

Abstract
To date, no targeted therapies are available to treat triple negative breast cancer (TNBC), while other breast cancer subtypes are responsive to current therapeutic treatment. Metabolomics was conducted to reveal differences in two hormone receptor-negative TNBC cell lines and two hormone receptor-positive Luminal A cell lines. Studies were conducted in the presence and absence of paclitaxel (Taxol®). TNBC cell lines had higher levels of amino acids, branched-chain amino acids, nucleotides and nucleotide sugars, and lower levels of proliferation-related metabolites like choline compared with Luminal A cell lines. In the presence of paclitaxel, each cell line showed unique metabolic responses, with some similarities by type. For example, in the Luminal A cell lines, levels of lactate and creatine decreased while certain choline metabolites and myo-inositol increased with paclitaxel. In the TNBC cell lines levels of glutamine, glutamate, and glutathione increased, whereas lysine, proline and valine decreased in the presence of drug. Profiling secreted inflammatory cytokines in the conditioned media demonstrated a greater response to paclitaxel in the hormone positive Luminal cells compared to a secretion profile that suggested greater drug resistance in the TNBC cells. The most significant differences distinguishing the cell types based on pathway enrichment analyses were related to amino acid, lipid and carbohydrate metabolism pathways, whereas several biological pathways were differentiated between the cell lines following treatment.
Keywords: metabolomics, triple negative breast cancer (TNBC), Luminal A, cell lines, paclitaxel/Taxol®, cytokines, health disparity
Graphical Abstract
Introduction
Triple negative breast cancer (TNBC), is a highly aggressive form of breast cancer (BCa) that derives its name from the absence of the estrogen receptor (ER+), progesterone receptor (PR+), and HER2/neu receptor (HER2+). These receptors are expressed differentially in other subtypes of BCa (i.e. Luminal A, Luminal B, HER2-positive) and have been successfully targeted with therapeutics such as Tamoxifen and Herceptin. TNBC represents as many as 20 % of current BCa cases,1 and continues to pose a disparate human health care problem for women diagnosed with this type of BCa when compared to other subtypes for which targeted therapeutics are available for treatment. Particularly for younger African American (AA) women who experience a disproportionate incidence and mortality from TNBC. For some BCa subtypes, five-year survival rates greater than 85% have been achieved, when the cancer is either detected early and/or targeted therapies have been incorporated into treatment plans.2 Unfortunately, there are still no approved therapeutics that specifically target TNBC available for clinical use and only a few validated novel targets are in drug development. This gap in available treatments is a contributing factor of recurrence and progression to metastatic disease3–5 for which currently there is no cure. Additionally, other characteristics, such as earlier age or later pathological stage at time of diagnosis, have led to TNBC being designated as some of the most aggressive forms of BCa.6 New targeted therapeutics are needed to improve the prognosis for patients who get a TNBC diagnosis.
Breast cancers characterized by the triple-negative phenotype typically present at a higher prevalence in premenopausal women under the age of 40, usually with a BMI greater than 30, and have higher incidences of mutations in BRCA1 or BRCA2,7–8 irrespective of ancestry.9–11 The disparity of the increased prevalence in AA women (~40–65%) compared to other racial ethnicities1, 12–17 is another troubling feature associated with the disease. Beyond genomic mutations, currently known, the increased incidence in younger women may also reflect the fact that without an established family history that results in earlier screening, many of these women have not started routine clinical monitoring programs18 that could facilitate earlier disease detection (i.e. Mammograms).
Within the past decade, one of the aggressive efforts in cancer research has been the ‘rediscovery’ of the critical role metabolic processes play in tumor etiology and progression. From the early 1950s Warburg effect observation of high anaerobic glycolysis in malignant cells, even when oxygen is plentiful,19 to recent years where differential metabolism has been coined “an emerging hallmark of cancer,”20 it is now well-accepted that cancer has unique metabolic signatures.21–25 The importance of these findings is the opportunity to leverage these divergences from normal cellular functions into targetable features for novel drug development. To this end, methods such as metabolomics is a valuable scientific tool in deciphering differential metabolic patterns and identifying their driving factors.26–27 Some examples include studies uncovering connections between proliferation of colon cancer cells and dietary polyphenols,28 BCa investigations using nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS) analytical methods to decipher pharmacodynamic mechanisms of adriamycin resistance29 and finding novel roles for chemokines such as CXCR4 in lipid and choline regulation.30
Treatment-dependent metabolites and pathways identified in the present study using BCa cell lines that have different hormone receptor expression profiles and originally derived from women of either AA or Caucasian American (CA) ethnicities (TNBC lines), suggests these differences may contribute to phenotypic characteristics in aggressiveness and poor chemotherapeutic treatment response outcomes. These results represent a significant starting point to further investigate the potential of identified markers to serve as new therapeutic targets for TNBC drug development. Further, useful data is provided from profiling changes that occurred in inflammatory signaling cytokines, as demonstrated in previous studies,31–34 to increase our understanding of treatment responses that occur in TNBC.
We set out to determine how TNBC cells derived from women of two different ethnicities respond to the taxane-based chemotherapeutic paclitaxel (Taxol®), which is commonly used in the clinic, compared to hormone-responsive BCa cell lines based on metabolic profiling to reveal novel biomarkers which may be leveraged for development of targeted therapeutics for high risk populations. We used TNBC cells derived from both an AA woman (MDA-MB-468) and a CA woman (MDA-MB-231),35–39 and compared their responses to paclitaxel with two hormone receptor-positive, Luminal A subtype-representative cell lines (CA-derived) with either a triple-positive ER+/PR+/HER2+ (BT474) or ER+/PR+/HER2− (MCF-7) phenotype. It is well-established that the cell lines used in this study are metabolically and phenotypically similar to clinical BCa subtypes,40–42 and thus were suitable for screening therapeutics to gauge representative therapy-dependent responses. Individual cell line and cell type (Luminal A versus TNBC) differences were initially assessed using 1H NMR broad spectrum metabolomics followed by comparison of treatment responsivity between the cell lines. We examined metabolic differences between the Luminal A and TNBC cell types, before and after treatment; as well as differences between the AA- and CA-derived TNBC cell lines and the two CA-derived Luminal A lines. Our study also confirmed several metabolite differences reported previously between the Luminal A and TNBC cell types. Chemotherapeutic treatment response was further assessed by examining inflammatory cytokines secreted into the media, revealing secretion profiles that suggests a pattern of increased drug sensitivity in the BT474 compared to MDA-MB-468 cells, reflected by the number and functional classification of significantly upregulated or downregulated cytokines. Integration of the differentially significant metabolites and cytokines into pathway enrichment analysis identified unique and potentially targetable pathways in the AA-derived versus the CA-derived TNBC cells suggesting metabolic differences in carbohydrate metabolism versus more significant changes in lipid metabolism pathways in the CA-derived TNBC cells. Further investigation into the treatment response-dependent metabolic characteristics that define the BCa subtypes across different ethnic populations may aid in identifying specific new markers for targeted drug development.
Experimental Procedures
Cell lines and culturing
Luminal A BCa cell lines (BT474 and MCF-7) and TNBC cell lines (MDA-MB-231 and MDA-MB-468) were purchased from ATCC and cultured in DMEM media (Gibco/Life Technologies) supplemented with 10% FBS and 1% antibiotic/antimycotic. All ATCC cell lines undergo authentication tests (i.e. for mycoplasma negativity) during the accessioning process, which is described in the online ATCC brochure Maintaining High Standards in Cell Culture (www.atcc.org). Cells were maintained in culture, incubated under humidified conditions at 37 °C in 5% CO2.
Drug treatment and sample preparation
Paclitaxel (Taxol®) from Taxus brevifolia (95%) was purchased from Sigma-Aldrich, Inc. Stock solutions [1 mg/mL] were prepared in molecular biology grade DMSO (Sigma), aliquoted and stored at −20 °C. Working solutions were prepared for treating cell lines [1 μg/mL] in sterile 1X PBS (Gibco) and stored for up to one week at 4 °C. Final dilutions were prepared at [10 nM] in DMEM treatment media, immediately prior to each treatment. The average LD50 concentration of paclitaxel across all four cell lines was determined to be 10 nM by cell viability assays using MTS-AQ reagent (Promega), with no noticeable effect of the vehicle (DMSO) on cell growth across the ranges tested, resulting in a 46% (BT474), 54% (MCF-7), 51% (MDA-MB-231) and 49% (MDA-MB-468) decrease in cell viability after 24 hr of drug exposure (Figure S-1, Supporting Information). To generate the samples for metabolomics analysis and cytokine profiling, each cell line was plated in 10 cm dishes with DMEM for 24 hr prior to treatment. Growth media was removed, cells were washed with sterile 1X PBS and treated for 48 hr in fresh media alone or containing 10 nM paclitaxel. Following treatment, 1 mL conditioned media aliquots were collected and stored at −80 °C. Treated cells on plates were washed with cold, 1X PBS, and extracted using a modified Folch method.43–44 Briefly, cells were quenched with 50:50 ice cold acetonitrile:water, scraped off dishes, and collected in to 15 mL tubes containing zirconia beads. Cold chloroform was added and each tube was vigorously vortexed on a multitube vortexer for three 30 sec pulses. Tubes were centrifuged at 3,700 rpm for 60 min at 4 °C, and the aqueous fractions were transferred to cryotubes, while the organic fractions were collected into glass vials. The remaining protein layer and residual aqueous & lipid layers were transferred to Lo-Bind Eppendorf tubes, cold chloroform:methanol (2:1) was added, and the tubes were quickly vortexed then centrifuged at 15,000 rpm for 20 min at 4 °C. The remaining aqueous and lipid fractions were transferred into collection tubes, while the protein pellets were dried for 20 min on a Speedvac (no heat) and weighed. All samples were stored at −80 °C except for the aqueous fractions which were lyophilized to dryness first then stored until NMR analysis.
NMR sample preparation and data acquisition
Lyophilized cellular extracts were reconstituted in 700 μL of a deuterium oxide (D2O, Aldrich) solution containing 0.6 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS-D6, Chemical Shift Indicator), 0.6 mM Imidazole (pH indicator), and 0.2 % NaN3. The samples were vortexed and centrifuged at 12,000 rcf for 3 min, then a 600 μL aliquot of each sample supernatant was transferred into 5 mm NMR tubes (Bruker-BioSpin, Switzerland) for data acquisition. 1H NMR spectra were acquired on a Bruker Avance III 600 MHz NMR spectrometer (Bruker-Biospin, Rheinstetten, Germany) using a cryogenically cooled 5mm ATMA probe at 25 °C. A 1D NOESY pulse sequence (noesypr1d) with water pre-saturation during the 2 sec relaxation delay and 100 ms mixing time was used, and 256 transients were collected into 16k data points with a spectral width of 6602.1 kHz (11 ppm) and an acquisition time of 2.48 sec.
NMR data analysis
Metabolomics analysis for 1H NMR spectra was performed on cellular extracts as described previously.45–51 Free induction decays (FIDs) were zero-filled to 64k and a line broadening factor of 0.5 Hz was applied before Fourier transformation. Spectra were manually phased, baseline corrected, and referenced to DSS. Spectra were binned (0.14–9.35 ppm) using intelligent bucketing integration with a 0.04 ppm bucket width and a 50% looseness factor in ACD NMR Processor 12.0 (ACD Labs Inc., Toronto Ontario, Canada) software. Chemical shift regions for water (4.66–4.86 ppm) imidazole (7.15–7.255 ppm), and region 2.03–2.08 ppm were excluded from binning. The bin integrals were normalized to the total integral of each spectrum. We performed parametric analysis using a 2-sample equal variance Student’s t-test to compare bins across cell lines in the absence of paclitaxel using the false discovery rate (FDR) to control for multiple comparisons, and the Benjamini–Hochberg procedure52 to control the FDR at ≤ 0.1, and for each cell line after paclitaxel treatment compared to its non-treated control, (Microsoft Excel). Principal components analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) of binned data was performed using Pareto-scaling and mean centering, in SIMCA 14.0 (Umetrics, Umeå, Sweden). All models used a 7-fold cross-validation to assess the predictive variation of the model (Q2) and model statistics are included in the figure legends in brackets. Variable Influence on Projection values (VIP ≥ 1.0 with a jack-knife confidence interval that did not include 0) were used to identify bins responsible for class separation based on multivariate analysis of treated versus non-treated groups/cell line and between the cell lines. These bins were matched to the metabolites using the metabolite library in Chenomx NMR Suite 8.1 Professional software. All plots represent three biological replicates per sample. Statistical analyses for the comparison between hormone-positive versus triple-negative BCa cell types were conducted using SAS 9.4 (SAS Institute Inc., Cary, NC), and the Wilcoxon Rank Sum Test was used to determine statistical differences in bin intensity between the cell types. In this exploratory analysis, p-values < 0.05 were considered to be statistically significant and were not adjusted for multiple testing.
Cytokine profiling
Secreted protein expression profiling of 80 inflammatory cytokines was performed with undiluted DMEM cultured media collected from control, untreated cells and paclitaxel-treated cells (10 nM) after 48 hr using Human Cytokine Antibody G Series 5 arrays (RayBiotech, Inc.) according to the manufacturer’s protocol. Cell media was collected into clean tubes in 1.0 mL aliquots and immediately frozen at −80 °C until further processing. For sample preparation, one aliquot was thawed on ice, centrifuged at 10,000 rpm for 5 min to pellet any remaining cellular debris then 100 μL was transferred to the glass slide array and incubated for 2 hr at room temperature, after the array was blocked for 30 min. The slide was washed and then incubated with secondary biotin-conjugated antibody overnight at 4 °C in the dark. The following day, the slide was washed and incubated with streptavidin antibody, washed again, air dried, and shipped to RayBiotech, Inc. for scanning on a GenePix 4000 scanner at a wavelength of 532 nm using GenePix Pro 6.0.1.25 software. The relative expression for each cytokine was calculated according to the manufacturer’s instructions by first subtracting the signal from the average of the Negative controls, then normalized to the average of the Positive controls for each sample. Cytokine signals for each paclitaxel-treated sample were then normalized to the untreated control for the corresponding sample/cell line. Results are presented as significantly upregulated if the signal intensity ≥ 1.50 fold, while those with an intensity ≤ 0.65 fold were considered significantly downregulated, based on the manufacturer’s criteria. Significant results are based on the average of three biological replicates per treatment condition for each cell line (Table 5). Selected cytokines are also highlighted for specific relevance and presented as the relative % change, calculated by subtracting 1 from the fold-change value and multiplying by 100% to better visualize the up- versus down-regulation (Figure 3).
Table 5.
Paclitaxel response-dependent inflammatory cytokine profiles in Luminal A and TNBC cell lines are highly heterogeneous.
| Cytokine Symbol | BT474 | MCF-7 | MDA-MB-231 | MDA-MB-468 |
|---|---|---|---|---|
|
| ||||
| CSF2/GM-CSF | 0.48 | 0.64 | 0.59 | 1.12 |
| GRO | 0.22 | 0.84 | 2.22 | 1.59 |
| IL-4 | 0.89 | 1.14 | 1.10 | 1.57 |
| IL-6 | 0.13 | 0.47 | 0.53 | 1.13 |
| MCP-1 | 0.46 | 0.78 | 1.34 | 1.46 |
| MDC | 0.76 | 2.38 | 2.07 | 1.11 |
| MIP-1δ/delta | 0.78 | 1.51 | 0.65 | 1.17 |
| RANTES | 0.47 | 0.52 | 0.87 | 1.68 |
| SCF | 0.64 | 1.27 | 0.85 | 1.17 |
| ANG/Angiogenin | 0.26 | 0.90 | 1.08 | 1.02 |
| VEGF-A | 0.28 | 0.82 | 1.08 | 0.92 |
| PDGF-BB | 0.27 | 2.67 | 1.42 | 1.29 |
| BDNF | 0.36 | 0.67 | 0.84 | 1.27 |
| FGF-4 | 0.81 | 1.67 | 1.09 | 0.97 |
| FGF-9 | 1.07 | 0.89 | 0.63 | 0.77 |
| CX3CL1/Fractalkine | 2.94 | 0.97 | 0.90 | 1.06 |
| IGFBP-1 | 0.02 | 0.37 | 0.61 | 1.13 |
| IGFBP-2 | 0.94 | 1.07 | 0.52 | 0.84 |
| IGFBP-4 | 0.60 | 0.92 | 0.90 | 1.25 |
| IL-16 | 0.64 | 1.34 | 0.86 | 1.71 |
| LIGHT | 1.84 | 0.70 | 0.89 | 1.02 |
| MIF | 0.81 | 2.77 | 1.03 | 1.04 |
| MIP-3 α/alpha | 0.60 | 1.50 | 1.60 | 1.37 |
| NAP-2 | 0.80 | 1.66 | 1.15 | 0.97 |
| OPG/Osteoprotegerin | 0.19 | 0.38 | 0.57 | 0.51 |
| TIMP-1 | 0.19 | 0.86 | 0.85 | 1.07 |
| TIMP-2 | 0.14 | 0.86 | 0.86 | 1.05 |
![]()
Relative expression of significantly heterogeneous profiles of secreted cytokines into media from BT474, MCF-7, MDA-MB-231 and MDA-MB-468 BCa cell lines after 48 hr treatment with paclitaxel (10 nM) normalized to their individual non-treated, time-matched control secretory signals. Relative expression levels are defined as significantly upregulated if ≥ 1.5 (highlighted in red and bold-print) or significantly downregulated if ≤ 0.65 (highlighted in green and italicized).
Figure 3. Secretome expression responses to paclitaxel treatment.

Bar graph depicts relative % change in expression of selected inflammatory cytokines secreted into the media of the four BCa cell lines tested, normalized to each cell line’s non-treated control.
Pathway analysis
GeneGo (MetaCore) pathway enrichment software was used to identify significantly perturbed biological canonical pathway and endogenous metabolic network differences between hormone-positive and triple-negative BCa cell lines and for each individual cell line in response to paclitaxel treatment. Integrative analysis was also performed using the significantly differential metabolite and cytokine markers across the two TNBC cell lines. The threshold and p-value criteria were included based on the calculated fold-changes (threshold) and p-values from the binned data for analyses using metabolites, unless otherwise stated in the table legend. For the analysis using cytokines, the direction and threshold were based on the significance criteria of the assay (upregulated ≥ 1.50 fold or downregulated ≤ 0.65) and a p-value ≤ 0.05.
Results and Discussion
Metabolomics distinguishes cell lines by ethnicity and receptor expression differences
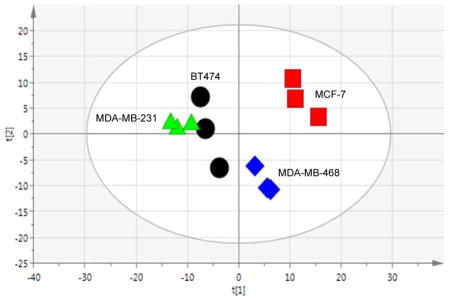
As a highly heterogeneous disease, with at least 5 clinically accepted subtypes,12, 53–54 BCa cell lines are highly useful research tools when studying BCa because they recapitulate many of the clinical characteristics such as expression of hormone receptors, invasiveness, or resistance to drug exposure. In that, we used broad spectrum 1H NMR metabolomics and showed that metabolic profiles reflected one of these primary clinical differences that distinguishes the cell lines, being the presence or absence of ER, PR and HER2/neu receptors. Although BT474 and MCF-7 are both CA-derived, hormone receptor-positive BCa cell lines (classified as Luminal A), another major difference is HER2 expression; where only BT474 cells express the receptor. MDA-MB-231 and MDA-MB-468 are both TNBC cell lines that are distinct based on the ethnicity of the patients from which they were derived (CA or AA, respectively). First, we performed a 2-sample equal variance Student’s t-test adjusted for false discovery rate to compare each bin across the cell lines to investigate metabolic differences between the cell lines in the absence of treatment (Table 1). Overall, in the analysis of 248 bins (249 total), the AA-derived TNBC cell line (MDA-MB-468) was very different from the three CA-derived cell lines (BT474, MCF-7 and MDA-MB-231), while the comparison of bin intensities between the MCF-7 and MDA-MDA-231 cell lines (126 bins) was also significant. The most significant difference was between the two TNBC cell lines (151 bins). Next unsupervised multivariate analysis (PCA) was used to visualize differences between the Luminal A and TNBC cell lines, and supervised multivariate analysis (OPLS-DA) was used to identify the metabolites that differentiate the Luminal A from the TNBC cell types. The PCA analysis (Figure 1A) demonstrates differences between the Luminal A cell lines (BT474 and MCF-7) cell lines, which both cluster by the second component, accounting for the variability between the replicates for BT474. Interestingly, they are separated along the first (primary) component, which provides a testable hypothesis that the difference in HER2 expression is related to metabolic changes. It is also an intriguing observation that the three CA-derived cell lines (BT474, MCF-7 and MDA-MB-231) appear to show more metabolic similarity in multivariate space when compared to the AA-derived cell line (MDA-MB-468), even though hormone-receptor expression strongly influences cell line responses and receptor profiles are associated with BCa patient outcomes. In a pairwise, supervised analysis to determine the significant signals (VIP ≥ 1) that distinguish the metabolic profiles of each cell type (Figure 1B), NMR library-matching (Chenomx) identified metabolites from the binned data, listed in Table 2, with corresponding fold-change and p-values. Most notably, the Luminal A cell type (BT474 and MCF-7) metabolic profiles are characterized by increased levels of proliferation-associated choline, O-acetylcholine and O-phosphocholine,55–57 lactate and myo-inositol versus the TNBC cell type. In contrast to the primarily proliferative signature in Luminal A cells, the two TNBC cell lines showed higher levels of metabolites across several compound classes including amino acids and tripeptide (i.e., alanine, histidine, glutamine, and glutathione), branched-chain amino acids (isoleucine and leucine), nucleotides (ADP, ATP) and nucleotide sugars (UDP-galactose, UDP-glucose, and UDP-glucuronate). We also saw comparatively higher levels of creatine and creatinine metabolites in the TNBC cells versus the hormone positive lines in the absence of any treatment. Interestingly, a recent report related creatine kinase and creatinine as oxidative stress indicators in BCa patients following chemotherapeutic treatment,58 and considering the primary function of creatine is to support energy production in the phosphorylation of ADP to make ATP, changes in these metabolites may be suggestive of cell sensitivity to treatment. The heterogeneity of increased levels of all these metabolites (Table 2) in TNBC cell lines as a cell type in comparison with Luminal A cells suggest a greater robustness and redundancy of breast cancers classified by this subtype to facilitate use from multiple pools of essential building blocks and nutrients to remain viable.
Table 1.
Number of bins that are significantly different between cell lines in the absence of paclitaxel.
| BT474 | MCF-7 | MDA-MB-231 | MDA-MB-468 | |
|---|---|---|---|---|
| BT474 | X | |||
| MCF-7 | 77 | X | ||
| MDA-MB-231 | 15 | 126 | X | |
|
MDA-MB-468
|
106 | 93 | 151 | X |
P<0.1 and FDR-corrected.
Figure 1. Multivariate analyses comparing breast cancer (BCa) cell lines.

(A.) Unsupervised multivariate analysis (PCA) of Luminal A BCa and TNBC cell lines in the absence of paclitaxel treatment. [R2X(cum) = 0.769, Q2(cum) = 0.211]. (B.) Supervised analysis (OPLS-DA) of Luminal A versus TNBC cells. [R2X(cum) = 0.901, R2Y(cum) = 0.993, Q2(cum) = 0.935].
Table 2.
NMR bins and library matched metabolites that distinguish hormone-positive versus triple-negative BCa cell types.
| bin region (ppm) | library matched metabolites | VIP | p-value* | Fold Change** | Cell Type Preference |
|---|---|---|---|---|---|
| [1.87 .. 1.92] | Acetate, Lysine | 1.7 | 0.017 | −1.2 | TNBC |
| [4.52 .. 4.58] | ADP, ATP, Glutathione | 1.4 | 0.040 | −1.4 | TNBC |
| [1.45 .. 1.50] | Alanine | 4.3 | 0.093 | −1.8 | TNBC |
| [3.78 .. 3.83] | Alanine, Cystathionine, Glucose, Glutathione, UDP-glucose, UDP-N-Acetylglucosamine | 1.2 | 0.397 | −1.2 | TNBC |
| [3.72 .. 3.78] | Alanine, Glutamine, Glutathione, Glucose, Leucine, Glutamate | 3.5 | 0.054 | −1.3 | TNBC |
| [2.80 .. 2.86] | Aspartate, Asparagine, Unknown | 1.1 | 0.093 | −1.3 | TNBC |
| [3.17 .. 3.23] | Choline, O-Acetylcholine, O-Phosphocholine, sn-Glycero-3-phosphocholine | 5.3 | 0.040 | 1.9 | luminal A |
| [3.01 .. 3.07] | Creatine, Creatinine, Cystathionine | 1.3 | 1.000 | −1.0 | TNBC |
| [3.90 .. 3.95] | Creatine, Glucose, Glycolate, UDP-N-Acetylglucosamine, Uridine, Tyrosine | 1.3 | 1.000 | −1.0 | TNBC |
| [2.68 .. 2.74] | Cystathionine | 1.1 | 0.071 | −1.3 | TNBC |
| [3.07 .. 3.10] | Cystathionine, Histidine | 0.7 | 0.017 | −1.1 | TNBC |
| [3.10 .. 3.15] | Cystathionine, Histidine, Cystine, Phenylalanine, Ethylmalonate | 1.4 | 0.017 | −1.4 | TNBC |
| [8.44 .. 8.46] | Formate | 0.3 | 0.017 | 1.5 | luminal A |
| [3.39 .. 3.44] | Glucose, Proline | 1.5 | 0.093 | 1.3 | |
| [3.44 .. 3.48] | Glucose, UDP-Glucose | 1.0 | 0.486 | −1.2 | TNBC |
| [2.42 .. 2.47] | Glutamine, Glutamate | 2.4 | 0.201 | −1.3 | TNBC |
| [2.17 .. 2.19] | Glutathione | 1.2 | 0.017 | −1.3 | TNBC |
| [2.51 .. 2.57] | Glutathione | 2.7 | 0.040 | −1.9 | TNBC |
| [2.94 .. 2.97] | Glutathione | 1.7 | 0.023 | −1.8 | TNBC |
| [2.90 .. 2.94] | Glutathione, Asparagine | 1.7 | 0.023 | −1.7 | TNBC |
| [3.50 .. 3.55] | Glycine, myo-Inositol, Glucose | 2.5 | 0.938 | −1.0 | |
| [1.40 .. 1.45] | Isoleucine, Alloisoleucine | 1.1 | 0.255 | −1.1 | TNBC |
| [0.91 .. 0.97] | Isoleucine, Leucine | 2.2 | 0.121 | −1.2 | TNBC |
| [3.65 .. 3.70] | Isoleucine, sn-Glycero-3-phosphocholine, Unknown | 1.2 | 0.697 | −1.1 | |
| [4.07 .. 4.13] | Lactate | 1.9 | 0.320 | 1.2 | luminal A |
| [1.29 .. 1.34] | Lactate, Threonine | 3.6 | 0.486 | 1.1 | |
| [0.86 .. 0.89] | Lipoproteins | 1.0 | 0.697 | −1.0 | TNBC |
| [1.66 .. 1.70] | Lysine | 1.1 | 0.017 | −1.1 | TNBC |
| [1.70 .. 1.76] | Lysine | 1.7 | 0.017 | −1.2 | TNBC |
| [2.97 .. 3.01] | Lysine, Glutathione | 1.4 | 0.121 | −1.2 | TNBC |
| [2.60 .. 2.62] | Methionine | 0.5 | 0.030 | −1.1 | TNBC |
| [4.02 .. 4.07] | myo-Inositol, Creatinine | 1.9 | 0.815 | 1.3 | luminal A |
| [3.60 .. 3.65] | myo-Inositol | 2.1 | 0.815 | 1.2 | luminal A |
| [3.23 .. 3.29] | myo-Inositol, Betaine, Taurine, Trimethylamine-N-oxide (TMAO) | 2.3 | 0.938 | 1.3 | |
| [3.55 .. 3.60] | myo-Inositol, Threonine, O-Phosphocholine, sn-Glycero-3-phosphocholine, Valine | 2.5 | 0.093 | 1.4 | luminal A |
| [9.12 .. 9.18] | NAD+ | 0.4 | 0.030 | 1.2 | luminal A |
| [9.31 .. 9.35] | NAD+ | 0.4 | 0.023 | 1.2 | luminal A |
| [9.26 .. 9.31] | NADP+ | 0.3 | 0.017 | 1.3 | luminal A |
| [8.46 .. 8.49] | Overlap | 0.2 | 0.040 | 1.3 | luminal A |
| [8.84 .. 8.90] | Overlap | 0.3 | 0.017 | 1.6 | luminal A |
| [8.94 .. 9.00] | Overlap | 0.4 | 0.023 | 1.6 | luminal A |
| [7.30 .. 7.34] | Phenylalanine | 1.2 | 0.093 | −1.6 | TNBC |
| [7.39 .. 7.45] | Phenylalanine | 1.1 | 0.156 | −1.6 | TNBC |
| [3.31 .. 3.37] | Proline, Cystine | 1.2 | 0.397 | 1.3 | |
| [1.99 .. 2.03] | Proline, N-Acetylamino acids, Glutamate | 1.3 | 0.697 | 1.0 | |
| [2.37 .. 2.42] | Pyroglutamate | 1.4 | 0.121 | 1.2 | luminal A |
| [4.20 .. 4.26] | Threonine, UDP-N-Acetylglucosamine, UDP-Glucuronate, UDP-Galactose, ATP | 2.3 | 0.023 | −1.3 | TNBC |
| [6.86 .. 6.92] | Tyrosine | 1.2 | 0.093 | −1.6 | TNBC |
| [4.13 .. 4.18] | UDP-Glucuronate, Pyroglutamate, O-phosphocholine, Proline | 2.7 | 0.054 | 1.7 | |
| [3.95 .. 4.00] | UDP-N-Acetylglucosamine, Asparagine, Histidine, Phenylalanine | 1.5 | 0.201 | −1.3 | TNBC |
| [5.95 .. 6.00] | UDP-N-Acetylglucosamine, UDP-Glucose, UDP-Glucuronate, UDP-Galactose | 1.3 | 0.815 | −1.2 | TNBC |
| [4.34 .. 4.38] | UDP-N-Acetylglucosamine, UDP-Glucose, UDP-Glucuronate, UDP-Galactose, ADP | 1.5 | 0.201 | −1.3 | TNBC |
| [4.26 .. 4.30] | UDP-N-Acetylglucosamine, UDP-Glucose, UDP-Glucuronate, UDP-Galactose, ATP | 1.6 | 0.156 | −1.2 | TNBC |
| [6.82 .. 6.86] | Unknown | 0.4 | 0.017 | −1.2 | luminal A |
| [8.28 .. 8.30] | Unknown | 0.3 | 0.017 | 1.4 | TNBC |
| [1.02 .. 1.07] | Valine | 1.7 | 0.093 | −1.3 | TNBC |
| [2.26 .. 2.30] | Valine | 1.1 | 0.071 | −1.2 | TNBC |
| [0.97 .. 1.02] | Valine, Isoleucine | 2.0 | 0.093 | −1.3 | TNBC |
The metabolites listed are important to differentiating the metabolic profiles between cell lines defined as hormone receptor-positive and Luminal A (BT474 and MCF-7) compared to triple hormone receptor-negative and TNBC (MDA-MB-231 and MDA-MB-468) BCa cell types, and are based on VIP ≥ 1 with a confidence interval not including 0 or a p-value < 0.05. VIP = variable influence on projection.
Wilcoxon Rank Sum Test.
A positive fold change indicates the median bin intensity of Luminal A cells > median bin intensity of TNBC cells. Cell type preference column lists the directionality of the predominant cell type within which the bin intensities of the library-matched metabolites were clearly higher. NMR bins identified as “Overlap” could not be assigned to metabolites distinctively using the NMR library and further 2D experiments are needed for confirmed identification.
Luminal A and TNBC cells demonstrate different metabolic treatment responses
Evaluating treatment response to paclitaxel, supervised (Figure S-2A, Supporting Information) and unsupervised (Figure S-2B, S-2C, Supporting Information) multivariate analyses enabled us to identify differences in metabolites between each BCa cell line (Table S-1, Supporting Information) following treatment. OPLS-DA plots for each treated cell line compared to its non-treated control (Figure 2) were used to identify drug response-dependent metabolites, listed in Table 3. Although there were some similarities between the two cell lines defined by the same cell type (Luminal or TNBC), each cell line demonstrated a unique metabolic profile, as evident in Table 3 listing how the identified metabolites changed based on cell line response to paclitaxel treatment. In both Luminal A cell lines, choline and associated metabolites (Table 2) were increased in the presence of drug, continuing to show proliferation as a dominant signature in this cell type. Interestingly, these metabolite levels (choline, o-acetylcholine, o-phosphocholine, sn-glycero-3-phosphocholine), also increased in the AA-derived TNBC (MDA-MB-468) cell line, but were reduced in the CA-derived TNBC (MDA-MB-231) cell line with the exception of sn-glycero-3-phosphocholine following treatment. The level of myo-inositol, shown to be higher in Luminal A cells compared to TNBC (Table 2), was also shown to be increased in the MCF-7 (Luminal A) cells but reduced in the MDA-MB-468 TNBC line with drug. Similarly, lactate was reduced in MDA-MB-468 and MCF-7 cells, but increased in MDA-MB-231 cells following treatment with paclitaxel. More individual changes included drug-responsive increases in glucose, glutamine, alloisoleucine, isoleucine, leucine and threonine in the BT474 cells; with decreases in most of the identified nucleotide sugars (i.e. UDP-glucose). Proline and valine also increased in the BT474 cell line after treatment, compared to a reduction of these amino acids in the other three cell lines. While glutamine was increased in BT474 following treatment, it was decreased in MCF-7 cells, but increased in both TNBC cell lines. In contrast, glutamate was increased in MCF-7 and MDA-MB-468 cells, but decreased in BT474 and MDA-MB-231 cells. Other notable TNBC cell line responses to paclitaxel treatment included increased glutathione in MDA-MB-231 cells and several decreases (acetate, lysine, cystathionine, histidine, cystine, phenylalanine, ethylmalonate and UDP-n-acetylglucosamine), with different decreases in MDA-MB-468 cells such as glycine, myo-inositol, betaine, taurine, trimethylamine-N-oxide and lipoproteins. Two significantly increased metabolites in MDA-MB-468 cells only included 1-methylhistidine and a metabolite currently unknown by library matching (Table 3).
Figure 2. Multivariate analyses comparing breast cancer (BCa) cell lines in the presence of paclitaxel treatment.

The panels are pairwise supervised analyses (OPLS-DA) of non-treated controls compared to cells treated for 48 hr with paclitaxel [10 nM]. (A.) BT474 (ER+/PR+/HER2+) Luminal A cells. [R2X(cum) = 1, R2Y(cum) = 1, Q2(cum) = 1]. (B.) MCF-7 (ER+/PR+/HER2−) Luminal A cells. [R2X(cum) = 1, R2Y(cum) = 1, Q2(cum) = 1]. (C.) MDA-MB-231 is the TNBC cell line derived from a Caucasian woman. [R2X(cum) = 1, R2Y(cum) = 1, Q2(cum) = 1] (D.) MDA-MB-468 is the TNBC cell line derived from an African-American woman. [R2X(cum) = 1, R2Y(cum) = 1, Q2(cum) = 1].
Table 3.
NMR bins and library matched metabolites that differentiate Luminal A and TNBC cell line responses to paclitaxel.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BT474 | MCF-7 | MDA-MB-231 | MDA-MB-468 | |||||||
|
| ||||||||||
| bin region | library matched metabolites | VIP | FC | VIP | FC | VIP | FC | VIP | FC | cell line response to paclitaxel |
| [7.64 .. 7.68] | 1-Methylhistidine | 1.1 | 1.6 | Increased in 468 | ||||||
| [1.87 .. 1.92] | Acetate, Lysine | 1.5 | −1.1 | Reduced in 231 | ||||||
| [1.45 .. 1.50] | Alanine | 2.2 | −1.1 | 3.3 | −1.1 | Reduced in 474 & 468 | ||||
| [3.78 .. 3.83] | Alanine, Cystathionine, Glucose, Glutathione, UDP-glucose, UDP-N-Acetylglucosamine | 2.4 | 1.1 | Increased in 474 | ||||||
| [3.72 .. 3.78] | Alanine, Glutamine, Glutathione, Glucose, Leucine, Glutamate | 4.5 | 1.1 | 2.5 | 1.0 | Increased in 231 & 468 | ||||
| [1.34 .. 1.38] | Alloisoleucine, Acetoin | 1.4 | 1.0 | 1.4 | −1.1 | Increased in 474; Reduced in 231 | ||||
| [2.62 .. 2.68] | Aspartate, Citrate, Unknown | 1.0 | 1.0 | Increased in MCF | ||||||
| [3.17 .. 3.23] | Choline, O-Acetylcholine, O-Phosphocholine, sn-Glycerophosphocholine | 3.7 | 1.1 | 1.8 | −1.0 | 2.7 | 1.1 | Increased in 474 & 468; Reduced in 231 | ||
| [3.90 .. 3.95] | Creatine, Glucose, Glycolate, UDP-N-Acetylglucosamine, Uridine, Tyrosine | 1.2 | −1.0 | Reduced in MCF | ||||||
| [3.10 .. 3.15] | Cystathionine, Histidine, Cystine, Phenylalanine, Ethylmalonate | 1.2 | −1.1 | Reduced in 231 | ||||||
| [3.70 .. 3.72] | Glucose | 2.1 | 1.3 | 1.3 | −1.0 | Increased in 474; Reduced in 468 | ||||
| [5.18 .. 5.24] | Glucose | 1.3 | 1.3 | Increased in 474 | ||||||
| [3.39 .. 3.44] | Glucose, Proline | 2.3 | 1.2 | 1.3 | 1.0 | Increased in 474 & 468 | ||||
| [3.44 .. 3.48] | Glucose, UDP-Glucose | 1.8 | −1.0 | Reduced in 468 | ||||||
| [3.83 .. 3.85] | Glucose, UDP-Glucose | 1.1 | 1.1 | Increased in 468 | ||||||
| [3.85 .. 3.90] | Glucose, UDP-Glucose | 3.2 | 1.2 | 2.3 | 1.1 | 1.5 | 1.0 | Increased in 474, 231 & 468 | ||
| [2.08 .. 2.11] | Glutamate | 1.4 | 1.1 | Increased in 468 | ||||||
| [2.30 .. 2.32] | Glutamate | 1.2 | −1.1 | Reduced in 231 | ||||||
| [2.32 .. 2.37] | Glutamate | 1.7 | −1.0 | 2.3 | 1.0 | 2.2 | −1.0 | 2.4 | 1.1 | Reduced in 474 & 231; Increased in MCF & 468 |
| [2.11 .. 2.17] | Glutamate, Glutamine | 2.4 | 1.1 | 2.2 | −1.0 | Increased in 474; Reduced in MCF | ||||
| [2.47 .. 2.49] | Glutamine | 1.0 | 1.0 | Increased in 231 | ||||||
| [2.42 .. 2.47] | Glutamine, Glutamate | 2.4 | 1.1 | 3.9 | 1.1 | Increased in 474 & 231 | ||||
| [3.50 .. 3.55] | Glycine, myo-Inositol, Glucose | 3.0 | −1.0 | Reduced in 468 | ||||||
| [1.18 .. 1.23] | Isoleucine, Overlap | 1.9 | −1.1 | Reduced in MCF & 468 | ||||||
| [1.23 .. 1.27] | Isoleucine | 1.5 | 1.1 | 1.7 | −1.1 | Increased in 474; Reduced in 468 | ||||
| [1.27 .. 1.29] | Isoleucine | 1.6 | −1.2 | Reduced in 468 | ||||||
| [1.40 .. 1.45] | Isoleucine, Alloisoleucine | 1.8 | 1.1 | 1.4 | −1.0 | Increased in 474; Reduced in 468 | ||||
| [0.91 .. 0.97] | Isoleucine, Leucine | 2.6 | −1.1 | 1.3 | 1.0 | 1.7 | −1.0 | Reduced in MCF & 468; Increased in 231 | ||
| [3.65 .. 3.70] | Isoleucine, sn-Glycero-3-phosphocholine, Unknown | 2.6 | 1.2 | Increased in 474 | ||||||
| [4.07 .. 4.13] | Lactate | 2.7 | −1.1 | 1.7 | 1.2 | 2.1 | −1.0 | Reduced in MCF & 468; Increased in 231 | ||
| [1.29 .. 1.34] | Lactate, Threonine | 5.7 | −1.1 | 5.3 | −1.1 | Reduced in MCF & 468 | ||||
| [0.70 .. 0.72] | Lipoproteins | 1.0 | −1.1 | Reduced in 468 | ||||||
| [0.72 .. 0.74] | Lipoproteins | 1.0 | −1.1 | Reduced in 468 | ||||||
| [0.74 .. 0.76] | Lipoproteins | 1.1 | −1.1 | Reduced in 468 | ||||||
| [0.76 .. 0.78] | Lipoproteins | 1.0 | −1.1 | Reduced in 468 | ||||||
| [0.78 .. 0.80] | Lipoproteins | 1.1 | −1.1 | Reduced in 468 | ||||||
| [0.89 .. 0.91] | Lipoproteins | 1.3 | −1.0 | Reduced in 468 | ||||||
| [1.70 .. 1.76] | Lysine | 1.1 | −1.0 | 1.0 | −1.0 | Reduced in MCF & 468 | ||||
| [1.92 .. 1.95] | Lysine | 1.2 | −1.1 | Reduced in 231 | ||||||
| [3.60 .. 3.65] | myo-Inositol | 4.0 | 1.1 | 1.8 | −1.0 | Increased in MCF; Reduced in 468 | ||||
| [3.23 .. 3.29] | myo-Inositol, Betaine, Taurine, Trimethylamine-N-oxide (TMAO) | 1.3 | −1.0 | Reduced in 468 | ||||||
| [4.02 .. 4.07] | myo-Inositol, Creatinine | 1.9 | −1.1 | Reduced in 468 | ||||||
| [3.55 .. 3.60] | myo-Inositol, Threonine, O-Phosphocholine, sn-Glycero-3-phosphocholine, Valine | 2.1 | 1.1 | Increased in 474 | ||||||
| [1.55 .. 1.60] | Overlap | 1.0 | −1.1 | Reduced in 231 | ||||||
| [4.94 .. 5.00] | Overlap | 0.7 | 2.2 | Increased in 474 | ||||||
| [5.63 .. 5.69] | Overlap | 0.3 | −2.2 | 0.7 | −2.2 | Reduced in 474 & 231 | ||||
| [5.69 .. 5.72] | Overlap | 0.2 | −2.1 | 0.4 | −2.0 | Reduced in 474 & 231 | ||||
| [5.72 .. 5.74] | Overlap | 0.3 | −2.2 | Reduced in 231 | ||||||
| [1.95 .. 1.97] | Proline | 1.1 | −1.1 | 1.0 | −1.0 | Reduced in 231 & 468 | ||||
| [1.97 .. 1.99] | Proline | 1.3 | −1.1 | 1.1 | −1.0 | 1.0 | −1.0 | Reduced in MCF, 231 & 468 | ||
| [1.99 .. 2.03] | Proline, N-Acetylamino acids, Glutamate | 2.9 | −1.1 | Reduced in MCF | ||||||
| [2.37 .. 2.42] | Pyroglutamate | 2.2 | 1.1 | Increased in 474 | ||||||
| [4.30 .. 4.34] | sn-Glycero-3-phosphocholine | 1.0 | 1.1 | Increased in 231 | ||||||
| [6.86 .. 6.92] | Tyrosine | 1.0 | −1.1 | Reduced in MCF | ||||||
| [4.13 .. 4.18] | UDP-Glucuronate, Pyroglutamate, O-phosphocholine, Proline | 1.7 | −1.0 | 1.3 | 1.0 | 1.0 | 1.1 | 1.3 | 1.0 | Reduced in 474; Increased in MCF, 231 & 468 |
| [5.48 .. 5.52] | UDP-N-Acetylglucosamine | 1.1 | −1.2 | Reduced in 231 | ||||||
| [4.26 .. 4.30] | UDP-N-Acetylglucosamine, UDP-Glucose, UDP-Glucuronate, UDP-Galactose, ATP | 1.0 | −1.1 | Reduced in 474 | ||||||
| [5.79 .. 5.85] | Unknown | 1.0 | −1.9 | Reduced in 231 | ||||||
| [9.21 .. 9.24] | Unknown | 0.5 | 4.1 | Increased in 468 | ||||||
| [1.02 .. 1.07] | Valine | 1.4 | −1.1 | 1.5 | −1.0 | Reduced in MCF & 468 | ||||
| [2.26 .. 2.30] | Valine | 1.1 | 1.1 | 1.2 | −1.0 | Increased in 474; Reduced in 231 | ||||
| [0.97 .. 1.02] | Valine, Isoleucine | 2.0 | −1.1 | 1.5 | −1.0 | Reduced in 468 | ||||
The metabolites listed were determined to be important in differentiating within a specific cell line between paclitaxel treated cells and the non-treated control at 48 hrs - based on VIP ≥ 1 with a confidence interval not including 0 (p-values were not calculated due to the small sample sizes).
A positive fold change indicates that the median bin intensity in cells treated with paclitaxel > median of signals in cells incubated in media only for each line. VIP = variable influence on projection. FC = fold change. Cell line names are abbreviated as 474 (BT474), MCF (MCF-7), 231 (MDA-MB-231) and 468 (MDA-MB-468) in the last column listing cell line responses with paclitaxel treatment. NMR bins identified as “Overlap” could not be assigned to metabolites distinctively using the NMR library and further 2D experiments are needed for confirmed identification.
Using the metabolites identified from the Luminal A versus TNBC cell type comparison before treatment, we also performed biochemical enrichment analysis in GeneGo (MetaCore) to determine if specific pathways could explain the differences in metabolic profiles between the BCa cellular phenotypes, and found that the hormone-positive and triple-negative cells are coordinated by different endogenous metabolic networks shown in Table 4. For example, Lipid metabolism is a significant pathway in both cell types, but TNBC cells appear to rely more on glycosphingolipids compared to a greater dependence on phospholipids in Luminal A cells. Overall, TNBC cells as a group were far more enriched for amino acid metabolic networks compared to several phosphocholine pathways in the Luminal A cell type. In terms of individual cell line treatment-responsive pathways, Table S-2, Supporting Information shows how different the profiles of each cell line’s response is to treatment. In the MDA-MB-231 cell line, for example, pathways related to sulfur and selenoamino acid metabolism significantly characterized treatment response; whereas taurine, hypotaurine and glycine pathways were significantly perturbed and unique in the MDA-MB-468 cells (Table S-2). In addition to the unique pathways across the cells, two of the most significantly impacted pathways common to all the BCa cell lines included Aminoacyl-tRNA biosynthesis in cytoplasm and in mitochondrion; demonstrating the ability of paclitaxel to disrupt protein biosynthesis and utilization of branched chain amino acids in BCa cells, which serve as essential nutrients and building blocks.
Table 4.
Luminal A and TNBC cell lines are driven by different endogenous metabolic networks.
| Pathway Enrichment by Metabolic Networks | Luminal A | TNBC | ||
|---|---|---|---|---|
| p-value | FDR | p-value | FDR | |
| Aminoacid metabolism_Alanine,Glycine,Cysteine metabolism and transport | 6.24E-03 | 2.26E-02 | 1.23E-06 | 1.38E-05 |
| Lipid metabolism_Phospholipid metabolism | 5.94E-05 | 1.72E-03 | ||
| 1-acyl-glycerol_3-phosphocholine pathway | 2.42E-03 | 1.75E-02 | ||
| 1-oleoyl-sn-glycero-3-phosphocholine pathway | 4.99E-03 | 2.26E-02 | ||
| 1-icosatrienoyl-sn-glycero-3-phosphocholine pathway | 5.23E-03 | 2.26E-02 | ||
| 1-docosahexaenoyl-glycerol_3-phosphocholine pathway | 5.98E-03 | 2.26E-02 | ||
| 2-arachidonoyl-glycerol_3-phosphocholine pathway | 7.67E-03 | 2.47E-02 | ||
| Glutamic acid pathways and transport | 1.74E-11 | 9.76E-10 | ||
| Aminoacid metabolism_Ala,Ser,Cys,Met,His,Pro,Gly,Glu,Gln metabolism and transport | 1.62E-09 | 3.02E-08 | ||
| Lipid metabolism_Glycosphingolipid metabolism | 1.62E-09 | 3.02E-08 | ||
| L-glutamate pathways and transport | 1.46E-07 | 2.04E-06 | ||
| Glutamic acid pathway | 1.30E-05 | 1.04E-04 | ||
| (L)-valine pathways and transport | 7.88E-04 | 3.15E-03 | ||
| Methionine pathways and transport | 1.05E-02 | 2.80E-02 | ||
| Carbohydrate metabolism_Galactose metabolism and transport | 2.58E-02 | 5.55E-02 | ||
GeneGo endogenous metabolic network analysis identified several significant biological pathways that vary between hormone receptor-positive (Luminal A) and triple-negative (TNBC) cell types based on group-differentiating metabolites. For the analysis the threshold was set to 0 with a p-value ≤ 0.05 filtering criteria, and only pathways with a p-value ≤ 0.05 and FDR ≤ 0.05 are reported. Highlighted rows distinguish pathways that are unique between the cell types defined as Luminal A versus lines defined as TNBC. FDR – False Discovery Rate.
Luminal A and TNBC cell lines display differential secretomes in response to paclitaxel
A relevant question concerning inflammatory profiles or signatures when considering their value is whether they are markers of cause or consequence? Many cancer-focused studies support the designation of cause or being causative,59–65 in spite of the fact that outside the conditions of malignancy, inflammation has been and is traditionally regarded as a consequence of injury. Even-so, because of the successes of drugs like the Denosumab, which inhibits RANKL signaling to treat breast and prostate cancers;66–67 Trabectedin, shown to reprogram tumor-associated macrophages in soft tissue sarcomas and ovarian cancer;68–71 or the recently FDA-approved Siltuximab72–75 which acts by neutralizing IL-6 mediated inflammation,76 implicated in the progression of numerous malignancies,77–78 our goal in profiling inflammatory cytokines in the context of treatment response was to determine if it is possible to shift the focus of what these mediators represent as symptomatic markers to ones of risk or responsivity prediction.
We profiled 80 inflammatory cytokines and chemokines in the media of cells before and after treatment to determine the impact of paclitaxel on cytokine secretion in each BCa cell line. Three corresponding media samples were analyzed per cell line and taken from the matched triplicate cellular extracted samples used for metabolomics analysis. Table 5 lists the significantly upregulated (≥1.50 fold) and downregulated (≤0.65 fold) cytokines following 48 hr paclitaxel treatment, normalized to the untreated control media samples. The patterns of cytokine expression also show a high degree of heterogeneity across the four different cell lines. We suggest the cytokine profiles reflect a pattern of chemotherapeutic sensitivity versus drug resistance based on the known aggressiveness of each cell line, such that a number of cytokines associated previously with metastatic progression of BCa to bone were only upregulated in the TNBC cell lines. Interestingly, osteoprotegerin (OPG) was the only cytokine with a uniform expression pattern, being downregulated in all four cell lines after treatment with paclitaxel, demonstrating the ability of the drug to reduce factors known to be involved in the metastatic progression of BCa to bone, where OPG plays a role by binding to TRAIL and blocking initiation of apoptosis.79–83 If this hypothesis is true, this reduction would be expected to be more significant in the Luminal A cell lines which represent the less aggressive clinical subtypes (Figure 3), and is what we observed. Additionally, LIGHT, a member of the TNF superfamily that can activate tumor infiltrating lymphocytes & induce apoptosis in cancer cells,84 is only upregulated in BT474 cells (triple hormone-positive, Table 5). This suggests perhaps a role for this mediator in paclitaxel sensitivity for BT474 cells compared to the other lines, as the immune responses within the microenvironment would subsequently remain detrimental to the tumor.85–86 Further support that the cytokine profiles reflect aggressiveness and treatment response resistance is evidenced by upregulation of the CXC chemokine, growth-regulated oncogene (GRO) only in the TNBC cell lines; and IL-4, IL-16 and RANTES in the AA-derived TNBC line. GRO has been directly implicated in tumor aggressiveness by promoting angiogenic activity and is linked to poorer prognosis in BCa patients;85,86,87 and was not only significantly upregulated in the MDA-MB-231 and MDA-MMB-468 cells, but also significantly downregulated in the least aggressive BT474 cell line (Figure 3, Table 5). Several interleukins participate in the tumor microenvironment facilitating the cross-talk between tumor cells and inflammation-driving immune mediators.88–92 In our data, IL-4 and IL-16 were the only significantly upregulated interleukins secreted and only from the MDA-MB-468 cell line in response to treatment (Table 5). In a previous study, using a coculture cell-based system to interrogate Luminal A and Basal-like BCa differences in response to immune mediators in the tumor microenvironment, it was shown that there is significantly more interplay between the more aggressive Basal-like cells, with tumor-associated macrophages.88 It is well accepted that a significant proportion of TNBC tumors have a Basal-like phenotype and are often classified as both.93 Interestingly, IL-6 secretion was downregulated in the three CA-derived cell lines and upregulated only in the AA-derived cell line, though not significantly based on the manufacturer’s threshold criteria. Finally, of importance to mention is the significant increase in RANTES/CCL5, again only in the most aggressive representative AA-derived cell line (Table 5, Figure 3), which has been recently implicated in TNBC progression and aggressiveness and is being investigated as a new potential therapeutic target for TNBC treatment.33
Pathway mapping analysis using GeneGo also revealed variability in the treatment-responsive, cytokine-associated pathways that may be related to aggressive and/or drug resistant phenotypes (Table S-3, Supporting Information). Immune response_T cell subsets: secreted signals and Multiple myeloma (general schema) were commonly enriched pathways across all four BCa cell lines, while Immune response_IL-17 signaling pathways and PDE4 regulation of cyto/chemokine expression in arthritis were significant to only the three CA-derived BCa cell lines (BT474, MCF-7 and MDA-MB-231). The Transcription_Role of VDR in regulation of genes involved in osteoporosis pathway was significantly enriched in the three HER2− cell lines (MCF-7, MDA-MB-231 and MDA-MB-468), while the AA-derived MDA-MB-468 cell line was enriched in several interleukin-related pathways (Immune response_IL-15 signaling via JAK-STAT cascade, Immune response_IL-4–anti-apoptotic action, and Signal transduction_PTMs in IL-23 signaling pathway) in response to paclitaxel treatment by comparison (Table S-3, Supporting Information). The full list of fold changes for all 80 cytokines profiled across the four BCa cell lines tested is shown in Table S-4, Supporting Information.
Conclusions
Our study was designed to demonstrate how TNBC cell lines derived from either AA or CA women respond to the chemotherapeutic paclitaxel compared to hormone receptor-responsive BCa cell lines, and reveal metabolic differences based on ethnicity, which could potentially provide a direction towards targets for validation studies and development of therapeutics. Although paclitaxel is a widely used and highly efficacious chemotherapeutic, as many as 20% of breast cancers can be refractory to it and other chemotherapeutics,33 and the causes are still poorly understood across all demographics of women diagnosed with BCa. As a fundamental hallmark of cancer19 metabolism of both the host and the malignancy are likely key contributors to this drug resistance. Metabolic differences may in particular help to explain the disparate response to chemotherapy outcomes for AA patients with TNBC compared to CA patients with this type of BCa.
Our data has shown that HER2+, Luminal A (BT474) cells are metabolically different from HER2−, Luminal cells (MCF-7) cells before and after treatment with paclitaxel. Also, CA-derived TNBC cells (MDA-MB-231) are metabolically different from AA-derived TNBC cells (MDA-MB-468). As demonstrated previously in both cell-based42, 94 or xenograft95 models and in human subjects samples,40–41 our data continue to confirm metabolomic profiling as a suitable technology to distinguish the clinical subtypes of BCa under non-treated conditions or following treatment. Biochemical pathway enrichment analyses revealed novel and potentially relevant pathways contributing to the biology that distinguishes the cell lines or that were perturbed in response to drug treatment depending on the cell line and type. In addition to those previously discussed, the Galactose metabolism pathway was also perturbed across all cell lines to varying degrees of significance (data not shown). This is interesting because in an integrated enrichment analysis, combining both metabolites and cytokines to determine their collective influence on how TNBC cells derived from women of different ethnicities respond to the drug, carbohydrate metabolism specifically related to Galactose metabolism was observed (Table S-5, Supporting Information) in the MDA-MB-468 cells as more significant compared to the MDA-MB-231 cells, but different metabolites (Network Objects) appear to be driving the pathway perturbations. The most significant pathways in the CA-derived (MDA-MB-231) cells compared to the AA-derived (MDA-MB-468) cells based on metabolites was Cannabinoid receptor signaling in nicotine addiction (Table S-5), pointing to a new potential avenue for further research. Although the significant differential cytokines were integrated into this overall enrichment analysis, no significant difference between the pathways was demonstrated by their contribution compared to the influence of the metabolites based on the integrated analysis, but looking at the contribution of the cytokines showed that pathways unique to the MDA-MB-231 cells included Regulation of lipid metabolism_Alpha-1 adrenergic receptors signaling via arachidonic acid and Regulation of lipid metabolism_Insulin regulation of fatty acid metabolism.
Secretome profiling of key inflammatory cytokines further distinguished the treatment response of the BCa subtype-representative cell lines suggesting that the hormone receptor-positive cells reflected a pattern of greater chemotherapeutic sensitivity to paclitaxel compared to potentially one of greater resistance in the TNBC cell lines, where more cytokines implicated previously in malignant progression (i.e. GRO, IL-4 and RANTES/CCL5), particularly in terms of TNBC aggressiveness33, 91, 96–97 were downregulated or not significantly expressed in the Luminal A lines, by comparison. Our data demonstrate these observations were most evident in the MDA-MB-468 cell line responses (Table 5; Figure 3), further support several in vitro and in vivo studies that suggest there is an ethnicity-based disparity in outcomes for AA women diagnosed with TNBC, in-part due to decreased therapeutic responsivity. These data strongly reiterate the need to identify suitable therapeutic targets for TNBC, and likely in a more personalized fashion, that may at the start take into account racial ethnicity during the rational design of drug discovery undertakings. The pathways defined as differentially perturbed following the integrative analysis (Table S-5, Supporting Information) provide biochemical roadmaps to facilitate such efforts. Some of our metabolic data showing distinctions between hormone-receptor positive versus triple hormone-negative BCa cells confirm other recent reports using cell-based systems or patient samples.40, 42 For ease of data interpretation, Table S-6, Supporting Information lists all metabolites input into GeneGo for pathway mapping.
Another speculation for the differences seen in metabolic drug response between the TNBC cell lines is based on the genes involved in paclitaxel/Taxol® metabolism. Figure 4 (Taxane Pathway), previously published by Oshiro and colleagues,98 nicely depicts many genes presently known to be involved in paclitaxel metabolism. It stands to reason that genetic (i.e. single nucleotide polymorphisms - SNPs) and epigenetic (i.e. methylation status)99 or genomic differences would impact paclitaxel treatment response, and based on numerous observations of inter-individual treatment response as a consequence of SNPs, in other populations100–102 or for other drugs,103–105 may also explain some of the disparities contributing to the poorer outcomes of AA women with TNBC. While several SNPs have been identified, no definitive evidence has been found linking non-synonymous SNPs in SLCO1B3102 or ABCB1, the primary transporter and efflux molecules, respectively, with inter-patient variation in paclitaxel clearance across several populations; whereas in vitro evidence for CYP2C8106 and in vivo for CYP3A4105, 107 have been shown for the two major metabolizing enzymes for paclitaxel108–111. Considering the other genes associated with xenobiotic metabolism of taxanes (e.g., CYP1B1, CYP2C8, and CYP3A4), only three of these previously reported studies looking for functional SNPs were conducted using samples from either healthy AA volunteers, AA BCa patients or both.110–112 To date, we found that none have been conducted with sufficient sample sizes to provide strong enough statistical power to draw definitive conclusions. Even-so, enzymes from these genes and the metabolites they result in provide an invaluable research path concerning how outcomes are impacted by therapeutic efficacy, which will be dictated in large part by host metabolism. Based on these published findings; and BCa-risk-associated data,113–118 we suggest there are as yet identified AA-specific SNPs involved in the way paclitaxel, as well as other commonly used chemotherapeutics, is metabolized that do significantly influence drug efficacy and ultimately treatment responsivity.
Figure 4. Taxane Pathway.

Representation of known candidate genes involved in the pharmacokinetics and pharmacodynamics of taxanes. Permission to reproduce diagram has been given by PharmGKB© and Stanford University https://www.pharmgkb.org/pathway/PA154426155.98
In considering a genetically-driven ethnic disparity, an understudied and plausible set of genes implicated in a number of malignancies,119–124 that should be mentioned includes those in the glutathione S-transferase (GST) superfamily, involved in cellular detoxification by conjugating chemotherapeutics (and other endogenous or xenobiotic compounds) to glutathione (GSH) for urinary clearance.122 These Phase-II enzymes are also known to act on apoptotic pathways.123 Because we found very few to no published reports of studies looking for relevant functional SNPs in GSTT1/2, GSTM1-5, GSTP1 or GSTO1/2 in the AA ethnic population,107 it represents a pressing knowledge gap for further investigation, especially in light of our findings that GSH was shown increased in the TNBC cell type when compared to Luminal A cells at steady state, and GSH metabolism pathways were most significantly perturbed in the MDA-MB-468 (AA-derived) cell line compared to the other three (data not shown). This observation could reflect a metabolic advantage for these cancer cells to eliminate chemotherapeutic drugs and thus contribute to resistance and ultimately be further investigated to determine an influence on poorer patient outcomes; whereby these cells potentially make more GSH, representing an advantage, or possibly have the GSH increased GST activity, which would also be beneficial for the cancer and not the patient.
Integrative analysis and prediction modeling tools will be essential to making the best use of different types of biomarkers, from genetic SNPs, genomic signatures, metabolic profiles and to inflammatory markers; to develop biomarker suites that best reflect the status of disease that can hopefully and eventually be used in the clinic. We believe metabolomics methods will continue to be instrumental in providing those signals that are most relevant for data integration on disease and population-based levels. Specific to our study is how these signatures hold the potential to inform on treatment responsiveness, drug resistance and potentially on relapse after treatment.125 Additionally, this would further a promise of omics-based methodologies to deliver on tools to implement personalized medicine strategies to individuals most at risk, who may in-tern be the best benefactors of having these types of data collections/screens incorporated into their treatment care plans.
Our current data demonstrates the ability of the metabolomics method in conjunction with secretory cytokine profiling to distinguish hormone-responsive phenotypes of BCa cells (HER2+ vs. HER2−) from each other and from TNBC cells that differ in their ethnic origin of women from which they were collected. Using enrichment analysis, different biological pathways related to carbohydrate and lipid-associated metabolism were shown utilized by AA-derived TNBC cells, suggesting a means through which they might overcome chemotherapeutic insult by overcompensating to ensure sufficient biomolecules and energy sources are available from the microenvironment; or potentially become resistant by down-regulating myo-inositol to activate autophagy of damaged products and impair apoptosis.126 Current studies are underway to follow up on these initial finding and better interrogate energy utilization differences in metabolic responses to paclitaxel and other frequently used chemotherapeutics (e.g., doxorubicin and cisplatin).
Supplementary Material
Acknowledgments
This work was supported by a RTI International Internal Research and Development award (Stewart, PI) and the NIH Common Fund Eastern Regional Comprehensive Metabolomics Resource Core at RTI International (1U24DK097193 - Sumner, PI). All raw and normalized analytical data and associated metadata have been uploaded to the public accessible NIH Common Fund Metabolomics Data Repository (www.metabolomicsworkbench.org). The authors thank Mr. Zachery Acuff for his contribution to this study and Mr. Kevin Knagge and Dr. Jason Burgess for their assistance with the manuscript.
Abbreviations
- BCa
breast cancer
- TNBC
triple negative breast cancer
- AA
African American
- CA
Caucasian American
- 1H NMR
proton nuclear magnetic resonance spectroscopy
- MS
mass spectrometry
- ER
estrogen receptor
- PR
progesterone receptor
- HER2
HER2/neu, human epidermal growth factor 2 receptor
- GRO
growth-regulated oncogene (CXC chemokine)
- IL-4
interleukin-4
- IL-6
interleukin-6
- DMEM
Dulbecco’s Modified Eagle Medium
- OPG
osteoprotegerin
- RANTES
Regulated upon Activation, Normal T-cell Expressed, and Secreted/CCL5
- RANK
receptor activator of nuclear factor kappa B
- RANKL
receptor activator for nuclear factor kappa B ligand
- TRAIL
TNF-related apoptosis inducing ligand
- LIGHT
Lymphotoxins exhibiting Inducible expression and competing with herpes simplex virus Glycoprotein D for Herpes virus entry mediator, a receptor expressed by T lymphocytes
- TNF
tumor necrosis factor
- SNP
Single nucleotide polymorphism
- GST
glutathione S-transferase
- T2D
Type 2 Diabetes
- NSAID
non-steroidal anti-inflammatory drug
Footnotes
The authors declare no competing financial interest.
Material supplied includes graph of Taxol/paclitaxel dose curves (Figure S-1), figure of multivariate analyses distinguishing BCa cell lines following paclitaxel treatment (Figure S-2), table of metabolites and corresponding bins distinguishing in Luminal A and TNBC cell line paclitaxel responses (Table S-1), pathway mapping analyses done in GeneGo software differentiating metabolite-dependent Luminal A from TNBC cell line responses to paclitaxel treatment (Table S-2), cytokine-dependent cell line responses to paclitaxel treatment (Table S-3), relative expression based on fold changes in 80 inflammatory cytokines profiled in the media of all paclitaxel-treated BCa cell lines normalized to untreated controls (Table S-4), pathway enrichment analysis comparing AA-derived (MDA-MB-468) and CA-derived (MDA-MB-231) TNBC cell line metabolite and cytokine responses to paclitaxel (Table S-5), and a table listing all metabolites input in to GeneGo for pathway analyses (Table S-6).
References
This article references 126 other publications.
- 1.LBBC Living Beyond Breast Cancer Guide To UnderstandingTriple-Negative Breast Cancer. http://www.tnbcfoundation.org/LBBCGuideUnderstandingTripleNegPrint2013.pdf.
- 2.Surveillance, E., and End Results (SEER) Program. [accessed 26 April 2015];SEER Stat Fact Sheets: Breast Cancer. http://seer.cancer.gov/statfacts/html/breast.html.
- 3.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–34. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 4.Ademuyiwa FO, Ellis MJ, Ma CX. Neoadjuvant therapy in operable breast cancer: application to triple negative breast cancer. J Oncol. 2013;2013:219869. doi: 10.1155/2013/219869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.den Hollander P, Savage MI, Brown PH. Targeted Therapy for Breast Cancer Prevention. Front Oncol. 2013;3:250. doi: 10.3389/fonc.2013.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 7.Greenup R, Buchanan A, Lorizio W, Rhoads K, Chan S, Leedom T, King R, McLennan J, Crawford B, Kelly Marcom P, Shelley Hwang E. Prevalence of BRCA mutations among women with triple-negative breast cancer (TNBC) in a genetic counseling cohort. Ann Surg Oncol. 2013;20(10):3254–8. doi: 10.1245/s10434-013-3205-1. [DOI] [PubMed] [Google Scholar]
- 8.Mowad R, Chu QD, Li BD, Burton GV, Ampil FL, Kim RH. Does obesity have an effect on outcomes in triple-negative breast cancer? J Surg Res. 2013;184(1):253–9. doi: 10.1016/j.jss.2013.05.037. [DOI] [PubMed] [Google Scholar]
- 9.Kurian AW, Fish K, Shema SJ, Clarke CA. Lifetime risks of specific breast cancer subtypes among women in four racial/ethnic groups. Breast Cancer Res. 2010;12(6):R99. doi: 10.1186/bcr2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keegan TH, DeRouen MC, Press DJ, Kurian AW, Clarke CA. Occurrence of breast cancer subtypes in adolescent and young adult women. Breast Cancer Res. 2012;14(2):R55. doi: 10.1186/bcr3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keegan TH, Press DJ, Tao L, DeRouen MC, Kurian AW, Clarke CA, Gomez SL. Impact of breast cancer subtypes on 3-year survival among adolescent and young adult women. Breast Cancer Res. 2013;15(5):R95. doi: 10.1186/bcr3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 13.Millikan RC, Newman B, Tse CK, Moorman PG, Conway K, Dressler LG, Smith LV, Labbok MH, Geradts J, Bensen JT, Jackson S, Nyante S, Livasy C, Carey L, Earp HS, Perou CM. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat. 2008;109(1):123–39. doi: 10.1007/s10549-007-9632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Setiawan VW, Monroe KR, Wilkens LR, Kolonel LN, Pike MC, Henderson BE. Breast cancer risk factors defined by estrogen and progesterone receptor status: the multiethnic cohort study. Am J Epidemiol. 2009;169(10):1251–9. doi: 10.1093/aje/kwp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham JE, Montero AJ, Garrett-Mayer E, Berkel HJ, Ely B. Racial differences in the incidence of breast cancer subtypes defined by combined histologic grade and hormone receptor status. Cancer Causes Control. 2010;21(3):399–409. doi: 10.1007/s10552-009-9472-2. [DOI] [PubMed] [Google Scholar]
- 16.Amirikia KC, Mills P, Bush J, Newman LA. Higher population-based incidence rates of triple-negative breast cancer among young African-American women : Implications for breast cancer screening recommendations. Cancer. 2011;117(12):2747–53. doi: 10.1002/cncr.25862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson BE, Lee NH, Seewaldt V, Shen H. The influence of race and ethnicity on the biology of cancer. Nat Rev Cancer. 2012;12(9):648–53. doi: 10.1038/nrc3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. [accessed 28 April 2015];ACS Guidelines for the Early Detection of Cancer. http://www.cancer.org/healthy/findcancerearly/cancerscreeningguidelines/american-cancer-society-guidelines-for-the-early-detection-of-cancer.
- 19.Warburg O. The effect of hydrogen peroxide on cancer cells and on embryonic cells. Acta Unio Int Contra Cancrum. 1958;14(1):55–7. [PubMed] [Google Scholar]
- 20.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, Xu X, Li C, Wang RH, Lee J, Csibi A, Cerione R, Blenis J, Clish CB, Kimmelman A, Deng CX, Haigis MC. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–63. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 23.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 24.Xie J, Wu H, Dai C, Pan Q, Ding Z, Hu D, Ji B, Luo Y, Hu X. Beyond Warburg effect - dual metabolic nature of cancer cells. Sci Rep. 2014;4:4927. doi: 10.1038/srep04927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finley LW, Zhang J, Ye J, Ward PS, Thompson CB. SnapShot: cancer metabolism pathways. Cell Metab. 2013;17(3):466–466. e2. doi: 10.1016/j.cmet.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 26.Lane AN, Fan TW, Higashi RM, Tan J, Bousamra M, Miller DM. Prospects for clinical cancer metabolomics using stable isotope tracers. Exp Mol Pathol. 2009;86(3):165–73. doi: 10.1016/j.yexmp.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan TW, Lorkiewicz PK, Sellers K, Moseley HN, Higashi RM, Lane AN. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol Ther. 2012;133(3):366–91. doi: 10.1016/j.pharmthera.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibanez C, Simo C, Garcia-Canas V, Gomez-Martinez A, Ferragut JA, Cifuentes A. CE/LC-MS multiplatform for broad metabolomic analysis of dietary polyphenols effect on colon cancer cells proliferation. Electrophoresis. 2012;33(15):2328–36. doi: 10.1002/elps.201200143. [DOI] [PubMed] [Google Scholar]
- 29.Cao B, Li M, Zha W, Zhao Q, Gu R, Liu L, Shi J, Zhou J, Zhou F, Wu X, Wu Z, Wang G, Aa J. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics. 2013;9:960–973. doi: 10.1007/s11306-013-0517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vermeer LS, Fruhwirth GO, Pandya P, Ng T, Mason AJ. NMR metabolomics of MTLn3E breast cancer cells identifies a role for CXCR4 in lipid and choline regulation. J Proteome Res. 2012;11(5):2996–3003. doi: 10.1021/pr300111x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rose DP, Komninou D, Stephenson GD. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes Rev. 2004;5(3):153–65. doi: 10.1111/j.1467-789X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 32.Morris PG, Hudis CA, Giri D, Morrow M, Falcone DJ, Zhou XK, Du B, Brogi E, Crawford CB, Kopelovich L, Subbaramaiah K, Dannenberg AJ. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila) 2011;4(7):1021–9. doi: 10.1158/1940-6207.CAPR-11-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lv D, Zhang Y, Kim HJ, Zhang L, Ma X. CCL5 as a potential immunotherapeutic target in triple-negative breast cancer. Cell Mol Immunol. 2013;10(4):303–10. doi: 10.1038/cmi.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar S, Donti TR, Agnihotri N, Mehta K. Transglutaminase 2 reprogramming of glucose metabolism in mammary epithelial cells via activation of inflammatory signaling pathways. Int J Cancer. 2014;134(12):2798–807. doi: 10.1002/ijc.28623. [DOI] [PubMed] [Google Scholar]
- 35.Brinkley BR, Beall PT, Wible LJ, Mace ML, Turner DS, Cailleau RM. Variations in cell form and cytoskeleton in human breast carcinoma cells in vitro. Cancer Res. 1980;40(9):3118–29. [PubMed] [Google Scholar]
- 36.Cailleau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14(11):911–5. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 37.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53(3):661–74. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pathak S, Siciliano MJ, Cailleau R, Wiseman CL, Hsu TC. A human breast adenocarcinoma with chromosome and isoenzyme markers similar to those of the HeLa line. J Natl Cancer Inst. 1979;62(2):263–71. [PubMed] [Google Scholar]
- 39.Siciliano MJ, Barker PE, Cailleau R. Mutually exclusive genetic signatures of human breast tumor cell lines with a common chromosomal marker. Cancer Res. 1979;39(3):919–22. [PubMed] [Google Scholar]
- 40.Budczies J, Brockmoller SF, Muller BM, Barupal DK, Richter-Ehrenstein C, Kleine-Tebbe A, Griffin JL, Oresic M, Dietel M, Denkert C, Fiehn O. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: alterations in glutamine and beta-alanine metabolism. J Proteomics. 2013;94:279–88. doi: 10.1016/j.jprot.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Kanaan YM, Sampey BP, Beyene D, Esnakula AK, Naab TJ, Ricks-Santi LJ, Dasi S, Day A, Blackman KW, Frederick W, Copeland RL, Sr, Gabrielson E, Dewitty RL., Jr Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genomics Proteomics. 2014;11(6):279–94. [PubMed] [Google Scholar]
- 42.Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, Ruller C, Lau E, Kim H, Brill LM, Jiang T, Rimm DL, Cardiff RD, Mills GB, Smith JW, Osterman AL, Kluger Y, Ronai ZA. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell. 2015;27(3):354–69. doi: 10.1016/j.ccell.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lorkiewicz P, Higashi RM, Lane AN, Fan TW. High information throughput analysis of nucleotides and their isotopically enriched isotopologues by direct-infusion FTICR-MS. Metabolomics. 2012;8(5):930–939. doi: 10.1007/s11306-011-0388-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan TW, Lane AN, Higashi RM, Yan J. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics. 2011;7(2):257–269. doi: 10.1007/s11306-010-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, Lindon JC, Nicholson JK. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc. 2007;2(11):2692–703. doi: 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- 46.Sumner S, Snyder R, Burgess J, Myers C, Tyl R, Sloan C, Fennell T. Metabolomics in the assessment of chemical-induced reproductive and developmental outcomes using non-invasive biological fluids: application to the study of butylbenzyl phthalate. Journal of Applied Toxicology. 2009;29(8):703–714. doi: 10.1002/jat.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sumner SC, Fennell TR, Snyder RW, Taylor GF, Lewin AH. Distribution of carbon-14 labeled C60 ([14C]C60) in the pregnant and in the lactating dam and the effect of C60 exposure on the biochemical profile of urine. J Appl Toxicol. 2010;30(4):354–60. doi: 10.1002/jat.1503. [DOI] [PubMed] [Google Scholar]
- 48.Sumner SJ, Burgess JP, Snyder RW, Popp JA, Fennell TR. Metabolomics of urine for the assessment of microvesicular lipid accumulation in the liver following isoniazid exposure. Metabolomics. 2010;6(2):238–249. doi: 10.1007/s11306-010-0197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pathmasiri W, Pratt KJ, Collier DN, Lutes LD, McRitchie S, Sumner SCJ. Integrating metabolomic signatures and psychosocial parameters in responsivity to an immersion treatment model for adolescent obesity. Metabolomics. 2012;8(6):1037–1051. [Google Scholar]
- 50.Banerjee R, Pathmasiri W, Snyder R, McRitchie S, Sumner S. Metabolomics of brain and reproductive organs: characterizing the impact of gestational exposure to butylbenzyl phthalate on dams and resultant offspring. Metabolomics. 2012;8(6):1012–1025. [Google Scholar]
- 51.Church RJ, Wu H, Mosedale M, Sumner SJ, Pathmasiri W, Kurtz CL, Pletcher MT, Eaddy JS, Pandher K, Singer M, Batheja A, Watkins PB, Adkins K, Harrill AH. A systems biology approach utilizing a mouse diversity panel identifies genetic differences influencing isoniazid-induced microvesicular steatosis. Toxicol Sci. 2014;140(2):481–92. doi: 10.1093/toxsci/kfu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57(1):289–300. [Google Scholar]
- 53.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 54.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aboagye EO, Bhujwalla ZM. Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Res. 1999;59(1):80–4. [PubMed] [Google Scholar]
- 56.Glunde K, Jie C, Bhujwalla ZM. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res. 2004;64(12):4270–6. doi: 10.1158/0008-5472.CAN-03-3829. [DOI] [PubMed] [Google Scholar]
- 57.Katz-Brull R, Seger D, Rivenson-Segal D, Rushkin E, Degani H. Metabolic markers of breast cancer: enhanced choline metabolism and reduced choline-ether-phospholipid synthesis. Cancer Res. 2002;62(7):1966–70. [PubMed] [Google Scholar]
- 58.Amin KA, Mohamed BM, El-Wakil MA, Ibrahem SO. Impact of breast cancer and combination chemotherapy on oxidative stress, hepatic and cardiac markers. J Breast Cancer. 2012;15(3):306–12. doi: 10.4048/jbc.2012.15.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finegan KG, Perez-Madrigal D, Hitchin JR, Davies CC, Jordan AM, Tournier C. ERK5 is a critical mediator of inflammation-driven cancer. Cancer Res. 2015;75(4):742–53. doi: 10.1158/0008-5472.CAN-13-3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grimm EA, Sikora AG, Ekmekcioglu S. Molecular pathways: inflammation-associated nitric-oxide production as a cancer-supporting redox mechanism and a potential therapeutic target. Clin Cancer Res. 2013;19(20):5557–63. doi: 10.1158/1078-0432.CCR-12-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Bone marrow fat: linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. 2014;33(2–3):527–43. doi: 10.1007/s10555-013-9484-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith JP, Solomon TE. Cholecystokinin and pancreatic cancer: the chicken or the egg? Am J Physiol Gastrointest Liver Physiol. 2014;306(2):G91–G101. doi: 10.1152/ajpgi.00301.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tu T, Budzinska MA, Maczurek AE, Cheng R, Di Bartolomeo A, Warner FJ, McCaughan GW, McLennan SV, Shackel NA. Novel aspects of the liver microenvironment in hepatocellular carcinoma pathogenesis and development. Int J Mol Sci. 2014;15(6):9422–58. doi: 10.3390/ijms15069422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whiteside TL. Regulatory T cell subsets in human cancer: are they regulating for or against tumor progression? Cancer Immunol Immunother. 2014;63(1):67–72. doi: 10.1007/s00262-013-1490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zambirinis CP, Pushalkar S, Saxena D, Miller G. Pancreatic cancer, inflammation, and microbiome. Cancer J. 2014;20(3):195–202. doi: 10.1097/PPO.0000000000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narayanan P. Denosumab: A comprehensive review. South Asian J Cancer. 2013;2(4):272–7. doi: 10.4103/2278-330X.119895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schieferdecker A, Voigt M, Riecken K, Braig F, Schinke T, Loges S, Bokemeyer C, Fehse B, Binder M. Denosumab mimics the natural decoy receptor osteoprotegerin by interacting with its major binding site on RANKL. Oncotarget. 2014;5(16):6647–53. doi: 10.18632/oncotarget.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, Olimpio CO, Bonardi C, Garbi A, Lissoni A, de Braud F, Jimeno J, D’Incalci M. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65(7):2964–71. doi: 10.1158/0008-5472.CAN-04-4037. [DOI] [PubMed] [Google Scholar]
- 69.Brodowicz T. Trabectedin in soft tissue sarcomas. Future Oncol. 2014;10(8 Suppl):s1–5. doi: 10.2217/fon.14.117. [DOI] [PubMed] [Google Scholar]
- 70.Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, Nebuloni M, van Rooijen N, Mortarini R, Beltrame L, Marchini S, Fuso Nerini I, Sanfilippo R, Casali PG, Pilotti S, Galmarini CM, Anichini A, Mantovani A, D’Incalci M, Allavena P. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 71.Ray-Coquard I. Biology of ovarian cancer and trabectedin mechanism of action. Future Oncol. 2013;9(12 Suppl):11–7. doi: 10.2217/fon.13.199. [DOI] [PubMed] [Google Scholar]
- 72.Cavarretta IT, Neuwirt H, Untergasser G, Moser PL, Zaki MH, Steiner H, Rumpold H, Fuchs D, Hobisch A, Nemeth JA, Culig Z. The antiapoptotic effect of IL-6 autocrine loop in a cellular model of advanced prostate cancer is mediated by Mcl-1. Oncogene. 2007;26(20):2822–32. doi: 10.1038/sj.onc.1210097. [DOI] [PubMed] [Google Scholar]
- 73.Li J, Hu XF, Xing PX. CNTO-328 (Centocor) Curr Opin Investig Drugs. 2005;6(6):639–45. [PubMed] [Google Scholar]
- 74.Mullard A. Green light for Janssen’s IL-6 blocker. Nat Biotechnol. 2014;32(7):607. doi: 10.1038/nbt0714-607b. [DOI] [PubMed] [Google Scholar]
- 75.Rossi JF, Lu ZY, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin Cancer Res. 2015;21(6):1248–57. doi: 10.1158/1078-0432.CCR-14-2291. [DOI] [PubMed] [Google Scholar]
- 76.Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15(2):425–30. doi: 10.1158/1078-0432.CCR-08-0149. [DOI] [PubMed] [Google Scholar]
- 77.Ahmed OI, Adel AM, Diab DR, Gobran NS. Prognostic value of serum level of interleukin-6 and interleukin-8 in metastatic breast cancer patients. Egypt J Immunol. 2006;13(2):61–8. [PubMed] [Google Scholar]
- 78.Kozlowski L, Zakrzewska I, Tokajuk P, Wojtukiewicz MZ. Concentration of interleukin-6 (IL-6), interleukin-8 (IL-8) and interleukin-10 (IL-10) in blood serum of breast cancer patients. Rocz Akad Med Bialymst. 2003;48:82–4. [PubMed] [Google Scholar]
- 79.Tometsko M, Armstrong A, Miller R, Jones J, Chaisson M, Branstetter D, Dougall W. RANK ligand directly induces osteoclastogenic, angiogenic, chemoattractive and invasive factors on RANK-expressing human cancer cells MDA-MB-231 and PC3. Journal of Bone and Mineral Research. 2004;19:S25–S25. [Google Scholar]
- 80.Casimiro S, Mohammad KS, Pires R, Tato-Costa J, Alho I, Teixeira R, Carvalho A, Ribeiro S, Lipton A, Guise TA, Costa L. RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS One. 2013;8(5):e63153. doi: 10.1371/journal.pone.0063153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weichhaus M, Segaran P, Renaud A, Geerts D, Connelly L. Osteoprotegerin expression in triple-negative breast cancer cells promotes metastasis. Cancer Med. 2014;3(5):1112–25. doi: 10.1002/cam4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, Zhong H, Vazquez A, Kang Y, Karantza V. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10(11):2036–52. doi: 10.4161/auto.34398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Azim HA, Jr, Peccatori FA, Brohee S, Branstetter D, Loi S, Viale G, Piccart M, Dougall WC, Pruneri G, Sotiriou C. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015;17:24. doi: 10.1186/s13058-015-0538-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rooney IA, Butrovich KD, Glass AA, Borboroglu S, Benedict CA, Whitbeck JC, Cohen GH, Eisenberg RJ, Ware CF. The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J Biol Chem. 2000;275(19):14307–15. doi: 10.1074/jbc.275.19.14307. [DOI] [PubMed] [Google Scholar]
- 85.Lim SG, Suk K, Lee WH. Reverse signaling from LIGHT promotes pro-inflammatory responses in the human monocytic leukemia cell line, THP-1. Cell Immunol. 2013;285(1–2):10–7. doi: 10.1016/j.cellimm.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 86.Qin JZ, Upadhyay V, Prabhakar B, Maker AV. Shedding LIGHT (TNFSF14) on the tumor microenvironment of colorectal cancer liver metastases. J Transl Med. 2013;11:70. doi: 10.1186/1479-5876-11-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 88.Stewart DA, Yang Y, Makowski L, Troester MA. Basal-like breast cancer cells induce phenotypic and genomic changes in macrophages. Mol Cancer Res. 2012;10(6):727–38. doi: 10.1158/1541-7786.MCR-11-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kovacs E. The serum levels of IL-12 and IL-16 in cancer patients. Relation to the tumour stage and previous therapy. Biomed Pharmacother. 2001;55(2):111–6. doi: 10.1016/s0753-3322(00)00023-8. [DOI] [PubMed] [Google Scholar]
- 90.Milke L, Schulz K, Weigert A, Sha W, Schmid T, Brune B. Depletion of tristetraprolin in breast cancer cells increases interleukin-16 expression and promotes tumor infiltration with monocytes/macrophages. Carcinogenesis. 2013;34(4):850–7. doi: 10.1093/carcin/bgs387. [DOI] [PubMed] [Google Scholar]
- 91.Perez R, Schally AV, Vidaurre I, Rincon R, Block NL, Rick FG. Antagonists of growth hormone-releasing hormone suppress in vivo tumor growth and gene expression in triple negative breast cancers. Oncotarget. 2012;3(9):988–97. doi: 10.18632/oncotarget.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Richmond J, Tuzova M, Cruikshank W, Center D. Regulation of cellular processes by interleukin-16 in homeostasis and cancer. J Cell Physiol. 2014;229(2):139–47. doi: 10.1002/jcp.24441. [DOI] [PubMed] [Google Scholar]
- 93.Rakha EA, Tan DS, Foulkes WD, Ellis IO, Tutt A, Nielsen TO, Reis-Filho JS. Are triple-negative tumours and basal-like breast cancer synonymous? Breast Cancer Res. 2007;9(6):404. doi: 10.1186/bcr1827. author reply 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Putluri N, Maity S, Kommagani R, Creighton CJ, Putluri V, Chen F, Nanda S, Bhowmik SK, Terunuma A, Dorsey T, Nardone A, Fu X, Shaw C, Sarkar TR, Schiff R, Lydon JP, O’Malley BW, Ambs S, Das GM, Michailidis G, Sreekumar A. Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia. 2014;16(5):390–402. doi: 10.1016/j.neo.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grinde MT, Moestue SA, Borgan E, Risa O, Engebraaten O, Gribbestad IS. 13C high-resolution-magic angle spinning MRS reveals differences in glucose metabolism between two breast cancer xenograft models with different gene expression patterns. NMR Biomed. 2011;24(10):1243–52. doi: 10.1002/nbm.1683. [DOI] [PubMed] [Google Scholar]
- 96.Gest C, Joimel U, Huang L, Pritchard LL, Petit A, Dulong C, Buquet C, Hu CQ, Mirshahi P, Laurent M, Fauvel-Lafeve F, Cazin L, Vannier JP, Lu H, Soria J, Li H, Varin R, Soria C. Rac3 induces a molecular pathway triggering breast cancer cell aggressiveness: differences in MDA-MB-231 and MCF-7 breast cancer cell lines. BMC Cancer. 2013;13:63. doi: 10.1186/1471-2407-13-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Strell C, Lang K, Niggemann B, Zaenker KS, Entschladen F. Neutrophil granulocytes promote the migratory activity of MDA-MB-468 human breast carcinoma cells via ICAM-1. Exp Cell Res. 2010;316(1):138–48. doi: 10.1016/j.yexcr.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 98.Oshiro C, Marsh S, McLeod H, Carrillo MW, Klein T, Altman R. Taxane pathway. Pharmacogenet Genomics. 2009;19(12):979–83. doi: 10.1097/FPC.0b013e3283335277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 100.Henningsson A, Marsh S, Loos WJ, Karlsson MO, Garsa A, Mross K, Mielke S, Vigano L, Locatelli A, Verweij J, Sparreboom A, McLeod HL. Association of CYP2C8, CYP3A4, CYP3A5, and ABCB1 polymorphisms with the pharmacokinetics of paclitaxel. Clin Cancer Res. 2005;11(22):8097–104. doi: 10.1158/1078-0432.CCR-05-1152. [DOI] [PubMed] [Google Scholar]
- 101.Leskela S, Jara C, Leandro-Garcia LJ, Martinez A, Garcia-Donas J, Hernando S, Hurtado A, Vicario JC, Montero-Conde C, Landa I, Lopez-Jimenez E, Cascon A, Milne RL, Robledo M, Rodriguez-Antona C. Polymorphisms in cytochromes P450 2C8 and 3A5 are associated with paclitaxel neurotoxicity. Pharmacogenomics J. 2011;11(2):121–9. doi: 10.1038/tpj.2010.13. [DOI] [PubMed] [Google Scholar]
- 102.Smith NF, Marsh S, Scott-Horton TJ, Hamada A, Mielke S, Mross K, Figg WD, Verweij J, McLeod HL, Sparreboom A. Variants in the SLCO1B3 gene: interethnic distribution and association with paclitaxel pharmacokinetics. Clin Pharmacol Ther. 2007;81(1):76–82. doi: 10.1038/sj.clpt.6100011. [DOI] [PubMed] [Google Scholar]
- 103.Aquilante CL, Wempe MF, Spencer SH, Kosmiski LA, Predhomme JA, Sidhom MS. Influence of CYP2C8*2 on the pharmacokinetics of pioglitazone in healthy African-American volunteers. Pharmacotherapy. 2013;33(9):1000–7. doi: 10.1002/phar.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Endo S, Fukahori A, Tokuhiro S, Shinagawa A, Walker J, Yoshihara K, Ishizuka H, Ieiri I, Sugiyama Y. Association study of genetic polymorphisms of drug transporters, SLCO1B1, SLCO1B3 and ABCC2, in African-Americans, Hispanics and Caucasians and olmesartan exposure. J Hum Genet. 2012;57(8):531–44. doi: 10.1038/jhg.2012.63. [DOI] [PubMed] [Google Scholar]
- 105.Hesselink DA, van Gelder T, van Schaik RH, Balk AH, van der Heiden IP, van Dam T, van der Werf M, Weimar W, Mathot RA. Population pharmacokinetics of cyclosporine in kidney and heart transplant recipients and the influence of ethnicity and genetic polymorphisms in the MDR-1, CYP3A4, and CYP3A5 genes. Clin Pharmacol Ther. 2004;76(6):545–56. doi: 10.1016/j.clpt.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 106.Gao Y, Liu D, Wang H, Zhu J, Chen C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug-drug interactions. Xenobiotica. 2010;40(7):467–75. doi: 10.3109/00498254.2010.487163. [DOI] [PubMed] [Google Scholar]
- 107.Petros WP, Hopkins PJ, Spruill S, Broadwater G, Vredenburgh JJ, Colvin OM, Peters WP, Jones RB, Hall J, Marks JR. Associations between drug metabolism genotype, chemotherapy pharmacokinetics, and overall survival in patients with breast cancer. J Clin Oncol. 2005;23(25):6117–25. doi: 10.1200/JCO.2005.06.075. [DOI] [PubMed] [Google Scholar]
- 108.Kumar GN, Walle UK, Walle T. Cytochrome P450 3A-mediated human liver microsomal taxol 6 alpha-hydroxylation. J Pharmacol Exp Ther. 1994;268(3):1160–5. [PubMed] [Google Scholar]
- 109.Sonnichsen DS, Liu Q, Schuetz EG, Schuetz JD, Pappo A, Relling MV. Variability in human cytochrome P450 paclitaxel metabolism. J Pharmacol Exp Ther. 1995;275(2):566–75. [PubMed] [Google Scholar]
- 110.Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, Goldstein JA. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11(7):597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 111.Marsh S, Somlo G, Li X, Frankel P, King CR, Shannon WD, McLeod HL, Synold TW. Pharmacogenetic analysis of paclitaxel transport and metabolism genes in breast cancer. Pharmacogenomics J. 2007;7(5):362–5. doi: 10.1038/sj.tpj.6500434. [DOI] [PubMed] [Google Scholar]
- 112.Lewis LD, Miller AA, Rosner GL, Dowell JE, Valdivieso M, Relling MV, Egorin MJ, Bies RR, Hollis DR, Levine EG, Otterson GA, Millard F, Ratain MJ Cancer; Leukemia Group B. A comparison of the pharmacokinetics and pharmacodynamics of docetaxel between African-American and Caucasian cancer patients: CALGB 9871. Clin Cancer Res. 2007;13(11):3302–11. doi: 10.1158/1078-0432.CCR-06-2345. [DOI] [PubMed] [Google Scholar]
- 113.Bailey LR, Roodi N, Verrier CS, Yee CJ, Dupont WD, Parl FF. Breast cancer and CYPIA1, GSTM1, and GSTT1 polymorphisms: evidence of a lack of association in Caucasians and African Americans. Cancer Res. 1998;58(1):65–70. [PubMed] [Google Scholar]
- 114.Haiman CA, Setiawan VW, Xia LY, Le Marchand L, Ingles SA, Ursin G, Press MF, Bernstein L, John EM, Henderson BE. A variant in the cytochrome p450 oxidoreductase gene is associated with breast cancer risk in African Americans. Cancer Res. 2007;67(8):3565–8. doi: 10.1158/0008-5472.CAN-06-4801. [DOI] [PubMed] [Google Scholar]
- 115.Kato I, Cichon M, Yee CL, Land S, Korczak JF. African American-preponderant single nucleotide polymorphisms (SNPs) and risk of breast cancer. Cancer Epidemiol. 2009;33(1):24–30. doi: 10.1016/j.canep.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee JS, Cheong HS, Kim LH, Kim JO, Seo DW, Kim YH, Chung MW, Han SY, Shin HD. Screening of Genetic Polymorphisms of CYP3A4 and CYP3A5 Genes. Korean J Physiol Pharmacol. 2013;17(6):479–84. doi: 10.4196/kjpp.2013.17.6.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McDaniel DO, Thurber T, Lewis-Traylor A, Berry C, Barber WH, Zhou X, Bigler S, Vance R. Differential association of cytochrome P450 3A4 genotypes with onsets of breast tumors in African American versus Caucasian patients. J Investig Med. 2011;59(7):1096–103. doi: 10.2310/JIM.0b013e3182277e3b. [DOI] [PubMed] [Google Scholar]
- 118.Zhou LP, Yao F, Luan H, Wang YL, Dong XH, Zhou WW, Wang QH. CYP3A4*1B polymorphism and cancer risk: a HuGE review and meta-analysis. Tumour Biol. 2013;34(2):649–60. doi: 10.1007/s13277-012-0592-z. [DOI] [PubMed] [Google Scholar]
- 119.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30(6):445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 120.O’Brien ML, Tew KD. Glutathione and related enzymes in multidrug resistance. Eur J Cancer. 1996;32A(6):967–78. doi: 10.1016/0959-8049(96)00051-2. [DOI] [PubMed] [Google Scholar]
- 121.Burg D, Mulder GJ. Glutathione conjugates and their synthetic derivatives as inhibitors of glutathione-dependent enzymes involved in cancer and drug resistance. Drug Metab Rev. 2002;34(4):821–63. doi: 10.1081/dmr-120015695. [DOI] [PubMed] [Google Scholar]
- 122.Townsend D, Tew K. Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics. 2003;3(3):157–72. doi: 10.2165/00129785-200303030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22(47):7369–75. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.L’Ecuyer T, Allebban Z, Thomas R, Vander Heide R. Glutathione S-transferase overexpression protects against anthracycline-induced H9C2 cell death. Am J Physiol Heart Circ Physiol. 2004;286(6):H2057–64. doi: 10.1152/ajpheart.00778.2003. [DOI] [PubMed] [Google Scholar]
- 125.Payne KK, Manjili MH. Adaptive immune responses associated with breast cancer relapse. Arch Immunol Ther Exp (Warsz) 2012;60(5):345–50. doi: 10.1007/s00005-012-0185-y. [DOI] [PubMed] [Google Scholar]
- 126.Cheng X, Liu H, Jiang CC, Fang L, Chen C, Zhang XD, Jiang ZW. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int J Mol Med. 2014;34(3):772–81. doi: 10.3892/ijmm.2014.1822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.