

Abstract

Asymmetric preparation of all-carbon quaternary stereocenters is an important goal. Despite advances in formation of highly enantioenriched products with quaternary stereocenters proximal to a functional group, methods to install quaternary stereocenters isolated from functional groups are limited. Transition metal catalysis offers a potential solution, but prior cross couplings are limited to allylic substrates or deliver little to no enantiomeric enrichment. We report a stereospecific, nickel-catalyzed Suzuki–Miyaura arylation of tertiary benzylic acetates to deliver products with diaryl and triaryl quaternary stereocenters in high yields and ee’s. This reaction employs an inexpensive, air-stable Ni(II) salt and commercially available phosphine ligand to transform tertiary alcohol derivatives, which are easily available in exceptional ee, into valuable products with stereoretention.
Benzylic, all-carbon quaternary stereocenters are present in a variety of bioactive molecules, including natural products and those developed in pharmaceutical and medicinal chemistry.1 Given their biomedical application, asymmetric synthesis of molecules with quaternary stereocenters is crucial, and great advances have been made to prepare several classes of quaternary stereocenters.2 Tremendous progress has been made in intramolecular reactions to deliver quaternary stereocenters.3 For intermolecular reactions, successful approaches focus largely on reactions at sp2-carbons, such as enolate allylations1b,4 or arylations,1c conjugate additions,1d,5 redox-relay Heck reactions,6 and hydrovinylations of styrenes.7
In contrast to these notable advances to deliver important carbonyl and vinyl products, synthesis of highly enantioenriched products with benzylic quaternary stereocenters isolated from functional groups remains a challenge. Stereospecific substitution (SN2) of tertiary electrophiles is outcompeted by elimination (E2).8 A transition metal-catalyzed cross coupling has the potential to overcome this limitation. However, the steric bulk of a tertiary alkyl group results in hindered oxidative addition (alkyl electrophiles) or transmetalation (alkyl nucleophiles). The presence of β-hydrogens leads to β-hydride elimination and isomerization. Thus, cross couplings to deliver quaternary stereocenters in high enantiomeric excess (ee) are limited to allylic electrophiles.9 Cross couplings of nonallylic tertiary alkyl substrates typically result in achiral or racemic products (Scheme 1A).10 In the single example of a cross coupling to form an enantioenriched quaternary stereocenter, Doyle showed a promising 27% ee in a Negishi cross coupling of an azirdine.11 Given these limitations, one of the best current methods to prepare quaternary stereocenters isolated from functional groups is Aggarwal’s enantiospecific, metal-free coupling of tertiary boronic esters with aryl lithium reagents.12
Scheme 1. Benzylic Stereocenters via Cross Couplings.

Unlike the limitations with tertiary electrophiles, extensive progress has been made in metal-catalyzed cross couplings of secondary electrophiles.10a In addition to enantioselective cross couplings pioneered by Fu,13 Jarvo, we, and others have demonstrated stereospecific cross couplings of secondary benzylic electrophiles (Scheme 1B).14 Given the high steric hindrance tolerated, including branched alkyl groups,14i we envisioned that a stereospecific cross coupling of tertiary electrophiles may be possible. When coupled with enantioselective ketone alkylation, this method would offer a powerful asymmetric synthesis of benzylic quaternary stereocenters from readily available, achiral ketone precursors (Scheme 1C). This strategy avoids differentiation of similar alkyl groups (R1 and R2) with a chiral catalyst, a highly challenging task at a saturated carbon of an acylic substrate. We now report the successful development of a stereospecific, nickel-catalyzed Suzuki–Miyaura arylation of tertiary benzylic acetates to deliver diaryl and triaryl quaternary stereocenters in high yields and ee’s. This reaction employs an inexpensive, air-stable Ni(II) salt and commercially available phosphine ligand to transform tertiary alcohol derivatives, which are easily available in exceptional ee’s, into valuable products with stereoretention. This reaction demonstrates that transition metal catalysis can be used with nonallylic tertiary electrophiles in stereospecific substitutions.
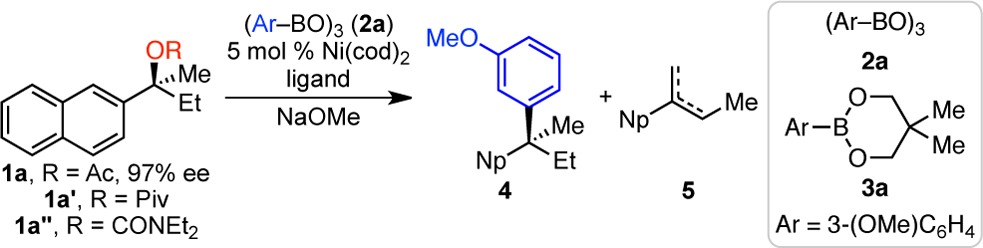
We selected carboxylates of 2-(naphthalen-2-yl)butan-2-ol (1) for initial investigation. Tertiary alcohols are easily prepared in 89–99% ee via Walsh’s enantioselective additions of alkyl,15 vinyl,16 and arylzinc reagents,17 as well as allyl stannanes,18 to acetophenones.19 Acylation then delivers acetate 1a, which is stable for >4 months when stored at −35 °C under N2. Notably, pivalate 1a′ or carbamate 1a″ did not form under standard acylation conditions, indicating how sterically congested the tertiary alcohol is.
Using 3-(methoxy)phenyl boroxine as the model nucleophile, under conditions similar to those for the cross coupling of secondary benzylic pivalates with aryl boroxines, high yield of desired product 4 was observed (Table 1, entry 1).14a However, the stereochemical fidelity was poor (21% es). Because the use of monodentate phosphines had proven helpful in other stereospecific cross couplings, we investigated the use of PhPCy2. High levels of stereochemical fidelity were achieved, particularly at lower reaction temperature and with THF as solvent (entries 2–4). However, alkenes 5 formed competitively. Although these alkenes may form via E2 elimination, they are more likely due to β-hydride elimination. Their formation depends on catalyst, and acetate 1a largely decomposes by hydrolysis (not E2) in the presence of only NaOMe. We thus examined Buchwald ligands, hoping that the hemilabile arene would block the open coordination site necessary for β-hydride elimination.20 Indeed, the use of 2-(dicylohexylphosphino)biphenyl, CyJohnPhos, resulted in less alkenes and even higher levels of stereochemical fidelity (entry 5). By using a 1:1 ratio of Ni(cod)2:CyJohnPhos, high yield of 4 (81%) was observed (entry 6). To maximize the method’s attractiveness, we prioritized the use of 2-methyl THF as a “greener” solvent (entries 6–8) and an air-stable nickel(II) precatalyst (NiCl2·DME, entry 9). Under these conditions, the use of boronate ester 3a in place of boroxine 2a resulted in a near perfect yield and stereochemical fidelity (entry 10). Use of the analogous aryl boronic acid and pinacol ester resulted in 86% yield with 98% es, and 80% yield with 98% es, respectively. However, cross coupling with the aryl potassium trifluoroborate resulted predominantly in hydrolysis of acetate 1a; no desired product 4 was observed.21 Although N-heterocyclic carbene ligands have been used in cross couplings of secondary benzylic carbamates,14h the use of 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazolium tetrafluoroborate (SIMes·HBF4) as the ligand precursor resulted in no desired product 4 or alkene 5; 90% recovered acetate was observed (entry 11). Given the potential for hydrolysis of acetate 1a in the presence of NaOMe, we also investigated the use of 2-(naphthalen-2-yl)butan-2-ol itself as substrate. However, neither 4 nor 5 were observed, and 75% of the alcohol was recovered.
Table 1. Optimization of Benzylic Arylationa.
| yield
(%)b |
||||||
|---|---|---|---|---|---|---|
| entry | ligand (mol %) | temp (° C) | solvent | 4 (ee, %)c | 5 | es (%)d |
| 1 | none | 80 | PhMe | 93 (20) | 2 | 21 |
| 2 | PhPCy2 (11) | 80 | PhMe | 74 (87) | 22 | 90 |
| 3 | PhPCy2 (11) | 60 | PhMe | 72 (90) | 25 | 93 |
| 4 | PhPCy2 (11) | 60 | THF | 63 (93) | 24 | 96 |
| 5 | CyJohnPhos (11) | 40 | THF | 57 (96) | 9 | 99 |
| 6 | CyJohnPhos (5) | 40 | THF | 81 (96) | 6 | 99 |
| 7 | CyJohnPhos (5) | 40 | 1,4-dioxane | 97 (97) | 5 | >99 |
| 8 | CyJohnPhos (5) | 40 | 2-Me-THF | 92 (96) | 8 | 99 |
| 9e | CyJohnPhos (5) | 40 | 2-Me-THF | 90 (95) | 6 | 98 |
| 10e,f | CyJohnPhos (5) | 40 | 2-Me-THF | 99 (97) | ≤3 | >99 |
| 11e,f | SIMes·HBF4 (5) | 40 | 2-Me-THF | 0 | 0 | |
Conditions: 1a (0.10 mmol), 2a (1.0 equiv), Ni(cod)2 (5 mol %), ligand, NaOMe (2.0 equiv), solvent (0.4 M), unless otherwise noted.
Determined by 1H NMR using an internal standard. Total yields over 100% reflect the error of 1H NMR yields, particularly for minor products.
Determined by HPLC using a chiral stationary phase.
es = enantiospecificity = (eeproduct)/(eestarting material).
NiCl2·DME in place of Ni(cod)2.
Boronate ester 3a (2.0 equiv) in place of boroxine 2a.
Under the optimized conditions, a range of aryl boronate esters underwent cross coupling (Scheme 2). Electron-rich (7, 8, 14, 15) and electron-poor (4, 9–13) aryl groups can be incorporated. A range of functional groups is well tolerated, including ether (4, 8), aniline (7), aryl chloride and fluoride (9, 13), amide (10), ester (11), trifluoromethyl (12), and dioxolane (14) groups. Aryl groups with increased steric hindrance can also be used successfully (15). In every case, near perfect levels of stereochemical fidelity were observed. Unfortunately, the use of heteroaryl boronic esters (pyridyl, furyl, indolyl) was not successful, and the use of vinyl boronate esters resulted in low yields. The absolute configuration of 10 was determined to be R via X-ray crystallography using Cu Kα radiation.22 The expected S configuration of acetate 1a, as well as the alcohol precursor to 18, was also confirmed via X-ray crystallography. The conversion of (S)-1a to (R)-10 demonstrates that this reaction proceeds with overall retention of absolute configuration.23 Absolute configurations of other products were assigned by analogy.
Scheme 2. Scope of Aryl Boronate Esters.

Conditions: 1a (0.40 mmol), 3 (2.0 equiv), NiCl2·DME (5 mol %), CyJohnPhos (5 mol %), NaOMe (2.0 equiv), 2-Me-THF (0.4 M), 40 °C, 22 h, unless noted. Average isolated yields (±9%) and ee’s (±1%, determined by HPLC or SFC using a chiral stationary phase) of duplicate reactions, unless otherwise noted.
Single experiment.
60 °C, 12 h.
3.0 equiv of 3.
A variety of tertiary acetates also successfully underwent arylation (Scheme 3). For the aryl substituent (Ar1), electron-rich naphthyl groups (16) and increased steric hindrance (17) are well tolerated. Heteroaryl groups can also be used (18). Various alkyl substituents (R1) can be incorporated, including alkyl chains capped with silyl ether (19), aryl (20, 22), and vinyl (21) groups. Notably, this reaction is not limited to formation of methyl-substituted quaternary stereocenters; ethyl-substituted product 22 was formed in 70% yield and 99% es. However, an aryl substituent with an extended π-system is required; the arylation of 2-([1,1′-biphenyl]-4-yl)butan-2-yl acetate resulted in no product.
Scheme 3. Scope of Tertiary Acetates.

Conditions: see Scheme 2. Average isolated yields (±7%) and ee’s (±1%).
Single experiment.
78%, 83% ee, 99% es (84% ee of 1).
0.3 mmol of 1.
2a (0.83 equiv) in place of 3a.
60 °C.
48 h.
Opposite enantiomer of 1 used.
10 mol % NiCl2·DME, 10 mol % CyJohnPhos, 60 °C, 48 h.
We also pursued the cross coupling of diaryl acetates to deliver triaryl quaternary stereocenters. These acetates are easily prepared via enantioselective addition of a diarylzinc to an acetophenone. Triarylethane 23 formed in 73% yield, 94% ee, and 98% es (Scheme 3). As previously reported in related reactions of secondary diaryl carbamates,14f−14h a naphthyl substituent is not required for diaryl acetates. By using 10 mol % Ni/CyJohnPhos catalyst and increased temperature and time, arylation resulted in 62% yield of triarene 24 with near perfect stereochemical fidelity. These conditions are not general for other non-naphthyl substrates, but this promising result suggests that broad scope of tertiary diaryl acetates may be possible with a future catalyst. Notably, the only other method that directly delivers triaryl quaternary stereocenters in high ee is Aggarwal’s metal-free coupling.12
With respect to mechanism, a directed SN2′ oxidative addition, which has been proposed for stereoretentive cross couplings of secondary benzylic and allylic electrophiles,9d,14h is consistent with our observed stereoretention, as well as the requirement for a naphthyl substituent. In a directed oxidative addition, the acetate binds the nickel catalyst, directing it to add in an SN2′ fashion (A, Scheme 4). This mechanism also suggests why the steric hindrance of a tertiary acetate is tolerated; this increased bulk is remote to the carbon attacked. Transmetalation and reductive elimination are well precedented to proceed with steroeoretention, ultimately resulting in net retention of configuration.24 Studies are underway to investigate this intriguing mechanism further.
Scheme 4. Putative Catalytic Cycle.

In summary, we developed a high-yielding Suzuki–Miyaura arylation of tertiary benzylic acetates to deliver products with diaryl and triaryl quaternary stereocenters in excellent stereochemical fidelity. By combining this stereospecific cross coupling with known enantioselective additions to acetophenones, this offers a highly efficient strategy for asymmetric synthesis of diaryl and triaryl quaternary stereocenters. This reaction overcomes traditional limitations in utilizing tertiary electrophiles in substitution reactions, and demonstrates that a transition metal-catalyzed cross coupling for the synthesis of enantioenriched quaternary stereocenters can go beyond allylic electrophiles. Efforts to expand the scope and to investigate the mechanism are ongoing.
Acknowledgments
NIH (R01 GM111820) is gratefully acknowledged. NMR and other data were acquired at UD on instruments obtained with assistance of NSF and NIH funding (NSF CHE0421224, CHE1229234, CHE0840401, and CHE1048367; NIH P20 GM104316, P20 GM103541, and S10 OD016267).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b08075.
Author Present Address
† Adesis, Inc., 27 McCullough Drive, New Castle, DE 19720.
The authors declare no competing financial interest.
Supplementary Material
References
- a Christoffers J.; Baro A. Adv. Synth. Catal. 2005, 347, 1473. 10.1002/adsc.200505165. [DOI] [Google Scholar]; b Hong A. Y.; Stoltz B. M. Eur. J. Org. Chem. 2013, 2013, 2745. 10.1002/ejoc.201201761. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burtoloso A. C. B. Synlett 2009, 2009, 320. 10.1055/s-0028-1087673. [DOI] [Google Scholar]; d Hawner C.; Alexakis A. Chem. Commun. 2010, 46, 7295. 10.1039/c0cc02309d. [DOI] [PubMed] [Google Scholar]; e Cozzi P. G.; Hilgraf R.; Zimmermann N. Eur. J. Org. Chem. 2007, 2007, 5969. 10.1002/ejoc.200700318. [DOI] [Google Scholar]
- Douglas C. J.; Overman L. E. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5363. 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Das J. P.; Marek I. Chem. Commun. 2011, 47, 4593. 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Jiang C. Synthesis 2006, 2006, 369. 10.1055/s-2006-926302. [DOI] [Google Scholar]; c Bella M.; Gasperi T. Synthesis 2009, 2009, 1583. 10.1055/s-0029-1216796. [DOI] [Google Scholar]
- a Krautwald S.; Sarlah D.; Schafroth M. A.; Carreira E. M. Science 2013, 340, 1065. 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]; b Liu W.-B.; Reeves C. M.; Stoltz B. M. J. Am. Chem. Soc. 2013, 135, 17298. 10.1021/ja4097829. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Trost B. M.; Miller J. R.; Hoffman C. M. J. Am. Chem. Soc. 2011, 133, 8165. 10.1021/ja2029602. [DOI] [PubMed] [Google Scholar]
- Dabrowski J. A.; Villaume M. T.; Hoveyda A. H. Angew. Chem., Int. Ed. 2013, 52, 8156. 10.1002/anie.201304035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei T.-S.; Patel H. H.; Sigman M. S. Nature 2014, 508, 340. 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.; RajanBabu T. V. J. Am. Chem. Soc. 2006, 128, 5620. 10.1021/ja060999b. [DOI] [PubMed] [Google Scholar]
- Pronin S. V.; Reiher C. A.; Shenvi R. A. Nature 2013, 501, 195. 10.1038/nature12472. [DOI] [PubMed] [Google Scholar]
- a Falciola C. A.; Alexakis A. Eur. J. Org. Chem. 2008, 2008, 3765. 10.1002/ejoc.200800025. [DOI] [Google Scholar]; b Zhang P.; Le H.; Kyne R. E.; Morken J. P. J. Am. Chem. Soc. 2011, 133, 9716. 10.1021/ja2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jung B.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 1490. 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nagao K.; Yokobori U.; Makida Y.; Ohmiya H.; Sawamura M. J. Am. Chem. Soc. 2012, 134, 8982. 10.1021/ja302520h. [DOI] [PubMed] [Google Scholar]; e Feng C.; Kobayashi Y. J. Org. Chem. 2013, 78, 3755. 10.1021/jo400248y. [DOI] [PubMed] [Google Scholar]; f Hojoh K.; Shido Y.; Ohmiya H.; Sawamura M. Angew. Chem., Int. Ed. 2014, 53, 4954. 10.1002/anie.201402386. [DOI] [PubMed] [Google Scholar]
- a Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang X.; Wang S.; Xue W.; Gong H. J. Am. Chem. Soc. 2015, 137, 11562. 10.1021/jacs.5b06255. [DOI] [PubMed] [Google Scholar]; c Joshi-Pangu A.; Wang C.-Y.; Biscoe M. R. J. Am. Chem. Soc. 2011, 133, 8478. 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Thapa S.; Kafle A.; Gurung S. K.; Montoya A.; Riedel P.; Giri R. Angew. Chem., Int. Ed. 2015, 54, 8236. 10.1002/anie.201502379. [DOI] [PubMed] [Google Scholar]; e Lohre C.; Dröge T.; Wang C.; Glorius F. Chem. - Eur. J. 2011, 17, 6052. 10.1002/chem.201100909. [DOI] [PubMed] [Google Scholar]; f Zultanski S. L.; Fu G. C. J. Am. Chem. Soc. 2013, 135, 624. 10.1021/ja311669p. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Tsuji T.; Yorimitsu H.; Oshima K. Angew. Chem., Int. Ed. 2002, 41, 4137. 10.1002/1521-3773(20021104)41:21<4137::AID-ANIE4137>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Huang C.-Y.; Doyle A. G. J. Am. Chem. Soc. 2015, 137, 5638. 10.1021/jacs.5b02503. [DOI] [PubMed] [Google Scholar]
- a Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Nat. Chem. 2014, 6, 584. 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]; b Llaveria J.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2015, 137, 10958. 10.1021/jacs.5b07842. [DOI] [PubMed] [Google Scholar]; c Odachowski M.; Bonet A.; Essafi S.; Conti-Ramsden P.; Harvey J. N.; Leonori D.; Aggarwal V. K. J. Am. Chem. Soc. 2016, 138, 9521. 10.1021/jacs.6b03963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Arp F. O.; Fu G. C. J. Am. Chem. Soc. 2005, 127, 10482. 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]; b Binder J. T.; Cordier C. J.; Fu G. C. J. Am. Chem. Soc. 2012, 134, 17003. 10.1021/ja308460z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Do H.-Q.; Chandrashekar E. R. R.; Fu G. C. J. Am. Chem. Soc. 2013, 135, 16288. 10.1021/ja408561b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhou Q.; Srinivas H. D.; Dasgupta S.; Watson M. P. J. Am. Chem. Soc. 2013, 135, 3307. 10.1021/ja312087x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Maity P.; Shacklady-McAtee D. M.; Yap G. P. A.; Sirianni E. R.; Watson M. P. J. Am. Chem. Soc. 2013, 135, 280. 10.1021/ja3089422. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shacklady-McAtee D. M.; Roberts K. M.; Basch C. H.; Song Y.-G.; Watson M. P. Tetrahedron 2014, 70, 4257. 10.1016/j.tet.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Taylor B. L. H.; Swift E. C.; Waetzig J. D.; Jarvo E. R. J. Am. Chem. Soc. 2011, 133, 389. 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]; e Taylor B. L. H.; Harris M. R.; Jarvo E. R. Angew. Chem., Int. Ed. 2012, 51, 7790. 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]; f Greene M. A. M.; Yonova I. M. I.; Williams F. J. F.; Jarvo E. R. E. Org. Lett. 2012, 14, 4293. 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]; g Wisniewska H. M.; Swift E. C.; Jarvo E. R. J. Am. Chem. Soc. 2013, 135, 9083. 10.1021/ja4034999. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Harris M. R.; Hanna L. E.; Greene M. A.; Moore C.; Jarvo E. R. J. Am. Chem. Soc. 2013, 135, 3303. 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Basch C. H.; Cobb K. M.; Watson M. P. Org. Lett. 2016, 18, 136. 10.1021/acs.orglett.5b03455. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Li C.; Zhang Y.; Sun Q.; Gu T.; Peng H.; Tang W. J. Am. Chem. Soc. 2016, 138, 10774. 10.1021/jacs.6b06285. [DOI] [PubMed] [Google Scholar]; k López-Pérez A.; Adrio J.; Carretero J. C. Org. Lett. 2009, 11, 5514. 10.1021/ol902335c. [DOI] [PubMed] [Google Scholar]
- a Jeon S.-J.; Li H.; Walsh P. J. J. Am. Chem. Soc. 2005, 127, 16416. 10.1021/ja052200m. [DOI] [PubMed] [Google Scholar]; b García C.; LaRochelle L. K.; Walsh P. J. J. Am. Chem. Soc. 2002, 124, 10970. 10.1021/ja026568k. [DOI] [PubMed] [Google Scholar]
- Li H.; Walsh P. J. J. Am. Chem. Soc. 2004, 126, 6538. 10.1021/ja049206g. [DOI] [PubMed] [Google Scholar]
- García C.; Walsh P. J. Org. Lett. 2003, 5, 3641. 10.1021/ol0352963. [DOI] [PubMed] [Google Scholar]
- Waltz K. M.; Gavenonis J.; Walsh P. J. Angew. Chem., Int. Ed. 2002, 41, 3697. 10.1002/1521-3773(20021004)41:19<3697::AID-ANIE3697>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for additional methods.
- Martin R.; Buchwald S. L. Acc. Chem. Res. 2008, 41, 1461. 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information.
- Parsons S.; Flack H. D.; Wagner T. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2013, 69, 249. 10.1107/S2052519213010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCDC 1424635 (10), 1502353 (1a), and 1424635 (S-1d) contain the supplementary crystallographic data for this paper. These can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- a Stille J. K. In The Chemistry of the Metal–Carbon Bond, Vol. 2; Hartley F. R.; Patai S., Eds.; John Wiley & Sons, Ltd.: New York, 1985; p 625. [Google Scholar]; b Netherton M. R.; Fu G. C. Angew. Chem., Int. Ed. 2002, 41, 3910. 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.