genetic principle components as covariates. Logistic regression was used to test the association between the rs1420101 and rs11685480 genotype and type 2-high vs. type 2-low status, including age, sex, steroid use, and racial/ethnic group (as defined above) as covariates. Between-group differences were assessed using a 2-tailed paired t test (cultured epithelial cell protein and gene expression unstimulated vs. IL-13–stimulated; distal vs. proximal promoter gene expression; EC50 IL-33 in whole blood vs. PBMC); 2-tailed unpaired t test (plasma sST2 protein, sST2 gene expression, or sST2 bronchial brush gene expression in healthy vs. asthma); ordinary 1-way ANOVA with Tukey’s multiple correction test (sST2 bronchial brush gene expression in healthy vs. type 2-high vs. type 2-low asthmatics; or ordinary 1-way ANOVA with post-test for linear trend (plasma sST2 protein in subjects carrying 0–5 risk alleles for rs1420101 or rs11685480). Linear regression was used to assess the relationship between whole blood EC50 for IL-33 and plasma sST2 protein LC-MS, as well as the relationship between plasma sST2 measured with LC-MS and critical diagnostics ELISA. A P value less than 0.05 was considered statistically significant. All statistical analyses were either performed in STATA/SE v11.0 or Graphpad Prism v6.0d.
Study approval
Research protocols were approved by the IRB at UCSF, and all subjects provided written informed consent for the study in which they originally participated. They also provided written informed consent for their biospecimens to be placed in the UCSF ATB for studies, in addition to the original protocol.
Author Contributions
EDG, MAS, and JVF conceived of the study. EDG, JP, MELS, RTS, KHN, JVF, and MAS designed experiments. RJM, JWL, and MAM provided reagents and technical support. EDG, JP, CLR, MELS, LZS, JLE, HJM, RTS, and MCP conducted experiments. EDG, AWA, LR, KHN, and MAS analyzed data. EDG, KHN, JVF, and MAS interpreted and synthesized the data. EDG generated figures. EDG and MAS wrote manuscript.
Supplementary Material
Acknowledgments
Funding sources included NIH 5K08HL114645-02, 5P01HL107202-02, R01HL128439A01; Pfizer Inc.; and Nina Ireland Program for Lung Health. We appreciate the work of Chris Gralapp in drawing Supplemental Figure 4. The GTEx Project was supported by the Common Fund of the Office of the Director of the NIH (www.commonfund.nih.gov/GTEx). Additional funds were provided by the NCI; National Human Genome Research Institute (NHGRI); National Heart, Lung, and Blood Institute (NHLBI); National Institute on Drug Abuse (NIDA); National Institute of Mental Health (NIMH); and National Institute of Neurological Disorders and Stroke (NINDS). Donors were enrolled at Biospecimen Source Sites funded by NCI\Leidos Biomedical Research Inc. subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care Inc. (X10S172). The Laboratory, Data Analysis, and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to the The Broad Institute Inc. Biorepository operations were funded through a Leidos Biomedical Research Inc. subcontract to Van Andel Research Institute (10ST1035). Additional data repository and project management were provided by Leidos Biomedical Research Inc. (HHSN261200800001E). Statistical methods development grants were made to the University of Geneva (MH090941 and MH101814), the University of Chicago (MH090951,MH090937, MH101825, and MH101820), the University of North Carolina - Chapel Hill (MH090936), North Carolina State University (MH101819), Harvard University (MH090948), Stanford University (MH101782), Washington University (MH101810), and the University of Pennsylvania (MH101822). The datasets used for the analyses described in this manuscript were obtained from dbGaP through dbGaP accession number phs000424.v6.p1.
Footnotes
Conflict of interest: JP, RTS, and KHN are employed by Pfizer Inc., and this research was partially funded by Pfizer Inc.
Reference information:JCI Insight. 2016;1(14):e87871. doi:10.1172/jci.insight.87871.
Contributor Information
Joe Palandra, Email: Joe.Palandra@pfizer.com.
Jamie L. Everman, Email: EvermanJ@NJHealth.org.
Robert J. Mason, Email: masonb@njhealth.org.
Michael A. Matthay, Email: michael.matthay@ucsf.edu.
References
- 1.Woodruff PG, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104(40):15858–15863. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodruff PG, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol. 2014;133(2):388–394. doi: 10.1016/j.jaci.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choy DF, et al. Gene expression patterns of Th2 inflammation and intercellular communication in asthmatic airways. J Immunol. 2011;186(3):1861–1869. doi: 10.4049/jimmunol.1002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenzel S, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455–2466. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 6.Corren J, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 7.Kondo Y, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20(6):791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 8.Schmitz J, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torgerson DG, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43(9):887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwahana H, et al. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur J Biochem. 1999;264(2):397–406. doi: 10.1046/j.1432-1327.1999.00615.x. [DOI] [PubMed] [Google Scholar]
- 12.Savenije OE, et al. Interleukin-1 receptor-like 1 polymorphisms are associated with serum IL1RL1-a, eosinophils, and asthma in childhood. J Allergy Clin Immunol. 2011;127(3):750–6.e1. doi: 10.1016/j.jaci.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Ho JE, et al. Common genetic variation at the IL1RL1 locus regulates IL-33/ST2 signaling. J Clin Invest. 2013;123(10):4208–4218. doi: 10.1172/JCI67119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akhabir L, et al. Lung expression quantitative trait loci data set identifies important functional polymorphisms in the asthma-associated IL1RL1 region. J Allergy Clin Immunol. 2014;134(3):729–731. doi: 10.1016/j.jaci.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 15.Gudbjartsson DF, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41(3):342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 16.Reijmerink NE, et al. Association of IL1RL1, IL18R1, and IL18RAP gene cluster polymorphisms with asthma and atopy. J Allergy Clin Immunol. 2008;122(3):651–4.e8. doi: 10.1016/j.jaci.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 17.Wu H, et al. Evaluation of candidate genes in a genome-wide association study of childhood asthma in Mexicans. J Allergy Clin Immunol. 2010;125(2):321–327.e13. doi: 10.1016/j.jaci.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimizu M, et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum Mol Genet. 2005;14(19):2919–2927. doi: 10.1093/hmg/ddi323. [DOI] [PubMed] [Google Scholar]
- 19.Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282(36):26369–26380. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- 20.Bhakta NR, et al. A qPCR-based metric of Th2 airway inflammation in asthma. Clin Transl Allergy. 2013;3(1):e87871. doi: 10.1186/2045-7022-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaya M, Finkbeiner WE, Chun SY, Widdicombe JH. Differentiated structure and function of cultures from human tracheal epithelium. Am J Physiol. 1992;262(6 Pt 1):L713–L724. doi: 10.1152/ajplung.1992.262.6.L713. [DOI] [PubMed] [Google Scholar]
- 22.Yin H, et al. Adenovirus-mediated delivery of soluble ST2 attenuates ovalbumin-induced allergic asthma in mice. Clin Exp Immunol. 2012;170(1):1–9. doi: 10.1111/j.1365-2249.2012.04629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfeffer PE, et al. Vitamin D enhances production of soluble ST2, inhibiting the action of IL-33. J Allergy Clin Immunol. 2015;135(3):824–7.e3. doi: 10.1016/j.jaci.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandara G, Beaven MA, Olivera A, Gilfillan AM, Metcalfe DD. Activated mast cells synthesize and release soluble ST2-a decoy receptor for IL-33. Eur J Immunol. 2015;45(11):3034–3044. doi: 10.1002/eji.201545501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HY, et al. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res. 2014;40(2):66–76. doi: 10.3109/01902148.2013.870261. [DOI] [PubMed] [Google Scholar]
- 26.Fahy JV. Type 2 inflammation in asthma--present in most, absent in many. Nat Rev Immunol. 2015;15(1):57–65. doi: 10.1038/nri3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melén E, et al. Analyses of shared genetic factors between asthma and obesity in children. J Allergy Clin Immunol. 2010;126(3):631–7.e1. doi: 10.1016/j.jaci.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bossé Y, et al. Asthma and genes encoding components of the vitamin D pathway. Respir Res. 2009;10:e87871. doi: 10.1186/1465-9921-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira MA, et al. Association between ORMDL3, IL1RL1 and a deletion on chromosome 17q21 with asthma risk in Australia. Eur J Hum Genet. 2011;19(4):458–464. doi: 10.1038/ejhg.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramasamy A, et al. Genome-wide association studies of asthma in population-based cohorts confirm known and suggested loci and identify an additional association near HLA. PLoS One. 2012;7(9):e87871. doi: 10.1371/journal.pone.0044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan YI, et al. Genome-wide association study to identify genetic determinants of severe asthma. Thorax. 2012;67(9):762–768. doi: 10.1136/thoraxjnl-2011-201262. [DOI] [PubMed] [Google Scholar]
- 33.Zhiguang X, et al. Over-expression of IL-33 leads to spontaneous pulmonary inflammation in mIL-33 transgenic mice. Immunol Lett. 2010;131(2):159–165. doi: 10.1016/j.imlet.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188(3):1503–1513. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim HY, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129(1):216–27.e1. doi: 10.1016/j.jaci.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Innes AL, et al. Epithelial mucin stores are increased in the large airways of smokers with airflow obstruction. Chest. 2006;130(4):1102–1108. doi: 10.1378/chest.130.4.1102. [DOI] [PubMed] [Google Scholar]
- 37.Diez D, et al. Network analysis identifies a putative role for the PPAR and type 1 interferon pathways in glucocorticoid actions in asthmatics. BMC Med Genomics. 2012;5:e87871. doi: 10.1186/1755-8794-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dougherty RH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol. 2010;125(5):1046–1053.e8. doi: 10.1016/j.jaci.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schroeder BW, et al. AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am J Respir Cell Mol Biol. 2012;47(2):178–185. doi: 10.1165/rcmb.2011-0421OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito Y, Correll K, Zemans RL, Leslie CC, Murphy RC, Mason RJ. Influenza induces IL-8 and GM-CSF secretion by human alveolar epithelial cells through HGF/c-Met and TGF-α/EGFR signaling. Am J Physiol Lung Cell Mol Physiol. 2015;308(11):L1178–L1188. doi: 10.1152/ajplung.00290.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, et al. Differentiated human alveolar epithelial cells and reversibility of their phenotype in vitro. Am J Respir Cell Mol Biol. 2007;36(6):661–668. doi: 10.1165/rcmb.2006-0410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Noorden R. ArXiv preprint server plans multimillion-dollar overhaul. Nature. 2016;534(7609):e87871. doi: 10.1038/534602a. [DOI] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):e87871. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen ES, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6:e87871. doi: 10.1038/ncomms9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7(3):500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.